

Abstract
The neurotrophin, brain derived neurotrophic factor (BDNF), exerts multiple effects on the development and maintenance of the nervous system, including regulating synaptic plasticity and promoting neuron survival. Here we report the selective failure of BDNF-dependent survival in cultured hippocampal neurons from the trisomy 16 (Ts16) mouse, an animal model of Down syndrome. This failure is accompanied by overexpression of a truncated, kinase-deficient isoform (T1) of the BDNF receptor tyrosine receptor kinase B (trkB). Adenovirus-mediated introduction of exogenous full-length trkB into Ts16 neurons fully restored BDNF-dependent survival, whereas exogenous truncated trkB expression in normal, euploid neurons reproduced the Ts16 BDNF signaling failure. Thus, the failure of Ts16 neurons to respond to BDNF is caused by dysregulation of trkB isoform expression. Such a neurotrophin signaling defect could contribute to developmental and degenerative disorders of the nervous system.
Keywords: adenovirus, BDNF, Down syndrome, neurodegeneration, neuron death, neurotrophin, trisomy 16, trkB
The neurotrophins comprise a class of polypeptide neuron survival factors that not only support the survival of postmitotic neurons (Lewin and Barde, 1996), but also regulate other neuronal functions, including axon growth and synaptic plasticity (Black, 1999; Lentz et al., 1999; Lu and Chow, 1999;McAllister et al., 1999; Schinder and Poo, 2000; Thoenen, 2000). The neurotrophins, which include nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and NT-4/5, act by activating tyrosine receptor kinases (trks) (Barbacid, 1994; Bothwell, 1995). NGF acts via trkA, BDNF and NT-4/5 via trkB, and NT-3 via trkC, although the specificity of these interactions is not absolute. Binding of neurotrophins to trk dimers initiates trans-autophosphorylation of specific tyrosine residues on the intracellular domain of the receptor (Segal and Greenberg, 1996; Kaplan and Miller, 2000). These phospho-tyrosine residues serve as docking sites for elements of intracellular signaling cascades that lead to the suppression of neuron death and other effects of the neurotrophins. TrkB and trkC are also present as truncated forms, which lack the intracellular kinase domain and are, therefore, incapable of normal tyrosine phosphorylation (Klein et al., 1990; Middlemas et al., 1991; Tsoulfas et al., 1993). The full-length and truncated trk isoforms are generated by alternative splicing of the primary trk message. Although there is some evidence that activation of truncated trk receptors can elicit cellular responses independently of normal tyrosine phosphorylation (Baxter et al., 1997; Hapner et al., 1998; Haapasalo et al., 1999), truncated trk receptors are generally thought to inhibit trk-mediated neurotrophin signaling by interacting with full-length receptors to form inactive heterodimers (Eide et al., 1996). The expression of truncated trk receptors is developmentally regulated (Fryer et al., 1996) and may represent a normal mechanism for modulating the cellular response to specific neurotrophins (Ninkina et al., 1996).
We have been studying the functional characteristics of hippocampal neurons from the trisomy 16 (Ts16) mouse (Coyle et al., 1988), which has a triplication of chromosome 16. Because a cassette of ∼185 genes on human chromosome 21 is located on mouse chromosome 16 (Hattori et al., 2000), Ts16 shares a common genetic defect with the human disorder, Down syndrome (trisomy 21; DS), although some mouse chromosome 16 genes that are not on human chromosome 21 are overexpressed in Ts16. DS is characterized by mental retardation and, in patients over 40 years of age, Alzheimer's disease (AD) (Mann et al., 1984). Neurons from embryonic Ts16 mice undergo accelerated death by apoptosis (Bambrick et al., 1995; Stabel-Burow et al., 1997; Hallam and Maroun, 1998; Bambrick and Krueger, 1999), as do cultured cortical neurons from DS fetuses (Busciglio and Yankner, 1995). CNS neurons produce BDNF in response to excitatory stimuli, and this endogenously produced BDNF mediates activity-dependent neuron survival (Ghosh et al., 1994). However, we have shown that Ts16 hippocampal neurons do not exhibit activity-dependent survival (Bambrick et al., 1995). Therefore, we investigated the possibility that the accelerated death of Ts16 neurons may be attributable to a failure of BDNF signaling.
MATERIALS AND METHODS
Reagents. Mouse monoclonal antibody to an extracellular epitope on trkB [anti-trkB(out)], which recognizes both full-length (trkB.FL) and truncated (trkB.T1) trkB, was obtained from BD Transduction Laboratories (Lexington, KY). Antibodies to the neuron-specific microtubule-associated protein MAP2ab and hemagluttin (HA) were obtained from Sigma (St. Louis, MO), and anti-p75 was fromChemicon (Temecula, CA). Rabbit polyclonal antibodies to an intracellular epitope on trkB.FL [trkB(in)] and to an extracellular epitope on trkC were provided by Dr. L. Reichardt (University of California San Francisco). Rabbit polyclonal antibody to an intracellular epitope on the T1 isoform of truncated trkB [trkB(T1)] (Yan et al., 1997) was a gift of Dr. S. C. Feinstein (University of California Santa Barbara). Polyclonal antibody specific for phospho-trk was obtained from New England BioLabs (Beverly, MA). Appropriate rhodamine-, fluorescein-, or peroxidase-conjugated secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, PA). BDNF and NT-3 were gifts of Regeneron Pharmaceuticals (Tarrytown, NY); basic fibroblast growth factor (FGF-2) was obtained from Upstate Biotechnology (Lake Placid, NY). TrkB-IgG (provided by Regeneron) is a soluble fusion protein consisting of the extracellular, BDNF binding domain of rat trkB coupled to an Fc fragment of human IgG (Croll et al., 1998), which decreases the free extracellular BDNF concentration and inhibits its effects. TrkA-IgG (Regeneron) had no effect on euploid neuron survival, demonstrating that there were no nonspecific effects of TrkB-IgG [hippocampal neurons do not respond to NGF (Ip et al., 1993)].
