

Abstract
Hereditary hyperekplexia is caused by disinhibition of motoneurons resulting from mutations in the ionotropic receptor for the inhibitory neurotransmitter glycine (GlyR). To study the pathomechanisms involvedin vivo, we generated and analyzed transgenic mice expressing the hyperekplexia-specific dominant mutant human GlyR α1 subunit 271Q. Tg271Q transgenic mice, in contrast to transgenic animals expressing a wild-type human α1subunit (tg271R), display a dramatic phenotype similar to spontaneous and engineered mouse mutations expressing reduced levels of GlyR. Electrophysiological analysis in the ventral horn of the spinal cord of tg271Q mice revealed a diminished GlyR transmission. Intriguingly, an even larger reduction was found for GABAA-receptor-mediated inhibitory transmission, indicating that the expression of this disease gene not only affects the glycinergic system but also leads to a drastic downregulation of the entire postsynaptic inhibition. Therefore, the transgenic mice generated here provide a new animal model of systemic receptor interaction to study inherited and acquired neuromotor deficiencies at different functional levels and to develop novel therapeutic concepts for these diseases.
Keywords: hyperekplexia, transgenic mouse model, neuromotor phenotype, glycine receptor, GABAA receptor, impaired postsynaptic inhibition
Human startle disease, or hereditary hyperekplexia, is a rare inborn neuromotor disease and one of the few examples of a syndrome linked to a defined mutation in a neuroreceptor ion-channel subunit gene (Andrew and Owen, 1997). The disease is characterized by an exaggerated startle reflex (i.e., overreaction to unexpected stimuli with myoclonic jerks and stiffness), often resulting in uncontrolled falling. In addition, uninduced nocturnal convulsive seizures occur occasionally. Enhanced startle reactions can already be detected in newborns in whom generalized hypertonia has also been observed (Suhren et al., 1966). Electromyographic studies reveal distinct overexcitability and diminished inhibition in patients with hyperekplexia (Matsumoto et al., 1992; Floeter et al., 1996).
The disease has been shown to result from mutations in the α1 subunit of the strychnine-sensitive glycine receptor (GlyR) (Shiang et al., 1993), which is the major inhibitory chloride channel in the spinal cord and the brainstem (Betz, 1992). In the adult mammal, the GlyR is composed of three copies of the ligand-binding α1 subunit and two copies of the structural β subunit, which targets the GlyR to the postsynaptic membrane by interacting with the anchoring protein gephyrin (Meyer et al., 1995; Feng et al., 1998).
The properties of mutant subunits have been studied in detail in vitro and in transfected heterologous cell systems (Langosch et al., 1994; Rajendra et al., 1994; Laube et al., 1995). However, it emerged from the study of mouse mutants with GlyR defects that in vivo mouse models were indispensable in understanding the pathomechanisms in patients. For example, although functional α1 subunit homomers are assembled in reconstituted Xenopus oocytes and in transfected non-neuronal tissue culture (Langosch et al., 1994; Rajendra et al., 1994), in the adult rodent the β subunit is essential for the formation of complexes containing functional α1subunit (Becker et al., 1992, 2000; Hartenstein et al., 1996). In addition, in vivo models are necessary to study possible interactions between different neurotransmitter systems in healthy and diseased states because the complexity of these interactions is not known and is therefore difficult to reconstitute. In patients with startle disease, such interactions were suggested by the therapeutic success of treatment with agonists of the other major inhibitory neurotransmitter receptor, the GABAA receptor (Ryan et al., 1992; Stayer and Meinck, 1998).
Although several genetically recessive mouse mutants exist, we generated a mouse model that more closely resembles a human dominant GlyR disease. The mutant human gene used to generate transgenic mice is associated with the most frequent genetically dominant form of hereditary hyperekplexia in humans (Shiang et al., 1993; Andrew and Owen, 1997). The mutation substitutes a glutamine for an arginine at position 271 in the extracellular domain of the GlyR α1 molecule; it has been shown in vitro and in tissue culture to strongly reduce receptor binding to the natural ligand glycine, although it does not significantly affect receptor binding to the antagonist strychnine (Langosch et al., 1994; Rajendra et al., 1994). We received transgenic animals displaying a characteristic neuromotor phenotype attributable to mutant transgene expression. Biochemical analyses confirmed thein vitro data, and electrophysiological studies revealed a novel interaction between the different inhibitory neurotransmitter systems in vivo.
MATERIALS AND METHODS
Constructs and transgenic mice. ASalI-fragment of 1.7 kb containing the human GlyR-subunit cDNA (Grenningloh et al., 1990b) was cloned into the uniqueXhoI site of a Thy-1 expression vector (Moechars et al., 1996). An EcoRI/PvuI fragment was used for microinjections into zygotes of C57BL/6/DBA/2 females and C57BL/6 males to generate tg271R lines. An analogous fragment in which codon 271 was mutated CGA to CAA (R→Q) was used to generate the tg271Q founders. Founders were bred with C57BL/6 to derive the experimental lines.
Expression analysis. Reverse transcriptase (RT)-PCR analysis on cortex and brainstem RNA was done using Superscript II (Invitrogen, Grand Island, NY) according to the manufacturer's instructions. GlyR α1-specific primers recognized endogenous and transgenic cDNA: 5′-CTCATCTTTGAGTGGCAGGA-3′ and 5′-GCATCCATGTTGATCC-AGAA-3′. The ratio of the GlyR α1-specific to the β-actin-specific signal was determined. Data (Fig.1B) are given as means ± SD. Statistical analysis was performed using ANOVA with the Bonferroni post hoc test.
Fig. 1.
