

Abstract
In contrast to the rapid regulation of AMPA receptors, previous evidence has supported the idea that the synaptic density of NMDA-type glutamate receptors is fairly static, modulated only over a long time scale in a homeostatic manner. We report here that selective activation of protein kinase C (PKC) with phorbol esters induces a rapid dispersal of NMDA receptors from synaptic to extrasynaptic plasma membrane in cultured rat hippocampal neurons. PKC activation induced a simultaneous translocation of calcium/calmodulin-dependent kinase II (CaMKII) to synapses but no change in spine number, presynaptic terminal number, or the distribution of AMPA receptors or the synaptic scaffolding protein PSD-95. PKC-induced accumulation of CaMKII was dependent on filamentous actin, whereas dispersal of NMDA receptors occurred by a different mechanism independent of actin or CaMKII. Consistent with the decrease in synaptic density of NMDA receptors, phorbol ester pretreatment reduced excitotoxicity. These results reveal a surprisingly dynamic nature to the molecular composition and functional properties of glutamatergic postsynaptic specializations.
Keywords: postsynaptic density, NMDA receptors, calcium/calmodulin-dependent kinase II, protein kinase C, synaptogenesis, synaptic plasticity, hippocampal neurons
Glutamatergic synapses are modifiable cell-to-cell junctions that mediate excitatory synaptic transmission in the mammalian brain. Underlying the glutamatergic postsynaptic membrane is a specialized, detergent-insoluble postsynaptic density (PSD). The NMDA ionotropic glutamate receptor (NMDAR) and calcium/calmodulin-dependent kinase II (CaMKII) are two major signaling components of the PSD and have been intensely studied for their roles in synaptic plasticity (Kennedy, 2000). Other major components of the PSD are structural scaffolds, typified by the PDZ domain protein PSD-95. PSD-95/SAP90 and related family members link NMDA receptors with a network of other PSD proteins, including nitric oxide synthase, the synaptic ras GTPase-activating protein SynGAP, the cell-adhesion molecule neuroligin, and GKAP scaffolding proteins (Sheng and Pak, 2000).
NMDA receptors are hetero-oligomers, consisting of the essential NR1 subunit and combinations of NR2A–NR2D and NR3 (Mori and Mishina, 1995). NMDA receptors function as molecular coincidence detectors, requiring both glutamate binding and depolarization-mediated removal of Mg2+ block to allow channel opening. The level of NMDAR function at the synapse critically regulates brain function. For example, mice expressing 5% of normal levels of NR1 exhibit increased motor activity and deficits in social interactions (Mohn et al., 1999). At the cellular level, the magnitude and kinetics of calcium entry through NMDA receptors is thought to be a major determinant of long-lasting potentiation or depression of synaptic efficacy in response to a given stimulus (Abraham and Bear, 1996). These long-term effects are mediated by calcium activation of CaMKII and protein kinase C (PKC) and other signaling pathways in the postsynaptic domain.
CaMKII is a holoenzyme composed of ∼12 monomers, primarily α and β subunits in neurons (Soderling et al., 2001). Autophosphorylation of CaMKIIα at Thr286 is required for normal spatial memory and place-cell representation, presumably through triggering of its calcium-independent kinase activity (Giese et al., 1998). CaMKII phosphorylates several key postsynaptic targets, including the NMDA receptor subunit NR2B, the AMPA receptor (AMPAR) subunit GluR1, and SynGAP (Kennedy, 2000; Soderling et al., 2001).
Rapid synaptic modification through regulation of the functional properties of ion channels and signal-transducing enzymes is a well established mode of plasticity. More recently, it has been appreciated that another mode of expression of synaptic plasticity is regulation of subcellular distribution of ion channels and kinases. Many studies support the idea that activity rapidly regulates the insertion and endocytosis of AMPA receptors at synapses (Ehlers, 2000). Translocation of exogenous CaMKII to synapses has also been visualized in response to glutamate stimulation (Shen and Meyer, 1999). In contrast, the synaptic localization of NMDA receptors is thought to be more static. Over a time course of several days, activity modulates the synaptic levels of NMDA receptors in a homeostatic direction (Rao and Craig, 1997). cAMP-dependent protein kinase mediates this long-term modulation of NMDA receptor targeting to synapses (Crump et al., 2001). In exploring the effects of other kinases, we found that NMDA receptor trafficking can be dynamically regulated on a time scale of minutes. Selective activation of PKC induces rapid dispersal of NMDA receptors from postsynaptic sites to the extrasynaptic plasma membrane. Furthermore, PKC activation induces a reciprocal translocation of CaMKII to postsynaptic sites but has no effect on the distribution of PSD-95.
MATERIALS AND METHODS
Cell culture. Rat hippocampal cultures were prepared from 18-d-old rat embryos by previously described methods (Goslin et al., 1998). Briefly, hippocampi were dissected and dissociated using trypsin and trituration through a Pasteur pipette. The neurons were plated on coverslips coated with poly-l-lysine in MEM with 10% horse serum and allowed to attach for 3–4 hr. After attachment, the neurons were transferred to a dish containing a glial monolayer and maintained for up to 4 weeks in serum-free MEM with N2 supplements. Standard cultures were plated at the low density of 2400–4800 cells/cm2. High-density cultures were plated at 14,300 cells/cm2. Neurons were chronically treated with 100 μmAPV or 10 μm MK-801 beginning on day 7, with renewal every 3–4 d (except for the −APV experiments described in Figs. 1 and 8). All analyses were performed at 18–28 d. Pharmacological agents were used at the following concentrations: 100 nm12-O-tetradecanoylphorbol-13-acetate (TPA) (Sigma, St. Louis, MO), 100 nm 4α-TPA (Sigma), 10 μm phorbol 12,13-diacetate (PDA) (Sigma), 100 nm phorbol didecanoate (PDD) (Sigma), 100 nm 4α-PDD (Sigma), 10–200 nm bisindolylmaleimide (LC Laboratories, Woburn, MA), 100 μm APV (Research Biochemicals, Natick, MA), 10 μm MK801 (Research Biochemicals), 2.5 μm NMDA (Alexis, San Diego, CA), 2 μm jasplakinolide (Molecular Probes, Eugene, OR), 5 μm latrunculin A (Biomol Research Laboratories, Plymouth Meeting, PA), 10 μm KN62 (Alexis), 3–30 μm KN93 (Calbiochem, La Jolla, CA), 10 μm lavendustin A (Calbiochem), 10 μm CNQX (Sigma), 50 μmpicrotoxin (Sigma), 200 μm AP-3 (Alexis), 5 μm nifedipine, and 1 μmTTX (Sigma). With the exception of chronic APV/MK801 and latrunculin A treatment, pharmacological agents were added to the culture media for 45–120 min before the addition of TPA.
Fig. 1.

PKC-induced synaptic remodeling: dispersal of NMDA receptors and synaptic translocation of CaMKIIα. Hippocampal neurons were cultured for 3 weeks at high density (−APV, a–d) or at low density in the presence of APV (e–h). Sister neurons were left untreated [controls (Con), a, c, e, g] or treated for 45 min with TPA (b, d, f, h), fixed, and immunolabeled for NR1 (green, a, b, e, f) or CaMKIIα (green, c, d, g, h) and synapsin (Syn; red). Areas of overlap between the presynaptic vesicle antigens and postsynaptic NR1 or CaMKIIα appear yellow. a, e, In high-density or APV-treated low-density control neurons, NR1 is found predominantly in synaptic clusters. b, f, TPA treatment resulted in dispersal of NR1 away from synapses to a diffuse dendritic shaft distribution. c, g, In high-density or APV-treated low-density control neurons, CaMKIIα is concentrated at some synapses but is also present at high levels throughout dendrites and axons.d, h, TPA treatment resulted in enhanced clustering of CaMKIIα at synapses and reduced levels in dendrite shafts. PKC-induced NR1 dispersal and CaMKIIα synaptic translocation occurred under conditions of spontaneous activity (a–d) and under conditions of NMDA receptor blockade (e–h).Boxed regions in e–h are shown below at higher magnification. Scale bars, 10 μm.
Fig. 8.

Neuroprotective effect of PKC-induced synaptic remodeling. Neurons were cultured at low density in the presence (gray bars) or absence (black bars) of APV and were treated with TPA for 45 min or untreated, as indicated. Neurons were then assayed for excitotoxicity by washout of the APV and TPA, exposure to high-K+ buffer, and determination of viability by Trypan blue exclusion. a, Neurons cultured chronically with APV exhibited greater toxicity than neurons cultured without APV (t test;p < 0.01), correlating with a higher level of synaptic NMDAR, as reported previously (Crump et al., 2001). This enhanced toxicity of the chronic APV group was mediated by NMDAR, as indicated by sensitivity to APV during the toxicity assay.b, TPA treatment of the APV group partially protected the neurons from excitotoxicity (p < 0.01) by an amount corresponding to the NMDAR-mediated component. TPA was not protective in the no-APV group with the much lower levels of synaptic NMDAR, suggesting that the protective effect of TPA was attributable to the dispersion of synaptic NMDAR in the chronic APV group.
