

Abstract
The dorsal striatum participates in motor function and stimulus–response or “habit” learning. Acetylcholine (ACh) is a prominent neurotransmitter in the striatum and exerts part of its actions through nicotinic cholinergic receptors. Activation of these receptors has been associated with the enhancement of learning and certainly is instrumental in habitual use of nicotine. Nicotinic receptors have also been suggested to be a possible therapeutic target for disorders of the basal ganglia. In this report we show that the activation of nicotinic acetylcholine receptors in the dorsal striatum contributes to dopamine (DA)- and activity-dependent changes in synaptic efficacy. High-frequency activation of glutamatergic synapses onto striatal neurons results in a long-term depression (LTD) of synaptic efficacy that is dependent on the activation of dopamine receptors. This stimulation also produces robust increases in extracellular dopamine concentration as well as strong activation of cholinergic striatal interneurons. Antagonists of nicotinic acetylcholine receptors inhibit striatal LTD. However, on coapplication of dopamine reuptake inhibitors with nicotinic receptor antagonists, activity-induced striatal LTD is restored. Dopamine release is modulated by activation of nicotinic receptors in the dorsal striatum, and activation of nicotinic receptors during high-frequency synaptic activation appears to be capable of interacting with dopaminergic actions that lead to striatal LTD. Our results suggest that stimulation of mechanisms involved in striatal synaptic plasticity is an important role for striatal nicotinic acetylcholine receptors and that these mechanisms may contribute to the enhancement of learning and habit formation produced by nicotine intake.
Keywords: striatum, synaptic plasticity, acetylcholine, dopamine, habit learning, drug addiction
The striatum plays key roles in forms of learning and memory including habit learning (Aosaki et al., 1994; Knowlton et al., 1996). Activity-dependent changes in synaptic efficacy are thought to underlie information storage in the nervous system. In the striatum, high-frequency activation of excitatory synapses onto striatal output neurons induces a long-term depression (LTD) of synaptic strength at glutamatergic synapses. Striatal LTD is dependent on activation of D1 and D2 dopamine (DA) receptors (Calabresi et al., 1992, 2000b; Choi and Lovinger, 1997a; Tang et al., 2001). This strong dependence on dopaminergic transmission is a shared feature of striatal synaptic plasticity and striatal-based learning in several species (Packard and White, 1991; Aosaki et al., 1994; Knowlton et al., 1996).
The striatum receives converging glutamatergic input from cortex and thalamus as well as dopaminergic input from the substantia nigra. Integration of these extrinsic inputs is modulated by the intrinsic actions of acetylcholine (ACh). Striatal ACh is supplied by large-sized cholinergic interneurons the functions of which are still not well characterized (Kawaguchi, 1993). At the cellular level, both striatal ACh and DA are potent neuromodulators that can affect activity-dependent changes in synaptic efficacy and may contribute to motor or habit learning (Calabresi et al., 1992, 2000a; Wickens et al., 1996; Tang et al., 2001).
Acetylcholine receptors (AchRs) are expressed at high levels in striatum. Muscarinic acetylcholine receptors are expressed both presynaptically and postsynaptically in striatum, and one of their actions is to decrease glutamatergic synaptic transmission (Malenka and Kocsis, 1988; Hersch et al., 1994). Nicotinic AChRs (nAChRs) are expressed on dopaminergic terminals in the dorsal striatum (Clarke and Pert, 1985). Acute activation of these receptors stimulates (3H)-DA release from striatal synaptosomes and in striatal slice preparations (Giorguieff et al., 1976; Kulak et al., 1997; Wonnacott et al., 2000). Chronic nicotine exposure alters the cell surface expression and function of certain kinds of neuronal nicotinic receptors, including those types expressed in rat striatum (Flores et al., 1992).
LTD in the developing striatum is initiated postsynaptically but is maintained presynaptically (Choi and Lovinger, 1997a,b; Tang et al., 2001), although plasticity in the adult striatum may involve additional mechanisms (Calabresi et al., 1994). The presynaptic expression of striatal LTD is supported by the observation that LTD is associated with decreased frequency, but not amplitude, of miniature EPSCs (mEPSCs) (Choi and Lovinger, 1997a). Striatal LTD expression is also strongly associated with an increase in the paired-pulse response (PPR) ratio (Choi and Lovinger, 1997b; Tang et al., 2001). It is less well understood whether parasynaptic elements, such as cholinergic transmission, modulate striatal LTD.
To determine whether ACh and DA interact in striatal LTD, we have examined the role of nAChRs in LTD in the rat striatum. Agents that block nicotinic receptor function prevent LTD, and blockade of DA reuptake could rescue LTD in the presence of nAChR antagonists. Our results suggest that nAChR activation contributes to the induction of striatal LTD by interacting with dopaminergic mechanisms.
MATERIALS AND METHODS
Materials
Dihydro-β-erythroidine (DHβE), hexamethonium, SKF 81297, SKF 38393, ascorbate, dopamine, quinpirole, fluoxetine, nomifensine, andl-sulpiride were obtained commercially from Sigma (St. Louis, MO).
Tissue preparation
Neonatal Sprague Dawley rats (16–21 d old) were decapitated, and brains were quickly removed and placed in ice-cold modified artificial CSF (ACSF) bubbled with 95% O2/5% CO2. After a 5 min equilibration period, the brain tissue was blocked at anterior and posterior ends and subsequently attached with cyanoacrylate to a Teflon pad. The tissue was then completely submerged in ice-cold modified ACSF containing the following (in mm): sucrose 194, NaCl 30, KCl 4.5, MgCl2 1, NaHCO3 26, NaH2PO4 1.2, andd-glucose 10. The tissue was sectioned coronally, 400 μm in thickness, with a manually driven vibroslice (Campden Instruments, Loughborough, UK). These slices were transferred to normal ACSF containing the following (in mm): NaCl 124, KCl 4.5, CaCl2 2, MgCl2 1, NaHCO3 26, NaH2PO4 1.2, andd-glucose 10, and allowed to equilibrate for at least 45 min at room temperature with bubbling from a mixture of 95% O2/5% CO2 gas.
One hemisphere of a slice was then transferred to a recording chamber attached to a heated superfusion system. The slices were submerged and constantly superfused with ACSF bubbled with a mixture of 95% O2/5% CO2 gas. A digital thermometer was placed in the recording chamber to monitor the temperature throughout the duration of an experiment. All experiments were performed between 30 and 32°C, with fluctuation of bath temperature being ≤1°C during any given experiment. Drugs from stock solutions were dissolved to their final concentrations in ACSF and delivered to the recording chamber through either a gravity-assisted superfusion system or a motor-driven minipump system (VWR Scientific Products). The superfusion rate was stable at 2.5 ml/min. Drug-containing solutions were allowed to equilibrate in the recording chamber for at least 5–10 min.
Electrophysiology
Synaptically driven potentials or currents were recorded from dorsolateral striatum as described previously (Partridge et al., 2000). A twisted bipolar pair of tungsten wires was placed in the white matter that overlies the striatum, and square-wave current pulses were passed into the white matter containing the afferent fibers that innervate the dorsal striatum. Synaptic responses were recorded at a frequency of 0.05 Hz during test periods, during drug application, and before and after high-frequency stimulation (HFS). The HFS protocol consisted of four trains of pulses at 100 Hz, with each train being 1 sec in duration, and an intertrain interval of 10 sec.