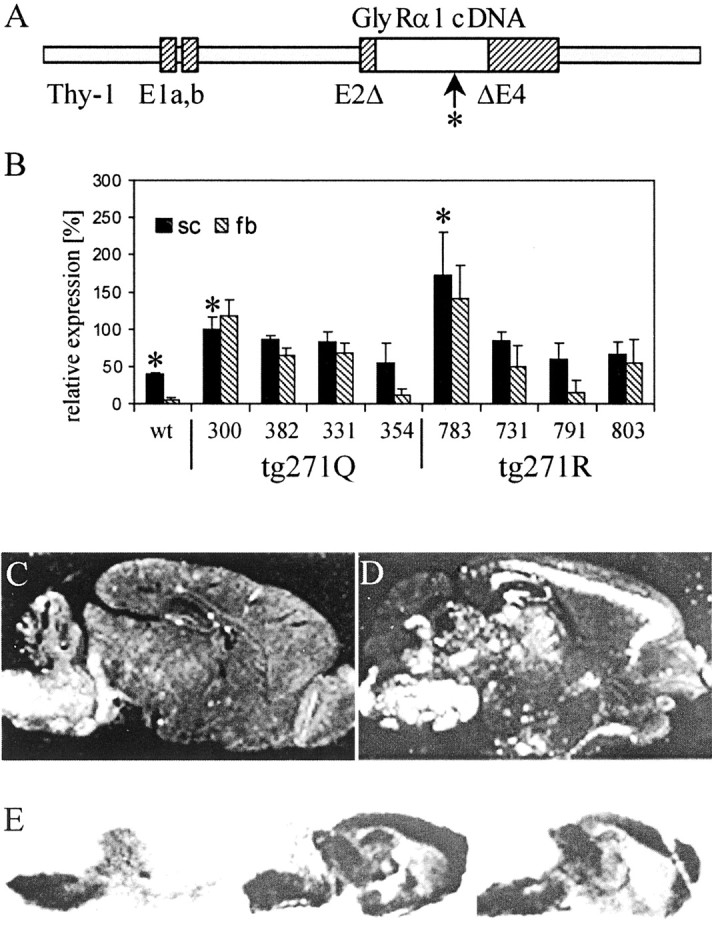
Transgene expression in GlyR transgenic mice is shown. A, Transgene constructs used. The human GlyR cDNAs (open bar) were cloned in a murine genomicThy-1 gene construct in which a region from exon 2 to exon 4 was deleted (Moechars et al., 1996) to confer neuron-specific expression (hatched bars, exon sequences; solid gray bars, intron sequences). The hyperekplexia-specific point mutation is marked by an arrow and anasterisk. B, Semiquantitative RT-PCR of brainstem and spinal cord (sc, black bars) and forebrain (fb,hatched bars) of different mouse lines. The expression of tg271Q-300 in spinal cord was set as 100%. The differences in spinal cord between wt and tg271Q-300 and tg271R-783 mice are significant (asterisks; p < 0.05)C, D, In situ hybridization of sagittal brain sections with GlyR α-specific RNA probes of a wt (C) and a tg271Q-300 (D) animal. E, 3H-strychnine binding to sagittal sections of wt (left), tg271Q-300 (center), and tg271R-783 (right) mice.
In situ hybridization was performed on sagittal sections of paraffin-embedded brains from mice of different genotypes. In vitro35S-labeled RNA transcripts from a 0.5 kb XhoI/SalI fragment of the human cDNA were used as a probe. Detection of transcripts was performed with LM-1 Photoemulsion (Amersham Biosciences, Arlington Heights, IL).
Ligand binding. Glycine-displaceable binding of3H-strychnine (DuPont NEN, Boston, MA) to crude membrane fractions was measured in triplicate as described previously (Becker et al., 1986). For radioligand displacement, 18 nm3H-strychnine was used. Data in Figure 2 are given as means ± SD. Statistical analysis was performed using ANOVA with the Bonferroni post hoc test (Fig. 2A) or the Mann–Whitney U test (Fig. 2B).
Fig. 2.
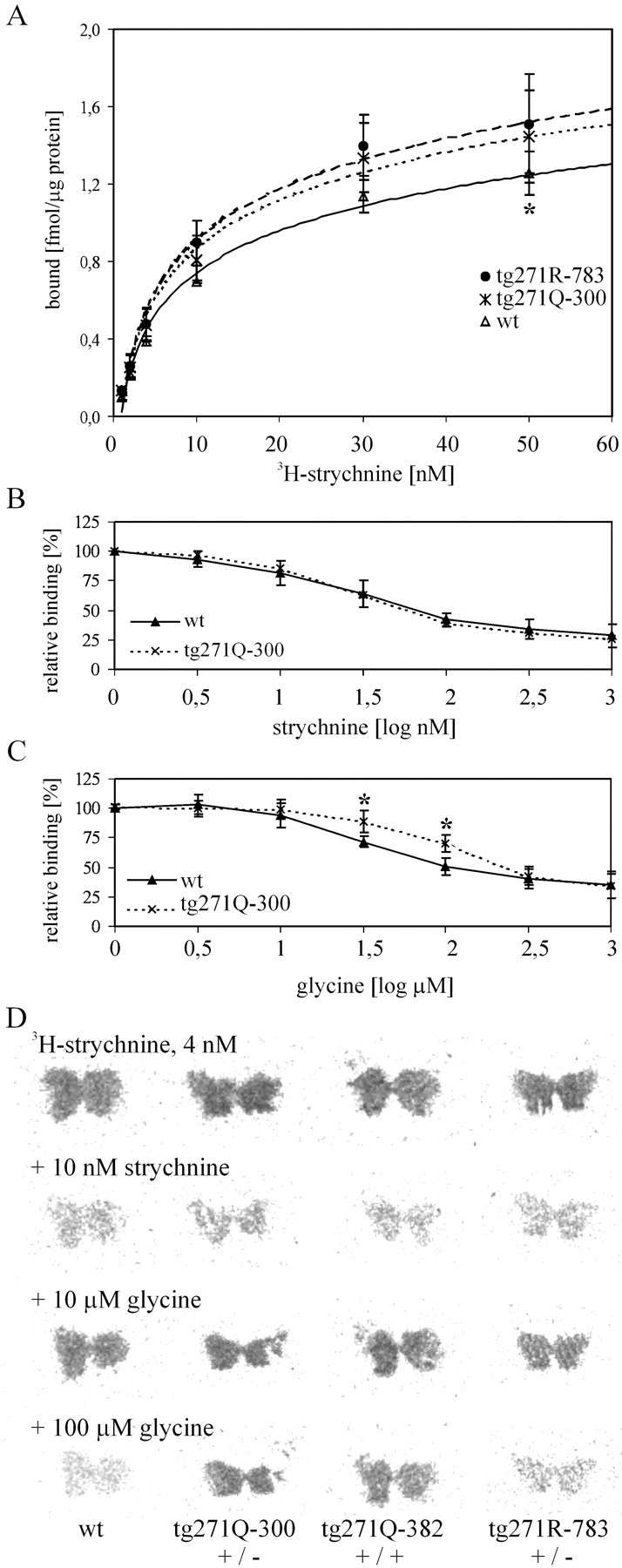
GlyR ligand binding to membranes from the brainstem and spinal cord is shown. A, Specific binding of 3H-strychnine in different genotypes (solid line, wt; dotted line, tg271Q-300; dashed line, tg271R-783). The asterisk at 50 nm marks a significant difference (p < 0.05) between the wt and the transgenic strains. B, C, An 18 nmconcentration of 3H-strychnine was displaced by increasing concentrations of either unlabeled strychnine (B) or glycine (C). Asterisks indicate significance (p < 0.05) D, Displacement of 3H-strychnine with unlabeled ligands on spinal cord sections from different genotypes is indicated; [Note the difference in binding in the tissue of the mutant transgenic animals (tg271Q-300 and tg271Q-382) compared with the wt transgenic (tg271R-783) and the wt line in the presence of 100 μmglycine (bottom).]
