

Abstract
The importance of postmenopausal estrogen replacement therapy in affording protection against the selective and delayed neuronal death associated with cardiac arrest or cardiac surgery in women remains controversial. Here we report that exogenous estrogen at levels that are physiological for hormone replacement in postmenopausal women affords protection against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1 of male gerbils. Global ischemia induced a marked increase in activated caspase-3 in CA1, evident at 6 hr after ischemia. Global ischemia induced a marked upregulation of the proapoptotic neurotrophin receptor p75NTR in CA1, evident at 48 hr. p75NTR expression was induced primarily in terminal deoxynucleotidyl transferase-mediated UTP nick-end labeling-positive cells, indicating expression in neurons undergoing apoptosis. Global ischemia also induced a marked downregulation of mRNA encoding the AMPA receptor GluR2 subunit in CA1. Caspase-3, p75NTR, and GluR2 were not significantly changed in CA3 and dentate gyrus, indicating that the ischemia-induced changes in gene expression were cell specific. Exogenous estrogen attenuated the ischemia-induced increases in activated caspase-3 and blocked the increase in p75NTR in post-ischemic CA1 neurons but did not prevent ischemia-induced downregulation of GluR2. These findings demonstrate that long-term estrogen at physiological levels ameliorates ischemia-induced hippocampal injury and indicate that estrogen intervenes at the level of apoptotic signaling cascades to prevent onset of death in neurons otherwise “destined to die.”
Keywords: estrogen, hormone replacement therapy, global ischemia, neuronal death, neurotrophin receptors, apoptosis
Transient, severe global ischemia arising in humans as a consequence of cardiac arrest or cardiac surgery or induced experimentally in animals leads to selective and delayed neuronal death, particularly of pyramidal neurons in the hippocampal CA1 (for review, see Tanaka et al., 2000). Global ischemia-induced neuronal death is not detected until 2–4 d after induction of global ischemia in rats and gerbils. The relative contributions of apoptotic and necrotic death to ischemia-induced neuronal loss remain controversial (for review, see MacManus and Buchan, 2000; Yamashima, 2000; Graham and Chen, 2001). Neurotrophins and their receptors play a critical role in receptor-activated apoptotic signaling cascades and in the onset and progression of apoptotic cell death (for review, seeKaplan and Miller, 2000). Global ischemia induces expression of p75NTR, a proapoptotic ligand-activated neurotrophin receptor and member of the tumor necrosis factor receptor superfamily, in CA1 neurons (Bagum et al., 2001). Ligand activation of p75NTR under conditions of low trkA expression is thought to trigger apoptosis (Kaplan and Miller, 2000;Yano and Chao, 2000). Global ischemia also induces expression of activated caspase-3 (Chen et al., 1998b; Namura et al., 1998), a cysteine protease and “terminator” protein implicated in the execution step of apoptosis, in CA1 (Cohen, 1997; Nicholson and Thornberry, 1997).
Estradiol and related ovarian steroids modify the structure and function of hippocampal neurons. Estradiol increases spine density (Woolley and McEwen, 1994; Murphy and Segal, 1996; Pozzo-Miller et al., 1999), synapse number (Woolley and McEwen, 1994), and NMDA receptor NR1 subunit expression (Gazzaley et al., 1996) and potentiates kainate-elicited currents in CA1 pyramidal neurons (Moss and Gu, 1999). The mechanisms underlying these actions are unclear, because intracellular estrogen receptors are not highly expressed in hippocampal pyramidal neurons (Shughrue et al., 1997; Weiland et al., 1997; Shughrue and Merchenthaler, 2000). Estrogen receptors are coexpressed with neurotrophins and neurotrophin receptors in neurons (Miranda et al., 1993; Toran-Allerand et al., 1999), and estrogen regulates their expression. Estrogen reduces p75NTR expression in rat forebrain (Gibbs and Pfaff, 1992; Gibbs, 1997), rat sensory neurons (Sohrabji et al., 1994b), and PC12 cells (Sohrabji et al., 1994a) and increases trkA mRNA expression in dorsal root ganglia (Sohrabji et al., 1994b), basal forebrain cholinergic neurons (McMillan et al., 1996), and PC12 cells (Sohrabji et al., 1994a).
Considerable evidence suggests that estrogen affords neuroprotection against brain injury and neurodegenerative diseases (for review, seeWise et al., 2001). Estrogen therapy in postmenopausal women reduces the incidence of stroke and the extent of ischemic neurodegeneration (Schmidt et al., 1996) and the onset and severity of Alzheimer's disease (Henderson et al., 1996; Tang et al., 1996; Kawas et al., 1997). The estrogen receptor antagonist tamoxifen is thought to increase incidence of stroke in premenopausal women (Gail et al., 1999). Estrogen administration to ovariectomized female (Dubal et al., 1998; Rusa et al., 1999; Dubal and Wise, 2001) and male (Toung et al., 1998) rats reduces brain injury after focal ischemia. Moreover, females with circulating estrogen are more resistant to focal ischemia than male counterparts (Hall et al., 1991; Alkayed et al., 1998; Zhang et al., 1998). The ability of estrogen to protect against global ischemia-induced neuronal death is, however, as yet unclear.
The present study was undertaken to examine the hypothesis that estrogen acts at the level of apoptotic signaling cascades to prevent onset of global ischemia-induced apoptotic death. Here we show that estrogen levels that are physiological for hormone replacement in postmenopausal women protects against delayed, ischemia-induced neuronal death in male gerbils. Ischemia increases caspase-3 expression and activation and increases p75NTR in CA1 neurons preceding neuronal death. Estrogen markedly attenuates the ischemia-induced activation of caspase-3 and increase in p75NTR expression. These findings suggest that neuroprotection by estrogen against ischemia-induced damage may occur via inhibition of apoptotic signaling cascades.
MATERIALS AND METHODS
Global ischemia. Animals were treated in accordance with the principles and procedures of the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals; all protocols were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine. Animals were maintained in a temperature- and light-controlled environment with a 14/10 hr light/dark cycle. Age-matched, adult male Mongolian gerbils (Tumblebrook Farms, Wilmington, MA) weighing 60–80 gm were administered placebo or 17β-estradiol (0.36 mg/pellet; 60 d, controlled time release; Innovative Research of America Inc., Sarasota, FL) by implantation in the subcutaneous tissue of the dorsal neck. These pellets use a biodegradable matrix for timed-release of hormones and have been tested extensively by both the vendor and a large number of research laboratories. They provide reliable, consistent plasma hormone levels for 60 d, and their use avoids the fluctuations in hormone levels produced by injections and reduces stress on the animals. Fourteen days after pellet implantation, animals were subjected to surgery (after fasting overnight). In brief, animals were anesthetized with halothane (4–1%) delivered by mask in a mixture of N2/O2 (70:30) by means of a Vapomatic anesthetic vaporizer (CWE Inc., Ardmore, PA). Animals were subjected to global ischemia by temporary bilateral occlusion of the carotid arteries (BCCO) (5 min) or to sham surgery, followed by reperfusion as described previously (Oguro et al., 1999; Opitz et al., 2000). Rectal temperature was maintained at 37°C by a heating lamp during the entire period of anesthesia.
Histological analysis. Neuronal damage was assessed by histological examination of brain sections at the level of dorsal hippocampus from animals killed at 7 d after global ischemia or sham operation. Animals were deeply anesthetized with pentobarbital (50 mg/kg, i.p.), and blood was collected by cardiac puncture for assay of plasma estradiol levels (see below). Animals were then fixed by transcardiac perfusion with 0.9% saline containing heparin (10 U/ml), followed by ice-cold 4% paraformaldehyde in PBS (0.1 m), pH 7.4. Brains were removed and immersed in fixative (4°C for 2 hr), transferred to PBS containing 30% sucrose (4°C for 48 hr), and then frozen. Coronal sections (15 μm) were cut with a cryotome and stained with toluidine blue. Hippocampal injury was assessed quantitatively by the grading scale of Pulsinelli and Brierley: 0, no neurons damaged; 1, a few (<30%) neurons damaged; 2, many (30–70%) neurons damaged; and 3, the majority of (>70%) neurons damaged (Pulsinelli et al., 1982). Neuronal damage scores from a minimum of four microscopic sections per animal were analyzed; comparisons among group means were made using an ANOVA, followed by Newman–Keuls test to determine significance and plotted as scatter graphs.
Serum estradiol assay. Tubes containing whole blood were placed on ice (20 min) and centrifuged (2000× rpm for 5 min). Serum was collected and stored at −20°C. Serum levels of estradiol were assessed by fluoro-immunoassay using the DELFIA estradiol assay (PerkinElmer Life Sciences, Wallac Oy, Turku, Finland). All samples were assayed in duplicate. The lower limit of detection was ∼13 pg/ml estradiol. Although this assay method is less sensitive than traditional radioimmunoassays, all serum samples fell within the range of the standard curve.