Immunocytochemistry. Immunolabeling was performed essentially as described previously (Rao and Craig, 1997; Allison et al., 2000). Briefly, for NR1, NR2A, NR2B, CaMKIIα, and PSD-95, neurons were fixed in methanol for 10 min at −20°C, blocked with 10% BSA for 30 min, and incubated overnight at room temperature with primary antibodies diluted in 3% BSA/PBS. For AMPA receptor and filamentous actin (F-actin) labeling, neurons were fixed in warm 4% paraformaldehyde/4% sucrose in PBS for 15 min at room temperature and permeabilized with 0.25% Triton X-100 for 5 min. The following mouse monoclonal antibodies were used: NR1 (clone 54.1; PharMingen, San Diego, CA; 1:1500), CaMKIIα (clone 6G9; Affinity Bioreagents, Golden, CO; 1:200), and PSD-95 (clone 6G6-1C9; Affinity Bioreagents; 1:2000; interacts with other members of the PSD/SAP family). For double labeling of NR1 and PSD-95 or NR2A, mouse NR1 (clone 54.1; PharMingen) and 1 μg/ml guinea pig anti-PSD-95 antiserum (gift from M. Sheng, Massachusetts Institute of Technology, Cambridge, MA) or 1 μg/ml rabbit anti-NR2A antiserum (gift from M. Sheng) were used. Other rabbit antibodies used were anti-NR2B (Upstate Biotechnology, Lake Placid, NY; 4.2 μg/ml), anti-GluR1 (gift from R. McIlhinney, Oxford University, Oxford, UK; 1:2000), and anti-MAP2 (gift from S. Halpain, Scripps Research Institute, La Jolla, CA; 1:10,000). F-actin was labeled using Texas Red-conjugated phalloidin (Molecular Probes; 1:200). Presynaptic terminals were revealed with rabbit antibodies to synaptophysin (G95; gift from P. DeCamilli, Yale University, New Haven, CT; 1:8000) or synapsin (Chemicon, Temecula, CA; 1:5000) or with mouse anti-SV2 (Developmental Studies Hybridoma Bank, Iowa City, IA; 1:40). Appropriate secondary antibodies conjugated to Texas Red, FITC, or biotin (Jackson ImmunoResearch, West Grove, PA, or Vector Laboratories, Burlingame, CA; 2.5–7.5 μg/ml) were added and incubated for 45 min at 37°C, followed by extensive washes with PBS. In cases in which biotin-conjugated secondary antibodies were used, either FITC or Texas Red/streptavidin (Jackson ImmunoResearch; 1:2000) was used as a tertiary reagent. Coverslips were mounted in elvanol (Tris-HCl, glycerol, polyvinyl alcohol with 2% 1,4-diazabicyclo[2,2,2]octane).
DiI labeling. The lipophilic dye DiI (Molecular Probes) was used to label the plasma membrane outline of random subsets of neurons (Hasbani et al., 2001). Neurons were fixed for 30 min in warm 4% paraformaldehyde/4% sucrose in PBS, washed in PBS, incubated in a freshly made suspension of DiI (0.4 μg/ml in PBS) for 30 sec, and washed further in PBS. Neurons were mounted and imaged immediately.
Microscopy and immunofluorescence quantification.Fluorescent and phase-contrast images of neurons were obtained on a PhotoMetrics (Roper Scientific, Tucson, AZ) Sensys cooled CCD camera mounted on a Zeiss (Thornwood, NY) Axioskop microscope with a 63×, 1.4 numerical aperture lens using Oncor or Metamorph (Universal Imaging, West Chester, PA) software. Before quantification, CCD images were processed by dark-field subtraction and correction for any nonuniformity in illumination. To define synaptic NR1, CaMKII, or PSD-95 clusters, thresholds for individual neurons and channels were chosen manually and corresponded to two times the average intensity of fluorescence in the dendritic shaft. Binary images of clusters of each postsynaptic marker (NR1, CaMKII, or PSD-95) were compared with binary images of synapsin or synaptophysin clusters. Any postsynaptic cluster that had at least one pixel of overlap with a presynaptic cluster was defined as synaptic. Data analysis was performed using Metamorph, Microsoft (Seattle, WA) Excel, Statview, and Delta Graph (Chicago, IL). Values indicate mean ± SEM. Group comparisons were made byt test. Images were processed and prepared for print using Adobe Photoshop (Adobe Systems, San Jose, CA).
Chymotrypsin treatment and Western blot analysis. Neurons were subjected to chymotrypsin protease treatment before harvesting and Western blot analysis to determine surface NR1 expression (Hall and Soderling, 1997). Briefly, 3 week, high-density (14,300 cells/cm2) dishes of cultured neurons were washed twice with warm HEPES-buffered saline solution, incubated in 1 mg/ml chymotrypsin in saline solution for 10 min, and then washed three times in saline solution containing 2 mm PMSF to inactivate chymotrypsin. Neurons were scraped into warm PBS, pelleted, and resuspended in Laemmli buffer. Samples were pooled from 15 to 20 coverslips per condition, and small aliquots were analyzed by SDS-PAGE to estimate total protein concentration. Equal amounts of protein between conditions were run on SDS-PAGE, transferred to a nitrocellulose membrane, blocked with 5% nonfat dried milk for 1 hr, and incubated overnight at 4°C with anti-NR1 antibody (clone 54.1; mouse anti-NR1; 1:4000). Blots were washed with TBS, incubated for 1 hr in HRP-conjugated secondary goat anti-mouse antiserum (Jackson ImmunoResearch; 1:5000), and visualized using chemiluminescent Super-Signal HRP substrate (Pierce, Rockford, IL) and exposure to x-ray film. As a control for protein loading between conditions, the blots were reprobed with rabbit anti-tau (Sigma; 1:10,000). The film signals were digitally scanned and analyzed using Metamorph or NIH Image densitometric analysis.
Triton X-100 extraction. Live neurons were detergent-extracted (Allison et al., 2000) with 1% Triton X-100 and 4% 40,000 kDa polyethylene glycol in BRB80 (80 mm PIPES, 1 mmMgCl2, 1 mm EDTA) for 5 min, rinsed twice with BRB80, fixed in methanol, and stained for rabbit anti-MAP2 and mouse anti-NR1 or mouse anti-CaMKIIα.
Excitotoxicity. For the toxicity assay (Crump et al., 2001), neurons were removed from APV and TPA pretreatments, placed in high-K+ buffer (in mm: 90 KCl, 31.5 NaCl, 2 CaCl2, 25 HEPES, 1 glycine, 30 glucose) for 3 min to induce the synaptic release of glutamate, and incubated for an additional 60 min in normal medium. Neurons were then incubated in 0.4% Trypan blue for 5 min and counted using the exclusion of dye as an indicator of cell viability. Approximately 150–200 neurons were scored per coverslip, and at least eight coverslips per group were scored from at least four independent cultures. Sister coverslips used for excitotoxicity analysis were fixed and immunostained to confirm the differential localization of NR1 with APV and TPA pretreatments.
NR1-green fluorescent protein expression and live imaging.The NR1-green fluorescent protein (GFP) and NR2A expression plasmids have been described previously (Crump et al., 2001). Neurons were transfected either at plating or 7–10 d after plating with GW1-NR1C-GFP and GW1-NR2A using Effectene reagent (Qiagen, Hilden, Germany) essentially according to the manufacturer's instructions. Low amounts of expression plasmid were used (0.1–0.5 μg per 60 mm dish) resulting in fairly dim but appropriately localized NR1-GFP. For colabeling of CaMKII, neurons were grown to day 21–28, fixed, and stained with mouse anti-CaMKIIα and donkey Texas Red anti-mouse antibodies. For live visualization of NR1-GFP, neurons were plated on poly-l-lysine-coated coverslips attached via silicone to a hole in the bottom of a tissue culture dish. Glia were grown on coverslips and suspended above the neurons by paraffin wax dots. The neurons were transfected at plating and grown in phenol red-free MEM with N2 supplements for 18–24 d. Imaging was performed after addition of the anti-oxidantsN-acetyl-cysteine (60 μm) and trolox (20 μm) to the medium. Imaging was performed on a Nikon (Tokyo, Japan) TE200 with a Prior XYZ stage, Sutter excitation and emission filter wheels (Sutter Instruments, Novato, CA), a transmitted light shutter, a Princeton Micromax 1300 YHS-cooled CCD camera (Roper Scientific), and Metamorph software. NR1-GFP-expressing cells were located, and images were acquired rapidly to prevent changes in the pH of the medium. TPA was added, neurons were returned to the 37°C CO2 incubator for 30 min, and the same cells were relocated and imaged again. Quantification of fluorescent intensity was performed in Metamorph. Spines did not exactly align between time points because of spine motility, so the pixel area for measuring spine fluorescence was manually centered over each spine for each image.
