

Abstract
Recombinant adeno-associated viral vectors display efficient tropism for transduction of the dopamine neurons of the substantia nigra. Taking advantage of this unique property of recombinant adeno-associated viral vectors, we expressed wild-type and A53T mutated human α-synuclein in the nigrostriatal dopamine neurons of adult rats for up to 6 months. Cellular and axonal pathology, including α-synuclein-positive cytoplasmic inclusions and swollen, dystrophic neurites similar to those seen in brains from patients with Parkinson's disease, developed progressively over time. These pathological alterations occurred preferentially in the nigral dopamine neurons and were not observed in other nondopaminergic neurons transduced by the same vectors. The degenerative changes were accompanied by a loss of 30–80% of the nigral dopamine neurons, a 40–50% reduction of striatal dopamine, and tyrosine hydroxylase levels that was fully developed by 8 weeks. Significant motor impairment developed in those animals in which dopamine neuron cell loss exceeded a critical threshold of 50–60%. At 6 months, signs of cell body and axonal pathology had subsided, suggesting that the surviving neurons had recovered from the initial insult, despite the fact that α-synuclein expression was maintained at a high level. These results show that nigral dopamine neurons are selectively vulnerable to high levels of either wild-type or mutant α-synuclein, pointing to a key role for α-synuclein in the pathogenesis of Parkinson's disease. Targeted overexpression of α-synuclein in the nigrostriatal system may provide a new animal model of Parkinson's disease that reproduces some of the cardinal pathological, neurochemical, and behavioral features of the human disease.
Keywords: Parkinson's disease, adeno-associated virus vector, neurodegeneration, dopamine, tyrosine hydroxylase, nigral inclusion
A growing body of evidence has implicated α-synuclein in the pathogenesis of Parkinson's disease (PD) (Polymeropoulos et al., 1997; Kruger et al., 1998; Spillantini et al., 1998; Takeda et al., 1998; Giasson et al., 2000b). The discovery that point mutations in the α-synuclein gene can be pathogenic in rare cases of familial PD (Polymeropoulos et al., 1997; Kruger et al., 1998) led to the identification of α-synuclein as a major component of Lewy bodies and dystrophic neurites, i.e., the pathological hall marks of PD (Spillantini et al., 1997, 1998; Baba et al., 1998;Irizarry et al., 1998; Takeda et al., 1998; Braak and Braak, 2000). Subsequent in vitro studies have shown that α-synuclein may interact with ubiquitin-proteosomal processing, oxidative injury, and/or mitochondrial dysfunction to induce neurodegeneration and cell death (Ostrerova et al., 1999; Giasson et al., 2000b; Hsu et al., 2000;Kanda et al., 2000; Tabrizi et al., 2000; McNaught et al., 2001;Shimura et al., 2001). In support of this concept, recent in vivo studies have shown that overexpression of wild-type (wt) or mutated α-synuclein in transgenic mice may lead to neuropathological changes, axonal degeneration, and occasionally also cell death (Kahle et al., 2000; Masliah et al., 2000; van der Putten et al., 2000).
However, in the transgenic mice generated to date, α-synuclein accumulation and signs of axonal pathology have been widespread and most prominent in structures outside the nigrostriatal system. AlthoughMasliah et al. (2000) observed accumulation of α-synuclein in neurons of the substantia nigra (SN) and reduction in tyrosine hydroxylase (TH)-positive axon terminals in the striatum (obtained only in the mice with the highest transgene expression), no degeneration or loss of nigrostriatal dopamine (DA) neurons was observed in any of these mice. Indeed, subsequent studies of transgenic mice expressing either the A30P (Matsuoka et al., 2001; Rathke-Hartlieb et al., 2001) or the A53T (Matsuoka et al., 2001) mutated form of α-synuclein have failed to find any nigral neuropathology. It remains unclear, therefore, what role the mutant forms of α-synuclein play or to what extent overexpression of α-synuclein is sufficient to cause Parkinson-like pathology and neurodegeneration in nigrostriatal DA neurons in the context of the rodent. Other rare forms of familial PD have been described that implicate the proteosomal processing pathway in the etiology of PD. Thus, in addition to mutations in α-synuclein, missense mutations in parkin, an E3 ubiquitin ligase (Shimura et al., 2000), and ubiquitin C-terminal-hydroxylase-L1, an E1 ubiquitin-activating enzyme (Leroy et al., 1998), all induce familial forms of PD. Indeed, a 22 kDa O-glycosylated isoform of α-synuclein has been found to be a specific substrate for the E3 ubiquitin ligase, parkin (Shimura et al., 2001). Mutated forms of the α-synuclein protein (both A53T and A30P) could not bind to parkin, thus abrogating their normal poly-ubiquitination and subsequent proteosomal processing. These data suggest therefore that accumulation of α-synuclein, as occurs in Lewy bodies present in idiopathic PD, either by inhibition of normal proteosomal processing, or by overexpression may be pathogenic to nigral dopamine neurons.