Receptor autoradiography. Frozen sections of brain were used for radioligand binding assays. Sections were incubated with 4 nm3H-strychnine alone and in the presence of unlabeled strychnine and glycine. For the benzodiazepine binding, 10 nm3H-RO15-4513 (kindly provided by D. Benke, Institute of Pharmacology, University of Zürich, Zürich, Switzerland) in the absence or presence of flumazenil was used. A 3H-sensitive imager plate with the Fujifilm Fluorescent image analyzer FLA2000 (Tokyo, Japan) was used for signal detection.
Phenotype analysis. Qualitatively, motor deficiencies in the animals were recognized with handling. In particular, tg271Q-300 as well as the homozygous tg271Q-382 and tg271Q-331 mice could be readily identified by a trained observer: When picked up by the tail they displayed obvious vibrations and/or showed the “hind feet clenching” phenotype. Sudden noise induced them to jump up and fall into tremor episodes, lasting for variable times even when held by the tail. Quantitative analysis was done as follows: Righting time was determined after bringing the animals into a supine position as described previously (Hartenstein et al., 1996; Becker et al., 2000) by twisting their tails. Tremor recording was performed by fixing mice by their tails to an F30 force transducer (Type 372) connected to a bridge amplifier (Type 336; both from Hugo Sachs Elektronik, March-Hugstetten, Germany). Electric signals were recorded by a Voltcraft Scope Card 220 (Voltcraft, Hirschau, Germany).
Slice preparation. Mice (both sexes) that were 14 to 21 d of age were anesthetized with ether and decapitated. The lumbar segments of the spinal cord were isolated and transferred to ice-cold standard external solution that contained (in mm): 120 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 5 HEPES, and 15 glucose, pH 7.4 (310 mOsm). The dorsal side of the spinal cord was glued onto a gelatin block and 250-μm-thick transverse slices were cut with a vibratome (Campden Instruments, Loughborough, UK). Slices were incubated for 1.5–7 hr after preparation in standard external solution at 32°C and bubbled continuously with carbogene (95% O2 and 5% CO2).
Electrophysiological recordings. Electrophysiological recordings were made from visually identified large-diameter neurons (>20 μm) in the ventral horn, presumptive α-motoneurons. They were visualized with the infrared gradient contrast technique (Dodt and Zieglgansberger, 1994) and monitored by a video microscopic system mounted on an upright microscope (Zeiss Axioskop FS, Jena, Germany; Infracontrast; Luigs & Neumann, Ratingen, Germany; video camera CF 8/1; Kappa Messtechnik, Gleichen, Germany). Slices were kept in position by a nylon mesh fixed to a U-shaped platinum wire. They were continuously superfused by standard external solution bubbled with carbogene. The experiments were performed at room temperature (20 ± 2°C). Patch pipettes were pulled from borosilicate glass capillaries (Kimax 51; Kimble, Vineland, NJ) by a DMZ Universal puller (Zeitz, Munich, Germany) and tip-polished by default. The pipette resistance ranged between 3 and 5 MΩ when filled with standard internal solution (in mm: 130 potassium gluconate, 20 KCl, 0.05 EGTA, 3 Na2ATP, 0.1 Na3GTP, 10 HEPES, pH 7.30) (290 mOsm). LidocaineN-ethyl bromide (QX-314) (5 μm) was added to the internal solution to block voltage-activated sodium currents in the recorded neuron. Postsynaptic currents (PSCs) were elicited via an ipsilaterally placed glass electrode (cathode, Kimax 51, inner tip diameter ∼2 μm), at a circumradial distance of ∼50 μm to the recording electrode, filled with 1 mmNaCl. Electrical stimulation (100 μsec; 1–5 V; AM Systems, Everett, MA) occurred at intervals of 10 sec. Recordings were performed in the whole-cell configuration of the patch-clamp technique with an EPC-7 patch-clamp amplifier (List Elektronik, Pfungstadt, Germany). Currents were monitored and stored with the Pulse Program (Heka Elektronik, Lambrecht/Pfalz, Germany) on a Macintosh Quadra 950 computer, linked to an ITC-16 interface (InstruTech, Port Washington, NY). To facilitate the establishment of a seal on the selected neuron, the external CaCl2 concentration was augmented to 3 mm during this phase. After breaking in, the concentration was lowered again to 2 mm. Currents were sampled at a frequency of 20 kHz and filtered at 3 kHz. Recordings were analyzed using the IgorPro 2.01 program (WaveMetrics Inc., Lake Oswego, OR). Drug solutions were applied by bath perfusion at a rate of 3–5 ml/min. All data in Figure 6 are given as means ± SEM. Statistical analysis was performed using the Mann–Whitney Utest.
Fig. 6.

Electrically evoked IPSCs from spinal cord neurons are shown. A, Representative traces from individual measurements on spinal cord neurons of wt (left) and tg271Q-300 (right) mice.Top, Glycinergic component of the IPSC.Bottom, GABAA receptor-mediated IPSC.B, Summary of the electrophysiological data. Mean amplitudes of the glycine-receptor-mediated (left) and GABAA-receptor-mediated IPSCs for tg271Q-300 animals (striped columns) and wt controls (black columns) are shown. In tg271Q-300 animals, the glycinergic component was reduced from −331.2 ± 124.8 pA to −100.1 ± 23.8 pA. The GABAA-receptor-mediated component was −4.1 ± 3.0 pA for tg271Q-300 mice, compared with −45.0 ± 25.4 pA in the controls. Asterisks indicate significance (p < 0.05).
