

Abstract
To elucidate mechanisms underlying mitochondrial dysfunctions induced by glutamate, we have examined the effects of in vivo treatment with the ionotropic glutamate receptor agonist kainate on localization of the transcription factor activator protein-1 (AP-1) in mitochondria as well as nuclei of murine brain. A systemic administration of kainate dramatically enhanced AP-1 DNA binding in both mitochondrial and nuclear extracts of mouse cerebral cortex and hippocampus 1 hr to 3 d later. Unlabeled AP-1 probe selectively competed for AP-1 DNA binding in mitochondrial extracts of cortex and hippocampus obtained from mice injected with kainate. Supershift and immunoblotting analyses revealed participation of c-Fos, Fos-B, and Jun-B proteins in potentiation by kainate of mitochondrial AP-1 DNA binding in cortex and hippocampus. An immunohistochemical study demonstrated marked expression by kainate of c-Fos protein in the pyramidal and dentate granular layers, whereas an immunoelectron microscopic analysis showed localization of c-Fos protein within mitochondria, as well as nuclei, of the CA1 pyramidal and dentate granular cells in hippocampus obtained 2 hr after the administration of kainate. Mitochondrial AP-1 DNA binding was inhibited by particular unlabeled oligonucleotides containing sequences similar to the AP-1 site found in the noncoding region of mitochondrial DNA. Kainate markedly potentiated binding of radiolabeled oligonucleotide probes containing sequences effective in competing for AP-1 DNA binding in hippocampal mitochondrial extracts. These results suggest that kainate may facilitate expression of the AP-1 complex and subsequent translocation into mitochondria to participate in mechanisms associated with transcriptional regulation of mitochondrial DNA in murine hippocampus.
Keywords: activator protein-1, DNA binding, c-Fos protein, kainate, mitochondria, mitochondrial DNA
The excitatory amino acidl-glutamate (Glu) participates in various neuronal processes from transient signal communication to long-term plasticity as well as neuropathology via interactions with its ionotropic and metabotropic receptors in the mammalian CNS (Greenamyre and Porter, 1994; Michaelis, 1998). Ionotropic Glu receptors are classified into NMDA, α-amino-3-hydroxy-5-methylisoxazole-4-propionate, and kainate receptor subtypes according to differential sensitivities to these exogenous agonists and to permeable cations (Hollmann and Heinemann, 1994; Schoepfer et al., 1994).
Several independent lines of evidence indicate that extracellular Glu signals modulate de novo synthesis of particular proteins through expression of certain transcription factors and subsequent translocation of these factors from the cytoplasm to the nucleus. In cultured cerebellar granular neurons, Glu signals lead to expression of both c-fos gene and c-Fos protein with marked potentiation of DNA binding activity of the transcription factor activator protein-1 (AP-1) that consists of Fos and Jun family member proteins (Szekely et al., 1989, 1990; Sakurai et al., 1992). Intraperitoneal (Sonnenberg et al., 1989) and intracerebroventricular (Ogita and Yoneda, 1994) injections of particular agonists at ionotropic Glu receptors result in marked potentiation of AP-1 DNA binding in nuclear extracts of the rodent brain. A systemic administration of kainate leads to rapid and prolonged enhancement of nuclear AP-1 DNA binding in rat (Pennypacker et al., 1994b) and mouse (Azuma et al., 1996) hippocampi, whereas that of NMDA induces a rapid but transient increase in nuclear AP-1 DNA binding in murine hippocampus (Yoneda and Ogita, 1994). Enhancement by these Glu agonists results at least in part from expression of Fos family proteins and subsequent translocation from the cytoplasm to the nucleus (Yoneda et al., 1999a,b). Activation of the NMDA receptor also increases nuclear DNA binding activity of nuclear factor κB after translocation into the nucleus in cultured cortical neurons (Ko et al., 1998).
There is accumulating evidence that activation of Glu receptors leads to different mitochondrial dysfunctions as a consequence of mitochondrial depolarization elicited by a loss of Ca2+ homeostasis (Schinder et al., 1996;White and Reynolds, 1996) and impairment of metabolic homeostasis by reduction of different enzyme activities within mitochondria (Atlante et al., 1999; Delgado-Esteban et al., 2000). In addition, recent studies suggest that some nuclear transcription factors may participate in regulation of mitochondrial functions through transcriptional regulation of mitochondrial DNA. Cammarota et al. (1999) demonstrated that the transcription factor cAMP-responsive element (CRE) binding protein (CREB), but not c-Fos protein, is located within mitochondria as well as nuclei in rat hippocampus. Mitochondrial CREB is phosphorylated on its serine-133 after one trial of inhibitory avoidance training procedures in rat hippocampus (Bevilaqua et al., 1999). Very little is known, however, about the responsiveness of transcription factors within mitochondria to activation of particular Glu receptors located on plasma membranes. In this article, we have attempted to demonstrate localization of the AP-1 complex with DNA binding activity after in vivo treatment with kainate in mitochondria of murine brain using different techniques, including electron microscopic analysis.
MATERIALS AND METHODS
Materials. [α-32P]deoxy-ATP (111 TBq/mmol) was purchased from NEN/DuPont (Boston, MA). All oligonucleotides were provided by Inter Tech Co. (Tokyo, Japan) and Amersham Biosciences (Buckinghamshire, UK). Kainate was purchased from Wako Pure Chemical Industries (Osaka, Japan). Polyclonal antibodies against histone H1 (AE-4), cytochrome c (H-104), c-Fos (H-125), Fos-B (H-75), Fra-1 (R-20), Fra-2 (Q-20), c-Jun (H-79), Jun-B (N-17), and Jun-D (329) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Biotinylated anti-rabbit IgG antibody was supplied by Vector Laboratories (Burlingame, CA). Gold (10 nm)-conjugated goat anti-rabbit IgG antibody (G7402) was provided by Sigma Chemicals (St. Louis, MO). All other chemicals used were of the highest purity that is available commercially.
Drug administration. The protocol used here meets the guidelines of the Japanese Society for Pharmacology and was approved by the Committee for Ethical Use of Experimental Animals at Setsunan University. All efforts were made to minimize animal suffering, to reduce the number of animals used, and to use alternatives to in vivo techniques. Adult male Std-ddY mice weighing 30–35 gm were housed in metallic breeding cages in a room with a 12 hr light/dark cycle and a humidity of 55% at 23°C, with ad libitumaccess to food and water for at least 4 d before use. Animals were injected intraperitoneally with kainate or NMDA dissolved in PBS at a dose of 30 or 100 mg/kg, respectively, followed by return to their home cages until the time of decapitation. The administration was performed between 10 A.M. and 1 P.M. to avoid possible diurnal rhythm as described previously (Azuma et al., 1996).