In the present study, we have used recombinant adeno-associated virus (rAAV) vectors to express wt and mutated α-synuclein at high levels in the nigrostriatal DA neurons in adult rats. The rAAV vectors integrate and stably express their transgene in nondividing cells, mostly neurons in the CNS. Because 96% of the viral genome has been removed, the vectors do not express any viral proteins, which minimize the risk of host immune responses (Muzyczka, 1992). This vector system has a particular affinity for the neurons of the SN pars compacta, which makes it possible to express proteins stably, and at high levels, in the nigrostriatal DA neurons in adult rats (Klein et al., 1998; Kirik et al., 2000). Moreover, the transduction can be restricted to one hemisphere, so that the contralateral side can serve as a control.
MATERIALS AND METHODS
Animals and surgery. Young adult Sprague Dawley rats (B & K Universal AB, Stockholm, Sweden) were housed six to a cage withad libitum access to food and water during a 12 hr light/dark cycle, according to the rules set by the Research Ethical Committee at Lund University. Under halothane anesthesia, 2 μl of the vector suspension was injected stereotaxically over the right SN at 5.2 mm posterior and 2.0 mm lateral to bregma, 7.2 mm ventral to dura. The needle was kept in place for an additional 5 min before slowly being withdrawn. rAAV-CBA-α-synuclein (8.2 × 1011 IU/ml), rAAV-CBA-μ-α-synuclein (1.4 × 1012 IU/ml), and rAAV-CBA-GFP (1.4 × 1011 IU/ml) were prepared and titered as described (McLaughlin et al., 1988; Conway et al., 1997;Zolotukhin et al., 1999). The CBA promoter used here is a chicken β-actin promoter with enhancer elements from the cytomegalovirus promoter, as previously described (Xu et al., 2001). For histological analysis, the animals were killed at 1, 3, 8, and 27 weeks after injection [green fluorescent protein (GFP) group,n = 5; α-synuclein group, n = 5–6; and mutant α-synuclein group, n = 5 for all time points], and for biochemical analysis at 3, 8, and 27 weeks after injection (n = 4–5 at 3 weeks and n = 5 per group at 8 and 27 weeks). A second group of animals (n = 18) were injected bilaterally with 0.2 μl of 2% fluorogold (FG) solution in the striatum (1.0 mm anterior, 3.0 mm lateral, and 5.0 mm ventral) followed 5 d later by unilateral vector injections, as above (n = 6 per group). These animals were killed at 3 and 8 weeks after the virus injection (n = 9 at each time point). A third group of animals (n = 7) were injected with rAAV-CBA-GFP vector bilaterally in the SN 5 d before a unilateral nigral injection of rAAV-CBA-α-synuclein on the right side. These animals were killed at 3 (n = 3) and 8 weeks (n = 4) after injection.
A separate group of animals (n = 30) were injected with the rAAV-CBA-GFP (n = 10), rAAV-CBA-α-synuclein (n = 10), or rAAV-CBA-mutant μ-α-synuclein (n = 10) vectors and were used in treatment with α-methyl-d,l-para-tyrosine methyl ester as described below.