Intracellular voltage-clamp recording. EPSCs were measured in single striatal neurons by whole-cell patch-clamp methods using an Axopatch 1-D amplifier. Neurons, either medium-sized or large, were identified with the use of differential interference contrast (DIC) optics and medium- or large-sized cells were identified and selected separately. In 66 medium-sized neurons, the whole-cell membrane capacitance ranged from 12 to 22 pF, and input resistance ranged from 110 to 275 MΩ. Paired t tests were performed across all experimental groups, and no significant differences were observed in any of the passive properties of the medium spiny neurons tested. Signals were filtered at 3 kHz, digitized at 6 kHz using a Digidata 1200 Interface, and recorded on a Pentium computer. Currents were measured in conventional ruptured-patch whole-cell mode using pipettes filled with a solution that consisted of the following (in mm): CsMeSO3 120, NaCl 5, TEA-Cl 10, HEPES 10, QX-314 5, EGTA 1.1, Mg-ATP 4, and Na-GTP 0.3. The intensity and duration of the stimuli, which averaged 43 ± 8 μsec and 0.36 ± 0.05 mA, was adjusted to generate synaptic currents ranging from 200 to 500 pA in amplitude, and all cells were clamped at −70 mV. During the HFS protocol, neurons were depolarized to −10 mV. In a few experiments (see Fig.2A), current-clamp recordings were performed from putative striatal cholinergic interneurons. The intracellular solution during these sets of experiments contained the following (in mm): 131 KMeSO4, 1 MgCl2, 0.1 CaCl2, 10 HEPES, 1 EGTA, 0.4 Na-GTP, and 2 Mg-ATP, and was adjusted to pH 7.3 with KOH.
Fig. 2.

Evidence for activation of cholinergic transmission in striatal slices. A, Current-clamp recording from a large-soma striatal cell chosen under DIC optics. Note the tonic firing in this cell. B, Extracellular field potentials recorded from a striatal slice in the presence of the cholinesterase inhibitor, neostigmine (3 μm), and the muscarinic receptor antagonist, scopolamine (500 nm). Each icon (black oval) represents the magnitude (in millivolts) of the evoked response as a function of time before, during, and after drug application. Bars above these icons indicate the duration of superfusion of drug-containing solution.C, EPSCs evoked from a large-soma striatal neuron voltage clamped at −70 mV. The stimulating electrode was placed in a position similar to that used to stimulate excitatory drive and synaptic plasticity from medium-sized neurons. Note the paired-pulse facilitation of the EPSC in this example. D, EPSCs evoked in a large-sized striatal neuron during HFS (100 Hz) of the white matter overlying the dorsal striatum.
Paired-pulse protocol. A stimulus protocol designed to test short-term plasticity associated with changes in presynaptic release probability was used during the majority of the experiments. This paired-pulse protocol consisted of identical stimuli delivered to afferent fibers with an interstimulus interval of 50 msec. In addition, a 3–5 mV hyperpolarizing pulse of voltage was applied 75 msec after the second stimulation to monitor the access and input resistance throughout the duration of an experiment. All cells included in the data analysis showed no significant change in the capacitative charging and discharging of their membrane throughout the duration of an experiment.
Extracellular field potential recording. Synaptically driven population spikes (PSs) were recorded with the use of a differential AC amplifier (model 1700, A-M Systems, Seattle, WA). The recording electrode was pulled from borosilicate glass filled with 0.9% NaCl solution and was placed at a distance of 1–2 mm ventral to the stimulating electrode. The intensity and duration of the stimulus used to evoke a population spike was chosen to generate a half-maximal response and averaged 90 ± 7 μsec in duration and 0.64 ± 0.08 mA in intensity. The stimulation parameters did not differ significantly in the presence or absence of drugs. During HFS, the intensity of the stimulus was set to evoke a maximal response and averaged 1.15 ± 0.15 mA.
High-speed chronoamperometry
DA release in slices was monitored by high-speed chronoamperometry using an IVEC-10 system (Medical Systems, Inc., Greenvale, NY) as described previously (Hoffman et al., 1998). Briefly, a single carbon fiber electrode (100 μm length × 30 μm outer diameter) was coated with Nafion (Sigma) and calibrated in vitro with DA (2–10 μm). Electrodes with selectivity of >300:1 for DA versus ascorbate were used in the experiment. To record electrochemical signals, the carbon fiber electrode was positioned ∼100 μm into the tissue, and square-wave pulses of 0.00 to +0.55 V (oxidation) or −0.55–0.00 V (reduction) were applied using IVEC-10.
Once a detectable electrochemical signal was achieved, the reduction/oxidation ratio was calculated by dividing the reduction current by the oxidation current at the peak of the electrochemical signal. Responses were digitized at a frequency of 5 Hz, and five such responses were averaged per point for presentation purposes. Increases in extracellular DA were evoked by afferent stimulation using the same stimulating electrode position and parameters used to evoke synaptic responses in the field-potential recording experiments. We examined the effect of single-pulse stimulation at 0.009–0.05 Hz and HFS (one train of 100 Hz stimulation, 1 sec duration).
Data and statistical analyses
Determination of the amplitude of EPSCs or PSs was achieved with Clampfit peak detection software. Values reported in the figures were normalized on a per recording basis and then plotted as the mean ± SE. In some experiments, the first 15 episodes were averaged and defined as “baseline transmission.” Evoked responses were compared with this value, and the ratio of subsequent responses to baseline responses was normalized. To determine whether HFS or drugs, or both, caused significant changes in synaptic responses, and whether the HFS effects differed between drug treatment groups, analysis was performed using two-way ANOVA. To determine whether drugs or HFS alone (time) altered synaptic responses, we used one-way ANOVA with post hoc Bonferroni tests or paired t tests, depending on the number of groups that were compared. The statistical criterion for significance was p < 0.05 for all analyses. Calculation of the paired-pulse response ratio was determined by dividing the amplitude of the second ESPC by the first evoked EPSC. The stimulus artifacts in the traces presented in all figures have been explicitly cut off to allow more detailed inspection of physiological responses without an interfering artifact. Apparent changes in stimulus artifacts are the product of the rapid rise and fall of this signal that leads to failed capture of every time point within the artifact at the digitization rates we have used. LTD magnitude was determined as reported previously by this group (Partridge et al., 2000) by averaging the normalized response amplitudes over time points 20–30 min after HFS.
RESULTS
Blockade of striatal LTD by nAChR antagonists
After high-frequency stimulation of the white matter dorsal to the lateral striatum, we regularly observe a decrease in the amplitudes of both the evoked PS obtained during field potential recording in striatum and the EPSC observed during whole-cell recording from striatal medium spiny projection neurons (Lovinger et al., 1993; Choi and Lovinger, 1997a,b; Partridge et al., 2000). In control slices for the present study, the magnitude of striatal LTD, as measured by the percentage of baseline PS or EPSC amplitude 20–30 min after HFS, averaged 64 ± 6% (n = 14) in field potential recordings and 68 ± 4% (n = 7) in whole-cell voltage-clamp recordings (Fig.1A–D). In field potential recordings, the initial negativity reflecting the amplitude of the fiber volley did not significantly change after HFS under any condition examined in this study.
Fig. 1.