Immunocytochemistry. Protein expression was assessed by immunolabeling as described previously (Kokaia et al., 1998; Roux et al., 1999). Animals were deeply anesthetized with pentobarbital (50 mg/kg, i.p.) and perfused transcardially with 4% paraformaldehyde in phosphate buffer (0.1 m), pH 7.4, at 6 hr, 12 hr, 24 hr, 48 hr, 72 hr, and 7 d after ischemia or sham operation (n = 3 for each time point and treatment group). Brains were removed, post-fixed (2 hr at 4°C), frozen, and cut into sections (40 μm) in the coronal plane of the dorsal hippocampus (3.3–4.0 mm posterior from bregma) by cryotome. For single immunolabeling experiments, free-floating sections were blocked in 10% normal serum, 5% bovine serum albumin, and 0.01% saponin in PBS (2 hr at room temperature) and processed for immunolabeling with either of the following: (1) anti-caspase-3 p20, an antibody that recognizes activated caspase-3, the large fragment that results from cleavage of procaspase (rabbit polyclonal antibody, directed against the C terminus of human caspase-3 p20; 1:100; Cell SignalingTechnology Inc., Beverly, MA); or (2) anti-p75NTR (rabbit polyclonal antibody directed to the cytoplasmic domain of human p75; 1:4000; gift of Dr. Moses V. Chao, New York University School of Medicine, New York, NY) overnight at 4°C, followed by biotinylated goat anti-rabbit IgG (1:200; Vector Laboratories, Burlingame, CA). Sections were then incubated with avidin–peroxidase complex (ABC kit; 1 hr at room temperature; Vector Laboratories), followed by 3–3′-diaminobenzidine (Vector Laboratories).
For double-immunolabeling studies, sections were blocked for 2 hr and incubated (overnight at 4°C) with anti-p75NTR (1:2000) and one of the following: (1) anti-β-tubulin (mouse monoclonal antibody; 1:1000; Sigma, St. Louis, MO), a neuronal marker; (2) anti-glutamic acid decarboxylase (GAD) (mouse monoclonal antibody; 1:200; Chemicon, Temecula, CA), a marker for inhibitory interneurons; or (3) anti-glial fibrillary protein (GFAP) (mouse monoclonal antibody; 1:4000; Sigma), a marker for astrocytes. Sections were then incubated (1 hr at room temperature) with biotinylated goat anti-rabbit IgG (1:200) and Texas Red-conjugated horse anti-mouse IgG (1:200; Vector Laboratories), followed by fluorescein–avidin (1:200), dry-mounted, and coverslipped with ProLong (Molecular Probes, Eugene, OR) to reduce fluorescence quenching. For p75NTR and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) colabeling, sections were directly blocked for 2 hr after the TUNEL reaction, followed by an overnight incubation with p75NTR antibody and processed as above (avidin-conjugated Texas Red, 1:200). In some experiments, nuclei were stained during the washes using Hoechst 33342 according to the protocol of the manufacturer (Molecular Probes). Images were collected with a Bio-Rad (Richmond, CA) MRC 600 krypton–argon laser scanning confocal microscope. Settings were held constant for imaging of sections from control and experimental animals. To assess specificity of immunofluorescent probes, separate control sections were processed with non-immune rabbit, mouse, or goat IgG in place of primary antibody. These control sections showed no labeling.
Detection of DNA cleavage. TUNEL was performed using anin situ cell death detection kit as per the instructions of the manufacturer (Roche Molecular Biochemicals, Mannheim, Germany). The kit contains terminal deoxynucleotidyl transferase, which catalyzes polymerization of fluorescein dUTP to free 3′-OH DNA ends in a template-independent manner. TUNEL-positive cells are identified directly by fluorescence of incorporated dUTP. Positive cells in 16 fields sampled from the hippocampal CA1 subfields from two different gerbils (eight fields each) at 72 hr after global ischemia were scored for p75NTR immunoreactivity, TUNEL reactivity, and their colocalization.
Western blot analysis. For quantitation of protein abundance in the hippocampal CA1, animals were deeply anesthetized and killed by decapitation at 24, 48, and 72 hr after ischemia or sham operation. Hippocampi were quickly dissected out, and thick (1 mm) transverse slices were cut on a McIllwain tissue chopper starting at the dorsal end of the hippocampus. For biochemical analysis, the CA1 subfield was rapidly separated from the CA3-dentate gyrus by microdissection, placed in ice-cold PBS supplemented with the protease inhibitor phenylmethylsulfonyl fluoride (PMSF) (1 mm; Sigma), and stored at −70°C until use. Tissue samples were homogenized by sonication in 200 μl of 1 mmNaHNO3 buffer, pH 6.8, containing PMSF and lysed (overnight at 4°C) in Laemmli's sample buffer (0.025m Tris-HCI, 5% glycerol, 1% SDS, 0.5% PBS, 0.1m dithiothreitol, 2.5 mmβ-mercaptoethanol, 1 mm PMSF, and 0.5 mm NaHNO3 buffer, pH 6.8). Protein concentration of samples was measured using the BCA protein assay kit (Pierce, Rockford, IL). Samples were diluted in Laemmli's sample buffer to achieve the same final protein concentration, after which 20 μg samples were loaded onto 10% polyacrylamide mini-gels (Bio-Rad) and subjected to gel electrophoresis.
Protein bands were transferred to nitrocellulose membranes (Bio-Rad) in blotting buffer containing 0.192 m glycine and 20% methanol. After blocking for 30 min with 25 mm Tris-HCl buffer, pH 8.0, 125 mm NaCl, 0.1% Tween 20, and 4% skim milk, membranes were incubated (1 hr at room temperature) with anti-p75NTR (1:1000), followed by incubation (1 hr at room temperature) with horseradish peroxidase-conjugated anti-rabbit IgG (1:3000; Amersham Biosciences, Arlington Heights, IL). After reaction, membranes were treated with enhanced chemiluminescence reagents (ECL; Amersham Biosciences) and apposed to XAR-5 x-ray film (Eastman Kodak, Rochester, NY).
To quantitate protein abundance, bands on Western blots were analyzed with a Scan Jet 4-C computing densitometer using NIH Image 1.61 software. Optical densities (ODs) were normalized to OD values for the corresponding brain region of control gerbils on the same membranes to enable comparisons of band densities of immunoblots apposed to different films. Data were expressed as grand means ± SEMs of individual means from a minimum of three animals. Statistical significance was assessed by means of the Student's unpairedt test.
Glutamate receptor 2 in situ hybridization.[35S]UTP-labeled RNA probe directed against the AMPA receptor subunit glutamate receptor 2 (GluR2) cDNA was transcribed by incubation of the corresponding cDNA (1 hr at 37°C) with T7 polymerase in the presence of labeled and unlabeled nucleotides using a transcription kit (Stratagene, La Jolla, CA). Radiolabeled probe was purified by phenol–chloroform extraction. To examine the effect of estrogen on ischemia-induced downregulation of GluR2 mRNA in CA1, estrogen-treated and control gerbils were subjected to ischemia or sham operation, anesthetized with pentobarbital, and decapitated at 72 hr after ischemia or sham operation. mRNA expression was assessed byin situ hybridization on coronal sections of gerbil brain at the level of hippocampus as described previously (Pellegrini-Giampietro et al., 1992; Gorter et al., 1997). In brief, brains were rapidly removed, frozen by immersion in 2-methylbutane at −4°C, and stored at −70°C until sectioning. Coronal sections (18 μm) were cut on a cryotome and thaw mounted onto slides. After fixation with 4% paraformaldehyde in 10 mm PBS containing 5 mm MgC12 (15 min at 4°C), sections were rinsed in PBS, dehydrated in graded ethanols, and stored in 95% ethanol (4°C) until use. For in situhybridization, sections were acetylated, incubated with prehybridization solution (2 hr at 50°C), and hybridized by incubation with [35S]-labeled RNA probe (106 cpm/section, 1 ng/ml; overnight at 50°C). Sections were washed, treated with RNase A (20 μg/ml, 30 min at room temperature), and dehydrated in graded ethanols. Slides were apposed to Kodak XAR-5 film for 5 d.
For quantitation of mRNA expression, autoradiograms were analyzed with a Scan Jet 4-C computing densitometer using NIH Image 1.61 image analysis software. Films were scanned at 2000 dpi resolution, and images (∼1 × 106 pixels) were created for each section. Mean OD values in regions of maximal labeling in hippocampal subfields were averaged for two sections per animal, and film background was subtracted to give the mean OD value for an animal. OD values were expressed as grand means ± SDs of individual means from a minimum of three animals per time point. OD values for subfields of experimental animals were normalized to OD values for the corresponding subfield of control animals on the same film to enable comparisons of sections apposed to different films. Statistical significance was assessed by the Student's unpaired t test.