Immunohistochemistry. Under pentobarbital anesthesia the animals were perfused through the ascending aorta with physiological saline, followed by 4% ice-cold paraformaldehyde. The brains were post-fixed in the same solution for 2 hr, transferred to 25% sucrose, and sectioned on a freezing microtome at 40 μm in the coronal plane. Immunohistochemical stainings were performed on free-floating sections using antibodies raised against TH (mouse IgG, 1:2000; Chemicon, Temecula, CA), GFP (chicken IgG, 1:5000; R & D systems, Minneapolis, MN), vesicular monoamine transporter-2 (VMAT-2) (rabbit IgG, 1:5000; Chemicon), Hu (mouse IgG, 1:1000; courtesy of Dr. Steven A. Goldman, Cornell University), and human α-synuclein (mouse IgG, 1:2000; courtesy of Dr. Virginia M. Lee, University of Pennsylvania). Sections were rinsed three times in potassium–phosphate buffer (KPBS) between each incubation period. All incubation solutions contained 0.25% Triton X-100 in KPBS. The sections were quenched for 10 min in 3% H2O2/10% methanol. Two hours of preincubation with 5% normal horse serum (NHS) was followed by incubation with the primary antibody in 2% NHS at room temperature and incubation with 1:200 dilution of biotinylated horse anti-mouse antibody (BA2001; Vector Laboratories, Burlingame, CA) in 2% NHS, followed with avidin–biotin–peroxidase complex (ABC Elite; Vector Laboratories, Burlingame, CA), and visualized using 3,3-diaminobenzidine as a chromogen, mounted on chrome–alum-coated glass slides, and coverslipped. For double fluorescent immunohistochemical analysis of α-synuclein and TH, Alexa 488 and Cy3-conjugated secondary antibodies were used.
Assessment of the total number of TH-positive neurons in the SN was made according to the optical fractionator principle, using the OlympusDenmark A/S (Albertslund, Denmark) CAST-Grid system, as described (Kirik et al., 1998). Every eighth section covering the entire extent of the SN was included in the counting procedure. A coefficient of error of < 0.10 was accepted. Striatal TH-positive fiber density was measured by densitometry at three coronal levels (+1.0, 0.0, −1.0) relative to bregma. Data are represented as the mean of the three levels and expressed as a percentage of the intact control side.
Biochemical analyses. Tissue DA and DOPAC content (Schmidt et al., 1982) and in vitro TH enzyme activity (Reinhard et al., 1986) were determined on tissue samples from striatum and SN (at all time points) and also from the nucleus accumbens and prefrontal cortex from the animals in the 27 week group. The tissue pieces were weighed and frozen separately on dry ice and kept at −80°C until assayed.
Behavioral testing. Drug-induced rotation was assessed at 8, 13, 19, and 27 weeks after vector injection and after a subcutaneous injection of 0.25 mg/kg apomorphine-HCl (Sigma, St. Louis, MO) and monitored for 40 min in automated rotometers. Data are expressed as net full turns per minute, with contralateral turns assigned a negative value.
Paw reaching in the staircase test was performed at 24–25 weeks after vector injection, as described (Kirik et al., 1998). After 2 d of food deprivation the animals were placed in the test boxes baited bilaterally with 10 food pellets on each of the four steps (45 mg). The test was conducted over 15 min for 9 consecutive days using the standard platform (27 mm), and an additional 5 d using a wider platform (34 mm). Average number of total successful pellet retrievals in the last 3 d of testing with the wide platform constituted the dependent variable.
The stepping test was performed as described (Kirik et al., 1998) at 8 weeks after injection of the viral vectors. The animals (n = 10 per group) were tested twice daily on 3 consecutive days to define the baseline performance. On the fourth day the animals were tested once before an intraperitoneal injection of 60 mg/kg α-methyl-dl-para-tyrosine methyl ester (dissolved in 0.02% ascorbate-saline; Sigma); and again 6, 24, and 48 hr after injection. The number of adjusting steps was counted while the rat was moved sideways along the table surface in the forehand direction (90 cm in 5 sec).
Statistical analysis. A two-way repeated-measures ANOVA was performed, and post hoc analysis was performed using simple main-effects with a Bonferroni correction of the acceptable α level of p = 0.05. For comparisons between groups and survival times for nigral cell numbers and striatal fiber density measurements, two-way factorial ANOVAs and post hoc analysis were performed using simple main effects with a Bonferroni correction of the acceptable α level of p = 0.05.
RESULTS
To investigate the effect of nigral overexpression of α-synuclein, the rats received a single 2 μl injection of a high-titer rAAV vector, containing either the wt or the A53T mutant of the human α-synuclein gene, unilaterally in the SN pars compacta (SNc). Control rats received identical injections of a rAAV vector encoding GFP. Immunohistochemistry, using an antibody recognizing human (wt and mutant), but not rat, α-synuclein (Giasson et al., 2000a), revealed expression of α-synuclein protein in virtually all neurons within the SNc, in a large number of neurons in the SN pars reticulata (SNr), and in neurons located within the mesencephalic reticular formation, dorsal to the SN (Fig. 2E–L). A variable number of cells in the ventral tegmental area (VTA) were also positive. A similar pattern of cellular expression was seen also in the rAAV-GFP-injected control animals (Fig.1). Double TH/α-synuclein immunofluorescence revealed that >90% of the TH-positive neurons in the SNc expressed the α-synuclein transgene (Fig.2M–O).