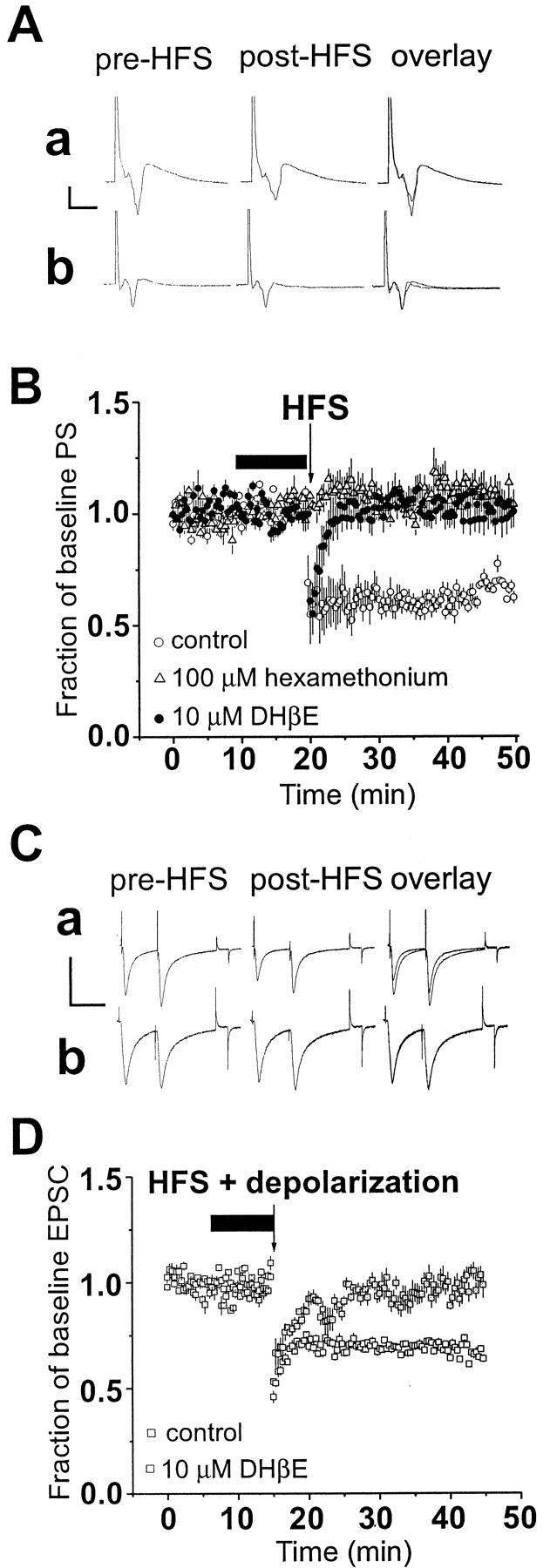
Striatal long-term depression is inhibited by nicotinic acetylcholine receptor antagonists. A, Average of 15 evoked field potentials before (pre-HFS) and after (post-HFS) high-frequency stimulation under control conditions showing a depression in the amplitude of the PS (a). On the right are the pre-HFS and post-HFS responses superimposed to show a more direct comparison (overlay). In part b, the same experiment was performed in the presence of 100 μmhexamethonium. Note the lack of change in the PS amplitude after HFS. Calibration (to left of traces): 0.3 mV, 5 msec. B, Time course plot of normalized mean ± SEM amplitude of the population spike (PS) during experiments examining the effect of HFS in dorsolateral striatum. A long-lasting depression of the PS was observed in control slices (n = 14) indicated by open circles. In the presence of hexamethonium (100 μm;n = 10), HFS did not reliably induce LTD, as indicated by the gray triangles. Application of dihydro-β-erythroidine (DHβE;n = 7) also greatly attenuated LTD induction, as indicated by the filled circles. Arrowindicates application of the HFS protocol. C, Average of 15–30 evoked EPSCs before (pre-HFS) and after (post-HFS) pairing HFS and depolarization under conditions in which no drug was added to the slice (a). In b are averaged EPSCs pre-HFS and post-HFS from a neuron exposed to 10 μmDHβE before and during HFS + depolarization. Calibration (toleft of traces): 300 pA, 50 msec.D, Plot of normalized EPSC (mean ± SEM) evoked from medium-sized striatal neurons under whole-cell conditions.Open squares are normalized responses from seven control cells. Pairing HFS and depolarization results in a decrease in the EPSC amplitude (p < 0.01; pairedt test). In the presence of 10 μm DHβE, as indicated by the filled squares, HFS and depolarization failed to evoke LTD (p > 0.05; paired t test). Arrow indicates application of HFS and depolarization. In B andD, drugs were added beginning 10 min before HFS and removed just after HFS.
When nicotinic receptor antagonists were present before and during the HFS protocol, the magnitude of LTD was severely reduced. Application of the use-dependent nicotinic receptor blocker hexamethonium (n = 10) (100 μm) prevented the induction of LTD in field potential experiments. The response amplitude was 109 ± 4% of pre-HFS response amplitude 20–30 min after HFS (Fig. 1A,B). LTD induction and expression were robust in the presence of a lower concentration of hexamethonium (10 μm) (percentage of baseline PS amplitude = 61%; n = 5), suggesting that the antagonist effect is concentration dependent. To more firmly establish that nAChRs are involved in LTD induction we examined the effect of the nAChR competitive antagonist DHβE (10 μm). In the presence of DHβE (n = 7), the average PS amplitude measured at 20–30 min post-HFS was 101 ± 6% of baseline, compared with 62 ± 5% in contralateral control hemislices (Fig. 1B). Analysis with two-way ANOVA indicated significant effects of drug and time when comparing the control, hexamethonium, and DHβE groups (Fdrug = 1268, df = 2, 4350,p < 0.0001; Ftime = 7.79, df = 149, 4350, p < 0.0001). One-way ANOVA analysis indicated a significant difference among groups (F = 189, df = 2, 447, p < 0.0001), and post hoc Bonferroni tests indicated significant differences between the control group and both the hexamethonium (100 μm) and DHβE groups (p < 0.001), but no significant difference between the two antagonist-treatment groups (p > 0.05).
To determine whether the change in PS amplitude correlated with efficacy changes at the level of the synapse, we repeated these experiments using whole-cell voltage-clamp recordings. In the presence of 10 μm DHβE (n = 7), evoked EPSC amplitude averaged 97 ± 2% of baseline values 20–30 min after HFS (Fig. 1C,D). Similarly, perfusion of 100 μm hexamethonium (n = 12) blocked the HFS-induced decrease in the amplitude of the EPSC (97 ± 3%). The average EPSC amplitude from control medium-sized neurons after HFS was 63 ± 5% of baseline. Analysis with two-way ANOVA indicated significant effects of drug and time when comparing the control, hexamethonium, and DHβE groups (Fdrug = 464.5, df = 2, 2700,p < 0.0001; Ftime = 7.37, df = 149, 2700, p < 0.0001). ANOVA analysis indicated a significant difference among groups (F = 96.8, df = 2, 447, p < 0.0001), and post hoc Bonferroni tests indicated significant differences between the control group and both the hexamethonium (100 μm) and DHβE groups (p< 0.001). In addition, none of these drugs, alone or in combination with other drugs used in this study, altered postsynaptic holding current levels or input resistance in these neurons during whole-cell recording (data not shown). To test for the reversibility of the effects of drugs, within-slice experiments were attempted but could not be performed because LTD could not be evoked in a slice when a second bout of HFS was delivered, regardless of the drug tested.
Nicotinic receptor antagonists do not alter the efficacy of glutamate-mediated synaptic transmission
It has been firmly established that glutamate is the major excitatory neurotransmitter in the striatum (Cherubini et al., 1988;Jiang and North, 1991; Lovinger et al., 1993). In the present study, neither hexamethonium (100 μm) nor DHβE (10 μm) affected the amplitude of either EPSCs (n = 19; paired t-test; p > 0.5) recorded from striatal cells during whole-cell voltage-clamp experiments or population spikes (n = 30, pairedt-test, p > 0.5) measured during field potential recordings from dorsolateral regions of the striatum (Fig.1). Furthermore, these agents did not alter the PPR ratio of EPSCs recorded from medium-sized striatal neurons. The PPR protocol was used to ascertain whether nicotinic receptor antagonists had any effect on presynaptic glutamate release. Together, these data suggest that the nicotinic receptor blockers used in these studies do not have any measurable effect on either stimulated glutamate release or the EPSC in striatum.