Chemicals. All inorganic salts, glucose, EGTA, Na2ATP, Na3GTP, potassium gluconate, QX-314, kynurenic acid, strychnine, and bicuculline were purchased from Sigma (St. Louis, MO). HEPES was purchased from Calbiochem-Novabiochem (Bad Soden, Germany). CNQX and AP-5 were obtained from Tocris Cookson (Bristol, UK). Bicuculline was solved in DMSO. The final concentration of the solvent in the experiments was 0.05%.
RESULTS
Human GlyRα1 subunits form chimeric receptors in transgenic mice
Two constructs were made (Fig. 1A), in which the human GlyR α1 cDNA [mutated (271Q) or unmutated (271R)] was under the control of the brain-specific Thy-1 promoter (Moechars et al., 1996). Four tg271Q strains and 10 tg271R control strains with different levels of total transgene expression in neuronal tissue were derived (data not shown); four of each were kept for additional analysis. RT-PCR was performed to examine transgene expression in spinal cord and forebrain relative to that of the endogenous gene (Fig. 1B). The primers used for this analysis amplified both the transgenic human and the endogenous murine GlyR α1-subunit mRNA. Compared with samples from the wild-type (wt) mice (left), we found strongly enhanced GlyR α1 mRNA levels in the spinal cord, indicating expression of the transgene in addition to the endogenous gene. In the forebrain, the endogenous GlyR α1-subunit mRNA is hardly expressed; therefore, the GlyR α1 mRNA detected in the transgenic mice must be predominantly derived from transgenes. In situ hybridization analysis confirmed strong transgene expression in tg271Q (Fig. 1D) and tg271R strains (data not shown) compared with wt animals (Fig. 1C). Because, in addition to the brainstem, transgene expression is also found in ectopic sites in the brain, we investigated whether GlyR was formed there as well. In light of the widespread endogenous expression of GlyR β mRNA in many brain regions in which no known complex partners of this subunit are expressed, this seemed to be possible (Grenningloh et al., 1990a; Malosio et al., 1991). Indeed,in situ binding studies with the competitive glycine receptor antagonist strychnine showed ectopic ligand binding in both wt 271R and mutant 271Q lines (Fig. 1E). This ligand-binding activity must involve recruitment of the endogenous β subunit, because GlyR α1-subunit homo-oligomers do not form functional receptors in vivo (Becker et al., 1992, 2000; Hartenstein et al., 1996). Furthermore, this implies the formation of interspecies complexes between the murine β and the human transgene encoded (wt as well as mutant) GlyR α1 subunits. These results confirm data derived from heterologous expression studies in vitro and from transfected cells (Langosch et al., 1994; Rajendra et al., 1994). Strychnine binding to brainstem appeared not to be increased in our receptor autoradiography, which is depicted in Figure1E. This raised the question of whether the GlyR in the spinal cord and brainstem of the transgenic animals contained transgene-derived α1 subunits.
Reduced glycine affinity in the spinal cord of tg271Q-300 mice
To measure the amount of receptor in the transgenic animals more accurately, we used quantitative ligand-binding assays on spinal cord membrane preparations of the strongest expressing strains of both transgene tg271Q-300 and transgene tg271R-783 (Fig.2A). When compared with wt animals, a slight but significant enhancement in 3H-strychnine binding was detected, assuming similar strychnine affinities of the mutated or nonmutated human or murine α1subunits (Langosch et al., 1994; Rajendra et al., 1994). This is consistent with a small increment of GlyR immunoreactivity detected in Western blot analyses (data not shown). Thus, in spinal cord, α1-subunit-specific mRNA expression was apparently not limiting the number of membranous receptor complexes formed. Therefore, the transgenic α1 subunits should compete here with the endogenous ones for the limiting component, most likely the endogenous β subunit.
If mutant transgene-derived subunits are incorporated into spinal cord GlyR, these should show altered pharmacological characteristics according to previous in vitro data (Langosch et al., 1994;Rajendra et al., 1994), in which different glycine but unchanged strychnine affinity of the mutant α1 subunits had been demonstrated. We analyzed this by means of a ligand competition assay, comparing spinal cord membrane preparations of tg271Q-300 mice with those of wt animals. Figure 2Bdemonstrates that 3H-strychnine binding (percentage of total) was unaltered in tg271Q transgenic mice when competing with increasing amounts of nonradioactive strychnine. However, competition with an excess of the physiological ligand glycine revealed a significant difference in glycine affinity in the tg271Q-300 transgenic mice (Fig. 2C); at 1.5 and 2 log μm glycine, the competition curve is shifted.
Furthermore, we investigated ligand binding at the histological level on tissue sections through the upper spinal cord region of transgenic and wt animals. We included two mutant tg271Q strains, tg271Q-300 and tg271Q-382, and one strain expressing the wt human α1 subunit (Fig. 1B). In accordance with the binding assays illustrated in Figure2A, 3H-strychnine binding in situ appeared not to be different between the different genotypes (Fig. 2D, top two rows). In contrast to the strychnine binding, in the animals carrying the human mutant transgene (tg271Q-300 and tg271Q-382) glycine competes less efficiently for GlyR binding compared with wt animals (Fig. 2D, bottom two rows). Mice with the human wt transgene (tg271R-783) show glycine competition similar to nontransgenic animals, despite their high transgene expression (Figs.1B, 2D).
In summary, the ectopic mRNA expression of wt and mutant human GlyR α1 subunit led to ectopic formation of ligand-binding activity. In spinal cord, in which the physiological sites of GlyR expression are located, the additional transgene-derived mutant human α1 subunit competes for and at least partially replaces the endogenous subunit in the GlyR complex, reducing its ability to bind the natural ligand glycine.