Fig. 2.
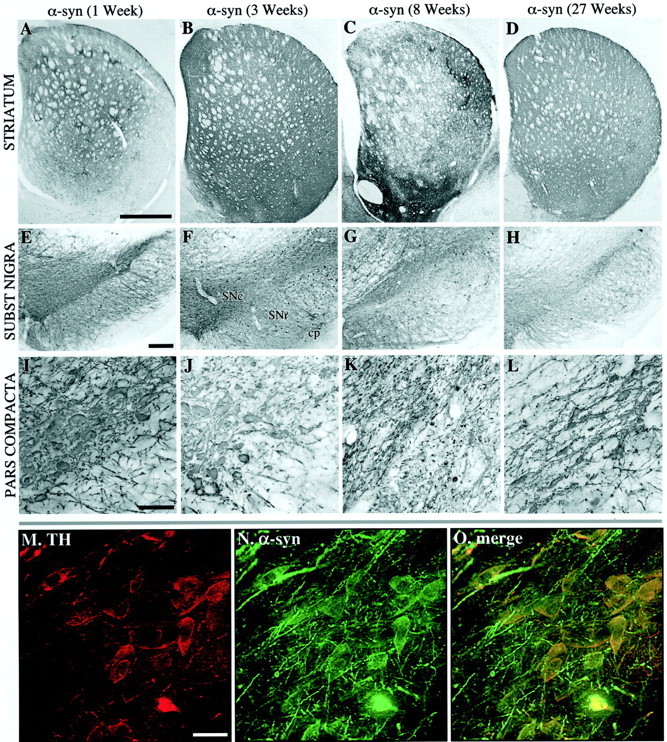
Overview of α-synuclein expression in the striatum (A–D) and SN (E–O) at 1 (A, E, I), 3 (B, F, J, M–O), 8 (C, G, K), and 27 (D, H, I) weeks after injection. The nigral DA cells were labeled with α-synuclein at 1 (E, I) and 3 (F, J) weeks after transduction. At 3 weeks, most of the TH-positive cell bodies in the pars compacta were also positive for α-synuclein (M–O). Similarly, nigrostriatal fiber terminals in the striatum were filled with transgenic human α-synuclein (compare A, B). In contrast to the rAAV-GFP-injected animals, the expression of α-synuclein led to the appearance of degenerative changes both in the nigral cell bodies (G, K) and in the striatal terminals (C). Degeneration of cell bodies in the pars compacta was seen as a reduction in the intensity of α-synuclein-immunoreactive cell bodies in the SN pars compacta at 8 weeks (K). The reduction in α-synuclein-positive cell bodies remained low at the 27 week time point (H, L) while some degree of recovery was seen at the striatal level (D). Scale bars:A, 1 mm (applies to A–C);E, 250 μm (applies to E–H);I, 200 μm (applies to I–L);M, 30 μm (applies to M–O).
Fig. 1.
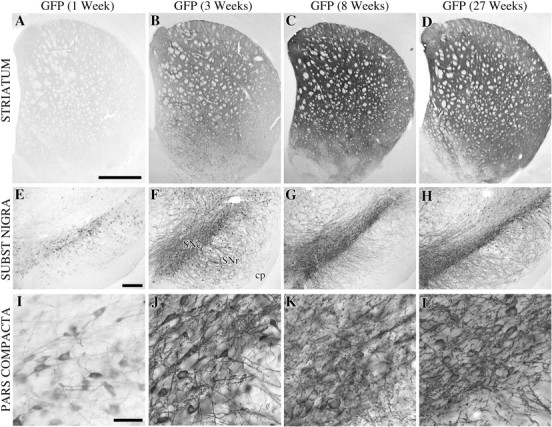
Overview of the extent of expression of GFP in the striatum (A–D) and SN (E–L) at 1 (A, E,I), 3 (B, F,J), 8 (C, G,K), and 27 (D, H,L) weeks after injection. GFP protein was detected at low levels at 1 week in the SN (B, C), with no or very low levels present in the terminals (A). At 3 weeks the immunoreactivity at the cell bodies was increased to cover the entire pars compacta and their dendritic tree in the pars reticulata (F, J) while the fiber terminals were becoming visible (B). At 8 weeks the expression was very high both in the striatal terminals (C) and the nigra, without any pathology (G, K). At the final end point of this experiment, i.e., 27 weeks after transduction, the GFP immunoreactivity was indistinguishable from the stainings obtained at 8 weeks, suggesting that the transgene expression was maintained at high levels between these two time points. Scale bars: A, 1 mm (applies to A–C); E, 250 μm (applies to E–H); I, 200 μm (applies to I–L).