Preparation and characterization of neuron cultures.Hippocampal neurons were cultured from euploid and Ts16 littermate fetuses on embryonic day 15.5 in minimal essential medium (MEM) supplemented with B27 as previously described (Bambrick et al., 1995). Neurons were plated at 104cells/cm2 on 12 mm glass coverslips etched with a lettered grid (Eppendorf AG, Hamburg, Germany) for survival experiments and at 5 × 105 cells/35 mm dish for Western blots. In initial experiments (see Fig. 2) coverslips and dishes were coated with poly-l-lysine (Sigma); in later experiments (see Figs. 1B, 6C–E), they were coated with poly-l-lysine and merosin. Neurons died approximately half as fast on merosin–poly-l-lysine substrate as compared with poly-l-lysine alone, however the relative differences between euploid and Ts16 neuron survival and the effects of neurotrophins were identical on the two substrates. Unless otherwise indicated, cell culture reagents were obtained from Invitrogen (Carlsbad, CA).
Fig. 2.

Survival of euploid (filled bars) and Ts16 (open bars) neurons 16 hr after withdrawal of B27 at 3 div. BDNF (100 ng/ml), NT-3 (100 ng/ml), FGF-2 (10 ng/ml), or vehicle (Control) was added at the time of B27 withdrawal. Survival is expressed as a percentage of cells present at the time of B27 withdrawal that were still alive 16 hr later. Error bars show SEM (n = 3), and theasterisk indicates where euploid and Ts16 survival were significantly different (*p < 0.01). Survival of euploid neurons in the presence of BDNF, NT-3, and FGF-2 was significantly different (*p < 0.05) from that in the absence of survival factors. Survival of Ts16 neurons in the presence of NT-3 and FGF-2, but not in the presence of BDNF, was significantly different (p < 0.05) from that in the absence of survival factors.
Fig. 1.
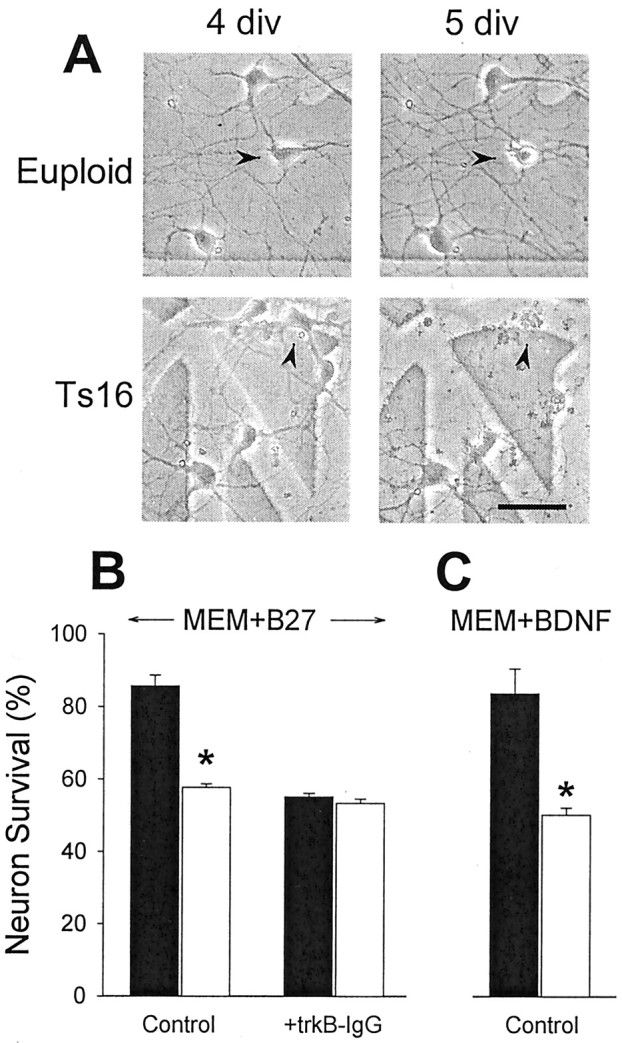
A, Phase-contrast images of euploid (top) and Ts16 (bottom) neurons prepared from E15.5 hippocampus. The same fields are shown at 4 div (left) and 5 div (right). One of the three euploid neurons present at 4 div had died by 5 div (arrowhead). Three of the five Ts16 neurons at 4 div had died by 5 div (the middle neuron is indicated by thearrowhead). Parts of the grid used to repeatedly locate the same fields can be seen in the background. Scale bar, 20 μm.B, Survival of euploid (filled bars) and Ts16 (open bars) hippocampal neurons at 5.5 div in the continuous presence of B27. TrkB-IgG (2 μg/ml) was added at 3 div as indicated. Survival is expressed as a percentage of cells present at 3 div that were still present at 5.5 div. In the absence of trkB-IgG, 14% of euploid neurons and 42% of Ts16 neurons died, a threefold increase in death of the Ts16 cells.C, Survival of euploid (filled bars) and Ts16 (open bars) hippocampal neurons at 4.5 div in the presence of 100 ng/ml BDNF. B27 was removed, and BDNF was added at 3 div. Survival is expressed as a percentage of cells present at 3 div that were still present at 4.5 div. InMEM+BDNF, 16% of euploid neurons, and 50% of Ts16 neurons died. Error bars show SEM (n = 3), and theasterisk indicates euploid and Ts16 survival were significantly different by t test (*p < 0.001).
Fig. 6.

Expression of exogenous trkB.FL restores BDNF survival signaling in Ts16 neurons. A, Euploid neurons were exposed to adenovirus carrying trkB.T1-HA DNA (AdTR), resulting in expression of trkB.T1 detected on Western blots using anti-trkB(out) (left). Anti-HA ICC revealed that the exogenous trkB.T1 was expressed in the plasma membrane and cytoplasm (right). Ts16 neurons were exposed to adenovirus carrying trkB.FL-HA DNA (AdFL) resulting in expression of trkB.FL detected on Western blots using anti-trkB(out) (left). Anti-HA ICC revealed that like exogenous trkB.T1, exogenous trkB.FL was expressed in the plasma membrane and cytoplasm (right). In A and B, the control virus, Ad−, did not contain trkB, and anti-HA was detected by confocal ICC with rhodamine-conjugated secondary antibody. Scale bar, 20 μm.C, Expression of trkB.T1 in euploid neurons inhibited BDNF survival signaling. Euploid neurons were either left untreated (●, Uninf) or treated with Ad− (▾), AdFL (▿), or AdTR (○) at 2 div. At 3 div, B27 was withdrawn from the cultures, and 100 ng/ml BDNF was added. Surviving neurons were repeatedly counted in five identified fields on each of two coverslips per condition. We counted 250–400 neurons for each data point. Solid line, Linear regression for uninfected data; dotted line, linear regression for AdTR-treated neurons. D, Expression of trkB.FL in Ts16 neurons restored BDNF survival signaling. Ts16 neurons were either untreated (●, Uninf) or treated with Ad− (▾), AdFL (▿), or AdTR (○) at 2 div. At 3 div, B27 was withdrawn from the cultures, and 100 ng/ml BDNF was added. Surviving neurons were repeatedly counted in five identified fields on each of two coverslips under each condition. We counted 250–400 neurons for each data point.Solid line, Linear regression for Uninf data;dotted line, linear regression for AdFL-treated neurons.E, Summary of the effect of trkB.FL expression on BDNF survival signaling. Data show mean ± SEM (n = 3 experiments) survival 36 hr after B27 withdrawal. *Significantly different from control, p < 0.01. Approximately half of the untreated euploid neurons died in the absence of 100 ng/ml BDNF, whereas <20% died in its presence. BDNF did not increase survival of untreated Ts16 neurons; however, in Ts16 neurons treated with AdFL, BDNF elicited a survival response that was indistinguishable from that of euploid neurons.