Electrical stimulation activates cholinergic neurons and DA release in the dorsal striatum
To determine whether ACh and DA are present at concentrations that can activate receptors during LTD induction in our striatal slice preparation, we conducted additional experiments in parallel with examining synaptic plasticity. It has been well documented that large aspiny cells in the striatum that are immunohistochemically positive for acetylcholinesterase are electrophysiologically distinct from their projection neuron counterparts (Wilson et al., 1990). Specifically, these neurons fire action potentials in a tonic yet irregular manner (Kawaguchi, 1993). We therefore wanted to determine whether identified large-sized cells in our slice preparation exhibited such properties. As shown in Figure 2A, tonic discharge of action potentials was observed during current-clamp recording from a large-sized striatal neuron. Similar firing patterns were observed in 10 of 14 striatal neurons with large somata. Spontaneous firing was never observed in striatal neurons with medium-sized somata. Although this result provides only indirect evidence that ACh is released in the slices, it is certainly consistent with previous results (Bennett and Wilson, 1999).
To more directly address the issue of cholinergic transmission in striatal slices, we examined the effects of the cholinesterase inhibitor, neostigmine (3 μm), on evoked responses. As shown in Figure 2B, PS amplitude was decreased (by 52 ± 5%; n = 3) when slices were exposed to neostigmine. The reduction in PS amplitude was reversed by coapplication of the muscarinic receptor antagonist scopolamine (500 nm), along with neostigmine. This result suggests that a basal level of ACh is released under those conditions when baseline excitatory transmission is monitored in our slice preparation. Afferent stimulation also activates cholinergic interneurons. In voltage-clamp experiments we observed that single-pulse afferent stimulation reliably evoked EPSCs in 96% (n = 24 of 25) of the large striatal neurons from which we recorded (Fig.2C). Repeated synaptic responses could be driven by high-frequency stimulation as shown in Figure 2D. Thus, acetylcholine release in striatal slices is driven both by the spontaneous activity of striatal cholinergic interneurons and by synaptic activation of these interneurons, and release may be especially robust during high-frequency afferent activation. These findings provide additional support for the idea that the striatal cholinergic system is activated under the stimulation and recording conditions used to examine LTD in our experiments.
To directly determine whether extracellular DA is increased after afferent stimulation in our experimental preparation, we used chronoamperometry with Nafion-coated carbon fiber electrodes. As shown in Figure 3, single-pulse afferent stimulation in striatal slices released a detectable amount of DA in five of eight slices that averaged 687 ± 249 nm. The stimulus parameters in these experiments were similar to those used for elicitation of field potentials. In the other three slices examined, we were unable to detect DA release; however, delivery of HFS trains, using parameters similar to those used to induce LTD, caused an increase in extracellular DA in every slice we examined, and the increases were much larger than those elicited by single-pulse stimulation, averaging 42.8 ± 21.3 μm(n = 8). We found that the amplitude and duration of these DA transients were quite variable from slice to slice. Thus it was not possible to compare stimulated DA release between slices as we did for LTD. It was equally difficult to perform the experiment using a within slice design (Schmitz et al., 2001), because the DA release appeared to be altered by the first set of HFS stimulus trains. Therefore, we were unable to examine increases in extracellular DA in the absence and presence of pharmacological agents.
Fig. 3.

Extracellular dopamine levels are elevated after direct stimulation of striatal slices. Representative time course of an experiment in which dopamine transients were evoked in the dorsal striatum by electrical stimulation of the white matter. Extracellular dopamine (DA) is plotted versus time.Asterisks indicate the application of single stimuli to the white matter overlying the striatum; arrows indicate the application of 100 Hz trains of stimuli. The oxidation curves are plotted for simplification purposes. Dashed linessignify responses to a single stimulus (left) and a high-frequency stimulus (right) at an enhanced level of time resolution for comparison purposes.
Restoration of LTD by dopamine reuptake inhibition in the presence of nAChR antagonists
Given the evidence that ACh and DA are released during high-frequency stimulation in striatum, it is possible that these two neurotransmitters might interact in some way to produce LTD induction. The nicotinic receptor antagonists hexamethonium and DHβE do not appear to affect evoked glutamatergic transmission at striatal synapses. Thus, it is possible that nicotinic receptors are functioning parasynaptically with respect to glutamate to alter LTD induction. In agreement with this idea, we observed that application of 10 μm nomifensine, a monoamine reuptake inhibitor, restored the expression of LTD in the presence of hexamethonium (Fig.4A,B). This result was observed in both field potential recordings (n = 8) and whole-cell voltage-clamp recordings from single neurons (n = 6). Similar results were observed on coapplication of DHβE and nomifensine (n = 9) (Fig. 4C). The HFS-induced decrease in striatal EPSCs after HFS was to 57 ± 7% of baseline under these pharmacological conditions. The magnitude of restored LTD was not significantly different from control LTD in the absence of any drug (one-way ANOVA comparing control with all conditions in which LTD was restored;F(3,26) = 0.19; p > 0.5).
Fig. 4.

LTD in the dorsal striatum is restored in the presence of nAChR antagonists and DA reuptake blockers.A, Blocking nicotinic receptors with 100 μm hexamethonium (n = 6) attenuates HFS-induced striatal LTD of evoked PS as indicated by the filled circles (paired t test; p > 0.05). Each point represents the normalized mean PS amplitude ± SEM. On coapplication of the dopamine reuptake inhibitor, nomifensine (10 μm), and hexamethonium (n = 8) in contralateral slices, HFS caused an LTD of the PS amplitude as indicated by the open circles(p < 0.05; paired t test). Two-way ANOVA indicated significant effects of drug and time in the experiment shown in B(Fdrug = 1680;Ftime = 8.89; df = 1, 149;p < 0.0001). Traces shown above the time course plot are the average of 15 field potential responses in the presence of 100 μm hexamethonium and 10 μmnomifensine before (left) and after (center) HFS. Calibration: 0.5 mV, 5 msec. Thefilled bar above baseline plot indicates the time course of drug-containing solution exposure to slice (where appropriate).B, Striatal LTD, measured with whole-cell recording from medium-sized striatal neurons, was inhibited by application of hexamethonium (hexameth) as indicated by the filled squares (n = 12). Superfusion of nomifensine along with hexamethonium restored LTD in single striatal neurons indicated by the open squares(n = 6; p < 0.05; pairedt test). Two-way ANOVA indicated significant effects of drug and time in the experiment shown in B(Fdrug = 662;Ftime = 8.70; df = 1, 149;p < 0.0001). Arrow indicates application of HFS and depolarization, and the filled bar represents the time course of the presence of drug(s).Traces shown above are averaged EPSCs before and after HFS in the presence of hexamethonium and nomifensine. Calibration: 300 pA, 20 msec. C, LTD was also observed on inclusion of nomifensine (nom) with DHβE (open squares;n = 9; p < 0.05) and when DHβE was coapplied with 3 μm cocaine (gray squares; n = 5) from intracellular recordings. However, when 1 μm l-sulpiride was included with this drug combination, HFS failed to induce LTD as indicated by the filled squares (n = 5). Two-way ANOVA indicated significant effects of drug and time in the experiment shown in B(Fdrug = 889;Ftime = 16.1; df = 2, 149;p < 0.0001). One-way ANOVA comparing all three groups yielded F = 100, df = 2, 447,p < 0.0001; post hoc Bonferroni tests indicate significant differences between all groups (p < 0.05). Arrow indicates application of HFS and depolarization. Traces are the averages of 15–30 EPSCs before (left) and after (center) HFS in the presence of 10 μmDHβE, 10 μm nomifensine plus 1 μmsulpiride. Calibration: 500 pA, 20 msec. D, Coapplication of exogenous dopamine receptor agonists with hexamethonium failed to restore LTD. As indicated by the gray circles, the PS amplitude did not decrease in the presence of hexamethonium and the dopamine receptor agonists after HFS (n = 11; p > 0.05; pairedt test). Note the lack of effect on baseline transmission of receptor agonists preceding HFS. In control hemislices from the contralateral hemisphere, inclusion of hexamethonium alone inhibited striatal LTD (n = 7).Traces are averages of 15 population spikes evoked in the presence of 100 μm hexamethonium plus 30 μm dopamine before and after HFS. Calibration: 0.3 mV, 5 msec.