Phenotypic characteristics of the tg271Q transgenic animals
When handling the transgenic lines described here, it became instantly apparent that tg271Q-300 animals developed spontaneous or handling-induced tremor episodes, which were most obvious in the extremities (Fig. 3A,B). The startle reaction was clearly exaggerated because these animals responded noticeably by uncontrolled jerks and jumps to sudden noise or touch. Furthermore, when picked up by the tail they displayed a hind feet clenching behavior (Fig. 3C). Their reproductive performance is also poor. Thus far, the phenotype was very similar to that described in the recessive mouse mutants spastic(spa) (Kingsmore et al., 1994; Mulhardt et al., 1994) orspasmodic (spd, spdot) (Buckwalter et al., 1994; Ryan et al., 1994; Saul et al., 1994), which carry mutations in the GlyR β- or α1-subunit gene, respectively. Compared with these, the phenotype in tg271Q-300 mice was genetically dominant because it was observed in heterozygous transgene carriers. Homozygous tg271Q-300 animals derived from double heterozygous crosses were not viable to adulthood. Two strains with less 271Q transgene expression, tg271Q-382 and tg271Q-331, (Fig.1B) also showed this spa-like phenotype but in an apparently gene-dosage-dependent manner (i.e., only animals homozygous for the symptoms displayed by transgenic mice) (Table1). In tg271Q-354 animals, the lowest expressing 271Q strain, no obvious phenotype was detected. Likewise, none of the 271R strains carrying the human wt transgene displayed this phenotype, regardless of the transgene expression level.
Fig. 3.

Phenotypic characteristics of tg271Q-300 mice.A, Tremor and disturbed righting when turned to the back. B, Handling-induced tremor, visible at the limbs.C, Hind feet clenching when picked up by the tail.
Table 1.
Transgene expression and phenotype in different GlyR α1 transgenic mice
| Transgenic mouse lines | Expression | Startle phenotype | Hind limb disorder | |
|---|---|---|---|---|
| tg +/+ | tg +/− | |||
| tg271Q-300 | +++++ | n.v.1-a | + | − |
| tg271Q-382 | +++ | + | − | − |
| tg271Q-331 | + | (+) | − | − |
| tg271Q-354 | + | − | − | − |
| tg271R-731 | +++ | − | − | + |
| tg271R-783 | +++++ | − | − | + |
| tg271R-791 | ++ | − | − | + |
| tg271R-803 | + | − | − | + |
n.v., Not viable.
To investigate the phenotype of tg271Q-300 mice in more detail, we used several methods established to characterize GlyR-deficient mouse mutants (Hartenstein et al., 1996; Becker et al., 2000). First, we measured the righting time, which can be easily quantified and has been shown to be a reliable marker for phenotype strength (Hartenstein et al., 1996; Becker et al., 2000). Animals with normal glycinergic neurotransmission instantly right themselves when brought to a supine position. However, in GlyR mutants this reflex is impaired, allowing for the simple measurement of the time it takes for them to get back on their feet. Figure 4Aillustrates that the righting time in tg271Q-300 mice is dramatically increased. Compared with previous studies of murine recessive GlyR mutants (Hartenstein et al., 1996; Becker et al., 2000) with respect to this task, the tg271Q-300 phenotype was similar to that ofspa mice with remnant GlyR protein expression and ligand binding of ∼10–20% of wt in the spinal cord and brainstem (Becker et al., 1992; Hartenstein et al., 1996). However, it was not as strong as the phenotype recorded from spdot(spasmodic oscillator) homozygotes (Hartenstein et al., 1996), which carry a null mutation in the GlyR α1 gene, leading to death within 4 weeks of birth (Buckwalter et al., 1994).
Fig. 4.

Phenotypic analysis of tg271Q-300 animals.A, The time required to right after being turned to the back (n = 65). B, The onset of inducible tremor (n = 42).
We also measured the onset of the inducible tremor in tg271Q-300 mice (Fig. 4B): Most animals started to display this at postnatal day 15, approximately the same time at which homozygousspdot mice start to show this motor deficiency. This implies that the mutant α1subunit interferes specifically with the endogenous adult α1 isoform, which takes over glycinergic control from the neonatal α2 isoform at approximately that time.
Finally, we recorded the tremor using an electromechanical transducer registering the vibrations, which can be sensed when picking up GlyR-deficient animals. We used an improved version of a setup described previously (Becker et al., 2000), which allows measurement of the frequency and amplitude of muscular contractions during tremor. Figure 5 shows oscilloscope traces derived from wt, tg271R-783, tg271Q-300, and homozygous spamice. The amplitude of the contractions recorded during tremor episodes (Fig. 3B) appeared stronger in tg271Q-300 (Fig.5D) than in spa (Fig. 5E) animals. As with partially GlyR-deficient homozygous spa animals but unlike completely GlyR-deficient homozygousspdot mice, tremor in tg271Q-300 mice was not permanent. During tremor-free periods, in which tg271Q-300 animals displayed the hind feet clenching phenotype (Fig. 3C), movements were recorded (Fig. 5C) that were similar to those seen in wt (Fig. 5A) or tg271R-783 (Fig. 5B) transgenic animals. Most interestingly, the tremor frequency in both tg271Q-300 and homozygous spa/spa mice was ∼25–30 Hz, which is similar to the frequency measured in hyperekplexia or stiff-man syndrome (SMS) (Stayer and Meinck, 1998).
Fig. 5.

Tremor recordings from transgenic and control mice. Traces of wt (A), and tg271R-783 wt transgenic (B) mice do not show any spastic motor disorder. The tremor in tg271Q-300 mice (D) shows a larger amplitude but a similar tremor frequency compared with homozygous spa mice (E).C, Trace taken from a tg271Q-300 animal during a nontremor period.
Although the 271Q transgene apparently caused a disease phenotype specific for GlyR deficiencies, expression of the tg271R construct did not. In contrast, breeding this construct into thespdot background could rescue the lethal phenotype of this complete loss-of-function mutation of the endogenous α1 subunit (data not shown). This provided genetic proof that the human α1 subunit was functional in interspecies hybrid receptors in mice. However, expression of the tg271R construct in addition to the endogenous murine α1 subunit caused a phenotype distinct from the one caused by the hyperekplexia transgene tg271Q (Table 1): An initially subtle but progressive limb malcoordination appeared most noticeable in the hind legs of adult tg271R transgenic mice. Preliminary analysis suggests that neurodegeneration is involved here (data not shown).
It should be noted that the transgenes were introduced into a hybrid genetic background (C57BL/6 × DBA/2). When comparing the phenotypes of transgenic animals within and between different litters of a particular transgenic strain, we did not notice an apparent contribution of genetic background variations in our animal population.