Measurement of neuron survival. At 3 d in vitro (div), all live neurons in each of five randomly selected, 175 × 175 μm fields per coverslip (identified by the etched grid) and at least two coverslips per condition were counted using phase-contrast microscopy. Cells that had assumed a globular, pyknotic appearance were scored as dead (see Fig. 1A, top). Separate studies have confirmed that cells scored as live by phase-contrast microscopy exclude trypan blue and are not undergoing DNA fragmentation (terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling-negative). In the experiments shown in Figure 1B, survival is expressed as the percentage of cells present at 3 div that remained at 5.5 div. In the experiments shown in Figures 1C, 2, and 5C–E, B27 was removed at 3 div, and the cultures were treated with neurotrophins or FGF-2; survival is expressed as the percentage of neurons present at the time of B27 withdrawal that remained at the end of the treatment period. The significance of differences between euploid and Ts16 cell counts for each condition was determined by Student's t test.
Fig. 5.

Reduced trkB phosphorylation in Ts16 neurons. Euploid and Ts16 neuron cultures were preincubated without B27 for 4 hr and then in the absence or presence of 100 ng/ml BDNF for 5 min. Cells were subjected to Western blot analysis using anti-phospho-trk (P-trkB) or trkB(out) (trkB). A, Representative result from a single experiment. B, Mean ± SEM of data from four experiments. Band intensity was determined as described in Materials and Methods. The euploid P-trkB:trkB ratio in the presence of BDNF is expressed as 100%. The dotted line shows the level of trkB phosphorylation predicted from the trkB.FL:trkB.T1 ratio in Ts16 neurons (see Results). *p < 0.05;n = 4.
Western blot analysis. SDS-solubilized cell extracts were incubated at 100°C for 5 min, fractionated on 4–12% NuPAGE bis-tris gels (Invitrogen) and transferred to a nitrocellulose membrane. After blocking in nonfat dried milk, membranes were incubated for 2–16 hr with primary antibody followed by incubation with appropriate peroxidase-conjugated secondary antibodies and visualized by chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ). Blots were quantified by scanning autoradiographs into NIH Image (version 1.62) to determine the optical density of each band.
Fluorescence immunocytochemistry. Cultures were fixed in 4% paraformaldehyde and incubated overnight with primary antibody at 4°C. Incubation with rhodamine- or fluorescein-conjugated secondary antibody was for 1 hr. Fluorescence images were acquired using a conventional microscope equipped with epifluorescence optics (Olympus, Melville, NY) or a confocal microscope (model LSM410; Carl Zeiss, Jena, Germany).
Replication-deficient recombinant adenoviruses. Adenoviruses were generated as described (Gonzalez et al., 1999). Viruses encoded TrkB isoforms epitope-tagged at the C terminus with HA and the fluorescent marker protein green fluorescent protein (GFP) under control of the cytomegalovirus promoter and an internal ribosome entry site sequence to allow translation of a bicistronic message. The virus designated AdTR encoded mouse truncated TrkB isoform TrkB.T1 (cDNA gift of Dr. M. Barbacid, Centro Nacional de Investigaciones Oncologicas, Madrid, Spain). The virus designated AdFL encoded mouse full-length TrkB. Virus designated Ad− encoded lacZ and GFP and served as a control for infection and overexpression of exogenous protein. Anti-HA immunostaining was used as an indicator of AdFL and AdTR infection in this study; GFP fluorescence was used to confirm infection by Ad− (75% of neurons were infected). Virally mediated transgene expression and function were evaluated by Western blot, immunocytochemistry (ICC), and in a pheochromocytoma (PC12) neurite outgrowth assay as described (Gonzalez et al., 1999). PC12 cells (which normally express TrkA but not TrkB) infected with AdFL extended neurites in response to recombinant BDNF as well as to NGF (M. Scott and R. Balice-Gordon, unpublished results).
RESULTS
Accelerated death of Ts16 neurons caused by failure of BDNF signaling
Cultures of normal (euploid) and Ts16 neurons were prepared from embryonic littermate hippocampi and maintained in serum-free medium containing the chemically defined supplement B27 (Brewer et al., 1993). The cultures contained almost exclusively postmitotic neurons. By 3 div, neurons from both genotypes took on the characteristics of differentiated neurons with extensive processes. At this time there were no differences in soma size or in neurite length or branching between the two genotypes. As illustrated in Figure1A, some cells in both euploid and Ts16 cultures died over 5 div. We previously reported that Ts16 neurons die about three times faster than euploid neurons (Bambrick et al., 1995; Bambrick and Krueger, 1999). Similarly, in the present study, ∼13% of euploid and ∼42% of Ts16 neurons died over a 2.5 d period (Fig. 1B). Addition of trkB-IgG (Croll et al., 1998) to deplete endogenous BDNF from the medium reduced the survival of euploid neurons to Ts16 levels without affecting Ts16 neuron survival (Fig. 1B). This demonstrates that BDNF is normally secreted in euploid hippocampal neuron cultures, where it promotes neuron survival and that this autocrine BDNF-mediated survival pathway is not functioning in Ts16 cultures.