Furthermore, cocaine, another DA transporter blocker, was capable of restoring LTD in the presence of a nicotinic receptor antagonist in the same manner as nomifensine (Fig. 4C). The percentage of baseline EPSC amplitude after HFS in the presence of cocaine and DHβE averaged 52 ± 8% (n = 5). Control experiments examining the expression of LTD under voltage clamp indicated that the magnitude of depression was normal when nomifensine (n= 6) or cocaine was applied alone, in the absence of nicotinic antagonists, before and during HFS (Fig.5C). Thus, DA transport blockers alone do not alter the induction or expression of LTD.
Fig. 5.
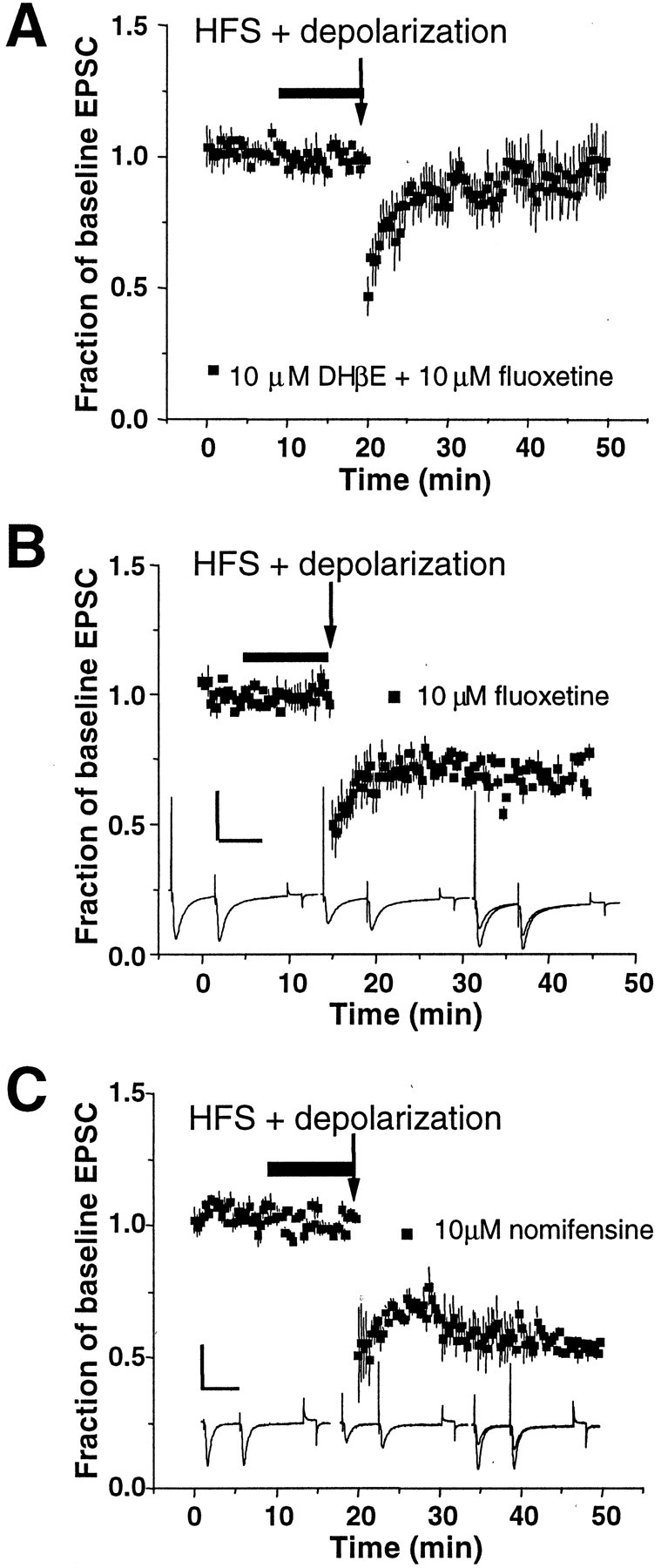
Serotonin transporters are not involved in the restoration of striatal LTD. A, Eachsquare represents normalized EPSC in the presence of 10 μm fluoxetine, a selective serotonin reuptake inhibitor, plus 10 μm DHβE (n = 5).Arrow indicates application of HFS and depolarization. Note the lack of rescue of LTD in the presence of fluoxetine.B, When applied alone, 10 μm fluoxetine (n = 4) does not alter the induction of striatal LTD as indicated by the filled squares.Traces are averages of 15 individual EPSCs. Calibration: 400 pA, 50 msec. C, Nomifensine (10 μm) when applied alone does not inhibit the induction of LTD in the dorsolateral striatum (n = 5). Two-way ANOVA analysis indicates significant drug and time effects (Fdrug = 1528;Ftime = 92.9; df = 2,149;p < 0.0001). Bonferroni post hoctests indicate significant differences between the DHβE + fluoxetine group and the two other groups (p < 0.001). Again, note the lack of an effect by any drug or drug combination on baseline transmission. Calibration: 400 pA, 50 msec.
Interestingly, coapplication of 30 μm dopamine (with 100 μm ascorbate) failed to restore LTD in the presence of hexamethonium (Fig. 4D) (n = 4). Additional attempts were made to rescue LTD expression with combined application of synthetic dopamine receptor agonists, including SKF 81297 [10 μm), SKF 38393 (3 μm, D1-DAR agonist], and quinpirole [10 μm, a D2-dopamine receptor (DAR) agonist] (n = 4). Application of these agonists together did not restore LTD expression when nAChRs were blocked. Furthermore, hexamethonium, dopamine receptor agonists, and (5)-3,5-dihydroxyphenylglycine (50 μm), a group I metabotropic glutamate (mGlu) receptor agonist, were coapplied in an attempt to activate both the DA and mGlu receptors that play roles in LTD (Calabresi et al., 1992; Lovinger et al., 1993). LTD was also not induced under these conditions (n = 3). It is important to note here that neither DA itself nor the synthetic dopamine receptor agonists had any effect on baseline excitatory transmission, consistent with most, but not all, previous reports (Calabresi et al., 1992; Umemiya and Raymond, 1997; Nicola and Malenka, 1998). These findings indicate that indiscriminate activation of DA receptors is not sufficient to restore LTD when nAChRs are blocked and suggests that the rescue of LTD most likely depends on spatially restricted activation of DA receptors near DA release sites, time-locked to HFS.
It has been shown previously that striatal LTD is dependent on D2-type dopamine receptor activation in the adult and developing striatum (Calabresi et al., 1992, 1997; Tang et al., 2001). We appliedl-sulpiride (1 μm), a D2-receptor antagonist, together with DHβE and nomifensine, and observed no long-term decrease in EPSC amplitude after HFS (Fig. 4C). The percent change in EPSC amplitude averaged 101 ± 3% after HFS and depolarization under these conditions (n = 5;p < 0.05; paired t test). Thus, the nomifensine-mediated rescue of LTD in the presence of nAChR antagonists does appear to involve enhanced synaptic DA release and activation of D2 receptors.
To ascertain whether nomifensine and cocaine are producing their actions via the dopamine reuptake system, as opposed to serotonin reuptake, experiments were performed in the presence of fluoxetine, a selective serotonin reuptake inhibitor. As shown in Figure5A, when coapplied with DHβE, fluoxetine (10 μm) did not restore LTD (n = 5; paired t test; p > 0.05). Application of fluoxetine alone had no apparent effect on baseline transmission or the expression of LTD (paired t test; p > 0.05;n = 4) (Fig. 5B). Thus, nomifensine and cocaine are most likely restoring LTD by blocking reuptake through the DA transporter. In addition, it is unlikely that norepinephrine is an important modulator at these synapses. Anatomical and biochemical studies have shown that adrenergic innervation to the dorsal striatum is sparse (Fuxe and Nilsson, 1965; Swanson and Hartman, 1975; Versteeg et al., 1976). Functionally, it has been shown that norepinephrine does not affect excitatory or inhibitory transmission in the dorsal striatum (Nicola and Malenka, 1998).