RESULTS
Synaptic remodeling by PKC: NMDA receptors and CaMKII
Effects of kinase activation on synaptic composition were assessed by immunocytochemistry in cultured neurons. Primary embryonic rat hippocampal neurons were grown under conditions in which NMDA receptors cluster at synapses, either at high density in the absence of pharmacological agents or at low density in the presence of NMDA receptor antagonists (Rao and Craig, 1997; Crump et al., 2001). To examine the role of PKC activity in the synaptic localization of NMDA receptors, pyramidal cells were treated with 100 nm TPA (a PKC agonist) for 45 min, fixed, and double labeled with antibodies to the essential NMDAR subunit NR1 and to synapsin or synaptophysin as markers for presynaptic terminals. In high-density neurons cultured for 16–18 d, NR1 was intensely clustered and highly apposed to presynaptic terminals (Fig. 1a). After TPA treatment for 45 min, NR1 dispersed away from the postsynaptic compartment and was no longer clustered (Fig. 1b). In the TPA-treated neurons, NR1 immunoreactivity appeared similar to that of uniformly distributed proteins in the dendritic plasma membrane. TPA treatment had no effect on the distribution of synaptic vesicle antigens as assessed by light microscopy. Puncta corresponding to presynaptic terminals were numerous in the control and TPA-treated cultures. Another phorbol ester that activates PKC, PDD, induced a similar dispersal of NR1, whereas 4α-TPA and 4α-PDD, which do not activate PKC, had no effect on NMDAR distribution (data not shown).
In contrast to the PKC-mediated dispersal of NMDA receptors, TPA had the opposite effect on localization of another major PSD protein, CaMKIIα. In control neurons, CaMKIIα immunoreactivity is concentrated in some spines but also present throughout dendritic shafts (Fig. 1c). Activation of PKC with TPA induced a dramatic translocation of CaMKIIα from the dendritic shaft to spine synapses (Fig. 1d). The translocation of CaMKIIα to synapses was specifically induced by active phorbol esters and not the inactive analog (data not shown). Thus, activation of PKC induces opposing effects on two major synaptic components, dispersal of NMDA receptors away from synapses and translocation of CaMKIIα to synapses.
To determine whether NMDA receptor activity was required for the TPA-induced dispersal of NMDA receptors or synaptic translocation of CaMKIIα, TPA treatment was performed in the presence of the NMDA receptor antagonist APV. For these experiments and most of the subsequent experiments, we used 3 week low-density neurons chronically pretreated with APV, which show strong synaptic clustering of NMDAR (Fig. 1e) and a partially spiny localization of CaMKIIα (Fig. 1g), similar to the high-density untreated cultures. Activation of PKC with TPA resulted in dispersal of NR1 and enhanced synaptic clustering of CaMKIIα even in the presence of NMDA receptor antagonist (Fig. 1f,h). Thus, NMDAR activity was not required and had no apparent effect on PKC-induced dispersal of NMDAR or clustering of CaMKIIα.
Quantitative immunofluorescence analysis was performed on randomly chosen populations of neurons immunolabeled for NR1 or CaMKIIα and synaptic vesicle antigens. Compared with control neurons, TPA treatment resulted in a significant decrease in synaptic NR1 (50.1 ± 1.8 synaptic NR1 clusters/100 μm dendrite in controls vs 9.8 ± 0.9 synaptic NR1 clusters/100 μm dendrite in TPA-treated neurons;t test; p < 0.01; n ≥ 85 neurons per group from seven independent cultures) and a significant increase in synaptic CaMKIIα (7.2 ± 0.6 synaptic CaMKIIα clusters/100 μm dendrite in controls vs 26.4 ± 1.1 synaptic CaMKIIα clusters/100 μm dendrite in TPA-treated neurons;t test; p < 0.01; n ≥ 79 neurons per group from seven cultures). These effects of TPA were blocked by coincubation with bisindolylmaleimide, a specific inhibitor of PKC (43.8 ± 3.8 synaptic NR1 clusters/100 μm dendrite and 9.4 ± 1.5 synaptic CaMKIIα clusters/100 μm dendrite in TPA/bisindolylmaleimide-treated neurons; p < 0.01 compared with TPA-treated neurons; p > 0.1 compared with control neurons; n ≥ 22 neurons per group from two cultures). Thus, the effects of TPA on redistribution of NMDAR and CaMKII are mediated by activation of PKC. PKC activation had no effect on the density of presynaptic inputs, quantified as puncta of synaptophysin or synapsin per dendrite length (82.2 ± 2.2 terminals/100 μm dendrite in controls vs 82.4 ± 2.3 terminals/100 μm dendrite in TPA-treated neurons; p> 0.1; n ≥ 136 neurons per group from eight cultures).
PKC effects on PSD-95, AMPA receptors, and spines
The effect of PKC activation on the localization of other prominent postsynaptic components was examined. Neurons were treated with TPA, fixed, and stained with combinations of antibodies to synapsin or synaptophysin, the NMDA receptor subunits NR1, NR2A, and NR2B, the AMPA receptor subunit GluR1, and the PDZ domain postsynaptic scaffolding protein PSD-95. As expected, the NR2A and NR2B subunits of the NMDA receptor dispersed away from synapses in concert with the NR1 subunit and appeared to be diffuse and membrane associated (Fig.2a,a′,b,b′; data not shown for NR2B). Although it binds NR2A and NR2B and colocalizes with NMDAR at synapses in control neurons, PSD-95 did not disperse after activation of PKC (Fig. 2c,c′,d,d′). In TPA-treated cells double labeled for NR1 and PSD-95, NMDAR was diffusely localized along dendrites, whereas PSD-95 remained punctate and closely apposed to presynaptic terminals. Quantification revealed no change in the density of synaptic PSD-95 clusters after PKC activation (67.1 ± 4.9 synaptic PSD-95 clusters/100 μm dendrite in controls vs 63.5 ± 3.3 synaptic PSD-95 clusters/100 μm dendrite in TPA-treated neurons;p > 0.1; n = 22 neurons per group from two cultures). The AMPA receptor subunit GluR1 also showed no obvious change in distribution after activation of PKC (Fig.2e,e′,f,f′). GluR1 was concentrated in some dendrite spines and detected at lower levels in dendrite shafts in both control and TPA-treated neurons. The lack of effect of PKC on GluR1 localization is in agreement with the results of Chung et al. (2000), who found no change in the density of synaptic GluR2 clusters after TPA treatment. Thus, PKC activation results in a dramatic selective dispersal of NMDA receptors and not AMPA receptors or PSD-95.
Fig. 2.

PKC-induced synaptic remodeling: effect on multiple NMDA receptor subunits and F-actin but not on AMPA receptor, PSD-95, or spine density. a, a′, c, c′, In control (Con) neurons, NR1 coclusters with NR2A and PSD-95.b, b′, d, d′, After 30 min of TPA treatment, PSD-95 remained clustered at synapses, whereas NR2A dispersed away from synapses along with NR1. e, e′, f, f′, There was no apparent change in the synaptic localization of GluR1 between control and TPA-treated neurons. g, h, TPA treatment increased the concentration of F-actin in dendritic spines and reduced filamentous actin in dendrite shafts, mirroring the rearrangement of CaMKIIα. i, j, TPA treatment had no effect on the overall number of spines but may alter their size and shape. Scale bar, 10 μm.
To assess the effects of PKC activation on more general features of spine structure and density, control and TPA-treated neurons were fixed and stained with Texas Red phalloidin to reveal filamentous actin or labeled with a suspension of DiI to reveal dendritic membrane profiles. As reported previously (Allison et al., 1998), F-actin appeared to be highly concentrated in dendritic spines in control neurons (Fig.2g). F-actin appeared to accumulate further in spines after PKC stimulation (Fig. 2h), however, and there appeared to be a slight rounding of spine heads in TPA-treated cells (Fig.2i,j). Despite the dynamic remodeling of postsynaptic components, the number of dendrite spines was not affected by PKC activation. Spine density quantified from dendrites randomly labeled with DiI was not significantly different between control and TPA-treated neurons (49.8 ± 3.8 protrusions/100 μm dendrite for controls vs 44.9 ± 3.4 protrusions/100 μm dendrite for TPA-treated cells; p > 0.1; n ≥ 19 neurons per group from four cultures).