RESULTS
Estradiol affords neuroprotection against global ischemia-induced neuronal death in males
Gerbils are ideal experimental animals for studies of global ischemia in that they lack the posterior communicating arteries; thus, the relatively simple two-vessel occlusion model can be used to induce forebrain ischemia. Because delayed, selective ischemia-induced death of CA1 neurons has been characterized extensively in male gerbils, we chose to first examine the neuroprotective actions of estrogen in male gerbils. Transient, severe forebrain or global ischemia (5 min) in adult, male gerbils induced selective death of hippocampal CA1 pyramidal neurons (Fig. 1). Examination of toluidine blue-stained brain sections at the level of the hippocampus revealed no detectable cell death at 24, 48, or 72 hr after ischemia (data not shown). At 1 week after ischemia, there was virtually complete loss of neurons in the CA1 pyramidal cell layer (Fig. 1C,D). Of the few surviving neurons, some exhibited pyknotic nuclei, indicative of early neurodegeneration. The CA3 and dentate gyrus exhibited no detectable neuronal death as late as 7 d (Fig. 1C). These data are in confirmation of others (Kirino, 1982; Gorter et al., 1997).
Fig. 1.

Estrogen pretreatment affords neuroprotection against global ischemia-induced neuronal death in male gerbils. Toluidine blue staining of coronal brain sections at the level of the dorsal hippocampus at 7 d after reperfusion from control (n = 8; A, B) and experimental male gerbils subjected to global ischemia (BCCO, 5 min;n = 9; C, D) or to estradiol (0.36 mg pellet, s.c.; 60 d controlled time release), followed by global ischemia (BCCO, 5 min; n = 12;E, F). Global ischemia induced significant cell loss in the CA1 pyramidal cell layer; little or no cell loss was apparent in CA3 or dentate gyrus (C,D). Estradiol treatment for 14 d afforded nearly complete neuroprotection against ischemia-induced damage (E, F). Hippocampal injury was assessed quantitatively by the grading scale of Pulsinelli and Brierley: 0, no neurons damaged; 1, a few (<30%) neurons damaged; 2, many (30–70%) neurons damaged; and 3, the majority (>70%) of neurons damaged (Pulsinelli et al., 1982). Neuronal damage scores from a minimum of four microscopic sections per animal were analyzed; comparisons among group means were made using an ANOVA, followed by Newman–Keuls test and plotted as scatter graphs (G). Neuronal damage scores from two estrogen-treated gerbils with low plasma estradiol levels, indicating likely loss of the implanted pellet, are shown asdiamonds. Global ischemia was induced in adult male gerbils by BCCO as described in Materials and Methods. Scale bar: lower magnification, 400 μm; higher magnification, 40 μm.DG, Dentate gyrus; so,stratum oriens;sp,stratum pyramidale; sr, stratum radiatum.
To examine neuroprotective actions of estrogen, adult male gerbils were pretreated for 14 d by implantation of pellets containing placebo or 17β-estradiol and then subjected to global ischemia or sham operation. Plasma estradiol levels at the time of death were 16.9 ± 1.6 and 74.7 ± 6.1 pg/ml in the placebo and estradiol implant groups, respectively. Although we do not know how these serum estradiol levels compare with those in gonadally intact female gerbils, they are within the physiological range for hormone replacement in postmenopausal women. Pretreatment with estradiol did not detectably alter morphology or number of neurons in brains of control (sham-operated) animals (data not shown) but afforded significant neuroprotection against ischemia-induced neuronal death (Fig.1E–G). Altogether, three estrogen-treated males failed to show significant neuroprotection; of these, two had estradiol levels comparable with placebo-treated animals (13.9 and 19.1 pg/ml, respectively), presumably attributable to loss of pellets or failure of implanted pellets to release estradiol. These animals were eliminated from the calculation of the mean neuronal damage score. Estradiol levels of the third male were not assayed. We have now replicated these experiments in ovariectomized female gerbils receiving placebo or estradiol implants, and we find that estradiol pretreatment also affords robust neuroprotection in females (data not shown). Pretreatment with placebo did not detectably alter neuronal survival or morphology (data not shown). These findings suggest that estrogen pretreatment may alter expression or functional activity of one or more signaling proteins in the molecular cascade between insult and neuronal death.
Global ischemia increases activated caspase-3 in CA1 neurons
To examine molecular mechanisms underlying global ischemia-induced neuronal death, we focused on two proteins implicated in apoptotic death. First, we examined global ischemia-induced activation of caspase-3 in the hippocampus of male gerbils. Activated caspase-3 is generated by cleavage of a larger proenzyme caspase-3 p32, which is related to the interleukin-1β-converting enzyme. Global ischemia induced a marked increase in expression of activated caspase-3 in CA1 pyramidal neurons, evident at 6 hr (Fig.2B), 12 hr (Fig.2C), and 24 hr (Fig. 2D) after ischemia, as assessed by immunolabeling (n = 3 for each time point and treatment group). Global ischemia induced a marked increase in expression of both procaspase and activated caspase-3 p20 at 3, 6, 12, and 24 hr after global ischemia, as assessed by Western blot analysis probed with a caspase-3 antibody that recognizes the procaspase and its cleavage products (p < 0.01;n = 4 for each time point and treatment group; data not shown). These data are in corroboration of others (Chen et al., 1998b;Namura et al., 1998). The effect of ischemia on caspase-3 activation was specific to CA1 in that activated caspase-3 was not detected in the resistant CA3 or dentate gyrus at any times examined. Estrogen treatment markedly attenuated the ischemia-induced increase of activated caspase-3 in CA1 at 24 hr (Fig. 2E).
Fig. 2.
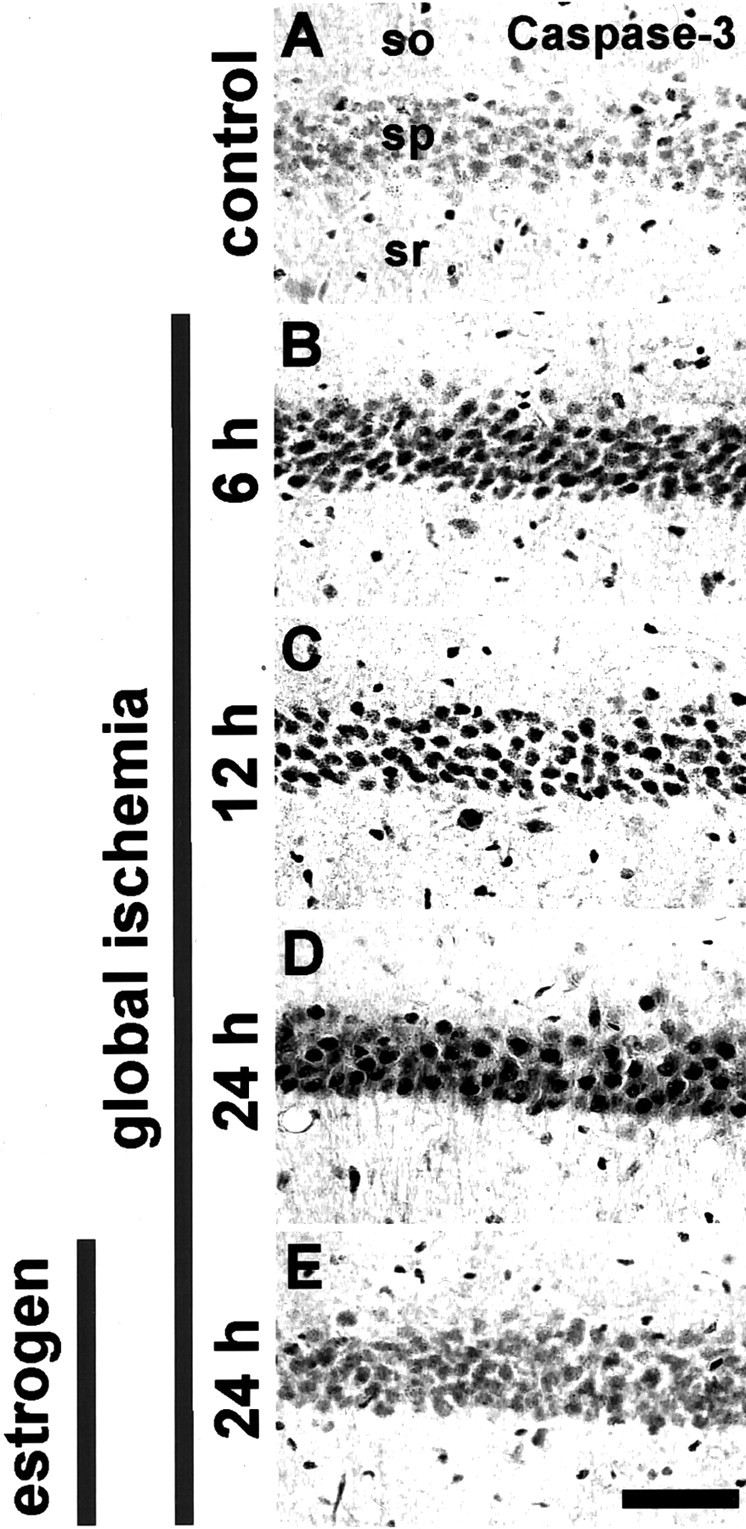
Estrogen attenuates ischemia-induced increase of activated caspase-3. Activated caspase-3 immunolabeling in the CA1 pyramidal cell layer in sections of brain from control (sham-operated) (A) and experimental animals subjected to global ischemia (B–E); male, adult Mongolian gerbils were implanted with pellets containing placebo (B,D) or 17β-estradiol (E) 14 d before induction of global ischemia by BCCO (5 min). Data are typical of three animals per treatment group. Global ischemia induced a marked increase in activated caspase-3 (assessed by immunolabeling with an antibody to activated caspase-3 p20), evident at 6, 12, and 24 hr primarily in the hippocampal CA1 pyramidal cell layer (B). Estradiol pretreatment markedly reduced the ischemia-induced increase in activated caspase-3 in the CA1 pyramidal layer at 24 hr (E). so, Stratum oriens; sp, stratum pyramidale; sr, stratum radiatum. Scale bar, 40 μm.