Glycine- and GABAA-receptor transmission are impaired in the spinal cord of tg271Q-300 mice
To test whether the GlyR-dependent neuromotor inhibition in the spinal cord is impaired in tg271Q mice, whole-cell patch-clamp studies were performed in visually identified large-diameter neurons, presumptive α-motoneurons, in the ventral horn of spinal cord slices. Tg271Q-300 mice and nontransgenic littermate controls were 14–19 d of age (i.e., around the onset of the phenotype or slightly later). The tissue was stimulated electrically to elicit postsynaptic responses in the recorded motoneurons (Fig.6A). The total PSC could be blocked completely by a combination of 1 mm kynurenic acid, 0.5 μmstrychnine, and 10 μm bicuculline, indicating that the PSC was mediated only by glutamate, glycine, and GABAA receptors in both genotypes (data not shown). The glycinergic component of the IPSC was isolated by a combination of 1 mm kynurenic acid and 10 μm bicuculline. In accordance with the biochemical analysis described above, this component was strongly reduced by 69.8% in tg271Q-300 mice compared with wt mice (n = 6 for each; p < 0.05) (Fig.6B, left). Interestingly, an even larger reduction was observed for the GABAA-receptor-mediated component of the IPSC. This component was isolated in a additional series of experiments by adding 1 mm kynurenic acid and 0.5 μm strychnine to the standard external solution. A reduction of 90.9% in tg271Q-300 animals was measured in comparison with wt animals (n = 6 for each;p < 0.05) (Fig. 6B,right). This modification of the GABAA-receptor-mediated transmission by the expression of the mutant human GlyR α1 subunit indicated that the entire inhibitory postsynaptic transmission is functionally downregulated by the expression of this transgene.
GABAA receptor binding in the brain of tg271Q and tg271R mice
To investigate whether the number of GABAA receptors was altered in the transgenic mice, we performed binding assays with3H-RO15-4513, a radioligand that binds to the benzodiazepine site of most GABAA receptor complexes. Brain sections of the mice with the two strongest mutant-transgene-expressing strains, tg271Q-300 and tg271Q-382, as well as those with the human wt-transgene-expressing strain tg271R-783, were analyzed in comparison with wt control animals (Fig.7). No obvious difference could be observed in the pattern and the amount of GABAA-receptor binding between the four tested genotypes at this level of resolution. Thus, these data are consistent with downregulation of GABA transmission on the functional rather than on the expression level, whereby analysis at a higher resolution to determine the intracellular distribution of the receptor remains to be done.
Fig. 7.

Autoradiographic studies of brains with benzodiazepine ligands. Distribution of 3H-RO15-4513 binding sites in the presence or absence of flumazenil in brain sections of wt, tg271Q-783, and tg271R-783 transgenic animals are shown.
DISCUSSION
The transgenic mouse model described here closely reproduces several symptoms found in human patients with hyperekplexia, such as inducible tremor development and exaggerated startle response. Although in humans a causal genotype–phenotype relationship cannot be established, our data provide formal proof of the capacity of this human GlyR mutation to induce a dominant neuromotor phenotype.
The hyperekplexia phenotype evoked in these mice is dependent on mutant transgene expression. It resembles mouse mutants with partially or completely obliterated endogenous GlyR function. Mice that topically and ectopically express a wt human GlyR gene form ectopic ligand-binding receptors but do not show this phenotype. In contrast, the wt transgene can rescue the mouse GlyR α1null mutation spdot, demonstrating that functional interspecies GlyR complexes are formed in vivoand thereby supporting the validity of this animal model. Thus, the hyperekplexia-like phenotype is specific for the hyperekplexia-associated mutated GlyR transgene.
The strength of the phenotype differed between strains carrying the tg271Q transgene. The strongest phenotype observed in these lines was similar to (and in terms of tremor even stronger than) the one described for homozygous spa mice, because of residual ligand binding of ∼10–20% in wt animals. The phenotype appears at approximately the same time as the GlyR α1isoform and becomes essential for glycinergic inhibition in mice. We conclude that the mutant α1 subunit interferes specifically with the endogenous adult α1isoform in spinal cord, in which it competes with endogenous α1 subunits for its essential partner β to form GlyR complexes.
Electrophysiological analyses on spinal cord motoneurons of tg271Q mice showed a strong reduction of the amplitude of the glycinergic component of the IPSC (69.8%). These data validate previous data derivedin vitro and in tissue culture (Langosch et al., 1994;Rajendra et al., 1994) and provide a physiological explanation for the symptoms observed in mice as well as in humans. In addition and unexpectedly, the GABAA-receptor-mediated transmission is also drastically reduced (90.9%) in the tg271Q-300 transgenic animals. This points to an adaptational interaction between the two inhibitory systems involved. Such interactions at the organism level are suggested by anatomical, clinical, and pharmacological evidence: (1) Both receptors often colocalize in rat spinal cord (Bohlhalter et al., 1994), and their transmitters are coreleased at many nerve endings (Jonas et al., 1998). (2) Both glycine and GABAA receptors are clustered at the synapses and require the presence of the receptor-associated protein gephyrin for correct synaptic localization of the receptor complexes (Essrich et al., 1998; Feng et al., 1998; Kneussel et al., 1999). (3) Genetic GlyR disease in humans can be treated with benzodiazepines, although with variable success (Ryan et al., 1992). (4) The symptoms in SMS, an autoimmune disease associated with antibodies against glutamic acid decarboxylase isoform 65, causing a dysfunction of the GABAergic system, resemble the symptoms of genetic GlyR defects with respect to electromyographic data (Stayer and Meinck, 1998). The recently identified autoimmune reactivity against gephyrin in a patient with SMS (Butler et al., 2000) might represent an example of an acquired GlyR malfunction.
Starting to address the mechanism by which the glycinergic and the GABAergic systems interact in our model, we found, at our limits of resolution, no influence of overexpression of mutated or unmutated GlyR α1 subunit on the binding of a GABAA-receptor ligand on brain tissue. This result is in line with previous observations on the mouse mutantspa, in which GABAA receptor was not reduced but rather slightly enhanced, suggesting compensatory mechanisms at the receptor level (White and Heller, 1982). It should be noted that our ligand-binding data do not exclude possible changes in the subunit composition of GABAA receptors or developmental changes in its subunit composition. They also do not exclude extrasynaptic localization of the GABAAreceptors. However, downregulation of GABAA-receptor transmission by the lack of GlyR function could take place at the level of physiology, whereby postsynaptic and presynaptic mechanisms are conceivable. Because it is hardly possible to address these questions directly, evidence for presynaptic changes can be gained by an analysis of miniature IPSCs and by an ANOVA of triggered postsynaptic currents (Bekkers and Stevens, 1989; Malinow and Tsien, 1990). Preliminary data point to a reduction of presynaptic release in GABAergic synapses as well as to regulatory alterations on the postsynaptic side (our unpublished data). Possible mechanisms affecting both the glycinergic- and GABAA-receptor-mediated transmission at the presynaptic level could involve factors important for the presynaptic transmitter release. An interesting candidate for a shared function important in both pathways could be the vesicular inhibitory amino acid transporter (Sagné et al., 1997), which is responsible for the uptake of glycine and GABA into synaptic vesicles (Burger et al., 1991; Dumoulin et al., 1999). It is conceivable that lack of GlyR function will affect the function of this transporter; this would also inhibit presynaptic GABA uptake.