Sample preparation. Brains were removed quickly and immersed in homogenizing buffer described below at 2°C, followed by dissection of hippocampus and the frontal part of cerebral cortex according to the procedures described by Glowinski and Iversen (1966). Tissues were homogenized in 1 ml of homogenizing buffer containing 10 mm Tris-HCl buffer, pH 7.5, 0.32m sucrose, 1 mm EDTA, 1 mm EGTA, 5 mmdithiothreitol (DTT), 10 mm each of phosphatase inhibitors (NaF and sodium β-glycerophosphate), and 1 μg/ml each of protease inhibitors [(p-amidinophenyl)methanesulfonyl fluoride, benzamidine, leupeptin, and antipain] using a Dounce homogenizer with a B-type pestle, followed by centrifugation at 1000 ×g for 10 min. Pellets (P1 fractions) and supernatants (S1 fractions) were used to prepare nuclear and mitochondrial fractions, respectively. For preparation of nuclear extracts, P1 fractions were resuspended in 1 ml of homogenizing buffer containing 0.5% Nonidet P-40, followed by centrifugation at 1000 × gfor 10 min to obtain nuclear fractions. These nuclear fractions were suspended in 0.3 ml of extraction buffer containing 50 mm Tris-HCl buffer, pH 7.5, 10% glycerol, 400 mm NaCl, 1 mm EDTA, 1 mm EGTA, 5 mm DTT, 0.5% Nonidet P-40, and the aforementioned inhibitors for phosphatases and proteases. Suspensions were kept on ice for 30 min, followed by centrifugation at 20,000 × g for 5 min. The resulting supernatants were pooled as nuclear extracts. Mitochondrial fractions were prepared using the method described by Clark and Nicklas (1970)with several modifications. Briefly, S1 fractions were centrifuged at 15,000 × g for 10 min. Pellets were gently suspended with 0.2 ml of 3% Ficoll in homogenizing buffer and then layered over 1 ml of 6% Ficoll in homogenizing buffer to produce a discontinuous density gradient for subsequent centrifugation at 11,500 × g for 30 min. After Ficoll solutions were removed, pellets (mitochondrial fractions) were washed with 1 ml of homogenizing buffer. Mitochondrial extracts were prepared by the addition of extraction buffer (0.15 ml) under the conditions used for preparation of nuclear extracts. Both extracts thus obtained were stored at −80°C until each use. Buffers and any solutions used in this study were sterilized by autoclave or filtration through a nitrocellulose membrane filter with a pore size of 220 nm before each use. Protein concentrations were measured by the method of Watanabe et al. (1986) using the Protein Assay Rapid kit (Wako Pure Chemical Industries, Osaka, Japan).
Electrophoretic mobility shift assay. Assays were performed using double-stranded oligonucleotides with a base length of 22 mer containing consensus core sequence for AP-1 (5′-CTAGTGATGAGTCAGCCGGATC-3′) as a probe for detection of DNA binding activity of AP-1. Of the double-stranded oligonucleotides (MT-1 to MT-10) containing each sequence similar to the AP-1 site in the noncoding region of mitochondrial DNA, MT-3 (5′-AGTTTATGACTGTATGGTG-3′) and MT-9 (5′-AAAATATGACTTATATTTT-3′) were also used as probes for detection of mitochondrial DNA binding activity. All of these probes were labeled with [α-32P]deoxy-ATP using Klenow fragment of DNA polymerase I in 10 mm Tris-HCl buffer, pH 7.5, containing 50 mm NaCl, 10 mmMgCl2, and 1 mm DTT at 25°C for 30 min in the presence of 50 μm each of deoxy-GTP, deoxy-CTP, and deoxy-TTP (Azuma et al., 1996). Aliquots of extracts were incubated with 50 fmol probe (0.5–5 × 106 cpm/pmol) in 20 μl of 50 mm Tris-HCl buffer, pH 7.5, containing 1 μg poly(dI-dC), 10% glycerol, 10 mmMgCl2, 160 mm NaCl, 1 mm EDTA, 1 mm EGTA, 5 mm DTT, 5 mm each of the aforementioned phosphatase inhibitors, and 1 μg/ml each of the protease inhibitors for 30 min at 2°C. Bound and free probes were separated by electrophoresis on a 5% polyacrylamide gel in buffer, pH 8.5, containing 50 mm Tris, 0.38m glycine, and 2 mm EDTA at a constant voltage of 11 V/cm for 1.5 hr in an ice bath. Gels were fixed and dried, followed by exposure to x-ray films for different periods to obtain autoradiograms most adequate for subsequent quantitative densitometry. Densitometric analysis for quantification of autoradiograms was performed with the aid of a densitograph AE-9600 (Atto Co. Tokyo, Japan) (Ogita and Yoneda, 1995).
Supershift assays were performed by the method described in our previous report (Azuma et al., 1999). Before the addition of a radiolabeled probe into incubation mixture, samples were incubated with each antibody against one of the Fos and Jun family proteins (c-Fos, Fos-B, Fra-1, Fra-2, c-Jun, Jun-B, and Jun-D) at 4°C overnight. Samples were then subjected to the electrophoretic mobility shift assay and subsequent autoradiography for determination of DNA binding activity. In supershift assays, antibodies are usually effective in either inhibiting DNA binding activity or altering the mobility of radioactive bands on the gel.
Immunoblotting assays. Samples were boiled at 100°C for 5 min in the presence of 2% SDS, 5% 2-mercaptoethanol, 10% glycerol, and 0.01% bromophenol blue immediately after preparation and then stored at −80°C until use. Immunoblotting assays were performed as described previously (Azuma et al., 1996). Briefly, aliquots were loaded on 7.5% polyacrylamide gel for detection of c-Fos protein or on 15% gel for detection of histone H1 and cytochrome c, respectively. Proteins were transferred to polyvinylidene fluoride membrane (Immobilon-P, Millipore, Bedford, MA) and blocked with 5% skim milk dissolved in washing buffer (Tris-buffered saline containing 0.05% Tween 20). Blots were incubated with each primary antibody (anti-c-Fos, 1:1000; anti-histone H1, 1:3000; anti-cytochromec, 1:3000) at 4°C overnight, followed by three cycles of washing procedures for 5 min with washing buffer and subsequent incubation with horseradish peroxidase-conjugated antibody against rabbit IgG (1:3000) for 1 hr at room temperature. Proteins reactive with the antibody were detected with the aid of Western Blot Chemoluminescence Reagent Plus (NEN Life Science Products Boston, MA), followed by exposure to x-ray films and subsequent quantification by densitometry.
Immunohistochemical analysis. Mice were deeply anesthetized with pentobarbital (250 mg/kg, i.p.) and perfused intracardially with saline, followed by 4% paraformaldehyde in 0.1 msodium phosphate buffer, pH 7.4. Brains were removed quickly and fixed further with the same fixation solution for an additional 2 hr at 4°C, followed by equilibration with 30% sucrose in 0.1m sodium phosphate buffer, pH 7.4, at 4°C overnight. Serial coronal sections were made at a thickness of 30 μm using a cryostat at −20°C. Immunoreactivity was determined by the avidin–biotin–peroxidase method (ABC kit, Vectastain, Vector Laboratories) on these sections. After being blocked with normal goat serum for 1 hr at room temperature, sections were incubated with the anti-c-Fos antibody (1:100) at 4°C overnight. Sections were then incubated with biotinylated anti-rabbit IgG antibody for 30 min at room temperature and subsequently with ABC solution for 1 hr at room temperature. The peroxidase reaction was visualized with diaminobenzidine/hydrogen peroxide solution (Histofine, Ninirei Co., Tokyo, Japan).
Electron microscopic analysis. Mice were deeply anesthetized with pentobarbital and then perfused intracardially with saline, followed by intracardial perfusion of 0.5% glutaraldehyde/4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.4. Thin blocks (∼1 mm3) of hippocampus were immediately cut out and prefixed with the same fixing solution for an additional 2 hr at 4°C, followed by equilibration with 8% sucrose in 0.1 m sodium phosphate buffer, pH 7.4, at 4°C overnight. Hippocampal blocks were then post-fixed with 1% osmium tetraoxide/1.5% potassium ferrocyanide in 0.1 m sodium phosphate buffer, pH 7.4. Samples were then dehydrated in ethanol and embedded in LR White (London Resin Company Laboratories, Derkshire, UK), followed by collection of ultrathin sections. After being blocked with normal goat serum for 1 hr at room temperature, ultrathin sections were incubated with an anti-c-Fos antibody (1:50) at 4°C overnight, followed by incubation with a gold (10 nm)-conjugated goat anti-rabbit IgG antibody for 4 hr at room temperature and subsequent counterstaining with uranyl acetate and lead citrate.
For analysis of mitochondrial fractions, resultant pellets were prefixed with 2% glutaraldehyde in 0.1 m sodium phosphate buffer, pH 7.4, for 2 hr at 4°C, followed by post-fixation with 1% osmium tetraoxide/1.5% potassium ferrocyanide in 0.1 msodium phosphate buffer, pH 7.4. These samples were dehydrated in ethanol and then embedded in epoxy resin (Oken Co., Tokyo, Japan), followed by collection of ultrathin sections for counterstaining with uranyl acetate and lead citrate before electron microscopic observation.