To determine whether Ts16 neurons were capable of responding to BDNF, B27 was removed, and the ability of exogenous BDNF alone to support neuron survival was determined. Removal of B27 caused approximately half of both euploid and Ts16 neurons to die within 1 d (data not shown). In euploid neurons, this death was blocked by BDNF (100 ng/ml), whereas the Ts16 neurons were not rescued by the exogenous BDNF (Fig.1C). BDNF failed to rescue Ts16 neurons even at 1 μg/ml, 10 times the maximally effective concentration for euploid neurons (data not shown).
To determine whether Ts16 neurons are capable of responding to other survival factors, B27 was withdrawn at 3 div and replaced with BDNF, NT-3, or FGF-2 (Fig. 2). Although BDNF was unable to promote the survival of Ts16 neurons, NT-3 and FGF-2 rescued both euploid and Ts16 neurons to the same extent. Thus, Ts16 neurons have a selective failure of the survival response to BDNF.
Ts16 neurons overexpress truncated trkB
The failure of BDNF-dependent survival in Ts16 neurons could be caused by a loss of the BDNF receptor, trkB. Therefore, we analyzed trkB expression in euploid and Ts16 cultures by Western blotting with an antibody [anti-trkB(out)] that recognizes the extracellular domain of the receptor (Fig. 3A). Euploid and Ts16 neurons expressed both a 145 kDa band corresponding to the full-length, functionally active isoform, trkB.FL, and a 95 kDa band. The 95 kDa band is a catalytically inactive, truncated isoform of trkB (Klein et al., 1990; Middlemas et al., 1991). The 95 kDa band was determined to be the T1 isoform of truncated trkB (trkB.T1) using an antibody (Yan et al., 1997) to the unique, intracellular domain present on that isoform (Fig. 3C). Truncated trkB isoforms have been proposed to inhibit BDNF signaling via trkB by a dominant-negative mechanism (Eide et al., 1996). Western blots using anti-trkB(out) showed that, compared with euploid neurons, Ts16 neurons expressed slightly less trkB.FL but substantially more trkB.T1 (Fig.3A). Analysis of these blots revealed that the ratio of trkB.FL to trkB.T1 expression was 3.8 in euploid neurons and only 1.5 in Ts16 neurons (Fig. 3B). The increased trkB.T1 immunoreactivity at 95 kDa in Ts16 neurons (Fig. 3C) demonstrates that the increased intensity of the 95 kDa band seen with anti-trkB(out) (Fig. 3A) reflects increased expression of the T1 isoform and not proteolytic degradation of trkB.FL.
Fig. 3.

Abnormal expression of trkB isoforms in Ts16 neurons. A, Western blot of euploid and Ts16 hippocampal neurons using anti-trkB(out), which labels a common epitope on the extracellular side of full-length (145 kDa) and truncated (95 kDa) trkB. Result is typical of seven experiments. B, The ratio of trkB.FL to trkB.T1 in euploid and Ts16 neurons. Error bars show SEM (n = 3; *p < 0.05).C, Western blot of euploid and Ts16 neurons using anti-trkB(T1), which labels an internal epitope on trkB.T1.D, Western blot of euploid and Ts16 neurons using an antibody to trkC that labels both full-length (150 kDa) and truncated (110 kDa) isoforms. Western blot of euploid and Ts16 neurons using anti-p75. Results in C–E are typical of three independent experiments.
The expression of the NT-3 receptor, trkC, and its truncated isoforms was the same in euploid and Ts16 neurons (Fig. 3D), consistent with the survival-promoting effect of NT-3 in both genotypes (Fig. 2). All of the neurotrophins also bind to the low-affinity neurotrophin receptor p75, which may facilitate neurotrophin activation of trk receptors (Chao and Hempstead, 1995) or may mediate neurotrophin-dependent neuron death in the absence of trk receptors (Majdan and Miller, 1999; Lee et al., 2001). However, p75 expression was the same in euploid and Ts16 neurons (Fig. 3E). Thus, the altered expression of truncated trkB is not due to a general defect in the expression of neurotrophin receptors in Ts16 neurons.
All Ts16 neurons express both full-length and truncated trkB
In order to rule out the possibility that Ts16 cultures contain a higher proportion of neurons that express only trkB.T1, we analyzed euploid and Ts16 cultures by fluorescence ICC using anti-trkB(in), which recognizes an intracellular domain present only in the full-length trkB isoform and anti-trkB(T1). All of the neurons in both euploid and Ts16 cultures expressed both trkB.FL and trkB.T1 (Fig. 4). The cellular distributions of the two isoforms were similar, with expression in the plasma membrane and cytoplasm as has been reported for trkB.FL (Meyer-Franke et al., 1998; Du et al., 2000). The distributions were indistinguishable between the two genotypes.
Fig. 4.

Fluorescence ICC of euploid (a, c) and Ts16 (b, d) neurons using anti-trkB(in) (a, b) or anti-trkB(T1) (c, d). Labeling was detected by fluorescein-coupled secondary antibodies. Scale bar, 20 μm.
We also considered the possibility that the results in Figure3A–C could be attributable to differences in the relative proportions of neurons and astrocytes in the euploid and Ts16 cultures, because astrocytes express primarily truncated trkB (Rudge et al., 1994). However, cultures from both genotypes contained >95% of MAP2ab-immunoreactive neurons (cf., Fig. 7) with the remainder being flat cells identified as astrocytes by GFAP ICC. The proportion of glial cells was the same in euploid and Ts16 cultures. Furthermore, cortical astrocytes, cultured from euploid and Ts16 littermate fetuses as previously described (Bambrick et al., 1996), contained the same amount of trkB.T1 by Western blot analysis, demonstrating that differences in trkB.T1 expression (Fig. 3A–C) were not attributable to differences in trkB.T1 levels in the small percentage of contaminating astrocytes (data not shown). Thus, the reduced trkB.FL:trkB.T1 ratio observed in Ts16 cultures (Fig. 3B) reflects a change in trkB isoform expression in the Ts16 hippocampal neurons.
Fig. 7.

Cultured neurons were incubated in the absence of B27 and the presence of 100 ng/ml BDNF for 36 hr and then immunostained for MAP2ab using a rhodamine-conjugated secondary antibody. Most euploid neurons had smooth neurites (a). In contrast, of the surviving Ts16 neurons, many had fragmented neurites (arrowheads) indicative of early neurodegeneration (b). Ts16 neurons treated with AdFL (c) had very few fragmented neurites, and the cultures were morphologically indistinguishable from euploid neurons. Scale bar, 20 μm.