The nomifensine- and cocaine-rescued LTD in the presence of nAChR blockade was mechanistically similar to LTD observed in the absence of pharmacological agents. Previously, our group has shown that the expression of striatal LTD in young animals is strongly associated with an increase in the PPR ratio (Choi and Lovinger, 1997b). The results of whole-cell voltage-clamp experiments under different pharmacological manipulations are summarized in Figure 6. In control cells, after HFS, the PPR ratio increased to 135 ± 9% of baseline ratio values. In the presence of nicotinic antagonists, the post-HFS PPR ratio was 105 ± 8% of baseline values. In the presence of nicotinic antagonists and dopamine reuptake blockers, the PPR ratio increased to 127 ± 5% of baseline values after HFS. These findings suggest that the presynaptic element contributing to the expression of LTD in the developing striatum is preserved under these pharmacological conditions. None of the pharmacological agents used in this study had any significant effect on the pre-HFS PPR ratio (Fig.6A). The baseline PPR ratio was close to 1.0 in all experiments, as we have observed previously in preparations from rats in this age range (Choi and Lovinger, 1997b), suggesting that release probability was similar at all glutamatergic synapses before HFS.
Fig. 6.
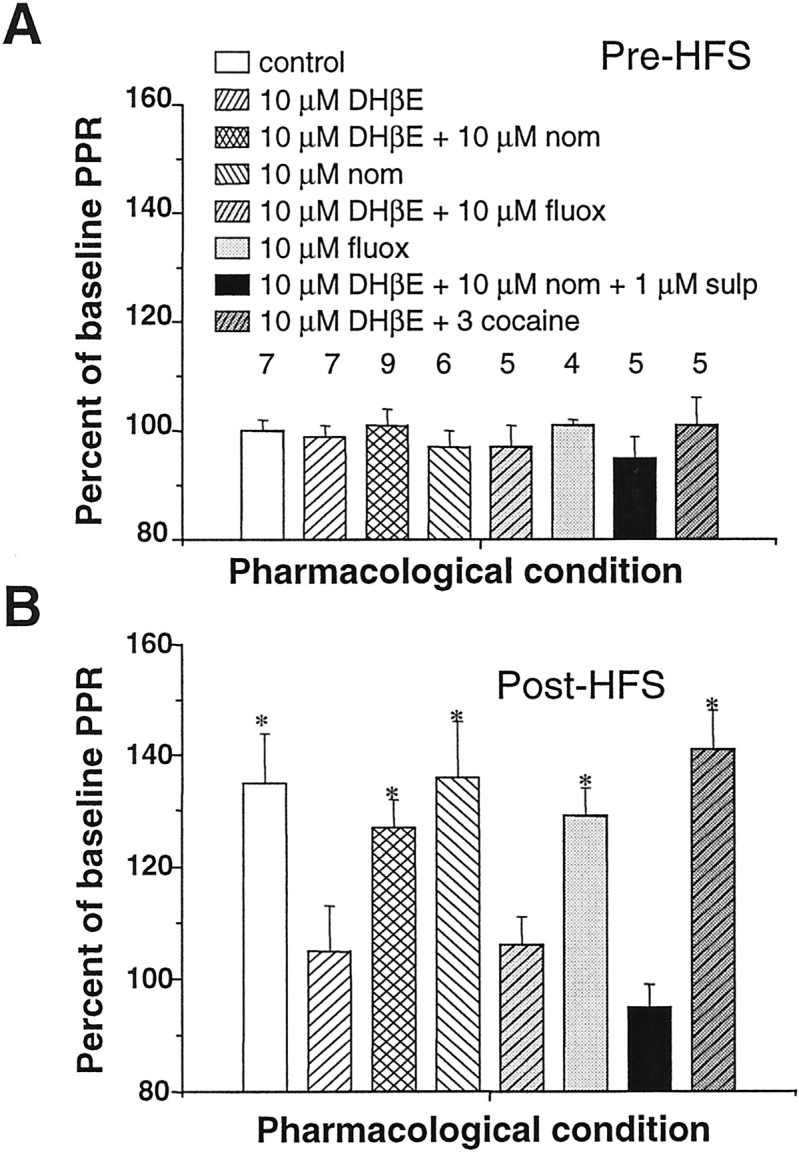
Restored LTD in the presence of nicotinic antagonists and DA reuptake inhibitors shares common features with LTD in control slices. A, Mean percentage (±SEM) of baseline paired-pulse response (PPR) ratio as a function of drug application before HFS and depolarization in cells under whole-cell voltage-clamp conditions. Baseline PPR was defined as the average of 15 PPR responses in the absence of drug. Under all pharmacological conditions, there was no significant change in the PPR in the pre-HFS drug exposure condition. The number above each bar represents the number of times an experiment in these conditions was repeated. nom, Nomifensine;fluox, fluoxetine; sulp, sulpiride.B, After HFS, PPR ratio increased significantly under those conditions when LTD was evoked but not when LTD was blocked (compare with Figs. 1, 4, and 5). Pattern code of histogram bars is identical to A. Asterisks indicate significant difference from pre-HFS baseline values (p < 0.05; paired ttest).
DISCUSSION
Our findings indicate that nicotinic acetylcholine receptor activation participates in induction of striatal LTD. Acetylcholine may thus act as an important modulator of striatal LTD, perhaps serving a “gating” role that facilitates plasticity under conditions of strong coactivation of extrinsic glutamatergic and dopaminergic afferents and intrinsic cholinergic neurons. The finding that LTD could be restored by DA reuptake blockers in the presence of nAChR antagonists suggests that nAChR activation is not absolutely required for LTD induction. Instead, it appears that nAChRs interact with striatal dopaminergic transmission to promote LTD induction. One mechanism by which this might occur is via nAChR stimulation of DA release from the terminals of nigral neurons. Indeed, it has recently been shown that nicotinic receptors play a prominent role in increased dopamine release produced by afferent stimulation in striatal slices (Zhou et al., 2001); however, we cannot yet exclude other possible mechanisms.
The observation that DA reuptake blockers were effective in restoring LTD in the presence of nAChR antagonists, whereas global DA receptor activation was not, suggests that a strict spatial and temporal pattern of DA release is required for LTD induction. It is likely that the dopamine receptors involved in LTD are located very near dopaminergic terminals, because this is where the DA would be highest during reuptake blockade (Gonon, 1997). Extracellular DA has been shown to be high during and just after HFS in slices that were not treated with receptor antagonists or uptake blockers (Calabresi et al., 1995), consistent with the chronoamperometric measures in the present study. However, the observation that nomifensine and cocaine do not induce LTD in the absence of HFS indicates that increased dopamine in the synaptic cleft is not sufficient to initiate synaptic plasticity.