Both types of α-synuclein, wt and mutant, were effectively transported intra-axonally to their respective terminal fields: from SNc to striatum, from VTA to limbic and cortical forebrain areas, from SNr to superior colliculus and thalamus, and from the reticular formation to widespread diencephalic regions. In the nigrostriatal pathway, the cell bodies and axons, including a portion of the striatal terminals, expressed a diffuse, cytoplasmic α-synuclein immunoreactivity at 1 week (Fig. 2A,E). At 3 weeks the entire striatal terminal network was filled (Fig.2B), including variable portions of the VTA projections to the olfactory tubercle, nucleus accumbens, lateral septum, and the anterior cingulate cortex. The transduction and transport of GFP in the rAAV-GFP-injected control animals was equally efficient, completely filling out the cell bodies, dendrites, axons, and axon terminals of the SNc neurons (Fig. 1). Expression increased progressively over the first few weeks; maximal expression of the GFP or α-synuclein proteins was observed at 3–8 weeks (Figs.1E–G, 2E–G) and was maintained at a high level in the 27 week animals (Figs. 1D,H,L,2D,H,L). Consistent with previous observations (cf.Kirik et al., 2000) no signs of toxic or inflammatory reactions were observed in the cresyl violet and hematoxylin-stained sections at any time point, with either of the vectors used.
α-Synuclein-induced neuropathology and cell death
Signs of neuronal pathology developed progressively over the first 2 months after vector injection. This included the appearance of α-synuclein-positive cytoplasmic inclusions and granular deposits (Fig. 3A–C, arrowheads), swollen α-synuclein-positive dystrophic and fragmented neurites (Fig.3B,D–F), and dense, shrunken neuronal perikarya with a pyknotic appearance and strong α-synuclein immunoreactivity in the cytoplasm (Fig. 3F). Some of the larger inclusions had a clear immunonegative core surrounded by a α-synuclein-positive halo (Fig. 3C,D). These changes were first observed in the SNc neurons at 3 weeks and were further increased in magnitude at 8 weeks after injection, and resembled the α-synuclein inclusions and dystrophic neurites seen in idiopathic PD (Hayashida et al., 1993;Arima et al., 1998; Braak and Braak, 2000; Duda et al., 2000; Giasson et al., 2000b). None of them, however, stained positively with the ubiquitin antibody.
Fig. 3.
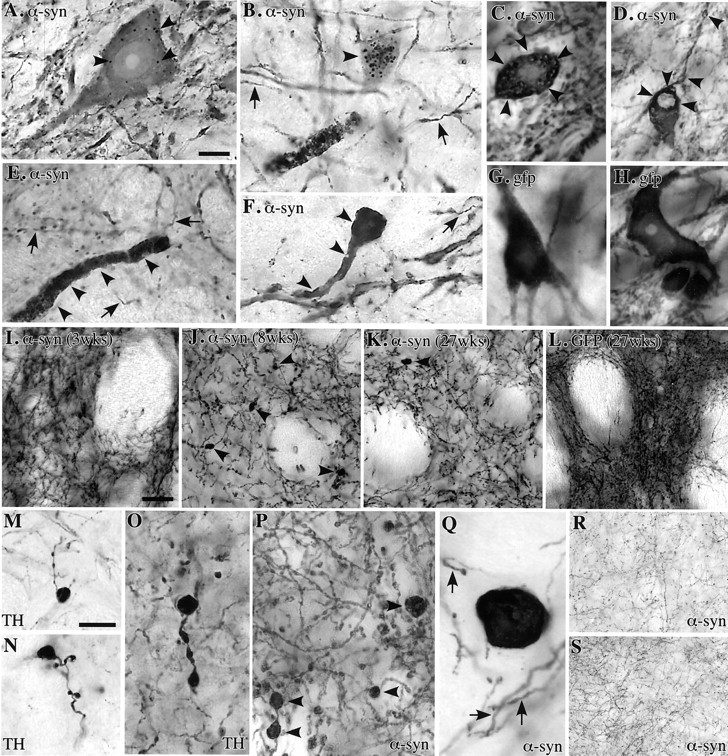
Eight weeks after injection α-synuclein-positive granular inclusions (A, B, arrowheads) and discrete intracytoplasmic inclusions where α-synuclein immunoreactivity occurred as a halo around a pale core (C, D, arrowheads) appear in large numbers in the cells of SNc, associated with swollen dystrophic α-synuclein-positive neurites (B,E, F) and atrophic perikarya (F). Arrows in B,E, F, and G indicate intact neurites of normal size; G, H, none of these changes occurred in GFP overexpressing SNc neurons;I–K, the α-synuclein-immunoreactive terminals in the striatum had a normal morphology at 3 weeks (I) but were reduced in number and displayed large numbers of swollen, dystrophic profiles at 8 weeks (J, P, arrowheads). At 27 weeks, fewer dystrophic fibers were seen, but the density of α-synuclein-positive terminals remained reduced (K); L, none of these axonal changes were observed in the GFP transduced controls, although GFP expression was maintained at high levels still by 27 weeks;M–Q, examples of swollen, dystrophic axon terminals in the striatum, revealed by TH (M–O) and α-synuclein antibodies (P, Q); R,S, expression of α-synuclein did not induce any pathological changes in nondopaminergic projections to the superior colliculus (R) and thalamus (S) (8 week survival). Scale bars: A,10 μm (applies to A–H, O–Q); I, 50 μm (applies to I–L, R, S); M, 20 μm (applies toM, N).