p75NTR protein expression increases in CA1 neurons after global ischemia
Second, we examined global ischemia-induced changes in expression of the neurotrophin receptor p75NTR in hippocampus. Recent evidence implicates p75NTR as a ligand-regulated proapoptotic receptor and suggests that elevated expression of neurotrophin receptors after ischemia could potentially promote cell death via p75NTR-dependent apoptotic mechanisms (for review, see Kaplan and Miller, 2000). In control brain, p75NTR content was very low throughout the pyramidal and granule cell layers of the hippocampus (Fig.3). Global ischemia induced a detectable increase in p75NTR abundance in CA1 at 48 hr (Fig. 3C,D), a greater increase at 72 hr (Fig.3E,F), and a pronounced increase at 7 d (Fig. 3G,H) after ischemia, as indicated by immunolabeling of brain sections at the level of the hippocampus (n = 3 per time point and treatment group). There was no change in p75NTR at 24 hr (data not shown). p75NTRimmunolabeling was unchanged in CA3 and dentate gyrus at all times examined.
Fig. 3.
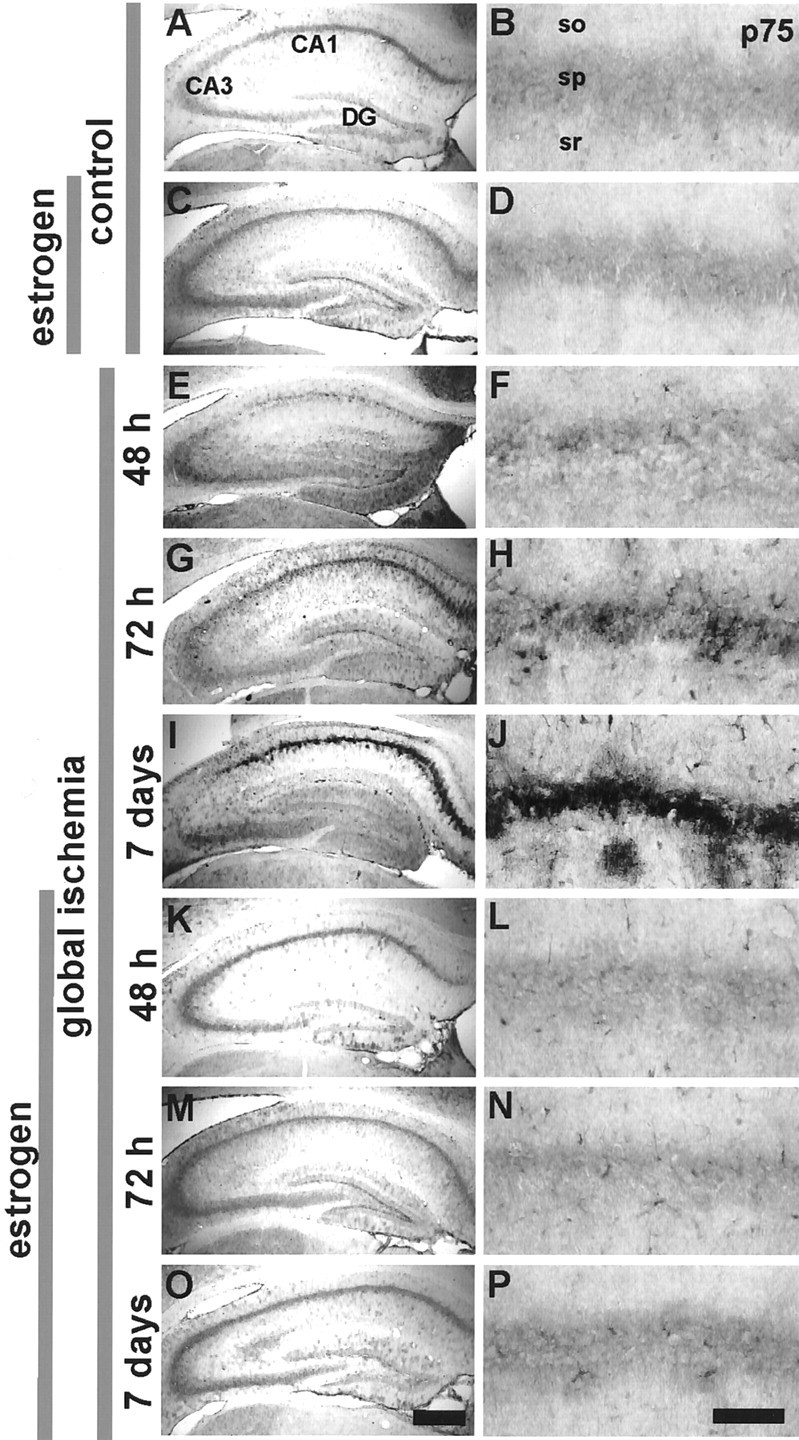
Estrogen attenuates ischemia-induced upregulation of p75NTR protein expression in CA1. p75NTR immunolabeling in the CA1 pyramidal cell layer in brain sections from control (A,B) and experimental animals at 48 hr (E,F), 72 hr (G,H), and 7 d (I,J) after global ischemia (BCCO, 5 min). Male, adult gerbils were implanted with pellets containing placebo (A, B, E–J) or 17β-estradiol 14 d before sham operation (C,D) or global ischemia (K–P). Data are typical of a minimum of three animals per time point and treatment group. Estradiol attenuated the ischemia-induced upregulation of p75NTR in the CA1 pyramidal layer. p75NTR immunolabeling was not altered in the CA3 pyramidal or dentate gyrus granule cell layer. so, Stratum oriens; sp, stratum pyramidale;sr, stratum radiatum. Scale bars: lower magnification, 400 μm; higher magnification, 40 μm.
To determine whether p75NTR immunolabeling is neuron specific, we examined the coincidence of p75NTR with β-tubulin (marker for neurons), GAD (marker for GABAergic interneurons), and GFAP (marker for astrocytes). p75NTR immunolabeling was essentially undetectable in control neurons (Fig.4B). At 72 hr after ischemia, p75NTR immunofluorescence was markedly increased in the hippocampal CA1 and distributed as small puncta around the cell somata and dendrites of some interneurons and a large number of pyramidal neurons (Fig. 4G). Double-label immunofluorescence showed that, at 72 hr, a moderate number (∼40%) of p75NTR-positive cells were GAD positive (Fig. 4H) and β-tubulin positive (Fig.4J). In contrast, essentially no p75NTR-positive cells were positive for GFAP (data not shown). These findings indicate that p75NTR is induced in neurons that die and also in GABAergic inhibitory interneurons of the CA1, which are known to survive the ischemic insult. At 7 d, p75NTR immunofluorescence was intense but finely granular (Fig. 4L) and did not entirely colocalize with the small number of surviving interneurons or pyramidal neurons (Fig. 4M,O) or with astrocytes labeled with GFAP (data not shown). Possible explanations are that the p75NTR immunofluorescence was in fine neuronal processes, phagocytosed by microglia or extracellular.
Fig. 4.
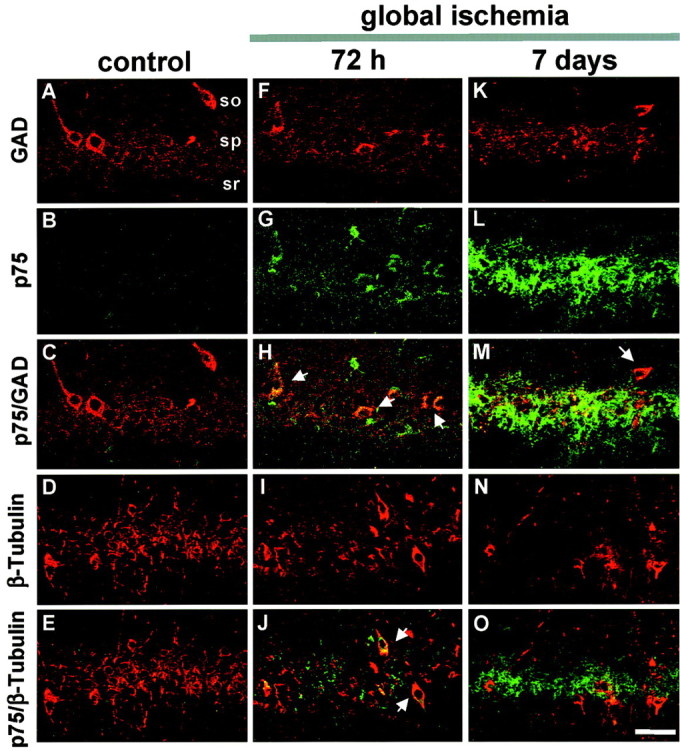
p75NTR colocalizes with GAD and β-tubulin in post-ischemic brain. Immunolabeling and merged images of p75NTR and GAD (C, H,M) or β-tubulin (E,J, O) in the CA1 pyramidal cell layer in brain sections from control (A–E) and experimental animals at 72 hr (F–J) and 7 d (K–O) after global ischemia. Data are typical of three control and three ischemic animals at each time point. At 72 hr, p75NTR immunolabeling was punctate over cell bodies within the pyramidal cell layer, suggestive of synaptic sites (G), and colocalized with GAD-positive (H) and a subpopulation of β-tubulin-positive (J) neurons. At 7 d, punctate p75NTR immunolabeling was reduced in GAD-positive (M) and β-tubulin-positive (O) cells; increased diffuse labeling was observed over the pyramidal cell layer, which was primarily devoid of neurons (L). p75NTRimmunolabeling was visualized in green and GAD or β-tubulin in red. so, Stratum oriens;sp, stratum pyramidale; sr, stratum radiatum. Scale bar, 40 μm.