Data analysis. Densitometric data were subjected to calculation of the area under the curve (AUC) by a PC computer. Results are all expressed as the mean ± SE, and the statistical significance was determined by one-way ANOVA with the Bonferroni/Dunnett post hoc test.
RESULTS
Animal models and preparations of mitochondria
After a single intraperitoneal injection of kainate at a dose of 30 mg/kg, >38 of 50 mice showed forelimb tremor and facial twitch within 30 min. The remaining animals developed wild running and jumping, but no animals died before use. Of these animals, three animals were chosen for evaluation of histological observations of neurons in hippocampus after the administration of kainate (data not shown). Kainate at 30 mg/kg did not induce marked alterations in microscopic histology of hippocampus 3 d afterward. Ten days after the injection of kainate, severe neuronal losses were observed in both the CA1 and CA3 subfields of the pyramidal neuronal layers, but not in the granule cell layer of the dentate gyrus. NMDA did not induce marked cell losses in the aforementioned hippocampal neuronal layers within 10 d after administration (data not shown).
To analyze the validity of isolation procedures used here, marker proteins were determined on immunoblotting analysis in isolated nuclear and mitochondrial fractions obtained from cerebral cortex and hippocampus of untreated animals using antibodies against histone H1 and cytochrome c (Fig.1a). In both brain regions tested, histone H1 immunoreactivity was detected exclusively in nuclear fractions but not in mitochondrial fractions (left panel). Even when films were overexposed to detect a small amount of histone H1, no detectable immunoblots were seen in mitochondrial fractions of either regions (data not shown). By contrast, cytochrome c immunoreactivity was observed only in mitochondrial fractions but not in nuclear fractions in both regions (right panel). Nuclear fractions showed a little immunoreactivity against the anti-cytochrome c antibody on overexposed films (data not shown). Electron microscopic analysis showed that no nuclear particles were seen in mitochondrial fractions obtained from hippocampus of untreated animals, although a large number of mitochondria and small organelles other than the nucleus were found in these mitochondrial fractions (Fig. 1b). In addition, immunoblotting and electron microscopic analyses revealed that no nuclear materials were seen in mitochondrial fractions prepared from hippocampus of mice killed 6 hr after kainate treatment (data not shown).
Fig. 1.

Identification of nuclear and mitochondrial fractions obtained from cerebral cortex and hippocampus of untreated animals. Cerebral cortex and hippocampus were homogenized individually, followed by centrifugation for preparation of nuclear and mitochondrial fractions. a, Histone H1 and cytochrome cimmunoreactivities in nuclear and mitochondrial fractions. An aliquot (50 μg of protein) of each fraction was subjected to immunoblotting analysis using each antibody. b, Electron micrograph of hippocampal mitochondrial fractions. Scale bar, 200 nm. These experiments were repeated at least three times with similar results.
Enhancement of mitochondrial AP-1 DNA binding after kainate and NMDA treatment
A systemic administration of NMDA and kainate resulted in more potent enhancement of AP-1 DNA binding in nuclear extracts of hippocampus than those of cerebral cortex 2 hr after administration (Fig. 2a, left panel). Both Glu agonists markedly potentiated AP-1 DNA binding in mitochondrial extracts of hippocampus and cerebral cortex 2 hr later (Fig. 2a, right panel), although quantitative analysis revealed that mitochondrial AP-1 DNA binding was less potent than nuclear binding in both brain regions of animals treated with kainate and NMDA (Fig. 2b). Neither NMDA nor kainate markedly affected AP-1 DNA binding in either nuclear or mitochondrial extracts of cerebellum (data not shown).
Fig. 2.

Enhancement of AP-1 DNA binding in nuclear and mitochondrial extracts of cerebral cortex and hippocampus after treatment with NMDA and kainate. Animals were given saline, NMDA, or kainate, followed by dissection of cerebral cortex and hippocampus 2 hr after administration for subsequent preparation of nuclear and mitochondrial extracts. An aliquot (5 μg of protein) was incubated with the radiolabeled probe for AP-1 and then subjected to the electrophoretic mobility shift assay. a, The left two panels show typical autoradiograms exposed for 6 hr, and each lane corresponds to nuclear extracts from one animal. The right two panels show typical autoradiograms exposed for 24 hr, and each lane corresponds to mitochondrial extracts from one animal. b, Quantitative densitometric data are shown here. Values are the mean ± SE from three animals. *p < 0.05, **p< 0.01; significantly different from each control value obtained in animals injected with saline alone.
Figure 3 shows the time course of enhancement of mitochondrial AP-1 DNA binding after the treatment with kainate. A systemic administration of kainate led to dramatic enhancement of AP-1 DNA binding in mitochondrial extracts of both cerebral cortex and hippocampus 1 hr later, with a sustained elevation of binding thereafter up to 6 hr after administration. At no time after kainate administration was any significant difference in mitochondrial AP-1 DNA binding seen between cerebral cortex and hippocampus (Fig.3a). Sustained potentiation was seen for AP-1 DNA binding in hippocampal mitochondria up to 3 d after the administration of kainate, whereas mitochondrial AP-1 DNA binding returned to the control levels found in untreated animals within 7 d after administration (Fig. 3b).
Fig. 3.

Time course of AP-1 DNA binding in mitochondrial extracts of cerebral cortex and hippocampus after kainate treatment.a, Animals were given kainate and decapitated at various times after administration for preparation of mitochondrial extracts from cerebral cortex and hippocampus. b, Hippocampus was also dissected for preparation of mitochondrial extracts on different days after the administration of kainate. An aliquot (10 μg of protein) was incubated with the radiolabeled probe for AP-1 and then subjected to the electrophoretic mobility shift assay as described in Materials and Methods. The right panels show typical autoradiograms, and each lane corresponds to a sample from one animal. Quantitative densitometric data are shown in the left panel, and each value is the mean ± SE from four to eight animals. *p < 0.05, **p < 0.01; significantly different from each control value obtained in untreated animals (time = 0).
To test the selectivity of AP-1 DNA binding in mitochondrial extracts, unlabeled probes for AP-1 and c-Myc were added into incubation mixture at different concentrations. As seen with nuclear extracts, unlabeled AP-1 probe was effective in competing for AP-1 DNA binding in mitochondrial extracts of hippocampus and cerebral cortex from kainate-treated animals in a concentration-dependent manner (Fig.4). Mitochondrial AP-1 DNA binding was completely abolished by the addition of unlabeled AP-1 probe at a molar concentration ratio of 10. The addition of excess unlabeled Myc probe with nucleotide sequence identical to that of AP-1 probe except for the core element did not significantly affect mitochondrial AP-1 DNA binding in cerebral cortex and hippocampus at molar ratios similar to those of the AP-1 probe.
Fig. 4.

Competition for mitochondrial AP-1 DNA binding. Animals were given kainate and decapitated 4 hr after administration for preparation of mitochondrial extracts from hippocampus. An aliquot (10 μg of protein) of mitochondrial extracts was incubated with the radiolabeled probe for AP-1 in either the absence or presence of various concentrations of an unlabeled double-stranded probe with the consensus element for AP-1 or Myc at molar concentration ratios of 1:100 as a competitor. Unlabeled double-stranded competitors were prepared by annealing the single-stranded oligonucleotide indicated at the bottom with the respective complementary single-stranded oligonucleotide. Nucleotide sequences of these oligonucleotides were identical to each other except for the respective consensus element written in bold letters. These experiments were repeated at least three times with similar results.