In the striatum, single α-synuclein-positive swollen axons occurred at 3 weeks after injection, and by 8 weeks abundant α-synuclein-positive dystrophic neurites were scattered throughout the striatum, most densely in the dorsolateral part (Fig. 3J,P,Q, arrowheads), accompanied by a marked reduction in the density of α-synuclein-positive striatal axon terminals (Figs.2B,C, 3I,J). Distorted axonal profiles also occurred in varying numbers in the olfactory tubercle and nucleus accumbens, and occasionally in septum and anterior cingulate cortex, i.e., in areas that receive projections from DA neurons in the VTA. Virtually all dystrophic neurites were positive for the DA-synthesizing enzyme TH (Fig. 3M–O), but they did not stain for either ubiquitin or the VMAT-2.
These pathological changes were similar, both quantitatively and qualitatively, in animals given either wt or mutant α-synuclein. No cellular or axonal pathology was seen at any time point in the rAAV-GFP-expressing animals (Figs. 1, 3G,H,L), showing that neither nonspecific impact of viral infection nor high expression of any cytoplasmic protein were sufficient to induce pathological changes of the kind seen in the α-synuclein overexpressing SNc neurons.
To analyze the impact of α-synuclein overexpression on nondopaminergic neurons, we studied three other projection systems that were efficiently transduced in the vector-injected animals: the nigrotectal and nigrothalamic projections, and the projections from the mesencephalic reticular formation to the diencephalon. α-Synuclein (both types) was diffusely expressed in cell bodies, axons, and terminals in all three projection systems, but apart from single swollen axonal profiles in the thalamus, α-synuclein-positive inclusions or dystrophic neurites were conspicuously absent in these nondopaminergic systems (Fig. 3R,S).
TH immunohistochemistry demonstrated a gradual loss of TH-positive cell bodies in the SN, similar for both wt and mutant α-synuclein, amounting to an average of 23% at 3 weeks and 55% at 8 weeks (Fig.4A,D–H) (data from the two α-synuclein groups combined). The TH-positive cell loss was notably variable, however, from 30–40% in some animals (Fig.4G) to 70–80% in others (Fig. 4H). Striatal TH-positive innervation was only marginally affected at 3 weeks after transduction (approximately −10%) (Fig.5B) but substantially reduced throughout the striatum (by ∼50%) at 8 weeks (Fig.5C,F). A similar reduction in the nigral DA neurons and striatal DA innervation was revealed by the VMAT-2 antibody (data not shown), as well as by the α-synuclein staining (Fig.2C). Analysis of adjacent sections stained for TH and α-synuclein revealed that part of the remaining fibers (identified through their α-synuclein content) had a very weak TH immunoreactivity. Some variable TH-positive cell loss occurred also in the VTA, and this was accompanied by a similarly variable reduction in the TH-positive innervation in nucleus accumbens and the olfactory tubercle (data not shown). TH-positive cell numbers and striatal TH-positive innervation were not significantly affected in the rAAV-GFP-injected animals at any time point, as observed in both the TH- and GFP-immunostained sections (Figs. 1C,D,4B,I, 5E,F).