The lack of rescue of LTD in the presence of nAChR antagonists during constant, global stimulation of DA receptors may seem surprising; however, there are several possible explanations for this observation. It should be emphasized that prolonged exposure to agonists could induce receptor desensitization that would reduce the signaling needed for LTD induction during HFS. Another possibility is that activation of DA receptors at sites distant from the locus of DA release, as would be the case in the experiment in which dopamine receptor agonists are bath applied, might activate signaling mechanisms that override or counteract the mechanisms involved in LTD induction. It is well known that dopamine receptor activation produces various modulatory effects on the excitability of striatal neuronal somata (Nicola et al., 2000). Thus, application of agonist to the neuronal cell body might interfere with restoration of LTD. Under those circumstances in which the DA transporter was blocked, one might expect that only dopamine receptors near DA reuptake sites would be activated. The idea of differential roles in plasticity of monoamine receptors located on different neuronal elements is not without precedent. A study examiningAplysia sensory to motor neuron synapses suggested that monoamine application at the soma of a sensory neuron produced different cooperative effects on plasticity than did serotonin-induced modulation at the synaptic cleft (Sherff and Carew, 1999). Our results might seem especially surprising in light of the observation of Calabresi et al. (1992) that LTD could be restored by acute application of DA in DA-depleted rat striatum. However, it is well known that DA depletion induces supersensitivity of DA receptors (Araki et al., 2000), and this may facilitate activation of receptors that play key roles in LTD induction. The difference in age of the animals in the present study in relation to that of Calabresi et al. (1992) might also contribute to the different results we have obtained.
It was reported previously that DA receptors can participate in striatal long-term potentiation (LTP) (Wickens et al., 1996;Calabresi et al., 1997; Centonze et al., 1999), and this observation suggests that activation of DA receptors plays a role in synaptic changes that may run counter to LTD. It is interesting to note that induction of LTP required local, transient DA application in theWickens et al. (1996) study. The necessity for tight temporal and spatial linkage of DA release with other events involved in the induction of plasticity is intuitively attractive because the depression of synaptic transmission should only occur during coordinated activation of glutamatergic and dopaminergic transmission and possibly cholinergic transmission as well.
If nACh receptors facilitate LTD induction by stimulating DA release, then the receptor subtypes that most likely play this role are the α3β2 and α4β2 subtypes that are known to enhance striatal dopamine release (Kulak et al., 1997; Sharples et al., 2000). The observation that DHβE inhibits LTD induction is consistent with this idea, because this antagonist blocks α3-containing receptors in preference to those containing α7 (Brioni et al., 1997). However, more extensive pharmacological studies will be needed to determine the role of different nAChR subtypes in striatal LTD. Interestingly, it has been reported that α7-containing nicotinic receptor activation can induce long-term potentiation in the ventral tegmental area (Mansvelder and McGehee, 2000). In this study, the α7 nicotinic receptors were suggested to reside on presynaptic glutamatergic terminals. Recent evidence suggests the presence of α7-containing nicotinic receptors on glutamatergic terminals in striatum, and it has been suggested that these receptors indirectly modulate dopamine release in striatal slices by enhancing glutamate release (Kaiser and Wonnacott, 2000). However, we did not observe any evidence for participation of this mechanism in striatal synaptic transmission in our preparation, because nAChR antagonists did not alter pre-HFS glutamatergic transmission or PPR (Fig. 6) in the present study.
Our findings may have significant clinical relevance, because intake of nicotine may inadvertently reinforce the learning and performance of motor sequences associated with smoking or tobacco intake. Acute activation of striatal nAChRs during an initial smoking session could lead to the onset of plasticity leading to the reinforcement of smoking behaviors. In these individuals, motor habits learned during a smoking session could be associated with increased release of DA and a stronger “reward” signal. Alternatively, desensitization of nAChRs during continuous smoking could disrupt normal LTD induction and affect normal habit formation. Furthermore, our findings suggest potential cooperative effects between tobacco and cocaine on the expression of synaptic plasticity in the dorsal striatum in humans.
It is important to note that we have examined the modulatory role of nAChRs in LTD in the developing striatum and thus our findings have implications for the onset and progression of habit learning. Increases in nAChR density, in areas of the brain including the striatum, occur in human smokers and rats chronically exposed to nicotine (Flores et al., 1992; Breese et al., 1997). Alterations of cholinergic or dopaminergic transmission during development, for example in teenaged humans, could lead to faulty LTD and dysfunctional positive reinforcement-driven habit learning that would last a lifetime.
These data suggest one mechanism by which striatal cholinergic interneurons participate in the reward-mediated initiation of action repertoires involving motivational drive (Graybiel et al., 1994). This study supports the idea that endogenous nAChR activation in striatum has a profound impact on the integration of information at the level of synaptic transmission.
Footnotes
This work was supported by grants from the National Institutes of Health (NS30470) and the Tourette Syndrome Association. We thank Drs. R. Blakely, D. J. Linden, K.-C. Tang, and D. Winder for discussion and comments on earlier versions of this manuscript. We also thank R. Blakely for the generous donation of cocaine for this study. In addition, we acknowledge Francois Pomerleau for excellent assistance with the chronoamperometry experiments.
Correspondence should be addressed to Dr. David M. Lovinger, Laboratory of Integrative Neuroscience, National Institute on Alcohol Abuse and Alcoholism, Room 158H Park 5 Building, 12420 Parklawn Drive, Rockville, MD 20852. E-mail: lovindav@mail.nih.gov.
REFERENCES
- 1.Aosaki T, Graybiel AM, Kimura M. Effect of the nigrostriatal dopamine system on acquired neural responses in the striatum of behaving monkeys. Science. 1994;265:412–415. doi: 10.1126/science.8023166. [DOI] [PubMed] [Google Scholar]
- 2.Araki T, Tanji H, Kato H, Imai Y, Mizugaki M, Itoyama Y. Temporal changes of dopaminergic and glutamatergic receptors in 6-hydroxydopamine-treated rat brain. Eur Neuropsychopharmacol. 2000;10:365–375. doi: 10.1016/s0924-977x(00)00094-8. [DOI] [PubMed] [Google Scholar]
- 3.Bennett BD, Wilson CJ. Spontaneous activity of neostriatal cholinergic interneurons in vitro. J Neurosci. 1999;19:5586–5596. doi: 10.1523/JNEUROSCI.19-13-05586.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC, Leonard S. Effect of smoking history on [3H]nicotine binding in human postmortem brain. J Pharmacol Exp Ther. 1997;282:7–13. [PubMed] [Google Scholar]
- 5.Brioni JD, Decker MW, Sullivan JP, Arneric SP. The pharmacology of (−)-nicotine and novel cholinergic channel modulators. Adv Pharmacol. 1997;37:153–214. doi: 10.1016/s1054-3589(08)60950-3. [DOI] [PubMed] [Google Scholar]
- 6.Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calabresi P, Pisani A, Mercuri NB, Bernardi G. Post-receptor mechanisms underlying striatal long-term depression. J Neurosci. 1994;14:4871–4881. doi: 10.1523/JNEUROSCI.14-08-04871.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calabresi P, Fedele E, Pisani A, Fontana G, Mercuri NB, Bernardi G, Raiteri M. Transmitter release associated with long-term synaptic depression in rat corticostriatal slices. Eur J Neurosci. 1995;7:1889–1894. doi: 10.1111/j.1460-9568.1995.tb00710.x. [DOI] [PubMed] [Google Scholar]