To examine alterations in nuclear morphology and induction of p75NTR in neurons undergoing apoptosis, we performed triple labeling of p75NTR, TUNEL, and Hoechst 33342 (a nuclear stain) on sections of hippocampus from experimental and control animals 72 hr after global ischemia (Fig.5). In sections from control animals, p75NTR and TUNEL were undetectable in neurons in CA1 (Fig. 5B,C) or other subfields (data not shown). Global ischemia induced a marked increase in the incidence of TUNEL-positive and p75NTR-positive neurons in CA1, evident at 72 hr after ischemia (Fig.5E,F). There was a marked overlap between p75NTR immunoreactivity and TUNEL in CA1 neurons; 70.2% of p75NTR-positive CA1 neurons were TUNEL positive, and 68.5% of TUNEL-positive neurons were p75NTR positive (Fig.5E,F,H; Table1).
Fig. 5.

Estrogen attenuates ischemia-induced increase in p75NTR expression in CA1 neurons undergoing apoptosis. Triple-labeling of p75NTR(B, E, H,K), TUNEL (C, F,I, L), and Hoechst 33342 (A, D, G,J) in the CA1 pyramidal cell layer in sections of control (A–C) and experimental male gerbils pretreated with estradiol (J–L) or placebo (D–I), followed by global ischemia.G is the merge of an inset ofD and E at high magnification,H is the merge of an inset ofE and F at high magnification, andI is the merge of an inset ofD and F at high magnification. p75NTR immunolabeling was visualized inred (Texas Red), TUNEL reaction in green(fluorescein), and Hoechst-stained nuclei in blue. A high coincidence of p75NTR immunolabeling and TUNEL was observed. so, Stratum oriens; sp, stratum pyramidale; sr, stratum radiatum. Scale bars:first, second, and fourth rows, 40 μm; third row, 10 μm.
Table 1.
Proportion of TUNEL and p75NTR colabeled cells in gerbil hippocampal CA1 72 hr after global ischemia
| TUNEL positive | p75NTRpositive | p75NTR negative |
| 933 (100%) | 639 (68.5 ± 2.9%) | 294 (31.5 ± 2.9%) |
| p75NTR positive | TUNEL positive | TUNEL negative |
| 910 (100%) | 639 (70.2 ± 3.1%) | 271 (29.7 ± 3.1%) |
Three days after global ischemia, brain sections were assayed for nicked DNA using TUNEL, followed by p75NTRimmunocytochemistry. TUNEL-positive (fluorescein) and p75NTR-positive (Texas Red) cells were counted, and the overlap between the two groups was determined. Values represent the percentage of each group compared with either the total p75NTR or total TUNEL-positive cells ± SE of 16 separate fields derived from two different animals.
Estradiol attenuates ischemia-induced upregulation of p75NTR in CA1
We next examined the effects of estrogen administration on ischemia-induced expression of p75NTR in the hippocampal CA1. Estrogen did not detectably alter p75NTR expression in hippocampal subfields of sham-operated males, as assessed by immunolabeling (Figs.3C,D, 5B), but markedly attenuated the ischemia-induced increase in p75NTR in CA1, evident at 48 hr, 72 hr, and 7 d after ischemia (Figs.3K–P, 5K).
Global ischemia increases p75NTR protein abundance in CA1
To quantitate ischemia-induced alterations in p75NTR protein abundance, we performed Western blot analysis. p75NTR expression was low but detectable in protein samples isolated from the CA1 of control (placebo-treated, sham-operated) male gerbils (Fig.6). Global ischemia markedly increased p75NTR abundance in CA1. At 48 hr after ischemia, p75NTR abundance in CA1 was increased to 140 ± 18% of the control value (p < 0.05 for ischemic vs control;n = 4); at 72 hr after ischemia, p75NTR abundance in CA1 was increased to 170 ± 15% of the control value (p < 0.01 for ischemic vs control; n = 4). Estrogen alone did not detectably alter p75NTR abundance in CA1 of control (sham-operated) animals but prevented the ischemia-induced elevation in p75NTR observed in samples of post-ischemic CA1 at 72 hr (p < 0.01;n = 4) (Fig. 6).
Fig. 6.

Estrogen attenuates ischemia-induced increase in p75NTR abundance in the hippocampal CA1. Film autoradiograms of representative Western blots probed with an antibody to p75NTR (top) and mean band densities (bottom) for protein samples extracted from the microdissected hippocampal CA1 of placebo- and estradiol-pretreated male gerbils subjected to sham operation (control) or at 24, 48, and 72 hr after global ischemia. Band densities are normalized to that of control gerbils. Data are means of four control and four experimental animals at each time point. Global ischemia induced an upregulation in p75NTR protein abundance at 48 hr (p < 0.05) and 72 hr (p < 0.01) relative to control. Estrogen pretreatment attenuated ischemia-induced upregulation of p75NTR protein levels in the CA1 pyramidal layer.
Estradiol does not alter ischemia-induced downregulation of GluR2 mRNA in CA1
A mechanism implicated in global ischemia-induced neuronal death is suppression of the AMPA receptor GluR2 subunit, leading to expression of Ca2+-permeable AMPA receptors, Ca2+ influx, and excitotoxicity at CA1 synapses (for review, see Tanaka et al., 2000). To examine the effect of estradiol on global ischemia-induced downregulation of GluR2, we performed in situ hybridization with an RNA probe directed to GluR2 mRNA. In sections of control (placebo-treated, sham-operated) brain, GluR2 mRNA expression was intense in the pyramidal cell layers of CA1 and CA3 and in the granule cell layer of the dentate gyrus (Fig. 7). In animals subjected to global ischemia, GluR2 mRNA expression was dramatically reduced to 49.4 ± 12.1% of control in the CA1 pyramidal cell layer at 72 hr, a time before onset of neuronal death (n = 5; p < 0.01) (Fig. 7). GluR2 mRNA expression was not significantly altered by ischemia in CA3 and dentate gyrus (Fig. 7). These data are in confirmation of Pellegrini-Giampietro et al. (1992) and Gorter et al. (1997).
Fig. 7.

Estradiol does not attenuate ischemia-induced decrease in GluR2 mRNA expression. Representative in situ hybridization for GluR2 mRNA expression (top) and mean optical density values (bottom) for the CA1 pyramidal cell layer of control (placebo-treated, sham-operated) animals (far left) and experimental animals subjected to global ischemia (middle left), estradiol treatment, followed by global ischemia (middle right), or to estradiol treatment, followed by sham operation (far right) at 72 hr after reperfusion. Mean optical density values for experimental animals were normalized to the mean value for control gerbils. The relative abundance of GluR2 mRNA did not differ significantly in placebo-treated (decline to 49.46 ± 12.17% of control) versus estradiol-treated (decline to 35.93 ± 8.43% of control) ischemic animals at 72 hr. Estradiol did not detectably alter GluR2 mRNA expression in CA1 pyramidal neurons of control animals. n.s., Not significant.
We next examined the effect of estradiol pretreatment on ischemia-induced downregulation of GluR2 mRNA expression in the hippocampal CA1. Estradiol pretreatment did not significantly alter GluR2 mRNA expression in the CA1 of sham-operated animals (Fig. 7, fourth autoradiogram, fourth bar) or the ischemia-induced downregulation of GluR2 mRNA at 72 hr (reduction to 35.9 ± 8.4% of control in estrogen-treated animals subjected to global ischemia (n = 6; p< 0.01) (Fig. 7, third autoradiogram,third bar). This finding indicates that the actions of estrogen did not interfere with post-ischemic changes in glutamate receptor expression.