Rapid time course and reversal of synaptic remodeling by PKC
Given the striking molecular rearrangement of NR1 and CaMKII in opposite directions within the postsynaptic compartment, we asked whether movement of these two PSD core components occurred on the same time course and thus might be related (Fig.3a,b). Even 5 min of TPA treatment resulted in a partial dispersal of NR1 and clustering of CaMKIIα. The redistributions of both NR1 and CaMKIIα were approximately half-maximal within 10 min and maximal by 45 min of TPA treatment. These results reveal unexpectedly rapid dynamics in the molecular organization of the hippocampal postsynaptic compartment.
Fig. 3.

Rapid time course and reversal of synaptic remodeling. a, b, Neurons were exposed to TPA for the indicated times, fixed, and immunolabeled for NR1 (a) or CaMKIIα (b) and synapsin (Syn). An initial observable effect on NR1 and CaMKIIα localization was seen as early as 5 min after addition of TPA. At 10 min, the effect of TPA on NR1 and CaMKIIα translocation was approximately half-maximal, and at 45 min the effect was complete.c, The effect of PDA on the distributions of NR1 and CaMKIIα was similar to that of TPA. A 1 hr washout of PDA resulted in significant reversal; NR1 again became more localized to synapses and CaMKII more diffuse. Reversal also occurred after an 8 hr washout in the presence of the protein synthesis inhibitor cycloheximide. Scale bar, 10 μm.
Furthermore, these effects of PKC activation on synaptic remodeling were rapidly reversible. The effect of a 1 hr treatment with PDA (a relatively water-soluble activator of PKC) in dispersing NMDA receptors and clustering CaMKII was similar to that of treatment with TPA (Fig.3c). After only 1 hr of washout of the PDA, NR1 and CaMKII showed a significant reversal in distribution; NR1 returned to the highly synaptic pattern and CaMKII to the partially spiny pattern. An 8 hr reversal in the presence of the protein synthesis inhibitor cycloheximide also showed reaccumulation of NR1 at synapses. Thus, the reappearance of synaptic NR1 suggests that the translocation of previously dispersed NMDA receptor contributed to the synaptic pool of NMDA receptors.
Mechanisms of PKC-induced synaptic remodeling
Possible mediators of the PKC-induced redistribution of NMDA receptors and CaMKII include actin reorganization, other kinases, and synaptic activity; therefore, we tested these possibilities pharmacologically. NMDA receptors and CaMKII hetero-oligomers have close associations to the F-actin cytoskeleton (Wyszynski et al., 1997;Shen et al., 1998), and we observed a PKC-induced increase in synaptic F-actin (Fig. 2g,h). We used jasplakinolide, a cell-permeable molecule that binds to and stabilizes F-actin (Halpain et al., 1998), to prevent rearrangements in the actin cytoskeleton. We tested whether CaMKII activity is necessary for PKC-induced NMDAR dispersal or for its own translocation by treatment of cultured hippocampal neurons with the CaMKII inhibitor KN62 before and during the addition of TPA. Another kinase that has been implicated in NMDA receptor modulation and has associations with the NMDA receptor complex is Src. Activation of PKC by phorbol esters results in the tyrosine phosphorylation of NR2A/B (Grosshans and Browning, 2001), and Src mediated NMDA receptor modulation has been shown to be downstream of PKC activation (Lu et al., 1999). Therefore, we examined the participation of Src in postsynaptic remodeling by treating hippocampal neurons with the tyrosine kinase inhibitor lavendustin A before and during TPA treatment. We found no significant difference between neurons treated with TPA alone, jasplakinolide/TPA, KN62/TPA, or lavendustin A/TPA with respect to the number of synaptic clusters of NR1 or CaMKIIα (Fig. 4). The role of CaMKII activity was tested further using a separate inhibitor, KN93 (3–30 μm range), which again did not block the PKC-induced redistribution (26.6 ± 1.8 synaptic clusters of CaMKII per 100 μm dendrite in neurons treated with TPA/KN93 at 30 μm;p > 0.1 compared with TPA-treated neurons). Thus, PKC-induced NMDA receptor dispersal and CaMKII translocation occur independently of F-actin dynamics, CaMKII activity, and tyrosine kinase activity.
Fig. 4.

Mechanisms of NMDAR and CaMKII synaptic remodeling by PKC: independence from actin reorganization, CaMKII, tyrosine kinase, and synaptic activity. TPA decreased the number of synaptic NR1 clusters per dendritic length and increased the number of synaptic CaMKIIα clusters per dendrite length compared with control neurons (n ≥ 79 neurons from 7 separate cultures;p < 0.01). Cotreatment with bisindolylmaleimide (Bis), a specific inhibitor of PKC, blocked these effects of TPA (n ≥ 22 neurons from 2 cultures;p < 0.01 compared with TPA; p> 0.1 compared with control). Cotreatment with jasplakinolide (Jasplak), KN62, lavendustin A (Lav A), or blockade (a combination of TTX, nifedipine, picrotoxin, CNQX, APV, and AP-3) did not significantly alter the degree of NR1 dispersal or CaMKIIα synaptic translocation induced by TPA (n≥ 19 from at least 2 cultures; p > 0.09).
In addition to direct postsynaptic effects, phorbol esters enhance presynaptic vesicle fusion (Malenka et al., 1986; Parfitt and Madison, 1993). We thus determined whether the synaptic remodeling of NMDAR and CaMKII was attributable to postsynaptic PKC activity rather than presynaptic vesicle release. Pharmacological presynaptic and postsynaptic blockade was mediated by a cocktail of inhibitors to voltage-gated sodium channels (tetrodotoxin), voltage-gated L-type calcium channels (nifedipine), inhibitory GABA receptors (picrotoxin), and glutamate receptors of the AMPA, NMDA, and group I metabotropic glutamate receptors subtypes (CNQX, APV, and AP-3). We again found no significant effect of these agents on TPA-induced dispersal of NMDAR or clustering of CaMKII (Fig. 4), suggesting a direct postsynaptic effect of PKC.
PKC modulation of postsynaptic density and cytoskeletal associations
We used detergent extraction after TPA treatment to determine the strength of associations of NR1 and CaMKIIα to the cytoskeleton. Insolubility of postsynaptic density components in the detergent Triton X-100 supports the tight associations of NR1, NR2, CaMKIIα/β, and PSD-95 among themselves and/or the cytoskeleton (Kennedy, 2000). Detergent extraction has classically been used for isolation of postsynaptic densities from biochemical synaptosome fractions (Kennedy, 2000) and can also be used to assess molecular interactions at the PSD in cultured hippocampal neurons (Allison et al., 1998). Treatment of living neurons with Triton X-100 had little effect on the distribution of NMDAR in control neurons; most of the NR1 was associated with the postsynaptic site and remained associated with the PSD after extraction (Fig. 5a,b) (Allison et al., 1998). After TPA treatment, however, much of the NR1 had dispersed out of the PSD region and was now extracted, whereas the small amount of NR1 remaining within the PSD region was unextracted (Fig.5c,d). Conversely, in control neurons, much of the cellular CaMKII was extrasynaptic and was extracted by Triton X-100, leaving only small amounts at the postsynaptic site (Fig.5e,f) (Allison et al., 2000). After TPA treatment, much of the cellular CaMKIIα had translocated to the PSD region and now became resistant to Triton-100 extraction (Fig. 5g,h). Thus, PKC-induced dispersal of NMDAR involves breakage of Triton-resistant associations, whereas PKC-induced clustering of CaMKII involves formation of new Triton-resistant associations with postsynaptic density components.
Fig. 5.

PKC-induced change in cytoskeletal association of NMDAR and CaMKII. Neurons were fixed immediately after a 45 min treatment with TPA (+TPA) and/or after subsequent extraction of living neurons with Triton X-100 (+EXT). Neurons were immunolabeled for NR1 or CaMKIIα (green) and the dendritic microtubule-associated protein MAP2 (red). a, b, In control neurons, NR1 was primarily punctate and spiny and was not extracted by detergent. c, d, In TPA-treated neurons, most of the diffusely localized NR1 was extracted by detergent, leaving only small amounts in dendritic spines. e, f, In control neurons, most of the diffusely localized CaMKIIα was extracted by detergent, leaving only small amounts in dendritic spines. g, h, In TPA-treated neurons, CaMKIIα was largely punctate and spiny and was not extracted by detergent. MAP2 localization and cytoskeletal association were not affected by TPA treatment. Boxed regions ina–h are shown below at higher magnification. Scale bar, 10 μm.