- 9.Calabresi P, Saiardi A, Pisani A, Baik JH, Centonze D, Mercuri NB, Bernardi G, Borrelli E. Abnormal synaptic plasticity in the striatum of mice lacking dopamine D2 receptors. J Neurosci. 1997;17:4536–4544. doi: 10.1523/JNEUROSCI.17-12-04536.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G. Acetylcholine-mediated modulation of striatal function. Trends Neurosci. 2000a;23:120–126. doi: 10.1016/s0166-2236(99)01501-5. [DOI] [PubMed] [Google Scholar]
- 11.Calabresi P, Gubellini P, Centonze D, Picconi B, Bernardi G, Chergui K, Svenningsson P, Fienberg AA, Greengard P. Dopamine and cAMP-regulated phosphoprotein 32 kDa controls both striatal long-term depression and long-term potentiation, opposing forms of synaptic plasticity. J Neurosci. 2000b;20:8443–8451. doi: 10.1523/JNEUROSCI.20-22-08443.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Centonze D, Gubellini P, Picconi B, Calabresi P, Giacomini P, Bernardi G. Unilateral dopamine denervation blocks corticostriatal LTP. J Neurophysiol. 1999;82:3575–3579. doi: 10.1152/jn.1999.82.6.3575. [DOI] [PubMed] [Google Scholar]
- 13.Cherubini E, Herrling PL, Lanfumey L, Stanzione P. Excitatory amino acids in synaptic excitation of rat striatal neurones in vitro. J Physiol (Lond) 1988;400:677–690. doi: 10.1113/jphysiol.1988.sp017143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi S, Lovinger DM. Decreased frequency but not amplitude of quantal synaptic responses associated with expression of corticostriatal long-term depression. J Neurosci. 1997a;17:8613–8620. doi: 10.1523/JNEUROSCI.17-21-08613.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi S, Lovinger DM. Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapses. Proc Natl Acad Sci USA. 1997b;94:2665–2670. doi: 10.1073/pnas.94.6.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clarke PB, Pert A. Autoradiographic evidence for nicotine receptors on nigrostriatal and mesolimbic dopaminergic neurons. Brain Res. 1985;348:355–358. doi: 10.1016/0006-8993(85)90456-1. [DOI] [PubMed] [Google Scholar]
- 17.Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol. 1992;41:31–37. [PubMed] [Google Scholar]
- 18.Fuxe K, Nilsson BY. Mechanoreceptors and adrenergic nerve terminals. Experientia. 1965;21:641–642. doi: 10.1007/BF02144053. [DOI] [PubMed] [Google Scholar]
- 19.Giorguieff MF, Le Floc'h ML, Westfall TC, Glowinski J, Besson MJ. Nicotinic effect of acetylcholine on the release of newly synthesized (3H)dopamine in rat striatal slices and cat caudate nucleus. Brain Res. 1976;106:117–131. doi: 10.1016/0006-8993(76)90077-9. [DOI] [PubMed] [Google Scholar]
- 20.Gonon F. Prolonged and extrasynaptic excitatory action of dopamine mediated by D1 receptors in the rat striatum in vivo. J Neurosci. 1997;17:5972–5978. doi: 10.1523/JNEUROSCI.17-15-05972.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graybiel AM, Aosaki T, Flaherty AW, Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265:1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- 22.Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1–m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14:3351–3363. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoffman AF, Lupica CR, Gerhardt GA. Dopamine transporter activity in the substantia nigra and striatum assessed by high-speed chronoamperometric recordings in brain slices. J Pharmacol Exp Ther. 1998;287:487–496. [PubMed] [Google Scholar]
- 24.Jiang ZG, North RA. Membrane properties and synaptic responses of rat striatal neurones in vitro. J Physiol (Lond) 1991;443:533–553. doi: 10.1113/jphysiol.1991.sp018850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaiser S, Wonnacott S. alpha-Bungarotoxin-sensitive nicotinic receptors indirectly modulate [(3)H]dopamine release in rat striatal slices via glutamate release. Mol Pharmacol. 2000;58:312–318. doi: 10.1124/mol.58.2.312. [DOI] [PubMed] [Google Scholar]
- 26.Kawaguchi Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci. 1993;13:4908–4923. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knowlton BJ, Mangels JA, Squire LR. A neostriatal habit learning system in humans. Science. 1996;273:1399–1402. doi: 10.1126/science.273.5280.1399. [DOI] [PubMed] [Google Scholar]
- 28.Kulak JM, Nguyen TA, Olivera BM, McIntosh JM. α-Conotoxin MII blocks nicotine-stimulated dopamine release in rat striatal synaptosomes. J Neurosci. 1997;17:5263–5270. doi: 10.1523/JNEUROSCI.17-14-05263.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lovinger DM, Tyler EC, Merritt A. Short- and long-term synaptic depression in rat neostriatum. J Neurophysiol. 1993;70:1937–1949. doi: 10.1152/jn.1993.70.5.1937. [DOI] [PubMed] [Google Scholar]
- 30.Malenka RC, Kocsis JD. Presynaptic actions of carbachol and adenosine on corticostriatal synaptic transmission studied in vitro. J Neurosci. 1988;8:3750–3756. doi: 10.1523/JNEUROSCI.08-10-03750.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- 32.Nicola SM, Malenka RC. Modulation of synaptic transmission by dopamine and norepinephrine in ventral but not dorsal striatum. J Neurophysiol. 1998;79:1768–1776. doi: 10.1152/jn.1998.79.4.1768. [DOI] [PubMed] [Google Scholar]
- 33.Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- 34.Packard MG, White NM. Dissociation of hippocampus and caudate nucleus memory systems by posttraining intracerebral injection of dopamine agonists. Behav Neurosci. 1991;105:295–306. doi: 10.1037//0735-7044.105.2.295. [DOI] [PubMed] [Google Scholar]
- 35.Partridge JG, Tang KC, Lovinger DM. Regional and postnatal heterogeneity of activity-dependent long-term changes in synaptic efficacy in the dorsal striatum. J Neurophysiol. 2000;84:1422–1429. doi: 10.1152/jn.2000.84.3.1422. [DOI] [PubMed] [Google Scholar]
- 36.Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharples CG, Kaiser S, Soliakov L, Marks MJ, Collins AC, Washburn M, Wright E, Spencer JA, Gallagher T, Whiteaker P, Wonnacott S. UB-165: a novel nicotinic agonist with subtype selectivity implicates the α4β2* subtype in the modulation of dopamine release from rat striatal synaptosomes. J Neurosci. 2000;20:2783–2791. doi: 10.1523/JNEUROSCI.20-08-02783.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sherff CM, Carew TJ. Coincident induction of long-term facilitation in Aplysia: cooperativity between cell bodies and remote synapses. Science. 1999;285:1911–1914. doi: 10.1126/science.285.5435.1911. [DOI] [PubMed] [Google Scholar]
- 39.Swanson LW, Hartman BK. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J Comp Neurol. 1975;163:467–505. doi: 10.1002/cne.901630406. [DOI] [PubMed] [Google Scholar]
- 40.Tang KC, Low MJ, Grandy DK, Lovinger DM. Dopamine-dependent synaptic plasticity in striatum during in vivo development. Proc Natl Acad Sci USA. 2001;98:1255–1260. doi: 10.1073/pnas.031374698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Umemiya M, Raymond LA. Dopaminergic modulation of excitatory postsynaptic currents in rat neostriatal neurons. J Neurophysiol. 1997;78:1248–1255. doi: 10.1152/jn.1997.78.3.1248. [DOI] [PubMed] [Google Scholar]
- 42.Versteeg DH, Van Der Gugten J, De Jong W, Palkovits M. Regional concentrations of noradrenaline and dopamine in rat brain. Brain Res. 1976;113:563–574. doi: 10.1016/0006-8993(76)90057-3. [DOI] [PubMed] [Google Scholar]
- 43.Wickens JR, Begg AJ, Arbuthnott GW. Dopamine reverses the depression of rat corticostriatal synapses which normally follows high-frequency stimulation of cortex in vitro. Neuroscience. 1996;70:1–5. doi: 10.1016/0306-4522(95)00436-m. [DOI] [PubMed] [Google Scholar]
- 44.Wilson CJ, Chang HT, Kitai ST. Firing patterns and synaptic potentials of identified giant aspiny interneurons in the rat neostriatum. J Neurosci. 1990;10:508–519. doi: 10.1523/JNEUROSCI.10-02-00508.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wonnacott S, Kaiser S, Mogg A, Soliakov L, Jones IW. Presynaptic nicotinic receptors modulating dopamine release in the rat striatum. Eur J Pharmacol. 2000;393:51–58. doi: 10.1016/s0014-2999(00)00005-4. [DOI] [PubMed] [Google Scholar]
- 46.Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]