BDNF-stimulated trkB phosphorylation is reduced in Ts16 neurons
If trkB.T1 acts by a dominant-negative mechanism to reduce trkB signaling, there should be less BDNF-stimulated tyrosine phosphorylation of trkB in Ts16 neurons. To test this prediction we measured phosphorylation of trkB by Western blot analysis using antibodies specific for phosphotyrosine in position Y490 in trkB.FL. This antibody was raised to phospho-trkA, and it also recognizes the corresponding phosphorylated tyrosine in trkB and trkC. Because there is no detectable trkA in mouse hippocampal neurons (data not shown) and any BDNF-stimulated phospho-trkC could be distinguished on the basis of molecular size on these gels, in mouse hippocampal neurons, the BDNF-induced increase in trk phosphorylation determined with this antibody is phospho-trkB. As shown in Figure5A, there was no detectable phosphorylation of trkB in the absence of BDNF, whereas 100 ng/ml BDNF caused a dramatic increase in trkB phosphorylation. There was ∼33% less trkB phosphorylation in Ts16 neurons (Fig. 5B). The predicted change in BDNF/trkB signaling via full-length homodimers for any reduction in the trkB.FL:trkB.T1 ratio can be computed assuming a dominant-negative mechanism of inhibition by the truncated isoform (Eide et al., 1996). Based on our observation that the trkB.FL:trkB.T1 ratio is 3.8 in euploid neurons and 1.5 in Ts16 neurons (Fig.3B), this calculation predicts a 37% decrease in full-length trkB homodimers and, therefore, in BDNF-stimulated trkB autophosphorylation, in the Ts16 neurons (Fig. 5B, dotted line). Thus, BDNF stimulation of trkB tyrosine phosphorylation is reduced in Ts16 neurons by an amount predicted from the measured decrease in the trkB.FL:trkB.T1 ratio.
Expression of exogenous trkB.FL in Ts16 neurons restores BDNF survival signaling
If trkB.T1 acts as a dominant negative inhibitor of BDNF signaling through trkB.FL, then overexpression of trkB.T1 relative to trkB.FL is a possible cause of the BDNF signaling failure in Ts16 neurons. To test this hypothesis, we used replication-deficient adenoviruses to introduce trkB.FL or trkB.T1 into the neurons to experimentally manipulate the proportions of the two trkB isoforms (cf., Gonzalez et al., 1999). The viruses contained DNA coding for trkB.FL (AdFL), trkB.T1 (AdTR), or no trkB DNA (Ad−). The latter virus served as a control for the effects of the viral infection itself. Both recombinant trkB genes contained an HA sequence at the C- (cytoplasmic) terminus to enable the detection of the exogenous trkB proteins, independently of endogenous trkB. Euploid and Ts16 neurons infected with AdTR expressed increased levels of trkB.T1 as detected by either anti-trkB(out) or anti-trkB(T1) (trkB.T1 in euploid neurons illustrated in Fig.6A). Similarly, euploid and Ts16 neurons infected with AdFL expressed increased amounts of trkB.FL (trkB.FL in Ts16 neurons illustrated in Fig.6B). ICC using anti-HA revealed that 75% of the neurons expressed exogenous trkB.T1 or trkB.FL, moreover, examination of expression of the HA tag by fluorescence confocal ICC revealed that most of the exogenous trkB.T1 and trkB.FL in infected neurons was located on the plasma membrane (Fig. 6A,B, right panels). Ad− did not affect levels or distribution of endogenous trkB.FL and trkB.T1 (data not shown).
If trkB.T1 acts as a dominant-negative inhibitor of BDNF signaling, raising the trkB.FL:trkB.T1 ratio in Ts16 neurons by expressing exogenous trkB.FL should restore BDNF survival signaling. To test this hypothesis, neuron survival was studied in cultures infected with Ad−, AdFL, and AdTR (Fig. 6C–E). Time courses of neuron survival in the presence of BDNF after B27 removal are shown for euploid (C) and Ts16 (D) neurons. Ad− and AdFL did not substantially affect the BDNF-induced survival of euploid neurons. In contrast, AdTR, which raised trkB.T1 expression (Fig.6A), increased the rate of euploid neuron death (Fig.6C, dotted line) to a level approximately equal to the rate of death of uninfected Ts16 neurons in the presence of BDNF. When added to Ts16 cultures (Fig. 6D), AdTR slightly increased the rate of neuron death, whereas Ad− had no effect. In contrast, AdFL increased Ts16 neuron survival in the presence of BDNF to the level of survival of euploid neurons in the presence of BDNF (Fig.6D, dotted line). The essential findings are summarized in Figure 6E. BDNF reversed ∼65% of the euploid neuron death induced by B27 withdrawal but had no effect on Ts16 neuron survival. Infection of Ts16 neurons with AdFL, which raised expression of trkB.FL (Fig. 6B), completely restored the ability of BDNF to rescue the Ts16 neurons. Uninfected, BDNF-treated Ts16 neurons frequently had fragmented neurites (Fig.7b), characteristic of early stages of neuronal apoptosis (Martin et al., 1988). In contrast, AdFL-infected Ts16 neurons rarely had fragmented neurites in the presence of BDNF and were morphologically indistinguishable from euploid neurons (Fig. 7a,c)
DISCUSSION
We report here that BDNF promotes the survival of euploid hippocampal neurons, but not of Ts16 neurons, and that the Ts16 defect may be caused by altered expression of the BDNF receptor trkB. Ts16 neuron survival was not supported by exogenous BDNF, and removal of endogenous BDNF by the addition of trkB-IgG reduced euploid neuron survival to that of Ts16 neurons without affecting Ts16 survival (Fig.1). This failure is specific to BDNF because survival signaling by NT-3 and FGF-2 is unaffected in Ts16 neurons (Fig. 2). Thus, a chromosomal abnormality in mice (Ts16) with considerable similarity to human Ts21 (DS) (Hattori et al., 2000) results in the selective failure of BDNF-induced survival signaling.
These results provide an explanation for an observation we had previously made on the apparent absence of activity-dependent survival of Ts16 neurons (Bambrick et al., 1995). We showed that, in contrast to euploid neurons, low concentrations of glutamate (≈1 μm) do not promote the survival of Ts16 neurons. Because activity-dependent neuron survival was shown to be mediated by the synthesis and release of endogenous BDNF (Ghosh et al., 1994), we evaluated the possibility that BDNF mediated-survival may be defective in Ts16 neurons. The failure of BDNF survival signaling demonstrated here is sufficient to explain the absence of activity-dependent survival in Ts16 neurons (Bambrick et al., 1995).