Localization of Fos family proteins in mitochondria after kainate treatment
Figure 5 shows effects of the addition of antibodies against Fos and Jun family proteins on AP-1 DNA binding in mitochondrial extracts prepared from cerebral cortex and hippocampus of animals injected with kainate. As shown in Figure 5(asterisks), antibodies against c-Fos and Fos-B proteins were effective in inhibiting binding in mitochondrial extracts of both brain regions. As shown by black arrows, in addition, the anti-Jun-B antibody induced an upward shift of the mobility of the probe/protein complex on gels. No marked changes were seen in the intensity and mobility of mitochondrial AP-1 DNA binding after the addition of antibodies against other member proteins, including Fra-1, Fra-2, c-Jun, and Jun-D.
Fig. 5.

Supershift analysis of AP-1 DNA binding in mitochondrial extracts obtained from animals treated with kainate. Animals were given kainate and decapitated 4 hr after administration for preparation of mitochondrial extracts from hippocampus. An aliquot (10 μg of protein) was incubated with each antibody (300 ng) at 4°C for >12 hr, followed by further incubation with the AP-1 probe to run electrophoretic mobility shift assays. Black arrowsindicate the probe/protein complex shifted by the addition of the anti-Jun-B antibody. Asterisks indicate the probe/protein complex inhibited by the addition of antibodies against c-Fos and Fos-B proteins. These experiments were repeated at least three times with similar results.
Next, an attempt was made to determine endogenous levels of these proteins in mitochondrial extracts obtained different times after kainate administration by means of immunoblotting analysis (Fig.6). In mitochondrial extracts of cerebral cortex and hippocampus from untreated animals (time = 0), Jun-B protein existed, but not c-Fos or Fos-B protein. The administration of kainate led to a dramatic increment of c-Fos protein level in mitochondrial extracts of hippocampus and cerebral cortex 2–6 hr later, with complete abolition within 24 hr after administration. Kainate administration also induced a delayed increment of Fos-B protein level in mitochondrial extracts compared with that of c-Fos protein. Fos-B protein was detected slightly in mitochondrial extracts of both telencephalic structures 2–4 hr after the administration of kainate, with an increased level 6 hr later and subsequent elimination within 24 hr. No marked changes were seen in the level of Jun-B protein in mitochondrial extracts in either brain region.
Fig. 6.

Immunoblotting analysis of c-Fos, Fos-B, and Jun-B in mitochondrial extracts of cerebral cortex and hippocampus in animals treated with kainate. Animals were given kainate and decapitated at various times after administration for preparation of mitochondrial extracts from hippocampus. An aliquot (50 μg of protein) was subjected to immunoblotting analysis using each antibody indicated as described in Materials and Methods. CBB represents proteins stained by Coomassie brilliant blue after the analysis. These experiments were repeated at least four times with similar results.
Mitochondrial fractions were isolated from hippocampus 4 hr after the administration of kainate, followed by treatment with trypsin at different concentrations and subsequent preparation of mitochondrial extracts to analyze AP-1 DNA binding and c-Fos protein. Trypsin treatment failed to markedly affect AP-1 DNA binding at concentrations below 100 ng/ml, with broadening of the radioactive band of probe/protein complex at 100 ng/ml (Fig.7a). Trypsin was much less potent in decreasing the c-Fos protein level in mitochondrial extracts than in cytosolic fractions at 100 ng/ml (Fig. 7b).
Fig. 7.

Effects of trypsin treatment on mitochondrial AP-1 DNA binding (a) and c-Fos protein (b) in cytosol and mitochondria. Animals were given kainate and decapitated 4 hr after administration for preparation of mitochondrial fractions from hippocampus. Mitochondrial fractions were incubated with trypsin at different concentrations at 30°C for 30 min, followed by the addition of 10 μg/ml (p-amidinophenyl)methanesulfonyl fluoride and subsequent centrifugation at 20,000 × g for 5 min. Aliquots of extracts from trypsin-treated mitochondrial fractions were subjected to electrophoretic mobility shift (10 μg of protein) and immunoblotting (40 μg of protein) assays, respectively. An aliquot (15 μg of protein) of cytosolic fractions treated with trypsin under the same conditions was also subjected to immunoblotting assay.
Immunohistochemical analysis of c-Fos protein expressed by kainate treatment
Figure 8 shows c-Fos immunoreactivity in hippocampus excised 2 hr after the administration of saline or kainate. In animals injected with kainate (Fig.8aB), c-Fos immunoreactivity was observed in pyramidal cell layers of the CA1–CA3 subfields and in the granular cell layer of the dentate gyrus, but not in those injected with saline (Fig.8aA). Electron microscopic analysis allowed us to observe c-Fos immunoreactivity within the nuclei and mitochondria of neuronal cells in the dentate gyrus and the CA1 subfield (Figs.8b,c). In saline-treated animals, slight c-Fos immunoreactivity was found in both the cytoplasm and the nucleus of dentate neurons (Fig. 8bA), with no immunoreactivity in the mitochondria of dentate (Fig. 8bB) and CA1 pyramidal (Fig.8bC) neurons. Kainate treatment not only led to marked expression of c-Fos immunoreactivity in the nucleus and cytoplasm of dentate neurons (Fig. 8bD), but also resulted in localization of c-Fos immunoreactivity in the mitochondria of both dentate (Fig. 8bE) and CA1 pyramidal neurons (Fig.8bF). Evident localization of c-Fos protein was seen in many mitochondria of dentate granular neurons from mice injected with kainate (Fig. 8cA–cC), although c-Fos protein was not always detected in other mitochondria of granule neurons on an identical section obtained from the same animals (Fig.8cD). In dentate granular neurons of kainate-treated animals, c-Fos protein was found in the nucleus at a concentration much higher than that in the cytoplasm, with mitochondrial c-Fos protein being not concentrated in the mitochondrial matrix. No immunoreactivity was found in control sections stained in the absence of the primary antibody (data not shown).
Fig. 8.
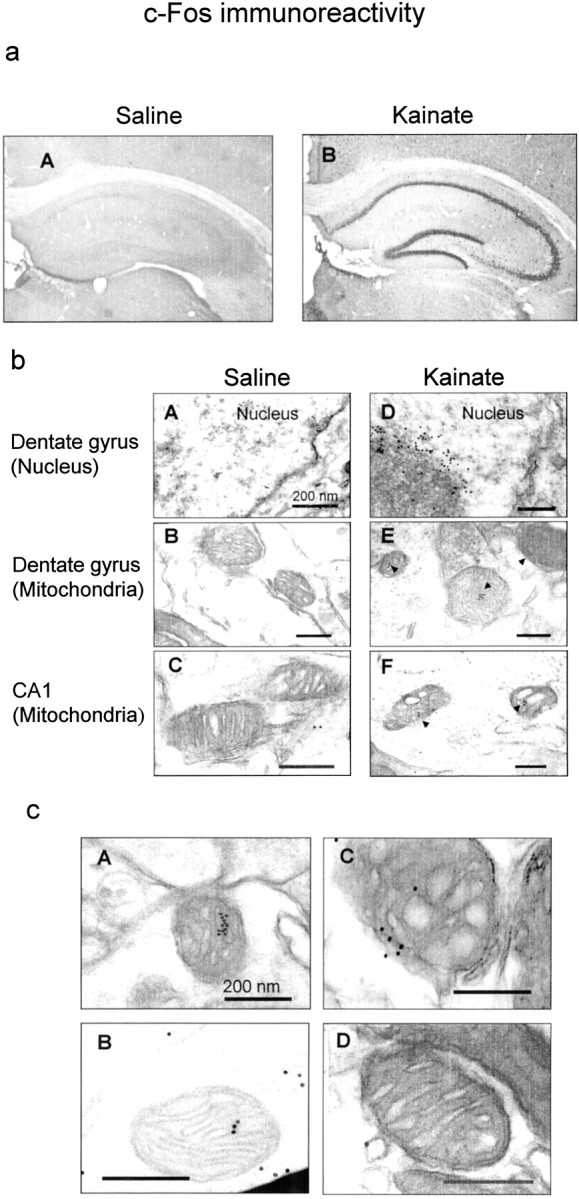
Immunohistochemical analysis of c-Fos protein expressed by kainate treatment. Animals were given saline or kainate, followed by perfusion with 4% paraformaldehyde or 0.5% glutaraldehyde/4% paraformaldehyde for fixation 2 hr after administration. Sections were prepared from fixed brains and subjected to immunohistochemical analysis of c-Fos protein by light (a) and electron (b, c) microscopy as described in Materials and Methods. a, Light micrographs of c-Fos immunoreactivity in sections obtained from animals treated with saline (A) or kainate (B). Note that c-Fos immunoreactivity was expressed in the pyramidal cell layer of CA1–CA3 subfields and the granule cell layer of the dentate gyrus in hippocampus after kainate treatment. b, Electron micrographs of c-Fos immunoreactivity in sections obtained from animals treated with saline (A–C) or kainate (D–F).Black arrowheads show immunoreactive gold particles within mitochondria (E, F). Note that kainate induced increases in gold particles within mitochondria of the granule cells of the dentate gyrus (E) and CA1 pyramidal cells (F), in addition to those within the nucleus of the dentate neurons (D).c, Electron micrographs of c-Fos immunoreactivity in mitochondria of the granule cells of the dentate gyrus from kainate-treated animals. Typical micrographs are shown from a section of the granule cells in a mouse treated with kainate. These experiments were repeated at least four times with similar results. Note that one of four mitochondria did not show immunoreactive gold particles (D) in contrast to other mitochondria (A–C).