DISCUSSION
The present study demonstrates for the first time that long-term estrogen at levels considered physiological for hormone replacement in postmenopausal women affords robust neuroprotection against global ischemia-induced neuronal damage in the hippocampal CA1 of male gerbils. Findings from the study also provide new insight into possible molecular mechanisms underlying estrogen-induced neuroprotection. Global ischemia induced a marked increase in expression of activated caspase-3 (the mammalian homolog of Caenorhabditis elegans ced-3), terminator protein implicated in the execution step of apoptosis (Cohen, 1997; Nicholson and Thornberry, 1997); enhanced caspase-3 activation was evident by 6 hr in CA1 pyramidal neurons, as assessed by immunolabeling. Ischemia also induced a marked increase in the proapoptotic neurotrophin receptor p75NTR; this change was significant at 48 hr in CA1 pyramidal neurons, as assessed by immunolabeling and Western blot analysis. The high incidence of p75NTR expression in TUNEL-positive cells in CA1 indicates that p75NTR expression occurs in post-ischemic neurons undergoing apoptosis. Global ischemia also induced a marked downregulation of mRNA encoding the AMPA receptor GluR2 subunit in CA1, suggesting that necrotic, as well as apoptotic, mechanisms are activated after ischemia. Activated caspase-3 and p75NTR protein expression, as well as GluR2 mRNA expression, were not significantly changed in CA3 and dentate gyrus at all times examined, indicating that ischemia-induced changes in proapoptotic proteins and GluR2 mRNA are cell specific.
Exogenous estrogen attenuated the ischemia-induced increases in activated caspase-3 and p75NTR in post-ischemic CA1 pyramidal neurons. These findings suggest that estrogen affords neuroprotection against global ischemia-induced death at least in part by intervening at the level of apoptotic signaling cascades in neurons otherwise “destined to die.” Studies involving focal ischemia in rats indicate that estrogen promotes survival of cortical neurons by upregulation of Bcl-2, a survival factor that can block both necrotic and apoptotic cell death (Dubal et al., 1999). Bcl-2 acts upstream to prevent the activation of caspases, inhibit free radical formation, promote calcium sequestration, and block the proapoptotic actions of other members of the Bcl-2 family, including Bax and Bad (Bredesen, 1995; Merry and Korsmeyer, 1997). In contrast, estrogen did not block ischemia-induced downregulation of GluR2 mRNA expression in CA1. The finding in the present study that estrogen at physiological concentrations affords robust neuroprotection, but does not prevent GluR2 downregulation, together with our finding thatN-naphthyl-acetyl-spermine, a selective blocker of Ca2+-permeable AMPA receptors affords robust protection in the same model (M. Noh, H. Yokota, M. V. L. Bennett, and R. S. Zukin, personal communication), implicates involvement of multiple signaling cascades in global ischemia-induced neuronal death.
Caspase-3 activation precedes induction of p75NTR
Our finding that caspase-3 induction and activation occurs 2–3 d before the onset of histologically detectable neuronal death is in confirmation of findings of others (Chen et al., 1998b; Namura et al., 1998) and suggests that neurons become “committed” to die early in the post-ischemic period. The importance of early caspase-3 activation in the delayed neurodegeneration after global ischemia is underscored by the finding that z-DEVD-FMK, a selective caspase-3 inhibitor, is neuroprotective if administered at the time of ischemia but not at 24, 48, or 66 hr after ischemia (Chen et al., 1998b) (our unpublished observations). Thus, early caspase activation is essential to neuronal death. Why then the long delay before histologically detectable cell death? During activation, caspase-3 promotes neuronal death by cleaving nuclear and cytosolic proteins, most notably poly (ADP-ribose) polymerase (an enzyme involved in DNA repair, genome surveillance, and integrity, predominantly in response to environmental stress), laminin cleaving enzyme Mch2α, DNA-dependent protein kinase, and DNA fragmentation factor (Nicholson and Thornberry, 1997). The precise timing of these events remains to be established.
Our finding that caspase-3 activation precedes p75NTR induction by 1–2 d indicates that ligand-bound p75NTR does not play a causal role in caspase activation. Rather, our observation is consistent with findings that the caspase-3 cascade can be initiated by a death receptor-independent pathway. Recent studies indicate that the terminator caspase apoptotic signaling cascade can be initiated either by ligand-bound death receptors that recruit caspase proenzymes via adaptor proteins or more directly by a protein signaling complex or apoptosome, which forms during release of cytochrome c into the cytoplasm (for review, see Cryns and Yuan, 1998). The apoptosome is comprised of cytochrome c, (d)ATP, the mammalian CED-4 homolog Apaf-1, and caspase-9 (Liu et al., 1996; Li et al., 1997; Zou et al., 1997) and enables cytochrome c to “jump-start” the self-amplifying caspase cascade. Although ligand-activated p75NTR does activate the caspase cascade in post-ischemic neurons, it may contribute to apoptotic death via other downstream signaling cascades. p75NTR acts via its cytoplasmic juxtamembrane region (Coulson et al., 2000a) to promote apoptotic death cascades, including generation of ceramide, activation and translocation of NF-κB from the cytoplasm to the nucleus, and enhancement of Jun kinase activity (Cryns and Yuan, 1998; Coulson et al. 2000b). The finding that caspase-3 activation precedes p75NTR induction is novel and, we believe, important to our understanding of molecular mechanisms that underlie global ischemia-induced cell death.
Apoptosis versus necrosis in global ischemia-induced cell death
The relative contributions of apoptotic and necrotic mechanisms to global ischemia-induced cell death remain controversial (Choi, 1996;MacManus and Buchan, 2000; Yamashima, 2000; Graham and Chen, 2001). Ultrastructural studies indicate that ischemia induces many of the hallmarks of necrotic cell death in CA1 neurons, including early proliferation of endoplasmic reticulum (Kirino, 1982; Kirino and Sano, 1984; Deshpande et al., 1992), disaggregation of polyribosomes (Kirino and Sano, 1984; Deshpande et al., 1992), selective swelling of dendrites (Johansen et al., 1984), and dilation of organelles and intranuclear vacuoles (Colbourne et al., 1999), but fail to detect apoptotic bodies containing chromatin, a critical hallmark of apoptosis (Colbourne et al., 1999). Strong evidence in support for apoptosis, defined as activation of specific intracellular signaling cascades that result in cellular suicide (for review, see Banasiak et al., 2000), comes from molecular studies that indicate activation of death receptors and terminator proteins such as caspase-3 (Chen et al., 1998b; Namura et al., 1998; present study) and p75NTR (Lee et al., 1995; Bagum et al., 2001; present study) and of TUNEL, a marker for apoptotic cell death (Bagum et al., 2001; present study).
Neuroprotection by estrogen in experimental models of stroke
The importance of postmenopausal estrogen replacement therapy for protection against the neuronal death induced by global ischemia associated with cardiac arrest or by stroke remains controversial. A critical issue is whether long-term estrogen exposure and/or previous availability alters the severity and/or duration of ischemic insults. A number of studies report that estrogen affords neuroprotection in experimental models of stroke, although the effectiveness of long-term estrogen replacement at levels used in estrogen replacement therapy in humans is less clear (for review, see Green and Simpkins, 2000; Hurn and Macrae, 2000; Roof and Hall, 2000; Wise et al., 2001). Acute exogenous estrogen at physiologic levels protects against focal ischemia-induced cortical and striatal injury in estrogen-deprived female (Alkayed et al., 1998; Dubal et al., 1998; Rusa et al., 1999;Hurn and Macrae, 2000; Dubal and Wise, 2001) and male (Toung et al., 1998; Culmsee et al., 1999; Dubal et al., 2001) rats and female mice subjected to cerebral artery occlusion but was ineffective against hippocampal injury (Dubal et al., 2001). Acute exogenous estrogen at levels outside the physiological range has been reported to protect against global ischemia-induced hippocampal injury (Sudo et al., 1997;Chen et al., 1998a) and to improve behavioral outcome after ischemia (Kondo et al., 1997) in male gerbils. Our findings extend those of previous studies in that we show for the first time that long-term estrogen administration at levels commonly used for hormone replacement in postmenopausal women affords robust protection against ischemia-induced hippocampal injury and that neuroprotection by estrogen is associated with block of caspase-3 activation and p75NTR upregulation.
Is neuroprotection by estrogen mediated by estrogen receptors?
The cellular targets that mediate actions of estrogen on hippocampal neurons under physiological and pathological conditions are as yet unclear. Estrogens regulate the expression of a number of target genes by binding the estrogen receptors ER-α and -β, which function as ligand-activated transcription factors. Estrogen receptors are expressed in the hippocampus, in which they are thought to subserve a number of functions, including regulation of spine density (Murphy and Segal, 1996; Pozzo-Miller et al., 1999), synapse number (Woolley and McEwen, 1994), and NMDA receptor NR1 subunit expression (Gazzaley et al., 1996). Studies involving mice with targeted deletions in the ER-α or ER-β gene indicate that ER-α is critical for estrogen protection against neuronal injury in a focal ischemia model (Dubal et al., 2001). In addition to direct actions on hippocampal neurons, it has been suggested that estrogen can affect hippocampal neurons indirectly via estrogen receptors on basal forebrain cholinergic neurons, which innervate the hippocampal CA1 (Toran-Allerand et al., 1992) or via a receptor-independent anti-oxidative mechanism to inhibit oxygen radical-induced lipid peroxidation (Culmsee et al., 1999).