Additional studies of this animal model are necessary to clarify these points. However, the data obtained thus far might have significant consequences on therapeutic strategies not only of the rare genetic GlyR defects, but also of the more frequent acquired neuromotor deficiency caused by the dysfunction of GABAAreceptors (SMS).
Considering that, as we have shown previously (Hartenstein et al., 1996; Becker et al., 2000), low expression of functional GlyR is sufficient for glycinergic inhibition, gene therapy for genetic GlyR defects should be possible, provided appropriate gene delivery technology is used. However, because of adverse effects of overexpression or ectopic expression of a “healthy” gene, as seen in the tg271R animals, such approaches require precise expression control. These tg271R animals might finally be useful to investigate the recently raised issue of a potential role of glycinergic transmission in neurodegeneration (Chen et al., 1999).
Footnotes
This work was supported by grants from the Bundesministerium für Forschung und Bildung (H.W.), the Deutsche Forschungsgemeinschaft (D.S.), and the Fritz Thyssen Stiftung.
Correspondence should be addressed to Hans Weiher, Institut für Diabetesforschung, Kölner Platz 1, 80804 Munich, Germany. E-mail:hans.weiher@lrz.uni-muenchen.de.
REFERENCES
- 1.Andrew M, Owen MJ. Hyperekplexia: abnormal startle response due to glycine receptor mutations. Br J Psychiatry. 1997;170:106–108. doi: 10.1192/bjp.170.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Becker CM, Hermans-Borgmeyer I, Schmitt B, Betz H. The glycine receptor deficiency of the mutant mouse spastic: evidence for normal glycine receptor structure and localization. J Neurosci. 1986;6:1358–1364. doi: 10.1523/JNEUROSCI.06-05-01358.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker CM, Schmieden V, Tarroni P, Strasser U, Betz H. Isoform-selective deficit of glycine receptors in the mouse mutant spastic. Neuron. 1992;8:283–289. doi: 10.1016/0896-6273(92)90295-o. [DOI] [PubMed] [Google Scholar]
- 4.Becker L, Hartenstein B, Schenkel J, Kuhse J, Betz H, Weiher H. Transient neuromotor phenotype in transgenic spastic mice expressing low levels of glycine receptor β-subunit: an animal model of startle disease. Eur J Neurosci. 2000;12:27–32. doi: 10.1046/j.1460-9568.2000.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bekkers JM, Stevens CF. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- 6.Betz H. Structure and function of inhibitory glycine receptors. Q Rev Biophys. 1992;25:381–394. doi: 10.1017/s0033583500004340. [DOI] [PubMed] [Google Scholar]
- 7.Bohlhalter S, Mohler H, Fritschy JM. Inhibitory neurotransmission in rat spinal cord: co-localization of glycine- and GABAA-receptors at GABAergic synaptic contacts demonstrated by triple immunofluorescence staining. Brain Res. 1994;642:59–69. doi: 10.1016/0006-8993(94)90905-9. [DOI] [PubMed] [Google Scholar]
- 8.Buckwalter MS, Cook SA, Davisson MT, White WF, Camper SA. A frameshift mutation in the mouse α1 glycine receptor gene (Glrα1) results in progressive neurological symptoms and juvenile death. Hum Mol Genet. 1994;3:2025–2030. doi: 10.1093/hmg/3.11.2025. [DOI] [PubMed] [Google Scholar]
- 9.Burger PM, Hell J, Mehl E, Krasel C, Lottspeich F, Jahn R. GABA and glycine in synaptic vesicles: storage and transport characteristics. Neuron. 1991;7:287–293. doi: 10.1016/0896-6273(91)90267-4. [DOI] [PubMed] [Google Scholar]
- 10.Butler MH, Hayashi A, Ohkoshi N, Villmann C, Becker CM, Feng G, De Camilli P, Solimena M. Autoimmunity to gephyrin in stiff-man syndrome. Neuron. 2000;26:307–312. doi: 10.1016/s0896-6273(00)81165-4. [DOI] [PubMed] [Google Scholar]
- 11.Chen Q, Moulder K, Tenkova T, Hardy K, Olney JW, Romano C. Excitotoxic cell death dependent on inhibitory receptor activation. Exp Neurol. 1999;160:215–225. doi: 10.1006/exnr.1999.7179. [DOI] [PubMed] [Google Scholar]
- 12.Dodt HU, Zieglgansberger W. Infrared videomicroscopy: a new look at neuronal structure and function. Trends Neurosci. 1994;17:453–458. doi: 10.1016/0166-2236(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 13.Dumoulin A, Rostaing P, Bedet C, Lévi S, Isambert M-F, Henry J-P, Triller A, Gasnier B. Presence of the vesicular inhibitory amino acid transporter in GABAergic and glycinergic synaptical terminal boutons. J Cell Sci. 1999;112:811–823. doi: 10.1242/jcs.112.6.811. [DOI] [PubMed] [Google Scholar]
- 14.Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the γ2 subunit and gephyrin. Nat Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- 15.Feng G, Tintrup H, Kirsch J, Nichol MC, Kuhse J, Betz H, Sanes JR. Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science. 1998;282:1321–1324. doi: 10.1126/science.282.5392.1321. [DOI] [PubMed] [Google Scholar]
- 16.Floeter MK, Andermann F, Andermann E, Nigro M, Hallett M. Physiological studies of spinal inhibitory pathways in patients with hereditary hyperekplexia. Neurology. 1996;46:766–772. doi: 10.1212/wnl.46.3.766. [DOI] [PubMed] [Google Scholar]
- 17.Grenningloh G, Pribilla I, Prior P, Multhaup G, Beyreuther K, Taleb O, Betz H. Cloning and expression of the 58 kd β subunit of the inhibitory glycine receptor. Neuron. 1990a;4:963–970. doi: 10.1016/0896-6273(90)90149-a. [DOI] [PubMed] [Google Scholar]