The selective failure of BDNF signaling in Ts16 neurons appears to be attributable to abnormal expression of full-length and truncated isoforms of the BDNF receptor trkB (Fig. 3A), resulting in a 60% decrease in the trkB.FL:trkB.T1 ratio (Fig. 3B). Expression of the T1 truncated isoform of trkB is elevated in Ts16 neurons (Fig. 3C). Levels of the NT-3 receptor trkC and its truncated isoforms were similar in euploid and Ts16 neurons (Fig.3D), consistent with normal NT-3-promoted survival of Ts16 neurons (Fig. 2). Expression of the low-affinity neurotrophin receptor p75 was also normal in Ts16 neurons (Fig. 3E). The increased levels of trkB.T1 expressed in Ts16 neurons would be expected to reduce BDNF signaling by forming heterodimers with trkB.FL that are incapable of signaling to downstream effectors. Indeed, BDNF-stimulated trkB phosphorylation is reduced in Ts16 neurons (Fig. 5). This reduction in trkB-mediated signaling may account for the failure of BDNF to promote survival of Ts16 neurons. To directly test this hypothesis, we used replication-deficient adenoviruses to manipulate the levels of trkB.T1 and trkB.FL in the neurons. Our results are consistent with the hypothesis that BDNF survival signaling through trkB is dependent on the ratio of trkB.FL:trkB.T1, because increasing trkB.T1 expression eliminates BDNF survival signaling in euploid neurons and increasing trkB.FL expression restores BDNF survival signaling in Ts16 neurons (Fig. 6).
BDNF-induced tyrosine phosphorylation of trkB is reduced by ∼35% in Ts16 neurons (Fig. 5), as would be predicted from the reduced trkB.FL:trkB.T1 ratio observed in Ts16 neurons (Fig. 3B). This provides further support for the conclusion that altered trkB isoform expression is the primary cause of the BDNF signaling failure in Ts16 neurons. Interestingly, there is complete BDNF signaling failure with ∼65% of the downstream signal remaining. This suggests that BDNF-mediated survival in Ts16 neurons is particularly vulnerable to small decreases in trkB signaling.
The mechanism underlying the abnormal trkB.FL:trkB.T1 ratio in Ts16 neurons is not known. Because trkB.FL and trkB.T1 are generated from the same gene by alternative splicing (Klein et al., 1990; Middlemas et al., 1991), abnormal trkB splicing in Ts16 neurons may generate more trkB.T1 and less trkB.FL. Alternatively, the synthesis or degradation of trkB mRNA or protein may be altered in Ts16 neurons to produce more trkB.T1 and less trkB.FL. Any of these mechanisms may be directly attributable to overexpression of one or more genes on chromosome 16 or may be secondary to another Ts16 phenotype such as elevated cytoplasmic [Ca2+], which has been observed in both Ts16 neurons (Schuchmann et al., 1998) and Ts16 astrocytes (Bambrick et al., 1997).
The levels of both trkB.FL and trkB.T1 are developmentally regulated in the normal nervous system (Fryer et al., 1996; Ninkina et al., 1996), reflecting continuing control of BDNF responses in specific cell types throughout development and adulthood. The present study is the first report of neuropathological consequences of overexpression of trkB.T1. It is important to note that, in vivo, failure of BDNF-dependent neuron survival may not always result in neuron death because of the redundancy of neurotrophic factors in the brain. For example, because both NT-3 and FGF-2 are fully effective survival factors for Ts16 neurons (Fig. 2), there may be no increase in neuron death if sufficient levels of these factors are present, even if BDNF signaling is not functioning. However, if neurodegenerative stressors, including genetic mutations, environmental toxins, oxidative stress, and aging, reduce the levels of other trophic factors, neurons lacking a functional BDNF-dependent survival mechanism would be expected to selectively to undergo accelerated death.
It has been suggested that neurodegenerative disorders such as AD and Parkinson's disease (PD) may arise from decreased production of endogenous neurotrophins and, consequently, may be treated by application of exogenous neurotrophins to the brain (Eide et al., 1993;Hefti, 1994; Lindsay, 1994; Yuen and Mobley, 1996; Salehi et al., 1998). Clinical trials of intracranial injections of BDNF and other neurotrophic substances for the treatment of AD, PD, amyotrophic lateral sclerosis, and other disorders have been largely unsuccessful, possibly because of a failure of the neurotrophins to reach the target neurons (Aebischer and Ridet, 2001). The present results raise the alternative possibility that in some neurodegenerative disorders, affected neurons may lose the ability to respond to neurotrophins because of abnormal trk isoform expression and, therefore, therapies designed to raise neurotrophin levels in the brain would be ineffective. It is of interest that, as in Ts16 mouse neurons, trkB.T1 is elevated in hippocampal and cortical neurons of AD patients (Ferrer et al., 1999), demonstrating that dysregulation of trk isoform expression occurs in a human neurodegenerative disorder. The demonstration here of the ability to reverse a spontaneous, naturally occurring failure to respond to a neuron survival factor by introducing a particular isoform of its receptor suggests a new potential therapeutic strategy for treatment of neurodegenerative disorders.
BDNF regulates other neural functions besides survival, including the generation and differentiation of neurons during development, axon growth and growth cone mobility, and synaptic plasticity (Black, 1999;Lentz et al., 1999; Lu and Chow, 1999; McAllister et al., 1999;Schinder and Poo, 2000; Thoenen, 2000). It was recently shown that BDNF also promotes neurogenesis from adult stem cells in vivo(Benraiss et al., 2001; Pencea et al., 2001). If one or more of these BDNF-mediated responses were also affected by aberrant trkB expression, cognitive function could be compromised because of errors in connectivity and the failure to properly modulate synaptic plasticity, even in the absence of neuronal loss. Such deficits could contribute to mental retardation and premature AD in DS.
S.G.D. and L.L.B. contributed equally to this work.