Conclusions
In conclusion, the present study shows that estradiol treatment within the physiological range for hormone replacement in postmenopausal women affords robust neuroprotection against global ischemia-induced CA1 injury in male gerbils. The molecular mechanisms underlying estrogen action involve intervention at the level of apoptotic signaling cascades, including caspase-3 activation and induction of p75NTR. Our findings provide strong evidence that estrogen replacement therapy may be clinically useful for ameliorating neuronal death arising after cardiac arrest or cardiac surgery.
Footnotes
This work was supported by National Institutes of Health Grants NS 20752, NS 31282 (to R.S.Z.), and MH 41414 (to A.M.E.), a grant from the F. M. Kirby Foundation, and a grant from Wyeth-Ayerst. M.V.L.B. is the Sylvia and Robert S. Olnick Professor of Neuroscience. We thank Roodland Regis, Judy Wong, and Tovaghgol Adel for technical support and acknowledge Drs. Sonja Y. Grooms and Nanette Santoro and the Analytical Imaging Facility of the Albert Einstein College of Medicine (Michael Cammer, Director).
Correspondence should be addressed to Dr. R. Suzanne Zukin, Department of Neuroscience, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461. E-mail: zukin@aecom.yu.edu.
REFERENCES
- 1.Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998;29:159–165. doi: 10.1161/01.str.29.1.159. [DOI] [PubMed] [Google Scholar]
- 2.Bagum MA, Miyamoto O, Toyoshima T, Masada T, Nagahata S, Itano T. The contribution of low affinity NGF receptor (p75NGFR) to delayed neuronal death after ischemia in the gerbil hippocampus. Acta Med Okayama. 2001;55:19–24. doi: 10.18926/AMO/32030. [DOI] [PubMed] [Google Scholar]
- 3.Banasiak KJ, Xia Y, Haddad GG. Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog Neurobiol. 2000;62:215–249. doi: 10.1016/s0301-0082(00)00011-3. [DOI] [PubMed] [Google Scholar]
- 4.Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Adachi N, Liu K, Arai T. The effects of 17beta-estradiol on ischemia-induced neuronal damage in the gerbil hippocampus. Neuroscience. 1998a;87:817–822. doi: 10.1016/s0306-4522(98)00198-5. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998b;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6:667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- 8.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colbourne F, Sutherland GR, Auer RN. Electron microscopic evidence against apoptosis as the mechanism of neuronal death in global ischemia. J Neurosci. 1999;19:4200–4210. doi: 10.1523/JNEUROSCI.19-11-04200.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coulson EJ, Reid K, Baca M, Shipham KA, Hulett SM, Kilpatrick TJ, Bartlett PF. Chopper, a new death domain of the p75 neurotrophin receptor that mediates rapid neuronal cell death. J Biol Chem. 2000a;275:30537–30545. doi: 10.1074/jbc.M005214200. [DOI] [PubMed] [Google Scholar]
- 11.Coulson EJ, Reid K, Murray SS, Cheema SS, Bartlett PF. Role of neurotrophin receptor p75NTR in mediating neuronal cell death following injury. Clin Exp Pharmacol Physiol. 2000b;27:537–541. doi: 10.1046/j.1440-1681.2000.03295.x. [DOI] [PubMed] [Google Scholar]
- 12.Cryns V, Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- 13.Culmsee C, Vedder H, Ravati A, Junker V, Otto D, Ahlemeyer B, Krieg JC, Krieglstein J. Neuroprotection by estrogens in a mouse model of focal cerebral ischemia and in cultured neurons: evidence for a receptor-independent antioxidative mechanism. J Cereb Blood Flow Metab. 1999;19:1263–1269. doi: 10.1097/00004647-199911000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Deshpande J, Bergstedt K, Linden T, Kalimo H, Wieloch T. Ultrastructural changes in the hippocampal CA1 region following transient cerebral ischemia: evidence against programmed cell death. Exp Brain Res. 1992;88:91–105. doi: 10.1007/BF02259131. [DOI] [PubMed] [Google Scholar]
- 15.Dubal DB, Wise PM. Neuroprotective effects of estradiol in middle-aged female rats. Endocrinology. 2001;142:43–48. doi: 10.1210/endo.142.1.7911. [DOI] [PubMed] [Google Scholar]
- 16.Dubal DB, Kashon ML, Pettigrew LC, Ren JM, Finklestein SP, Rau SW, Wise PM. Estradiol protects against ischemic injury. J Cereb Blood Flow Metab. 1998;18:1253–1258. doi: 10.1097/00004647-199811000-00012. [DOI] [PubMed] [Google Scholar]
- 17.Dubal DB, Shughrue PJ, Wilson ME, Merchenthaler I, Wise PM. Estradiol modulates bcl-2 in cerebral ischemia: a potential role for estrogen receptors. J Neurosci. 1999;19:6385–6393. doi: 10.1523/JNEUROSCI.19-15-06385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci USA. 2001;98:1952–1957. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gail MH, Costantino JP, Bryant J, Croyle R, Freedman L, Helzlsouer K, Vogel V. Weighing the risks and benefits of tamoxifen treatment for preventing breast cancer. J Natl Cancer Inst. 1999;91:1829–1846. doi: 10.1093/jnci/91.21.1829. [DOI] [PubMed] [Google Scholar]
- 20.Gazzaley AH, Weiland NG, McEwen BS, Morrison JH. Differential regulation of NMDAR1 mRNA and protein by estradiol in the rat hippocampus. J Neurosci. 1996;16:6830–6838. doi: 10.1523/JNEUROSCI.16-21-06830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibbs RB. Effects of estrogen on basal forebrain cholinergic neurons vary as a function of dose and duration of treatment. Brain Res. 1997;757:10–16. doi: 10.1016/s0006-8993(96)01432-1. [DOI] [PubMed] [Google Scholar]
- 22.Gibbs RB, Pfaff DW. Effects of estrogen and fimbria/fornix transection on p75NGFR and ChAT expression in the medial septum and diagonal band of Broca. Exp Neurol. 1992;116:23–39. doi: 10.1016/0014-4886(92)90173-n. [DOI] [PubMed] [Google Scholar]
- 23.Gorter JA, Petrozzino JJ, Aronica EM, Rosenbaum DM, Opitz T, Bennett MV, Connor JA, Zukin RS. Global ischemia induces downregulation of Glur2 mRNA and increases AMPA receptor-mediated Ca2+ influx in hippocampal CA1 neurons of gerbil. J Neurosci. 1997;17:6179–6188. doi: 10.1523/JNEUROSCI.17-16-06179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graham SH, Chen J. Programmed cell death in cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:99–109. doi: 10.1097/00004647-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 25.Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int J Dev Neurosci. 2000;18:347–358. doi: 10.1016/s0736-5748(00)00017-4. [DOI] [PubMed] [Google Scholar]
- 26.Hall ED, Pazara KE, Linseman KL. Sex differences in postischemic neuronal necrosis in gerbils. J Cereb Blood Flow Metab. 1991;11:292–298. doi: 10.1038/jcbfm.1991.61. [DOI] [PubMed] [Google Scholar]
- 27.Henderson VW, Watt L, Buckwalter JG. Cognitive skills associated with estrogen replacement in women with Alzheimer's disease. Psychoneuroendocrinology. 1996;21:421–430. doi: 10.1016/0306-4530(95)00060-7. [DOI] [PubMed] [Google Scholar]
- 28.Hurn PD, Macrae IM. Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab. 2000;20:631–652. doi: 10.1097/00004647-200004000-00001. [DOI] [PubMed] [Google Scholar]
- 29.Johansen FF, Jorgensen MB, Ekstromv LD, Diemer NH. Selective dendrite damage in hippocampal CA1 stratum radiatum with unchanged axon ultrastructure and glutamate uptake after transient cerebral ischaemia in the rat. Brain Res. 1984;291:373–377. doi: 10.1016/0006-8993(84)91272-1. [DOI] [PubMed] [Google Scholar]
- 30.Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Cell Biol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 31. Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, Bacal C, Lingle DD, Metter E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer's disease: the Baltimore Longitudinal Study of Aging. Neurology 48 1997. 1517 1521 [Erratum (1998) 51:654] [DOI] [PubMed] [Google Scholar]
- 32.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 33.Kirino T, Sano K. Fine structural nature of delayed neuronal death following ischemia in the gerbil hippocampus. Acta Neuropathol (Berl) 1984;62:209–218. doi: 10.1007/BF00691854. [DOI] [PubMed] [Google Scholar]
- 34.Kokaia Z, Andsberg G, Martinez-Serrano A, Lindvall O. Focal cerebral ischemia in rats induces expression of p75 neurotrophin receptor in resistant striatal cholinergic neurons. Neuroscience. 1998;84:1113–1125. doi: 10.1016/s0306-4522(97)00579-4. [DOI] [PubMed] [Google Scholar]