Attempts to determine AP-1 binding sites in mitochondrial DNA
We next searched for AP-1 binding sites in the noncoding region of mitochondrial DNA. Although completely identical core nucleotide sequences for recognizing AP-1 (TGAC/GTCA) were not found anywhere in mitochondrial DNA, there were 10 sites with sequences similar to the AP-1 site in the noncoding region of mitochondrial DNA (Fig.9). One of these sites was also similar to CRE (MT-10). Such mitochondrial AP-1-like elements include five sites with TGAC(GTCA), three sites with TGACT(AGTCA), and two sites with TGAG(CTCA), respectively.
Fig. 9.

Particular nucleotide sequences of the noncoding region in mouse mitochondrial DNA. There are 10 sequences with similarity to the AP-1 site as written in bold letters underlined in the noncoding region (15417–16295) (Bibb et al., 1981). One of these sequences also shows similarity to CRE site as indicated by CRE like in parentheses. Double-stranded oligonucleotides (MT-1 to MT-10) with similarity to the AP-1 site were synthesized individually to evaluate binding sites for the AP-1 complex in mitochondria.
To assess the affinity and selectivity for these AP-1-like sites of mitochondrial AP-1 DNA binding, we prepared 10 pieces of double-stranded oligonucleotides containing each sequence similar to the AP-1 site (MT-1 to MT-10). Figure10a shows effects of the addition of these unlabeled double-stranded oligonucleotides on mitochondrial AP-1 DNA binding in hippocampus of kainate-treated animals. Unlabeled oligonucleotides with TGACT(AGTCA), including MT-3, MT-4, and MT-9, were most effective in competing for mitochondrial AP-1 DNA binding enhanced by kainate among 10 synthesized mitochondrial oligonucleotides. Of five oligonucleotides with TGAC(GTCA), both MT-6 and MT-7 exhibited an ability to compete moderately for AP-1 DNA binding in hippocampal mitochondrial extracts prepared from mice injected with kainate. However, other oligonucleotides had no marked effects on mitochondrial AP-1 DNA binding. These included MT-1, MT-2, MT-5, MT-8, and MT-10. As shown in Figure 10b, mitochondrial AP-1 DNA binding was not affected in hippocampus of kainate-treated animals by any of the mutant oligonucleotides of MT-3, MT-4, MT-6, MT-7, and MT-9 with nucleotide sequences almost identical to wild-type probes except for AP-1-like sites.
Fig. 10.

Competition for mitochondrial AP-1 DNA binding with synthesized nucleotides containing sequences similar to AP-1 sites in the noncoding region of mitochondrial DNA. Animals were given kainate and decapitated 4 hr after administration for preparation of mitochondrial extracts from hippocampus. An aliquot (10 μg of protein) of mitochondrial extracts was incubated with the radiolabeled probe for AP-1 in either the absence or presence of unlabeled double-stranded oligonucleotides at a molar concentration ratio of 100.a, Effects of oligonucleotides (MT-1 to MT-10) with similarity to AP-1 site on mitochondrial AP-1 DNA binding. Typical autoradiograms are shown in the left panel, and quantitative densitometric data are shown in the right panel, where each value is the mean ± SE from three independent experiments. **p < 0.01; significantly different from the control value obtained in the absence of any competitors. b, Effect of mutant oligonucleotides on the binding. Typical autoradiograms are shown in the left panel, and nucleotide sequences of wild and mutant oligonucleotides are shown in the right panel. Note that the binding was not affected by addition of mutant oligonucleotides (mMT-3, -4, -6, -7, and -9), of which wild types (MT-3, -4, -6, -7, and -9) markedly inhibited the binding.
To examine whether kainate indeed affects binding to AP-1-like sites in the noncoding region of mitochondrial DNA, an attempt was made to determine DNA binding on electrophoretic mobility shift assays using radiolabeled mitochondrial oligonucleotides containing sequences of a high competition potency, such as MT-3 and MT-9, as probes (Fig.11). No marked binding was detectable for either radiolabeled probe in mitochondrial extracts obtained in untreated animals, whereas marked potentiation was seen with binding of radiolabeled MT-9 probe in hippocampal mitochondrial extracts prepared 6 hr after the administration of kainate. Kainate was also effective in slightly potentiating binding of the radiolabeled MT-3 probe in hippocampal mitochondrial extracts 6 hr after administration.
Fig. 11.

Binding of radiolabeled probes with mitochondrial AP-1-like sites in mitochondrial extracts. Animals were given either saline or kainate and decapitated 6 hr after administration for preparation of mitochondrial extracts from hippocampus. An aliquot (10 μg of protein) was incubated with radiolabeled probes with a sequence of MT-3 or MT-9 under routine experimental conditions. Probes were double-stranded oligonucleotides containing MT-3 (5′-AGTTTATGACTGTATGGTG-3′) and MT-9 (5′-AAAATATGACTTATATTTT-3′). Typical autoradiograms are shown, and each lanecorresponds to a sample from one animal.
DISCUSSION
The essential importance of the present findings is that the systemic administration of kainate led to marked potentiation of AP-1 DNA binding through localization of particular Fos and Jun family member proteins in mitochondria of murine cerebral cortex and hippocampus. To our knowledge, this is the first direct demonstration of the localization of mitochondrial AP-1 complex in response to extracellular kainate signals, in addition to nuclear AP-1 complex, in these two telencephalic structures. The findings from trypsin treatment clearly indicate that mitochondrial AP-1 binding is not derived from simple adhesion of the AP-1 complex to outer mitochondrial membranes: indeed, the AP-1 complex seems to exist in the inner matrix of mitochondria. Point mutation experiments also suggest that mitochondria AP-1 complex expressed by kainate treatment may selectively recognize AP-1-like sites of the noncoding region in mitochondrial DNA.