To further understand the nature of these molecular associations, we determined the effect of depolymerization of F-actin on the distributions of NR1 and CaMKII and their redistribution by PKC (Fig.6). As reported previously (Allison et al., 1998, 2000), a 9 hr treatment with latrunculin A induced a loss of spine F-actin, a complete dispersal of CaMKII, and a modest conversion of NMDA receptors from spiny to nonsynaptic clusters. Subsequent treatment with TPA resulted in no additional change in CaMKII distribution, indicating that the PKC-induced accumulation of CaMKII involves an association with spine F-actin. In contrast, loss of F-actin did not inhibit the ability of PKC to disperse NMDA receptors. Thus, PKC-induced dispersal of NMDA receptors is independent of F-actin and independent of the translocation of CaMKII. The nonsynaptic clusters of NMDA receptor that are present in low numbers under our control conditions and at higher numbers in immature neurons or in latrunculin-treated neurons also largely dispersed with PKC activation (data not shown).
Fig. 6.
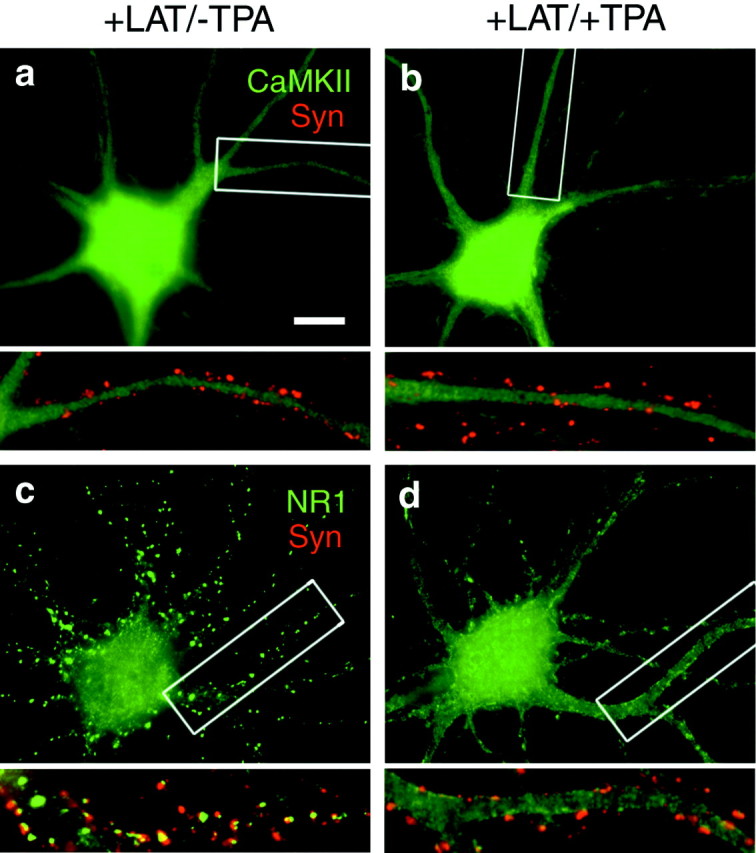
Dependence of CaMKII translocation but not NMDAR translocation on filamentous actin. Neurons were treated for 9 hr with latrunculin A (+LAT) to depolymerize actin, treated for 45 min with TPA as indicated (+TPA), fixed, and immunolabeled for CaMKIIα or NR1 (green) and synapsin (Syn; red). a, b, In latrunculin-treated neurons, CaMKII was diffusely localized, and activation of PKC was unable to induce synaptic accumulation. c, d, In latrunculin-treated neurons, NR1 was clustered at synaptic and nonsynaptic sites. PKC activation induced a loss of synaptic and nonsynaptic clusters and a redistribution to a dispersed plasma membrane-associated pattern even in the absence of detectable F-actin. Boxed regions in a–d are shown below at higher magnification. Scale bar, 10 μm.
Cell-surface association of NMDA receptors
To assess the level of surface expression of NR1 after TPA treatment, we subjected live neurons to the extracellular protease chymotrypsin before harvesting and Western blot analysis (Hall and Soderling, 1997). In neuron culture extracts, immunoblotting for NR1 reveals a strong band at ∼120 kDa (Fig.7). When intact neurons are subjected to the extracellular proteolytic activity of chymotrypsin, the extracellular region of any NR1 protein on the cell surface is cleaved and the immunoblot signal at ∼120 kDa is decreased proportionally to the percentage of NR1 on the cell surface. Tau, an intracellular cytoskeletal protein, is inaccessible to chymotrypsin. Previous results using this method indicate that NR1 is present in significant intracellular pools in immature or low-density neurons with a nonsynaptic immunostaining pattern (42% surface), but largely on the surface of neurons with a highly synaptic immunostaining pattern (87% surface) (Crump et al., 2001). Our finding here that 84% of the NR1 was cleaved and thus surface associated in neurons with the highly synaptic NMDA receptor pattern is in agreement with these previous results. TPA treatment did not change the percentage of surface NR1; 84% of total cellular NR1 was still accessible to extracellular protease (average of three experiments each). The total amount of NR1 also did not change with TPA treatment. Sister neurons from the TPA-treated group were immunolabeled to confirm the predominantly extrasynaptic localization of NR1. Thus, although TPA induces a redistribution of NR1 away from synapses, the redistributed NR1 is on the cell surface. These experiments are supported by the immunolabeling pattern for NR1 throughout the exposure to phorbol esters (Figs.1f, 3a). NR1 appeared to be associated with plasma membrane; at no time were nonsynaptic clusters characteristic of endosomes observed. Direct cell-surface labeling of live or nonpermeabilized neurons with commercially available antibodies against NR1 was not possible, perhaps because of limited accessibility of the native NMDA receptor complex in neurons.
Fig. 7.

Cell-surface association of clustered and dispersed NMDAR. Intact hippocampal neurons were treated with TPA (30 min) and then with extracellular protease chymotrypsin (10 min) as indicated before cultures were harvested for Western blot analysis. Immunoblotting for NR1 revealed a strong band at ∼120 kDa, as expected. After chymotrypsin treatment, the extracellular region of cell-surface NR1 is cleaved and the native ∼120 kDa signal is decreased. Tau, an intracellular cytoskeletal protein, is inaccessible to chymotrypsin and was used as a control for protein loading between conditions. The percentage of cleaved and thus surface-accessible NR1 was 84% for both control and TPA-treated neurons (average of 3 experiments each).
Neuroprotective effect of PKC-induced synaptic remodeling
Excessive calcium entry through NMDA receptors is a major trigger of neuronal cell death (Rothman and Olney, 1995). In low-density hippocampal cultures under conditions of spontaneous activity, NMDA receptors are primarily extrasynaptic and neurons exhibit limited toxicity in response to a high-potassium insult (Crump et al., 2001). Chronic blockade of NMDA receptors results in a large increase in synaptic targeting of NMDA receptors (the baseline condition for the experiments described above) and a concomitant increase in excitotoxicity after antagonist washout (Crump et al., 2001). In agreement with these previous studies, in response to a short pulse of high potassium to induce synaptic release of glutamate, APV-pretreated neurons exhibited greater toxicity (43.7 ± 2.8% cell viability) than neurons cultured without APV (62.2 ± 4.0% viability;t test; p < 0.01; n = 8 coverslips) (Fig. 8a). This enhanced toxicity was dependent on NMDA receptor function, as indicated by sensitivity to APV during the toxicity assay (Fig. 8a, compare columns 3 and 4: the decrease in viability from >90% in untreated neurons to ∼60% after treatment with high K+ was NMDAR-independent, but the additional decrease in viability of APV-pretreated cells to ∼40% was NMDAR-dependent). We subsequently asked whether dispersing NMDAR from postsynaptic sites by TPA treatment would be neuroprotective. In APV-pretreated neurons with highly synaptic NMDAR clusters, treatment with TPA and concomitant dispersal of NMDAR (as in Fig. 1) significantly reduced excitotoxicity (63.2 ± 2.6% cell viability; t test; p < 0.01 compared with APV group; n = 8) (Fig. 8, column 2 inb vs column 3 in a). In contrast, TPA treatment had no effect on excitotoxicity in low-density neurons cultured without APV and lacking synaptic NMDA receptors clusters (Fig.8, column 1 in b vs column 2 ina). These results suggest that the TPA-induced neuroprotection is attributable to the dispersal of NMDAR in APV-pretreated neurons rather than other effects of TPA that would presumably also occur in neurons cultured without APV.