- 35.Kondo Y, Suzuki K, Sakuma Y. Estrogen alleviates cognitive dysfunction following transient brain ischemia in ovariectomized gerbils. Neurosci Lett. 1997;238:45–48. doi: 10.1016/s0304-3940(97)00847-1. [DOI] [PubMed] [Google Scholar]
- 36.Lee TH, Abe K, Kogure K, Itoyama Y. Expressions of nerve growth factor and p75 low affinity receptor after transient forebrain ischemia in gerbil hippocampal CA1 neurons. J Neurosci Res. 1995;41:684–695. doi: 10.1002/jnr.490410515. [DOI] [PubMed] [Google Scholar]
- 37.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 38.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 39.MacManus JP, Buchan AM. Apoptosis after experimental stroke: fact or fashion? J Neurotrauma. 2000;17:899–914. doi: 10.1089/neu.2000.17.899. [DOI] [PubMed] [Google Scholar]
- 40.McMillan PJ, Singer CA, Dorsa DM. The effects of ovariectomy and estrogen replacement on trkA and choline acetyltransferase mRNA expression in the basal forebrain of the adult female Sprague Dawley rat. J Neurosci. 1996;16:1860–1865. doi: 10.1523/JNEUROSCI.16-05-01860.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merry DE, Korsmeyer SJ. Bcl-2 gene family in the nervous system. Annu Rev Neurosci. 1997;20:245–267. doi: 10.1146/annurev.neuro.20.1.245. [DOI] [PubMed] [Google Scholar]
- 42.Miranda RC, Sohrabji F, Toran-Allerand CD. Neuronal colocalization of mRNAs for neurotrophins and their receptors in the developing central nervous system suggests a potential for autocrine interactions. Proc Natl Acad Sci USA. 1993;90:6439–6443. doi: 10.1073/pnas.90.14.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moss RL, Gu Q. Estrogen: mechanisms for a rapid action in CA1 hippocampal neurons. Steroids. 1999;64:14–21. doi: 10.1016/s0039-128x(98)00092-0. [DOI] [PubMed] [Google Scholar]
- 44.Murphy DD, Segal M. Regulation of dendritic spine density in cultured rat hippocampal neurons by steroid hormones. J Neurosci. 1996;16:4059–4068. doi: 10.1523/JNEUROSCI.16-13-04059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 47.Oguro K, Oguro N, Kojima T, Grooms SY, Calderone A, Zheng X, Bennett MV, Zukin RS. Knockdown of AMPA receptor GluR2 expression causes delayed neurodegeneration and increases damage by sublethal ischemia in hippocampal CA1 and CA3 neurons. J Neurosci. 1999;19:9218–9227. doi: 10.1523/JNEUROSCI.19-21-09218.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Opitz T, Grooms S, Bennett MVL, Zukin RS. Remodeling of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit composition in hippocampal neurons after global ischemia. Proc Natl Acad Sci USA. 2000;97:13360–13365. doi: 10.1073/pnas.97.24.13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pellegrini-Giampietro DE, Zukin RS, Bennett MV, Cho S, Pulsinelli WA. Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc Natl Acad Sci USA 89 1992. 10499 10503[Erratum (1993) 90:780]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pozzo-Miller LD, Inoue T, Murphy DD. Estradiol increases spine density and NMDA-dependent Ca2+ transients in spines of CA1 pyramidal neurons from hippocampal slices. J Neurophysiol. 1999;81:1404–1411. doi: 10.1152/jn.1999.81.3.1404. [DOI] [PubMed] [Google Scholar]
- 51.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 52.Roof RL, Hall ED. Gender differences in acute CNS trauma and stroke: neuroprotective effects of estrogen and progesterone. J Neurotrauma. 2000;17:367–388. doi: 10.1089/neu.2000.17.367. [DOI] [PubMed] [Google Scholar]
- 53.Roux PP, Colicos MA, Barker PA, Kennedy TE. p75 neurotrophin receptor expression is induced in apoptotic neurons after seizure. J Neurosci. 1999;19:6887–6896. doi: 10.1523/JNEUROSCI.19-16-06887.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rusa R, Alkayed NJ, Crain BJ, Traystman RJ, Kimes AS, London ED, Klaus JA, Hurn PD. 17beta-estradiol reduces stroke injury in estrogen-deficient female animals. Stroke. 1999;30:1665–1670. doi: 10.1161/01.str.30.8.1665. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt R, Fazekas F, Reinhart B, Kapeller P, Fazekas G, Offenbacher H, Eber B, Schumacher M, Freidl W. Estrogen replacement therapy in older women: a neuropsychological and brain MRI study. J Am Geriatr Soc. 1996;44:1307–1313. doi: 10.1111/j.1532-5415.1996.tb01400.x. [DOI] [PubMed] [Google Scholar]
- 56.Shughrue PJ, Merchenthaler I. Evidence for novel estrogen binding sites in the rat hippocampus. Neuroscience. 2000;99:605–612. doi: 10.1016/s0306-4522(00)00242-6. [DOI] [PubMed] [Google Scholar]
- 57.Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol. 1997;388:507–525. doi: 10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 58.Sohrabji F, Greene LA, Miranda RC, Toran-Allerand CD. Reciprocal regulation of estrogen and NGF receptors by their ligands in PC12 cells. J Neurobiol. 1994a;25:974–988. doi: 10.1002/neu.480250807. [DOI] [PubMed] [Google Scholar]
- 59.Sohrabji F, Miranda RC, Toran-Allerand CD. Estrogen differentially regulates estrogen and nerve growth factor receptor mRNAs in adult sensory neurons. J Neurosci. 1994b;14:459–471. doi: 10.1523/JNEUROSCI.14-02-00459.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sudo S, Wen TC, Desaki J, Matsuda S, Tanaka J, Arai T, Maeda N, Sakanaka M. Beta-estradiol protects hippocampal CA1 neurons against transient forebrain ischemia in gerbil. Neurosci Res. 1997;29:345–354. doi: 10.1016/s0168-0102(97)00106-5. [DOI] [PubMed] [Google Scholar]
- 61.Tanaka H, Grooms SY, Bennett MVL, Zukin RS. The AMPAR subunit GluR2: still front, center-stage. Brain Res. 2000;886:190–207. doi: 10.1016/s0006-8993(00)02951-6. [DOI] [PubMed] [Google Scholar]
- 62.Tang MX, Jacobs D, Stern Y, Marder K, Schofield P, Gurland B, Andrews H, Mayeux R. Effect of oestrogen during menopause on risk and age at onset of Alzheimer's disease. Lancet. 1996;348:429–432. doi: 10.1016/S0140-6736(96)03356-9. [DOI] [PubMed] [Google Scholar]
- 63.Toran-Allerand CD, Miranda RC, Bentham WD, Sohrabji F, Brown TJ, Hochberg RB, MacLusky NJ. Estrogen receptors colocalize with low-affinity nerve growth factor receptors in cholinergic neurons of the basal forebrain. Proc Natl Acad Sci USA. 1992;89:4668–4672. doi: 10.1073/pnas.89.10.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Toran-Allerand CD, Singh M, Setalo GJ. Novel mechanisms of estrogen action in the brain: new players in an old story. Front Neuroendocrinol. 1999;20:97–121. doi: 10.1006/frne.1999.0177. [DOI] [PubMed] [Google Scholar]
- 65.Toung TJ, Traystman RJ, Hurn PD. Estrogen-mediated neuroprotection after experimental stroke in male rats. Stroke. 1998;29:1666–1670. doi: 10.1161/01.str.29.8.1666. [DOI] [PubMed] [Google Scholar]
- 66.Weiland NG, Orikasa C, Hayashi S, McEwen BS. Distribution and hormone regulation of estrogen receptor immunoreactive cells in the hippocampus of male and female rats. J Comp Neurol. 1997;388:603–612. doi: 10.1002/(sici)1096-9861(19971201)388:4<603::aid-cne8>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 67.Wise PM, Dubal DB, Wilson ME, Rau SW, Liu Y. Estrogens trophic and protective factors in the adult brain. Front Neuroendocrinol. 2001;22:33–66. doi: 10.1006/frne.2000.0207. [DOI] [PubMed] [Google Scholar]
- 68.Woolley CS, McEwen BS. Estradiol regulates hippocampal dendritic spine density via an N-methyl-d-aspartate receptor-dependent mechanism. J Neurosci. 1994;14:7680–7687. doi: 10.1523/JNEUROSCI.14-12-07680.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamashima T. Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog Neurobiol. 2000;62:273–295. doi: 10.1016/s0301-0082(00)00006-x. [DOI] [PubMed] [Google Scholar]
- 70.Yano H, Chao MV. Neurotrophin receptor structure and interactions. Pharm Acta Helv. 2000;74:253–260. doi: 10.1016/s0031-6865(99)00036-9. [DOI] [PubMed] [Google Scholar]
- 71.Zhang YQ, Shi J, Rajakumar G, Day AL, Simpkins JW. Effects of gender and estradiol treatment on focal brain ischemia. Brain Res. 1998;784:321–324. doi: 10.1016/s0006-8993(97)00502-7. [DOI] [PubMed] [Google Scholar]
- 72.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]