The AP-1 complex has been thought to modulate de novosynthesis of inducible target proteins at the level of gene transcription through selective recognition of the core sequence phorbol 12-myristate 13-acetate-response element (TCAC/GTCA) on stress-response genes in the nucleus (Curran and Franza, 1988;Pennypacker et al., 1994a). The AP-1 complex is also shown to transduce signals from a number of different biological mediators, such as cytokines, growth factors, and neurotransmitters, to these stress-response genes (Papaconstantinou, 1994; Tong et al., 1998). Among the biological mediators described above, both kainate (Sonnenberg et al., 1989; Pennypacker et al., 1994b; Azuma et al., 1996; Yoneda et al., 1999a) and NMDA (Ogita and Yoneda, 1994; Yoneda et al., 1999b) are well known to induce expression of the AP-1 complex with DNA binding activity in the nucleus of rodent hippocampus in vivo. In addition to these findings on nuclear gene expression, the present paper proposes a hypothesis that kainate signals may regulate expression of mitochondrial genes in neurons through facilitation of import of the AP-1 complex expressed in the cytoplasm. The AP-1 complex could be translocated into the mitochondrion and the nucleus from the cytoplasm in particular situations.
Eukaryotic mitochondrial biogenesis requires the coordinated expression of numerous genes encoded in two different genetic compartments. (1) Most proteins involved in mitochondrial functions are synthesized in the cytoplasm through transcriptional regulation of the nuclear genome. (2) Some of essential proteins involved in mitochondrial electron transport and oxidative phosphorylation are synthesized within mitochondria through encoding in the mitochondrial genome (Bibb et al., 1981; Chomyn et al., 1985). An essential process in this coordinated interaction between the nucleus and the mitochondrion is the transcription of mitochondrial DNA, which requires a nucleus-encoded mitochondrial RNA polymerase and other nuclear proteins to interact with specific elements on the noncoding region of mitochondrial DNA (Clayton, 1984; Suzuki et al., 1991). Several sites with nucleotide sequences similar to the AP-1 site are found in the noncoding region (bases from 15417 to 16295) of mouse mitochondrial DNA (Bibb et al., 1981). The present finding that kainate treatment led to marked potentiation of binding of radiolabeled oligonucleotides with sequences effective in competing for mitochondrial AP-1 DNA binding is favorable to the proposal that kainate signals indeed may be transduced into mitochondria as well as nuclei to modulate gene transcription through localization of the AP-1 complex in this organelle. The noncoding region is shown to contain promoters to regulate gene transcription in mitochondria (Chang and Clayton, 1984, 1986; Suzuki et al., 1991).
In other words, the transcription factor AP-1 complex may play an important role in mechanisms associated with coordinated expression of mitochondrial genes through binding to those AP-1-like sites in the noncoding region. Mitochondrial gene expression by the AP-1 complex may be one of triggers leading to mitochondrial dysfunctions mediated by particular Glu receptors. Indeed, expression of cytochrome coxidase subunit I mRNA in mitochondria is markedly decreased in hippocampal CA1 pyramidal neurons after transient brain ischemia, which is well known to cause CA1 neuronal damage through excessive activation of Glu receptors (Abe et al., 1993, 1996). Glu receptor stimulation is shown to cause persistent inhibition of ATP synthesis leading to necrosis in primary cultured cortical neurons (Delgado-Esteban et al., 2000; Almeida and Bolaños, 2001). This persistent inhibition of ATP synthesis would result from sustained inhibition of mitochondrial cytochrome c oxidase and succinate-cytochrome creductase activities by ONOO−, which is generated through activation by Glu signals of nitric oxide synthase and the formation of reactive oxygen species. However, the possibility that Glu-induced delayed inhibition of ATP synthesis could be caused by alterations in expression of mitochondria-encoded proteins through transcriptional regulation by the AP-1 complex in mitochondrial genome seems to be beyond the scope of this study.
What mechanisms are involved in translocation of the AP-1 complex from the cytoplasm to mitochondria? A considerable body of evidence suggests that mitochondria may play a critical role in mechanisms underlying neurotoxicity by Glu (Ankarcrona et al., 1995; Khodorov et al., 1996;White and Reynolds 1996; Almeida et al., 1998; Castilho et al., 1998;Duchen, 2000). The mechanism would involve glutamate-induced accumulation of Ca2+ within the mitochondrial matrix. Accumulation of Ca2+could dissipate membrane potentials across the mitochondria inner membrane to open the mitochondrial permeability transition pores that are inner membrane channels allowing the free passage of various solutes, such as cytochrome c, apoptosis-inducing factor, and procaspases (Liu et al., 1996; Atlante et al., 1999; Susin et al., 1999). Facilitation of the opening of these pores would be involved in the translocation of particular Fos and Jun family members into the mitochondria from the cytosol where the AP-1 complex is expressed at ribosomes in a large quantity after kainate treatment. The possibility that cytosolic AP-1 complex expressed by kainate may be translocated into mitochondria through common import systems of this organelle (Haucke and Schatz, 1996; Schatz and Dobberstein, 1996; Neupert, 1997) is also feasible.
The AP-1 complex is a homodimer or heterodimer between Fos, Jun, and other family member proteins with leucine-zipper motif (Schuermann et al., 1989). Fos and Jun family proteins include c-Fos, Fos-B, Fra-1, Fra-2, c-Jun, Jun-B, and Jun-D. Kainate treatment is shown to induce expression of nuclear AP-1 complex with different partner proteins in rodent hippocampus in a manner that is dependent on the time after treatment (Kaminska et al., 1994; Pennypacker et al., 1994b,c; Feng et al., 1997; Kitayama et al., 1999). Kainate treatment induces expression of the AP-1 complex composed of c-Fos, c-Jun, Jun-B, and Jun-D proteins in the nucleus at an early time window, followed by delayed expression of Fos-B, Fra-2, and Jun-D proteins afterward (Pennypacker et al., 1994c; Kitayama et al., 1999). At an early time window in this study, the AP-1 complex expressed by kainate differs between mitochondria and nucleus as follows. (1) Participation of Jun-D protein in AP-1 DNA binding: Nuclear AP-1 complex contains both Jun-D and c-Jun proteins after kainate treatment as mentioned above, whereas Jun-D protein does not participate in mitochondrial AP-1 complex expressed by kainate. In our preliminary experiments using the anti-Jun-D antibody, Jun-D protein is not detected in mitochondrial extracts from hippocampus obtained 4 hr after kainate treatment (our unpublished data). Supershift analysis using antibodies against Jun-D and c-Jun proteins reveals that both Jun-D and c-Jun proteins really participate in AP-1 DNA binding in nuclear extracts from hippocampus of kainate-treated mice (Kitayama et al., 1999). (2) Participation of Jun-B protein in AP-1 DNA binding: Kainate markedly increases expression of Jun-B protein in the nucleus (Kitayama et al., 1999; Won et al., 2000), but not in mitochondria as shown in this study. (3) Participation of Fra proteins in AP-1 DNA binding: Although Fra proteins, as well as c-Fos and Fos-B proteins, participate in AP-1 DNA binding in the nucleus of hippocampus from kainate-treated animals (Pennypacker et al., 1994b; Kitayama et al., 1999), our present data demonstrate involvement of both c-Fos and Fos-B, but not of Fra-1 and Fra-2 proteins, in enhancement of mitochondrial AP-1 DNA binding by kainate. Both c-Fos and Fos-B proteins are thus likely to form AP-1 complex mainly as a counterpart of Jun-B protein in mitochondria of hippocampus from kainate-treated animals. Dimerization with different partner proteins would be one of the important determinants for the AP-1 complex to recognize particular target genes in the mitochondria as well as in the nucleus. Moreover, the present finding that kainate treatment induced AP-1 complex formed by c-Fos, Fos-B, and Jun-B proteins in mitochondria at an early time window would indicate that translocation could participate in differential compositions of AP-1 complex expressed in response to kainate between mitochondria and nuclei. However, mechanisms underlying the selective translocation of particular Fos and Jun family members into mitochondria and nuclei remain to be elucidated in futures studies.
In conclusion, the AP-1 complex expressed by kainate signals would be translocated into mitochondria and bind to the noncoding region of mitochondrial DNA. Kainate may affect mitochondrial functions as a result of modulation of de novo synthesis of particular target proteins through gene transcriptional changes mediated by the AP-1 complex expressed in both mitochondria and nuclei of hippocampal neurons. Elucidation of the exact mechanism for translocation as well as the functional significance of mitochondrial AP-1 complex is now under way in our laboratories.