Live visualization of NMDAR dispersion by PKC
To visualize the effect of PKC activity on NMDAR distribution without constraints imposed by antibody-based methods, we used a GFP-tagged NR1 subunit. Coexpression of NR1-GFP and NR2A at low expression levels in APV-treated cultured hippocampal neurons results in synaptic clustering of the NR1-GFP (Fig.9a,a′) (Crump et al., 2001). In all experiments, the distribution of transfected NR1-GFP was similar to that of endogenous NR1 visualized by immunolabeling. Immunolabeling of NR1-GFP-expressing neurons for CaMKII shows clearly in a single neuron the differential distributions implied by the separate antibody labeling experiments: NR1-GFP was highly clustered on dendritic spines, whereas CaMKII showed some concentration in these spines but was also present diffusely throughout the neuron (Fig. 9b,b′). TPA treatment resulted in a reversal of the distribution patterns: NR1-GFP became diffusely distributed throughout the dendrite, with minor concentrations remaining at dendritic spines, whereas CaMKII became highly concentrated in dendritic spines and showed reduced levels in shafts (Fig. 9c,c′).
Fig. 9.
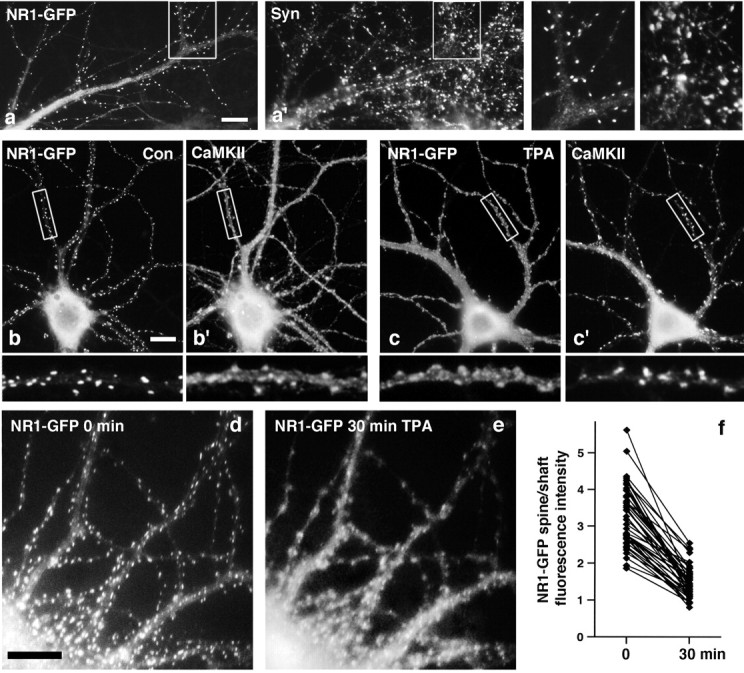
PKC-induced transposition of NMDAR and CaMKII distributions and visualization of NMDAR dispersal in live neurons.a–c, Neurons were transfected at 1 week with NR1-GFP and NR2A, cultured for another 2 weeks, treated with TPA as indicated, fixed, and immunolabeled for synaptophysin (Syn;a, a′) or CaMKIIα (b, b′, c, c′).a, a′, Recombinant NR1-GFP clustered on dendritic spines opposite synaptic terminals. b, b′, NR1-GFP was highly concentrated in dendritic spines in control (Con) neurons, and CaMKIIα was detected in the same spines but also at high levels throughout the dendrite shaft. c, c′, In TPA-treated neurons, the distribution patterns of NR1-GFP and CaMKIIα were reversed relative to controls. CaMKIIα now clustered strongly on dendritic spines, and NR1-GFP was present at high levels in both spines and shafts. d–f, Neurons were transfected with NR1-GFP and NR2A at plating and imaged at 3 weeks before and 30 min after addition of TPA. As expected on the basis of the population analyses, NR1-GFP dispersed after TPA treatment. f, Analysis of the spine/shaft fluorescence intensity ratio in individual dendritic spines revealed a consistent decrease after TPA treatment (control mean ± SEM, 3.2 ± 0.1; TPA mean ± SEM, 1.5 ± 0.1; paired t test; p < 0.01;n = 45 spines from 3 neurons). Scale bars, 10 μm.
NR1-GFP expression allowed us to visualize the effects of PKC activation on NMDAR distribution in single neurons over time. A 30 min treatment with TPA induced a dramatic dispersal of spiny NR1-GFP clusters visualized in the same neuron before and after TPA treatment (Fig. 9d,e), as expected on the basis of the population analyses. There appeared to be both a decrease in fluorescence in the dendritic spines and an increase in fluorescence in the dendrite shafts. The average NR1-GFP fluorescence intensity ratio in the same set of dendrite spines relative to the dendrite shaft decreased from 3.2 ± 0.1 before TPA addition to 1.5 ± 0.1 after TPA addition (paired t test; p < 0.01;n = 45 spines from three neurons) (Fig.9f). Thus, PKC induces dispersal of existing NMDA receptors away from synaptic sites to the extrasynaptic membrane. The spine/shaft intensity values indicate a more than twofold decrease in the ratio of synaptic to extrasynaptic tagged NMDA receptor after PKC activation.
DISCUSSION
We show here that CaMKII and NMDA receptors exhibit rapid and reciprocal redistribution to and from synapses in response to selective activation of one kinase, PKC. NMDA receptors dispersed and CaMKII clustered with approximately half-maximal effects at 10 min of treatment with phorbol esters, and both effects were rapidly reversible. PKC also induced a slight enhancement of filamentous actin at synapses but no change in the density of presynaptic terminals, dendritic spines, or postsynaptic clusters of the scaffolding molecule PSD-95 and no change in AMPA receptor localization. PKC-induced clustering of CaMKII was dependent on the presence of F-actin, whereas dispersal of NMDA receptors was independent of F-actin. Live cell visualization of PKC-induced redistribution of NR1-GFP revealed a decrease in NR1-GFP synaptic to extrasynaptic ratio in individual dendrites over time. NMDA receptors showed no net internalization but redistributed to the extrasynaptic plasma membrane after PKC activation. Consistent with the decrease in synaptic NMDA receptor density, pretreatment with phorbol esters protected neurons against NMDA-mediated excitotoxicity. These results reveal a surprisingly dynamic nature to the molecular composition of glutamatergic synapses, not just with respect to AMPA receptor as shown in many previous studies but also with respect to NMDA receptor and endogenous CaMKII.
Mechanisms of postsynaptic remodeling by PKC
The postsynaptic density is a highly interconnected network of proteins in which each component binds with high affinity to several other components. NMDA receptor subunits and CaMKIIα/β all have multiple interacting partners in the PSD. NR1 binds Ca2+/CaM, the actin-binding proteins α-actinin and spectrin, and the A-kinase anchoring protein (AKAP) yotiao (Wyszynski et al., 1997; Lin et al., 1998; Wechsler and Teichberg, 1998; Westphal et al., 1999). NR2A and NR2B bind spectrin and the PDZ domain proteins PSD-95/SAP90, SAP102, PSD-93/chapsyn-110, S-SCAM, and mLin7 (Wechsler and Teichberg, 1998; Hirao et al., 2000;Setou et al., 2000; Sheng and Pak, 2000). CaMKIIα, through interaction with CaMKIIβ, binds actin (Shen et al., 1998) and directly binds Ca2+/CaM, α-actinin, densin-180, and SynGAPβ (Strack et al., 2000b; Li et al., 2001;Walikonis et al., 2001). Furthermore, CaMKIIα binds NR2A/B with high affinity (Gardoni et al., 2000; Strack et al., 2000a). Thus, it is likely that a change in the affinity of multiple interactions is involved in the synaptic remodeling induced by PKC.
The mechanisms for localizing NMDA receptors to synapses are not well understood. Evidence from genetically targeted mice lacking the C termini of NR2A or NR2B indicates a partial role for the NR2A/B C termini in synaptic localization of NMDA receptors (Mori et al., 1998;Steigerwald et al., 2000). NR1, NR2A, and NR2B are all substrates of PKC in vitro, and all show increased phosphorylation after treatment of neurons with phorbol esters (Tingley et al., 1993; Hall and Soderling, 1997; Leonard and Hell, 1997). We attempted to determine whether PKC disrupts the interactions of recombinant NR2A and PSD-95 in heterologous systems, but we could find no consistent evidence for such a direct mechanism (data not shown). Thus, although we have ruled out any requirement for an intermediary tyrosine kinase (Fig. 4,TPA/Lav A column), there may be other intermediary signaling proteins specific to neurons that mediate PKC-induced disruption of NR2 binding to PSD-95. Alternatively, it may be that the interaction between NR2A and PSD-95 is not a major force for anchoring synaptic NMDA receptors, and that their dissociation occurs as a consequence of direct disruption of other associations of NR2 or NR1 with PSD components.