Fig. 4.
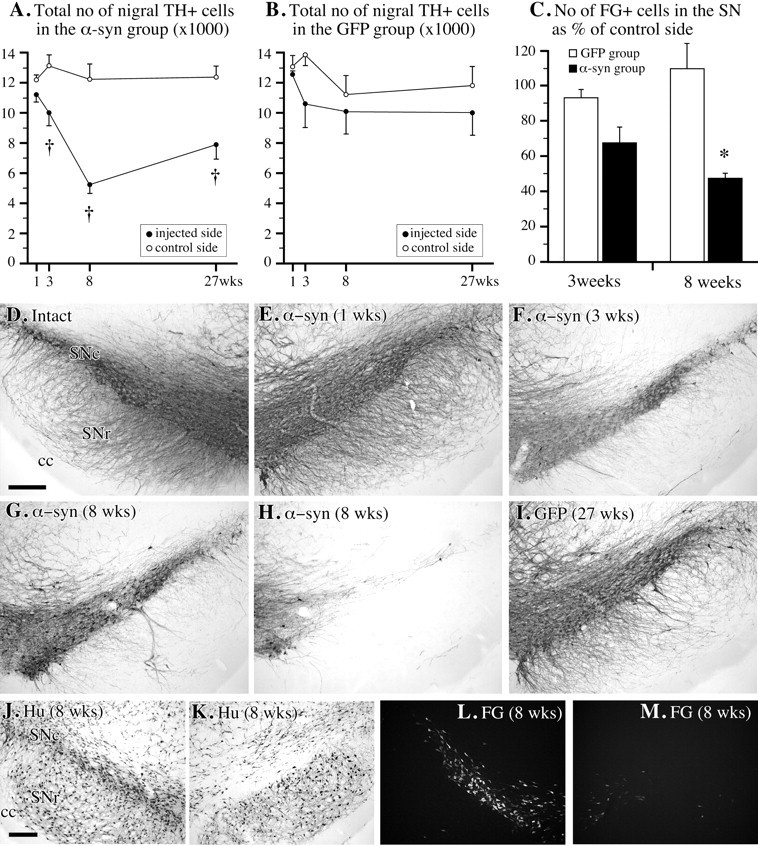
Cell loss in SN. A,B, Changes in total number of TH-positive cells in SNc, as determined by stereology in the rAAV-α-synuclein (A; n = 10–11 per time point) and rAAV-GFP-injected rats (B; n = 5 per time point); C, the number of SNc cells labeled with FG was reduced by ∼50% at 8 weeks in the rAAV-α-synuclein transduced animals but remained unchanged in the GFP transduced controls (M, injected side; L, contralateral intact side); D–H, TH immunohistochemistry showing the progressive loss of TH-positive cells in SNc over time, varying from ∼30–40% (G) to 70–80% (H); I, no TH-positive cell loss was observed in the rAAV-GFP-treated animals; J,K, the degeneration of neurons in the injected SNc (K) was confirmed by using the neuron specific antibody Hu. Scale bars: D, J, 250 μm (apply to D–I and J–M, respectively). † p < 0.0125 compared with intact side; *p < 0.05 compared with rAAV-GFP group.
Fig. 5.
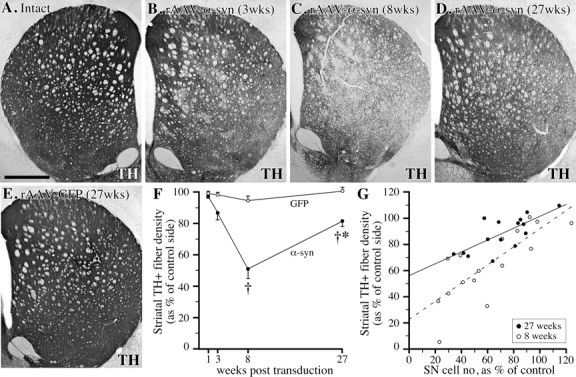
Loss of TH-positive innervation in the striatum. A–E, TH-immunostained sections showing the extent of loss of TH-positive striatal innervation at 3 (B), 8 (C), and 27 weeks (D) after intranigral rAAV-α-synuclein injection, as compared with the intact control side (A), and the innervation in the rAAV-GFP injected animals, which was unaffected (E).F, Densitometry revealed a significant reduction in TH-positive striatal innervation in the α-synuclein transduced animals at 8 and 27 weeks after injection († significant difference from both 1 week value and the GFP transduced control group atp < 0.0125). The recovery between 8 and 27 weeks in the α-synuclein group was significant (*p < 0.0125); G, further analysis of the correlation between nigral TH-positive cell numbers and striatal TH-positive fiber density (% of control side) showed that the best fit was obtained with a simple-linear regression analysis. At 8 weeks, the regression line (dashed) had a slope of 0.695 andy-intercept of 22.905 (p = 0.0003). However, at 27 weeks the slope of the line (solid) was reduced to 0.478, and y-axis intercept increased to 55.167 (p = 0.0016), suggesting that the loss of nigrostriatal projection neurons was in part compensated by increased innervation from the remaining cells. Scale bar: A, 1 mm (applies toA–E).