- 18.Grenningloh G, Schmieden V, Schofield PR, Seeburg PH, Siddique T, Mohandas TK, Becker CM, Betz H. Alpha subunit variants of the human glycine receptor: primary structures, functional expression and chromosomal localization of the corresponding genes. EMBO J. 1990b;9:771–776. doi: 10.1002/j.1460-2075.1990.tb08172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartenstein B, Schenkel J, Kuhse J, Besenbeck B, Kling C, Becker CM, Betz H, Weiher H. Low level expression of glycine receptor β subunit transgene is sufficient for phenotype correction in spastic mice. EMBO J. 1996;15:1275–1282. [PMC free article] [PubMed] [Google Scholar]
- 20.Jonas P, Bischofberger J, Sandkuhler J. Corelease of two fast neurotransmitters at a central synapse. Science. 1998;281:419–424. doi: 10.1126/science.281.5375.419. [DOI] [PubMed] [Google Scholar]
- 21.Kingsmore SF, Giros B, Suh D, Bieniarz M, Caron MG, Seldin MF. Glycine receptor β-subunit gene mutation in spastic mouse associated with LINE-1 element insertion. Nat Genet. 1994;7:136–141. doi: 10.1038/ng0694-136. [DOI] [PubMed] [Google Scholar]
- 22.Kneussel M, Brandstatter JH, Laube B, Stahl S, Muller U, Betz H. Loss of postsynaptic GABAA receptor clustering in gephyrin-deficient mice. J Neurosci. 1999;19:9289–9297. doi: 10.1523/JNEUROSCI.19-21-09289.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langosch D, Laube B, Rundstöm N, Schmieden V, Bormann J, Betz H. Decreased agonist affinity and chloride conductance of mutant glycine receptors associated with human hereditary hyperekplexia. EMBO J. 1994;2013:4223–4228. doi: 10.1002/j.1460-2075.1994.tb06742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laube B, Langosch D, Betz H, Schmieden V. Hyperekplexia mutations of the glycine receptor unmask the inhibitory subsite for β-amino-acids. NeuroReport. 1995;6:897–900. doi: 10.1097/00001756-199504190-00018. [DOI] [PubMed] [Google Scholar]
- 25.Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- 26.Malosio ML, Marqueze-Pouey B, Kuhse J, Betz H. Widespread expression of glycine receptor subunit mRNAs in the adult and developing rat brain. EMBO J. 1991;10:2401–2409. doi: 10.1002/j.1460-2075.1991.tb07779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsumoto J, Fuhr P, Nigro M, Hallett M. Physiological abnormalities in hereditary hyperekplexia. Ann Neurol. 1992;32:41–50. doi: 10.1002/ana.410320108. [DOI] [PubMed] [Google Scholar]
- 28.Meyer G, Kirsch J, Betz H, Langosch D. Identification of a gephyrin binding motif on the glycine receptor β subunit. Neuron. 1995;15:563–572. doi: 10.1016/0896-6273(95)90145-0. [DOI] [PubMed] [Google Scholar]
- 29.Moechars D, Lorent K, De Strooper B, Dewachter I, Van Leuven F. Expression in brain of amyloid precursor protein mutated in the α-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J. 1996;15:1265–1274. [PMC free article] [PubMed] [Google Scholar]
- 30.Mulhardt C, Fischer M, Gass P, Simon-Chazottes D, Guenet JL, Kuhse J, Betz H, Becker CM. The spastic mouse: aberrant splicing of glycine receptor β subunit mRNA caused by intronic insertion of L1 element. Neuron. 1994;13:1003–1015. doi: 10.1016/0896-6273(94)90265-8. [DOI] [PubMed] [Google Scholar]
- 31.Rajendra S, Lynch JW, Pierce KD, French CR, Barry PH, Schofield PR. Startle disease mutations reduce the agonist sensitivity of the human inhibitory glycine receptor. J Biol Chem. 1994;269:18739–18742. [PubMed] [Google Scholar]
- 32.Ryan SG, Sherman SL, Terry JC, Sparkes RS, Torres MC, Mackey RW. Startle disease, or hyperekplexia: response to clonazepam and assignment of the gene (STHE) to chromosome 5q by linkage analysis. Ann Neurol. 1992;31:663–668. doi: 10.1002/ana.410310615. [DOI] [PubMed] [Google Scholar]
- 33.Ryan SG, Buckwalter MS, Lynch JW, Handford CA, Segura L, Shiang R, Wasmuth JJ, Camper SA, Schofield P, O'Connell P. A missense mutation in the gene encoding the α1 subunit of the inhibitory glycine receptor in the spasmodic mouse. Nat Genet. 1994;7:131–135. doi: 10.1038/ng0694-131. [DOI] [PubMed] [Google Scholar]
- 34.Sagné C, El Mestikawy S, Isambert M-F, Hamon M, Henry J-P, Giros B, Gasnier B. Cloning of a functional vesicular GABA and glycine transporter by screening of genome databases. FEBS Lett. 1997;417:177–183. doi: 10.1016/s0014-5793(97)01279-9. [DOI] [PubMed] [Google Scholar]
- 35.Saul B, Schmieden V, Kling C, Mulhardt C, Gass P, Kuhse J, Becker CM. Point mutation of glycine receptor α1 subunit in the spasmodic mouse affects agonist responses. FEBS Lett. 1994;350:71–76. doi: 10.1016/0014-5793(94)00736-5. [DOI] [PubMed] [Google Scholar]
- 36.Shiang R, Ryan SG, Zhu YZ, Hahn AF, O'Connell P, Wasmuth JJ. Mutations in the α1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet. 1993;5:351–358. doi: 10.1038/ng1293-351. [DOI] [PubMed] [Google Scholar]
- 37.Stayer C, Meinck HM. Stiff-man syndrome: an overview. Neurologia. 1998;13:83–88. [PubMed] [Google Scholar]
- 38.Suhren O, Bruyn GW, Tuynman JA. Hyperekplexia: a hereditary syndrome. J Neurol Sci. 1966;3:577–605. [Google Scholar]
- 39.White WF, Heller AH. Glycine receptor alteration in the mutant mouse spastic. Nature. 1982;298:655–657. doi: 10.1038/298655a0. [DOI] [PubMed] [Google Scholar]