This work was supported by National Institutes of Health (NIH) Grants AG10686, NS40492 (B.K.K.), AG15207 (L.L.B.), and NS34373 (R.J.B.-G.), by National Science Foundation Grant MOD653 (R.G.B.-G.), and by NIH predoctoral National Research Service Award Fellowship F31NR07189 (S.G.D.). We thank Drs. C. Cisterni and S. Kraner and M. Scott and H.-Y. Zhou for recombinant adenoviruses and for sharing unpublished data on the functional characterization of recombinant adenovirus encoding full-length TrkB. We are grateful to Drs. S. Feinstein (University of California Santa Barbara) and L. Reichardt (University of California San Francisco) for providing antibodies and to Regeneron Pharmaceuticals Company (Tarrytown, NY), for providing trkB-IgG, BDNF, and NT-3. We thank Drs. M. Raff, L. Tessarollo, S. Wiegand, and T. Kingsbury for comments on this manuscript.
Correspondence should be addressed to B. K. Krueger, Department of Physiology, University of Maryland School of Medicine, 655 West Baltimore Street, Baltimore, MD 21201. E-mail:bkrueger@umaryland.edu.
S. G. Dorsey's present address: Neural Development Group, National Cancer Institute, Frederick Cancer Research and Development Center, Frederick, MD 21701.
REFERENCES
- 1.Aebischer P, Ridet J-L. Recombinant proteins for neurodegenerative diseases: the delivery issue. Trends Neurosci. 2001;24:533–540. doi: 10.1016/s0166-2236(00)01899-3. [DOI] [PubMed] [Google Scholar]
- 2.Bambrick LL, Krueger BK. Neuronal apoptosis in mouse trisomy 16: mediation by caspases. J Neurochem. 1999;72:1769–1772. doi: 10.1046/j.1471-4159.1999.721769.x. [DOI] [PubMed] [Google Scholar]
- 3.Bambrick LL, Yarowsky PJ, Krueger BK. Glutamate as a hippocampal neuron survival factor: an inherited defect in the trisomy 16 mouse. Proc Natl Acad Sci USA. 1995;92:9692–9696. doi: 10.1073/pnas.92.21.9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bambrick LL, de Grip A, Seenivasan V, Krueger BK, Yarowsky PJ. Expression of glial antigens in mouse astrocytes: species differences and regulation in vitro. J Neurosci Res. 1996;46:305–315. doi: 10.1002/(SICI)1097-4547(19961101)46:3<305::AID-JNR3>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 5.Bambrick LL, Golovina VA, Blaustein MP, Yarowsky PJ, Krueger BK. Abnormal calcium homeostasis in astrocytes from the trisomy 16 mouse. Glia. 1997;19:352–358. doi: 10.1002/(sici)1098-1136(199704)19:4<352::aid-glia8>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 6.Barbacid M. The Trk family of neurotrophin receptors. J Neurobiol. 1994;25:1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- 7.Baxter GT, Radeke MJ, Kuo RC, Makrides V, Hinkle B, Hoang R, Medina-Selby A, Coit D, Valenzuela P, Feinstein SC. Signal transduction mediated by the truncated trkB receptor isoforms, trkB.T1 and trkB. T 2. J Neurosci. 1997;17:2683–2690. doi: 10.1523/JNEUROSCI.17-08-02683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benraiss A, Chmielnicki E, Lerner K, Roh D, Goldman SA. Adenoviral brain-derived neurotrophic factor induces both neostriatal and olfactory neuronal recruitment from endogenous progenitor cells in the adult forebrain. J Neurosci. 2001;21:6718–6731. doi: 10.1523/JNEUROSCI.21-17-06718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Black IB. Trophic regulation of synaptic plasticity. J Neurobiol. 1999;41:108–118. [PubMed] [Google Scholar]
- 10.Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci. 1995;18:223–253. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 11.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 12.Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down's syndrome neurons in vitro. Nature. 1995;378:776–779. doi: 10.1038/378776a0. [DOI] [PubMed] [Google Scholar]
- 13.Chao MV, Hempstead BL. p75 and Trk: a two-receptor system. Trends Neurosci. 1995;18:321–326. [PubMed] [Google Scholar]
- 14.Coyle JT, Oster-Granite ML, Reeves RH, Gearhart JD. Down syndrome, Alzheimer's disease and the trisomy 16 mouse. Trends Neurosci. 1988;11:390–394. doi: 10.1016/0166-2236(88)90075-6. [DOI] [PubMed] [Google Scholar]
- 15.Croll SD, Chesnutt CR, Rudge JS, Acheson A, Ryan TE, Siuciak JA, DiStefano PS, Wiegand SJ, Lindsay RM. Co-infusion with a TrkB-Fc receptor body carrier enhances BDNF distribution in the adult rat brain. Exp Neurol. 1998;152:20–33. doi: 10.1006/exnr.1998.6836. [DOI] [PubMed] [Google Scholar]
- 16.Du J, Feng L, Yang F, Lu B. Activity- and Ca2+-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J Cell Biol. 2000;150:1423–1433. doi: 10.1083/jcb.150.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eide FF, Lowenstein DH, Reichardt LF. Neurotrophins and their receptors–current concepts and implications for neurological disease. Exp Neurol. 1993;121:200–214. doi: 10.1006/exnr.1993.1087. [DOI] [PubMed] [Google Scholar]
- 18.Eide FF, Vining ER, Eide BL, Zang K, Wang XY, Reichardt LF. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J Neurosci. 1996;16:3123–3129. doi: 10.1523/JNEUROSCI.16-10-03123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrer I, Marin C, Rey MJ, Ribalta T, Goutan E, Blanco R, Tolosa E, Marti E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J Neuropathol Exp Neurol. 1999;58:729–739. doi: 10.1097/00005072-199907000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Fryer RH, Kaplan DR, Feinstein SC, Radeke MJ, Grayson DR, Kromer LF. Developmental and mature expression of full-length and truncated trkB receptors in the rat forebrain. J Comp Neurol. 1996;374:21–40. doi: 10.1002/(SICI)1096-9861(19961007)374:1<21::AID-CNE2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 22.Gonzalez M, Ruggiero FP, Chang Q, Shi YJ, Rich MM, Kraner S, Balice-Gordon RJ. Disruption of TrkB-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron. 1999;24:567–583. doi: 10.1016/s0896-6273(00)81113-7. [DOI] [PubMed] [Google Scholar]