The NR1 subunit of the NMDA receptor has multiple possible interactions with PSD proteins, some of which could be regulated by PKC. In the NR1 subunit, the alternatively spliced C1 exon contains the major sites of PKC phosphorylation (Tingley et al., 1997). Homomeric NR1A containing the C1 exon is trapped in the endoplasmic reticulum (ER), and PKC phosphorylation adjacent to the ER retention signal promotes cell-surface delivery of fusion proteins containing the NR1 C1 exon (Scott et al., 2001). In our experiments, the native NMDA receptors were already efficiently localized to the cell surface, and PKC did not change the amount or percentage of NR1 on the cell surface. PKC reduces binding of NR1 to α-actinin (Lu et al., 2000) and to spectrin (Wechsler and Teichberg, 1998), but our experiments with latrunculin (Fig. 6) rule out a major role for actin-dependent proteins. Another interaction that is potentially important in NMDAR dispersal is the binding of NR1 via the C1 exon to the AKAP yotiao (Lin et al., 1998). Thus, PKC phosphorylation of NR1-C1 might release NR1 from binding to yotiao. Furthermore, because yotiao also binds PKA (Westphal et al., 1999) and PKA activity is necessary to maintain synaptic clusters of NMDAR (Crump et al., 2001), disruption of this interaction could lead to dispersal both through direct release of NR1 and through decreased local activity of PKA. In our experiments expressing NR1-GFP, the NR1C-GFP isoform lacking the C1 exon dispersed; however, obtaining synaptic clustering of any NR1-GFP isoform required a low expression level, such that the expressed subunits could be associating with endogenous NR1 containing the C1 exon; therefore, we were unable to directly test the role of the NR1 C1 exon in PKC dispersion.
Transient translocation of recombinant CaMKII-GFP to the PSD has been observed in response to stimulation of NMDA receptors in hippocampal neurons (Shen and Meyer, 1999; Shen et al., 2000). The initial translocation requires CaM binding to CaMKII, and association for several minutes requires CaMKII autophosphorylation on Thr286. The PKC-induced translocation of CaMKII to synapses reported here differs from these previous findings in several respects. First, the PKC-induced translocation appears to be more robust and longer-lasting. These previous studies did not report any effect on localization of endogenous CaMKII, and it is possible that the high levels of overexpression might have induced some nonphysiological associations. Second, PKC-induced translocation was not inhibited by KN62, a compound that inhibits CaMKII kinase activity and autophosphorylation competitively with CaM (Tokumitsu et al., 1990). Third, CaMKIIα can bind directly to NR2A and/or NR2B (Strack et al., 2000b; Gardoni et al., 2001), and it has been reported recently that CaMKII binding to NR2B is necessary for the glutamate-induced synaptic translocation of CaMKII-GFP (Bayer et al., 2001). The simultaneous dispersal of NR2B away from synapses in our experiments, however, would preclude such a mechanism for PKC-induced synaptic translocation of CaMKII. Thus, different molecular associations appear to underlie the synaptic accumulation of CaMKII in these two paradigms.
Physiological significance of postsynaptic remodeling by PKC
There are conflicting reports of the specific effects of PKC on the NMDAR-mediated component of synaptic transmission in hippocampal neurons. As predicted by our results, Markram and Segal (1992) found that phorbol esters had no effect on the fast AMPAR-mediated component of synaptic transmission but suppressed the NMDAR-mediated component of synaptic transmission in CA1 neurons in hippocampal slice. Other groups, however (Lozovaya and Klee, 1995), have reported that phorbol esters enhance EPSCs, including the NMDAR-mediated component, in CA1 neurons in slice. Part of this confusion may be attributable to the enhancement of presynaptic vesicle fusion by PKC (Malenka et al., 1986;Parfitt and Madison, 1993). In addition, PKC can increase whole-cell responses to NMDA in neurons. This increased agonist response has been ascribed variously to reduction of voltage-dependent Mg2+ block (Chen and Huang, 1992), increased probability of channel opening through a mechanism requiring activity of the tyrosine kinase src (Lu et al., 1999), and an increase in the number of functional channels on the neuronal surface (Lan et al., 2001). The PKC-induced dispersal of NMDA receptors reported here would be predicted to have no effect on the whole-cell response to NMDA or perhaps to mediate a slight increase caused by enhanced accessibility of the channels to agonist. The major effect of the PKC dispersal would be to decrease the NMDAR-mediated response to synaptic transmission, as supported by the enhanced excitotoxicity in response to high-K+ stimulation (Fig. 8).
PKC-mediated dispersal of synaptic NMDA receptors could be a mechanism underlying long-term depression (LTD) of the NMDA component of synaptic transmission. Excitatory synaptic activity activates PKC via calcium entry through NMDA receptors and via group I metabotropic glutamate receptor-mediated activation of phospholipase C. Low-frequency stimulation induces LTD of both AMPA- and NMDA-mediated components of synaptic transmission in hippocampal CA1 neurons (Selig et al., 1995). LTD of the AMPA-mediated component is accompanied by net endocytosis of AMPA receptors from synapses in hippocampal cultures (Carroll et al., 1999). Internalization is also promoted by phorbol esters (Chung et al., 2000). Our results suggest that LTD of the NMDA component might occur by PKC-mediated dispersal of NMDA receptors from the synaptic to extrasynaptic plasma membrane.
Considering the dramatic effects on localization of both NMDA receptors and CaMKII, which are major signal-transducing proteins of the PSD, PKC-mediated synaptic remodeling is likely to be a form of metaplasticity. Metaplasticity refers to the effects of previous activity in regulating the subsequent ability of a synapse to undergo potentiation or depression in response to a given stimulus (Abraham and Bear, 1996). Consistent with the effects of PKC on NMDAR distribution reported here, Stanton (1995)found that previous activation of PKC with phorbol esters enhanced LTD in response to low-frequency stimulation and inhibited induction of long-term potentiation in response to high-frequency stimulation in CA1 neurons. The PKC-induced increase in levels of CaMKII at the synapse, however, might function in an opposing manner to enhance potentiation. CaMKII increases AMPAR-mediated responses through direct phosphorylation of AMPA receptors (Barria et al., 1997) and through promotion of AMPA receptor insertion (Hayashi et al., 2000). Indeed, other more complex forms of metaplasticity activated by group I metabotropic receptors and requiring PKC activity have been described previously (Bortolotto and Collingridge, 2000). The precise form of metaplasticity may depend on the route of PKC activation and on cross-talk with other signaling pathways to regulate the kinetics and spatial redistribution of both NMDA receptors and CaMKII. Current evidence indicates that synaptic targeting of NMDA receptors is inhibited by PKC (this work) but promoted by PKA (Crump et al., 2001), whereas synaptic targeting of CaMKII is promoted both by PKC (this work) and by autophosphorylation (Shen et al., 2000). Defining the precise regulatory pathways for dynamic trafficking of kinases and neurotransmitter receptors, including AMPA and NMDA, will be a necessary part of understanding mechanisms of synaptic plasticity.
Footnotes
This work was supported by National Institutes of Health Grant NS33184 and the Pew Charitable Trust. We thank Huaiyang Wu for excellent preparation of neuron cultures.
Correspondence should be addressed to Ann Marie Craig, Department of Anatomy and Neurobiology, Washington University School of Medicine, 660 South Euclid Avenue, Box 8108, St. Louis, MO 63110. E-mail:acraig@thalamus.wustl.edu.