Footnotes
This work was supported in part by Grants-in-Aid for Scientific Research to Y.Y. from the Ministry of Education, Science, Sports, and Culture, Japan.
Correspondence should be addressed to Dr. Kiyokazu Ogita, Department of Pharmacology, Setsunan University Faculty of Pharmaceutical Sciences, 45-1 Nagaotoge-ho, Hirakata, Osaka 573-0101, Japan. E-mail: ogita@pharm.setsunan.ac.jp.
REFERENCES
- 1.Abe K, Kawagoe J, Aoki M, Kogure K. Changes of mitochondrial DNA and heat shock protein gene expressions in gerbil hippocampus after transient forebrain ischemia. J Cereb Blood Flow Metab. 1993;13:773–780. doi: 10.1038/jcbfm.1993.98. [DOI] [PubMed] [Google Scholar]
- 2.Abe K, Kawagoe J, Itoyama Y, Kogure K. Isolation of an ischemia-induced gene and early disturbance of mitochondrial DNA expression after transient forebrain ischemia. Adv Neurol. 1996;71:485–530. [PubMed] [Google Scholar]
- 3.Almeida A, Bolaños JP. A transient inhibition of mitochondrial ATP synthesis by nitric oxide synthase activation triggered apoptosis in primary cortical neurons. J Neurochem. 2001;77:676–690. doi: 10.1046/j.1471-4159.2001.00276.x. [DOI] [PubMed] [Google Scholar]
- 4.Almeida A, Heales SJR, Bolaños JP, Medina JM. Glutamate neurotoxicity is associated with nitric oxide-mediated mitochondrial dysfunction and glutathione depletion. Brain Res. 1998;790:209–216. doi: 10.1016/s0006-8993(98)00064-x. [DOI] [PubMed] [Google Scholar]
- 5.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 6.Atlante A, Gagliardi S, Marra E, Calissano P, Passarella S. Glutamate neurotoxicity in rat cerebellar granule cells involves cytochrome c release from mitochondria and mitochondrial shuttle impairment. J Neurochem. 1999;73:237–246. doi: 10.1046/j.1471-4159.1999.0730237.x. [DOI] [PubMed] [Google Scholar]
- 7.Azuma Y, Ogita K, Yoneda Y. Particular nuclear transcription factors responsive to systemic administration of kainic acid in murine brain. Neurochem Int. 1996;29:289–299. doi: 10.1016/0197-0186(95)00157-3. [DOI] [PubMed] [Google Scholar]
- 8.Azuma Y, Ogita K, Yoneda Y. Constitutive expression of cytoplasmic activator protein-1 with DNA binding activity and responsiveness to ionotropic glutamate signals in the murine hippocampus. Neuroscience. 1999;92:1295–1308. doi: 10.1016/s0306-4522(99)00090-1. [DOI] [PubMed] [Google Scholar]
- 9.Bevilaqua LR, Cammarota M, Paratcha G, de Stein ML, Izquierdo I, Medina JH. Experience-dependent increase in cAMP-responsive element binding protein in synaptic and nonsynaptic mitochondria of the rat hippocampus. Eur J Neurosci. 1999;11:3753–3756. doi: 10.1046/j.1460-9568.1999.00830.x. [DOI] [PubMed] [Google Scholar]
- 10.Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981;26:167–180. doi: 10.1016/0092-8674(81)90300-7. [DOI] [PubMed] [Google Scholar]
- 11.Cammarota M, Paratcha G, Bevilaqua LR, Levi de Stein M, Lopez M, Pellegrino de Iradi A, Izquierdo I, Medina JH. Cyclic AMP-responsive element binding protein in brain mitochondria. J Neurochem. 1999;72:2272–2277. doi: 10.1046/j.1471-4159.1999.0722272.x. [DOI] [PubMed] [Google Scholar]
- 12.Castilho RF, Hansson O, Ward MW, Budd SL, Nicholls DG. Mitochondrial control of acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 1998;18:10277–10288. doi: 10.1523/JNEUROSCI.18-24-10277.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang DD, Clayton DA. Precise identification of individual promoters for transcription of each strand of human mitochondrial DNA. Cell. 1984;36:635–643. doi: 10.1016/0092-8674(84)90343-x. [DOI] [PubMed] [Google Scholar]
- 14.Chang DD, Clayton DA. Precise assignment of the light-strand promoter of mouse mitochondrial DNA: a functional promoter consists of multiple upstream domains. Mol Cell Biol. 1986;6:3253–3261. doi: 10.1128/mcb.6.9.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chomyn A, Mariottini P, Cleeter MWJ, Ragan CI, Matsuno-Yagi A, Hatefi Y, Doolittle RF, Attardi G. Six unidentified reading frames of human mitochondrial DNA encode components of the respiratory-chain NADH dehydrogenase. Nature. 1985;314:592–597. doi: 10.1038/314592a0. [DOI] [PubMed] [Google Scholar]
- 16.Clark JB, Nicklas WJ. The metabolism of rat brain mitochondria. Preparation and characterization. J Biol Chem. 1970;245:4724–4731. [PubMed] [Google Scholar]
- 17.Clayton DA. Transcription of the mammalian mitochondria genome. Annu Rev Biochem. 1984;53:573–594. doi: 10.1146/annurev.bi.53.070184.003041. [DOI] [PubMed] [Google Scholar]
- 18.Curran T, Franza BR., Jr Fos and Jun: the AP-1 connection. Cell. 1988;55:395–397. doi: 10.1016/0092-8674(88)90024-4. [DOI] [PubMed] [Google Scholar]
- 19.Delgado-Esteban M, Almeida A, Bolaños JP. d-Glucose prevents glutathione oxidation and mitochondrial damage after glutamate receptor stimulation in rat cortical primary neurons. J Neurochem. 2000;75:1618–1624. doi: 10.1046/j.1471-4159.2000.0751618.x. [DOI] [PubMed] [Google Scholar]
- 20.Duchen MR. Mitochondria and calcium: from cell signaling to cell death. J Physiol (Lond) 2000;529:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng Z, Zhang W, Hudson P, Bing G, Feng W, Hong J-S. Characterization of the long-lasting activator protein-1 complex induced by kainic acid treatment. Brain Res. 1997;770:53–59. doi: 10.1016/s0006-8993(97)00744-0. [DOI] [PubMed] [Google Scholar]
- 22.Glowinski J, Iversen LL. Regional studies of catecholamines in the rat brain. I. The disposition of the [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. J Neurochem. 1966;13:655–669. doi: 10.1111/j.1471-4159.1966.tb09873.x. [DOI] [PubMed] [Google Scholar]
- 23.Greenamyre JT, Porter RH. Anatomy and physiology of glutamate in the CNS. Neurology. 1994;44:S7–S13. [PubMed] [Google Scholar]
- 24.Haucke V, Schatz G. Import of proteins into mitochondria and chloroplasts. Trends Cell Biol. 1996;7:103–106. doi: 10.1016/S0962-8924(96)10052-0. [DOI] [PubMed] [Google Scholar]
- 25.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 26.Kaminska B, Filipkowski RK, Zurkowska G, Lason W, Przewlocki G, Kaczmarek L. Dynamic changes in the composition of the AP-1 transcription factor DNA-binding activity in rat brain following kainate-induced seizures and cell death. Eur J Neurosci. 1994;6:1558–1566. doi: 10.1111/j.1460-9568.1994.tb00546.x. [DOI] [PubMed] [Google Scholar]
- 27.Khodorov B, Pinelis V, Vergun O, Storozhevykh T, Vinskaya N. Mitochondrial deenergization underlies neuronal calcium overload following a prolonged glutamate challenge. FEBS Lett. 1996;394:230–234. doi: 10.1016/s0014-5793(96)01139-8. [DOI] [PubMed] [Google Scholar]