- 23.Haapasalo A, Saarelainen T, Moshnyakov M, Arumae U, Kiema TR, Saarma M, Wong G, Castren E. Expression of the naturally occurring truncated trkB neurotrophin receptor induces outgrowth of filopodia and processes in neuroblastoma cells. Oncogene. 1999;18:1285–1296. doi: 10.1038/sj.onc.1202401. [DOI] [PubMed] [Google Scholar]
- 24.Hallam DM, Maroun LE. Anti-gamma interferon can prevent the premature death of trisomy 16 mouse cortical neurons in culture. Neurosci Lett. 1998;252:17–20. doi: 10.1016/s0304-3940(98)00541-2. [DOI] [PubMed] [Google Scholar]
- 25.Hapner SJ, Boeshore KL, Large TH, Lefcort F. Neural differentiation promoted by truncated trkC receptors in collaboration with p75(NTR). Dev Biol. 1998;201:90–100. doi: 10.1006/dbio.1998.8970. [DOI] [PubMed] [Google Scholar]
- 26.Hattori M, Fujiyama A, Taylor TD, Watanabe H, Yada T, Park HS, Toyoda A, Ishii K, Totoki Y, Choi DK, Soeda E, Ohki M, Takagi T, Sakaki Y, Taudien S, Blechschmidt K, Polley A, Menzel U, Delabar J, Kumpf K. The chromosome 21 mapping, sequencing consortium. The DNA sequence of human chromosome 21. Nature. 2000;405:311–319. doi: 10.1038/35012518. [DOI] [PubMed] [Google Scholar]
- 27.Hefti F. Neurotrophic factor therapy for nervous system degenerative diseases. J Neurobiol. 1994;25:1418–1435. doi: 10.1002/neu.480251109. [DOI] [PubMed] [Google Scholar]
- 28.Ip NY, Li Y, Yancopoulos GD, Lindsay RM. Cultured hippocampal neurons show responses to BDNF, NT-3, and NT-4, but not NGF. J Neurosci. 1993;13:3394–3405. doi: 10.1523/JNEUROSCI.13-08-03394.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 30.Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990;61:647–656. doi: 10.1016/0092-8674(90)90476-u. [DOI] [PubMed] [Google Scholar]
- 31.Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- 32.Lentz SI, Knudson CM, Korsmeyer SJ, Snider WD. Neurotrophins support the development of diverse sensory axon morphologies. J Neurosci. 1999;19:1038–1048. doi: 10.1523/JNEUROSCI.19-03-01038.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewin GR, Barde Y-A. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- 34.Lindsay RM. Neurotrophic growth factors and neurodegenerative diseases: therapeutic potential of the neurotrophins and ciliary neurotrophic factor. Neurobiol Aging. 1994;15:249–251. doi: 10.1016/0197-4580(94)90124-4. [DOI] [PubMed] [Google Scholar]
- 35.Lu B, Chow A. Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res. 1999;58:76–87. [PubMed] [Google Scholar]
- 36.Majdan M, Miller FD. Neuronal life and death decisions: functional antagonism between the Trk and p75 neurotrophin receptors. Int J Dev Neurosci. 1999;17:153–161. doi: 10.1016/s0736-5748(99)00016-7. [DOI] [PubMed] [Google Scholar]
- 37.Mann DMA, Yates PO, Marcyniuk B. Alzheimer's presenile dementia, senile dementia of Alzheimer type and Down's syndrome in middle age form an age related continuum of pathological changes. Neuropathol Appl Neurobiol. 1984;10:185–207. doi: 10.1111/j.1365-2990.1984.tb00351.x. [DOI] [PubMed] [Google Scholar]
- 38.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 40.Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG, Reichardt LF, Jr, Barres BA. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–693. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Middlemas DS, Lindberg RA, Hunter T. trkB, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol. 1991;11:143–153. doi: 10.1128/mcb.11.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ninkina N, Adu J, Fischer A, Pinon LG, Buchman VL, Davies AM. Expression and function of TrkB variants in developing sensory neurons. EMBO J. 1996;15:6385–6393. [PMC free article] [PubMed] [Google Scholar]
- 43.Pencea V, Bingaman KD, Wiegand SJ, Luskin MB. Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J Neurosci. 2001;21:6706–6717. doi: 10.1523/JNEUROSCI.21-17-06706.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rudge JS, Li Y, Pasnikowski EM, Mattsson K, Pan L, Yancopoulos GD, Wiegand SJ, Lindsay RM, Ip NY. Neurotrophic factor receptors and their signal transduction capabilities in rat astrocytes. Eur J Neurosci. 1994;6:693–705. doi: 10.1111/j.1460-9568.1994.tb00981.x. [DOI] [PubMed] [Google Scholar]
- 45.Salehi A, Verhaagen J, Swaab DF. Neurotrophin receptors in Alzheimer's disease. Prog Brain Res. 1998;117:71–89. doi: 10.1016/s0079-6123(08)64009-1. [DOI] [PubMed] [Google Scholar]
- 46.Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23:639–645. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- 47.Schuchmann S, Muller W, Heinemann U. Altered Ca2+ signaling and mitochondrial deficiencies in hippocampal neurons of trisomy 16 mice: a model of Down's syndrome. J Neurosci. 1998;18:7216–7231. doi: 10.1523/JNEUROSCI.18-18-07216.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 49.Stabel-Burow J, Kleu A, Schuchmann S, Heinemann U. Glutathione levels and nerve cell loss in hippocampal cultures from trisomy 16 mouse–a model of Down syndrome. Brain Res. 1997;765:313–318. doi: 10.1016/s0006-8993(97)00480-0. [DOI] [PubMed] [Google Scholar]
- 50.Thoenen H. Neurotrophins and activity-dependent plasticity. Prog Brain Res. 2000;128:183–191. doi: 10.1016/S0079-6123(00)28016-3. [DOI] [PubMed] [Google Scholar]
- 51.Tsoulfas P, Soppet D, Escandon E, Tessarollo L, Mendoza-Ramirez JL, Rosenthal A, Nikolics K, Parada LF. The rat trkC locus encodes multiple neurogenic receptors that exhibit differential response to neurotrophin-3 in PC12 cells. Neuron. 1993;10:975–990. doi: 10.1016/0896-6273(93)90212-a. [DOI] [PubMed] [Google Scholar]
- 52.Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997;378:135–157. [PubMed] [Google Scholar]
- 53.Yuen EC, Mobley WC. Therapeutic potential of neurotrophic factors for neurological disorders. Ann Neurol. 1996;40:346–354. doi: 10.1002/ana.410400304. [DOI] [PubMed] [Google Scholar]