A. Rao's present address: Neuron, Cell Press, 1100 Massachusetts Avenue, Cambridge, MA 02138.
REFERENCES
- 1.Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- 2.Allison DW, Gelfand VI, Spector I, Craig AM. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18:2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allison DW, Chervin AS, Gelfand VI, Craig AM. Postsynaptic scaffolds of excitatory and inhibitory synapses in hippocampal neurons: maintenance of core components independent of actin filaments and microtubules. J Neurosci. 2000;20:4545–4554. doi: 10.1523/JNEUROSCI.20-12-04545.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 5.Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- 6.Bortolotto ZA, Collingridge GL. A role for protein kinase C in a form of metaplasticity that regulates the induction of long-term potentiation at CA1 synapses of the adult rat hippocampus. Eur J Neurosci. 2000;12:4055–4062. doi: 10.1046/j.1460-9568.2000.00291.x. [DOI] [PubMed] [Google Scholar]
- 7.Carroll RC, Lissin DV, von Zastrow M, Nicoll RA, Malenka RC. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat Neurosci. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- 8.Chen L, Huang LY. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- 9.Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci. 2000;20:7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crump FT, Dillman K, Craig AM. cAMP-dependent protein kinase mediates activity-regulated synaptic targeting of NMDA receptors. J Neurosci. 2001;21:5079–5088. doi: 10.1523/JNEUROSCI.21-14-05079.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- 12.Gardoni F, Bellone C, Cattabeni F, Di Luca M. Protein kinase C activation modulates alpha-calmodulin kinase II binding to NR2A subunit of N-methyl-d-aspartate receptor complex. J Biol Chem. 2000;276:7609–7613. doi: 10.1074/jbc.M009922200. [DOI] [PubMed] [Google Scholar]
- 13.Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- 14.Goslin K, Asmussen H, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT; Cambridge, MA: 1998. pp. 339–370. [Google Scholar]
- 15.Grosshans DR, Browning MD. Protein kinase C activation induces tyrosine phosphorylation of the NR2A and NR2B subunits of the NMDA receptor. J Neurochem. 2001;76:737–744. doi: 10.1046/j.1471-4159.2001.00034.x. [DOI] [PubMed] [Google Scholar]
- 16.Hall RA, Soderling TR. Differential surface expression and phosphorylation of the N-methyl-d-aspartate receptor subunits NR1 and NR2 in cultured hippocampal neurons. J Biol Chem. 1997;272:4135–4140. doi: 10.1074/jbc.272.7.4135. [DOI] [PubMed] [Google Scholar]
- 17.Halpain S, Hipolito A, Saffer L. Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J Neurosci. 1998;18:9835–9844. doi: 10.1523/JNEUROSCI.18-23-09835.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasbani MJ, Schlief ML, Fisher DA, Goldberg MP. Dendritic spines lost during glutamate receptor activation reemerge at original sites of synaptic contact. J Neurosci. 2001;21:2393–2403. doi: 10.1523/JNEUROSCI.21-07-02393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 20.Hirao K, Hata Y, Yao I, Deguchi M, Kawabe H, Mizoguchi A, Takai Y. Three isoforms of synaptic scaffolding molecule and their characterization: multimerization between the isoforms and their interaction with N-methyl-d-aspartate receptors and SAP90/PSD-95-associated protein. J Biol Chem. 2000;275:2966–2972. doi: 10.1074/jbc.275.4.2966. [DOI] [PubMed] [Google Scholar]
- 21.Kennedy MB. Signal-processing machines at the postsynaptic density. Science. 2000;290:750–754. doi: 10.1126/science.290.5492.750. [DOI] [PubMed] [Google Scholar]
- 22.Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci. 2001;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- 23.Leonard AS, Hell JW. Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-d-aspartate receptors at different sites. J Biol Chem. 1997;272:12107–12115. doi: 10.1074/jbc.272.18.12107. [DOI] [PubMed] [Google Scholar]
- 24.Li W, Okano A, Tian QB, Nakayama K, Furihata T, Nawa H, Suzuki T. Characterization of a novel synGAP isoform, synGAP-beta. J Biol Chem. 2001;276:21417–21424. doi: 10.1074/jbc.M010744200. [DOI] [PubMed] [Google Scholar]
- 25.Lin JW, Wyszynski M, Madhavan R, Sealock R, Kim JU, Sheng M. Yotiao, a novel protein of neuromuscular junction and brain that interacts with specific splice variants of NMDA receptor subunit NR1. J Neurosci. 1998;18:2017–2027. doi: 10.1523/JNEUROSCI.18-06-02017.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lozovaya NA, Klee MR. Phorbol diacetate differentially regulates the N-methyl-d-aspartate (NMDA) and non-NMDA receptor-mediated components of the rat hippocampal excitatory postsynaptic currents. Neurosci Lett. 1995;189:101–104. doi: 10.1016/0304-3940(95)11463-7. [DOI] [PubMed] [Google Scholar]
- 27.Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci. 1999;2:331–338. doi: 10.1038/7243. [DOI] [PubMed] [Google Scholar]
- 28.Lu WY, Jackson MF, Bai D, Orser BA, MacDonald JF. In CA1 pyramidal neurons of the hippocampus protein kinase C regulates calcium-dependent inactivation of NMDA receptors. J Neurosci. 2000;20:4452–4461. doi: 10.1523/JNEUROSCI.20-12-04452.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature. 1986;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- 30.Markram H, Segal M. Activation of protein kinase C suppresses responses to NMDA in rat CA1 hippocampal neurones. J Physiol (Lond) 1992;457:491–501. doi: 10.1113/jphysiol.1992.sp019389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- 32.Mori H, Mishina M. Structure and function of the NMDA receptor channel. Neuropharmacology. 1995;34:1219–1237. doi: 10.1016/0028-3908(95)00109-j. [DOI] [PubMed] [Google Scholar]
- 33.Mori H, Manabe T, Watanabe M, Satoh Y, Suzuki N, Toki S, Nakamura K, Yagi T, Kushiya E, Takahashi T, Inoue Y, Sakimura K, Mishina M. Role of the carboxy-terminal region of the GluR epsilon2 subunit in synaptic localization of the NMDA receptor channel. Neuron. 1998;21:571–580. doi: 10.1016/s0896-6273(00)80567-x. [DOI] [PubMed] [Google Scholar]
- 34.Parfitt KD, Madison DV. Phorbol esters enhance synaptic transmission by a presynaptic, calcium-dependent mechanism in rat hippocampus. J Physiol (Lond) 1993;471:245–268. doi: 10.1113/jphysiol.1993.sp019900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- 36.Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor–still lethal after eight years. Trends Neurosci. 1995;18:57–58. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- 37.Scott DB, Blanpied TA, Swanson GT, Zhang C, Ehlers MD. An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci. 2001;21:3063–3072. doi: 10.1523/JNEUROSCI.21-09-03063.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Selig DK, Hjelmstad GO, Herron C, Nicoll RA, Malenka RC. Independent mechanisms for long-term depression of AMPA and NMDA responses. Neuron. 1995;15:417–426. doi: 10.1016/0896-6273(95)90045-4. [DOI] [PubMed] [Google Scholar]
- 39.Setou M, Nakagawa T, Seog DH, Hirokawa N. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science. 2000;288:1796–1802. doi: 10.1126/science.288.5472.1796. [DOI] [PubMed] [Google Scholar]
- 40.Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- 41.Shen K, Teruel MN, Subramanian K, Meyer T. CaMKIIbeta functions as an F-actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron. 1998;21:593–606. doi: 10.1016/s0896-6273(00)80569-3. [DOI] [PubMed] [Google Scholar]
- 42.Shen K, Teruel MN, Connor JH, Shenolikar S, Meyer T. Molecular memory by reversible translocation of calcium/calmodulin-dependent protein kinase II. Nat Neurosci. 2000;3:881–886. doi: 10.1038/78783. [DOI] [PubMed] [Google Scholar]
- 43.Sheng M, Pak DT. Ligand-gated ion channel interactions with cytoskeletal, signaling proteins. Annu Rev Physiol. 2000;62:755–778. doi: 10.1146/annurev.physiol.62.1.755. [DOI] [PubMed] [Google Scholar]
- 44.Soderling TR, Chang BH, Brickey DA. Cellular signaling through multifunctional Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2001;276:3719–3722. doi: 10.1074/jbc.R000013200. [DOI] [PubMed] [Google Scholar]
- 45.Stanton PK. Transient protein kinase C activation primes long-term depression and suppresses long-term potentiation of synaptic transmission in hippocampus. Proc Natl Acad Sci USA. 1995;92:1724–1728. doi: 10.1073/pnas.92.5.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steigerwald F, Schulz TW, Schenker LT, Kennedy MB, Seeburg PH, Kohr G. C-terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. J Neurosci. 2000;20:4573–4581. doi: 10.1523/JNEUROSCI.20-12-04573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 2000a;275:23798–23806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- 48.Strack S, Robison AJ, Bass MA, Colbran RJ. Association of calcium/calmodulin-dependent kinase II with developmentally regulated splice variants of the postsynaptic density protein densin-180. J Biol Chem. 2000b;275:25061–25064. doi: 10.1074/jbc.C000319200. [DOI] [PubMed] [Google Scholar]
- 49.Tingley WG, Roche KW, Thompson AK, Huganir RL. Regulation of NMDA receptor phosphorylation by alternative splicing of the C-terminal domain. Nature. 1993;364:70–73. doi: 10.1038/364070a0. [DOI] [PubMed] [Google Scholar]
- 50.Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-d-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–5166. doi: 10.1074/jbc.272.8.5157. [DOI] [PubMed] [Google Scholar]
- 51.Tokumitsu H, Chijiwa T, Hagiwara M, Mizutani A, Terasawa M, Hidaka H. KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1990;265:4315–4320. [PubMed] [Google Scholar]
- 52.Walikonis R, Oguni A, Khorosheva E, Jeng C, Asuncion F, Kennedy M. Densin-180 forms a ternary complex with the α-subunit of Ca2+/calmodulin-dependent protein kinase II, α-actinin. J Neurosci. 2001;21:423–433. doi: 10.1523/JNEUROSCI.21-02-00423.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wechsler A, Teichberg VI. Brain spectrin binding to the NMDA receptor is regulated by phosphorylation, calcium and calmodulin. EMBO J. 1998;17:3931–3939. doi: 10.1093/emboj/17.14.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- 55.Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]