- 28.Kitayama T, Ogita K, Yoneda Y. Sustained potential of AP1 DNA binding is not always associated with neuronal death following systemic administration of kainic acid in murine hippocampus. Neurochem Int. 1999;35:453–462. doi: 10.1016/s0197-0186(99)00088-1. [DOI] [PubMed] [Google Scholar]
- 29.Ko HW, Park K-Y, Kim H, Han P-L, Kim YU, Gwag BJ, Choi E-J. Ca2+-mediated activation of c-Jun N-terminal kinase and nuclear factor κB by NMDA in cortical cell cultures. J Neurochem. 1998;71:1390–1395. doi: 10.1046/j.1471-4159.1998.71041390.x. [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 31.Michaelis EK. Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog Neurobiol. 1998;54:369–415. doi: 10.1016/s0301-0082(97)00055-5. [DOI] [PubMed] [Google Scholar]
- 32.Neupert W. Protein import into mitochondria. Annu Rev Biochem. 1997;66:863–917. doi: 10.1146/annurev.biochem.66.1.863. [DOI] [PubMed] [Google Scholar]
- 33.Ogita K, Yoneda Y. Selective potentiation of DNA binding activities of both activator protein 1 and cyclic AMP response element binding protein through in vivo activation of N-methyl-d-aspartate receptor complex in mouse brain. J Neurochem. 1994;63:525–534. doi: 10.1046/j.1471-4159.1994.63020525.x. [DOI] [PubMed] [Google Scholar]
- 34.Ogita K, Yoneda Y. Detection of DNA binding activities of transcription factors with different protein motifs in nuclear extracts of murine brain using gel retardation electrophoresis. J Neurochem. 1995;64:1431–1439. doi: 10.1046/j.1471-4159.1995.64041431.x. [DOI] [PubMed] [Google Scholar]
- 35.Papaconstantinou J. Unifying model of the programmed (intrinsic) and stochastic (extrinsic) theories of aging, the stress response genes, signal transduction-redox-pathways and aging. Ann NY Acad Sci. 1994;719:195–211. doi: 10.1111/j.1749-6632.1994.tb56829.x. [DOI] [PubMed] [Google Scholar]
- 36.Pennypacker KR, Hong J-S, MacMillian MK. Pharmacological regulation of AP-1 transcription factor DNA binding activity. FASEB J. 1994a;8:475–478. doi: 10.1096/fasebj.8.8.8181665. [DOI] [PubMed] [Google Scholar]
- 37.Pennypacker KR, Thai L, Hong J-S, MacMillian MK. Prolonged expression of AP-1 transcription factors in the rat hippocampus after systemic kainate treatment. J Neurosci. 1994b;14:3998–4006. doi: 10.1523/JNEUROSCI.14-07-03998.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pennypacker KR, McMillian MK, Douglass J, Hong J-S. Ontogeny of kainate-induced gene expression in rat hippocampus. J Neurochem. 1994c;62:438–444. doi: 10.1046/j.1471-4159.1994.62020438.x. [DOI] [PubMed] [Google Scholar]
- 39.Sakurai H, Kurusu R, Sano K, Tsuchiya T, Tsuda M. Stimulation of cultured cerebellar granule cells via glutamate receptor induces TRE- and CRE-binding activities mediated by common DNA-binding complexes. J Neurochem. 1992;59:2067–2075. doi: 10.1111/j.1471-4159.1992.tb10096.x. [DOI] [PubMed] [Google Scholar]
- 40.Schatz G, Dobberstein B. Common principle of protein translocation across membranes. Science. 1996;271:1591–1526. doi: 10.1126/science.271.5255.1519. [DOI] [PubMed] [Google Scholar]
- 41.Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schoepfer R, Monyer H, Sommer B, Widen W, Sprengel R, Kuner T, Lomeli H, Herb A, Kohler M, Burnashev N, Günther W, Ruppersberg P, Seeburg P. Molecular biology of glutamate receptors. Prog Neurobiol. 1994;42:353–357. doi: 10.1016/0301-0082(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 43.Schuermann M, Neuberg M, Hunter JB, Jenuwein T, Ryseck RP, Bravo R, Muller R. The leucine repeat motif in Fos protein mediates complex formation with Jun/AP-1 and is required for transformation. Cell. 1989;56:507–516. doi: 10.1016/0092-8674(89)90253-5. [DOI] [PubMed] [Google Scholar]
- 44.Sonnenberg JL, Mitchelmore C, Macgregor-Leon PF, Hempstead J, Morgan JL, Curran T. Glutamate receptor agonists increase the expression of Fos, Fra, and AP-1 DNA binding activity in the mammalian brain. J Neurosci Res. 1989;24:72–80. doi: 10.1002/jnr.490240111. [DOI] [PubMed] [Google Scholar]
- 45.Susin S, Lorenzo H, Zamzami N, Marzo I, Brenner C, Larochette N, Prevost M, Alzari P, Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J Exp Med. 1999;189:381–393. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki H, Hosokawa Y, Nishikimi M, Ozawa T. Existence of common homologous element in the transcriptional regulatory regions of human nuclear genes and mitochondrial gene for the oxidative phosphorylation system. J Biol Chem. 1991;266:2333–2338. [PubMed] [Google Scholar]
- 47.Szekely AM, Barbaccia ML, Alho H, Costa E. In primary cultures of cerebellar granule cells the activation of N-methyl-d-aspartate-sensitive glutamate receptors induced c-fos mRNA expression. Mol Pharmacol. 1989;35:401–408. [PubMed] [Google Scholar]
- 48.Szekely AM, Costa E, Grayson DR. Transcriptional program coordination by N-methyl-d-aspartate-sensitive glutamate receptor stimulation in primary cultures of cerebellar neurons. Mol Pharmacol. 1990;38:624–633. [PubMed] [Google Scholar]
- 49.Tong L, Toliver-Kinsky T, Taglialatela G, Werrbach-Perez K, Wood T, Perez-Polo JR. Signal transduction in neuronal death. J Neurochem. 1998;71:447–459. doi: 10.1046/j.1471-4159.1998.71020447.x. [DOI] [PubMed] [Google Scholar]
- 50.Watanabe N, Kamei S, Ohkubo A, Yamanaka M, Ohsawa S, Makino K, Tokuda K. Urinary protein as measured with a pyrogallol red-molybdate complex manually and in a Hitachi 726 automated analyzer. Clin Chem. 1986;32:1551–1544. [PubMed] [Google Scholar]
- 51.White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Won JS, Song DK, Huh SO, Kim YH, Suh HW. Effect of melatonin on the regulation of proenkephalin and prodynorphin mRNA levels induced by kainic acid in the rat hippocampus. Hippocampus. 2000;10:236–243. doi: 10.1002/1098-1063(2000)10:3<236::AID-HIPO4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 53.Yoneda Y, Ogita K. Rapid and selective enhancement of DNA binding activity of the transcription factor AP1 by systemic administration of N-methyl-d-aspartate in murine hippocampus. Neurochem Int. 1994;25:263–271. doi: 10.1016/0197-0186(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 54.Yoneda Y, Ogita K, Azuma Y, Ikeda M, Tagami H, Manabe T. N-methyl-d-aspartate signaling to nuclear activator protein-1 through mechanisms different from those for kainate signaling in murine hippocampus. Neuroscience. 1999a;90:519–533. doi: 10.1016/s0306-4522(98)00647-2. [DOI] [PubMed] [Google Scholar]
- 55.Yoneda Y, Ogita K, Azuma Y, Kuramoto N, Manabe T, Kitayama T. Predominant expression of nuclear activator protein-1 complex with DNA binding activity following systemic administration of N-methyl-d-aspartate in dentate granule cells of murine hippocampus. Neuroscience. 1999b;93:19–31. doi: 10.1016/s0306-4522(99)00117-7. [DOI] [PubMed] [Google Scholar]