To determine whether the reduced number of TH-positive cells in the SN was attributable to an actual loss of cells or to a downregulation of the cellular markers used, i.e., the TH enzyme and the VMAT protein, we used three independent ways to label the nigral neurons. First, we used a retrograde tracer, FG, injected bilaterally in the striatum 5 d before the injection of the rAAV-α-synuclein vector, to label the cells in the SNc before the insult. The number of surviving FG-positive cells in the SNc was reduced by 32% at 3 weeks and by 52% at 8 weeks (Fig. 4C,L,M). Cell loss was similar in the wt and mutant α-synuclein groups (p > 0.05). Second, we used an antibody to the neuron-specific protein, Hu, as an independent marker of surviving SNc neurons. Consistent with the FG data, the Hu marker revealed a marked loss of SNc cells on the rAAV-α-synuclein-injected side that was fully developed at 8 weeks (Fig. 4J,K). Third, we used the rAAV-GFP vector to prelabel the SNc neurons before the α-synuclein vector injection. These animals received bilateral rAAV-GFP injections over the SN 5 d before the unilateral rAAV-α-synuclein injection. The number of GFP-labeled cells in the SNc (as assessed 8 weeks after injection) was markedly reduced on the α-synuclein transduced side (data not shown).
In the long-term surviving animals, analyzed 6 months after vector injection, the signs of cell body and axonal pathology were greatly reduced (Fig. 3K), suggesting that the damaged nigral cell bodies and axons had either degenerated (and disappeared), or recovered from the initial insult. α-Synuclein protein expression, however, was still maintained at a high level in the cell bodies and axons of the surviving nigrostriatal neurons after 6 months (Fig.3K). Signs of recovery were also observed in the TH-immunostained sections: the density of TH-positive fibers in the striatum had increased, from ∼50% of normal at 8 weeks to ∼80% of normal at 6 months (p < 0.0125) (Fig.5C,D,F). A similar degree of recovery was also observed in the VMAT-2-stained sections (data not shown). Further analysis indicated that the recovery of striatal TH-positive fibers, in part at least, was attributable to recovery of TH expression in a portion of the striatal axon terminals that were weakly TH immunoreactive at 8 weeks. Although the number of TH-positive neurons in the SNc showed a trend to an increase (from 46 to 65% of normal) between 8 and 27 weeks (Fig. 4A), this did not reach significance. Thus, the recovery of striatal TH-positive innervation was not matched by a similar recovery of TH-positive neurons in SNc, suggesting that axonal sprouting may have contributed to the recovery of striatal TH-positive innervation seen in the long-term survival animals (Fig. 5G).
Functional decline in the nigrostriatal dopamine system
The impact of α-synuclein overexpression on DA synthesis, storage, and turnover was assessed neurochemically in tissue samples from striatum and SN. Striatal DA levels (Fig.6A) and striatal TH enzyme activity (Fig. 6B) were reduced by 40–50% at 3 weeks, and these reductions remained unchanged at 8 and 27 weeks after α-synuclein transduction, similar for both wt and mutated α-synuclein (p > 0.05). No significant changes were seen in the rAAV-GFP-injected control rats (Fig.6A,B). DA levels in SN were reduced by 27.3 ± 7.9% at 3 weeks, 41.8 ± 7.0% at 8 weeks, and 28.2 ± 10.1% at 27 weeks in the rAAV-α-synuclein animals, whereas DA levels in other forebrain areas (nucleus accumbens and prefrontal cortex) were unchanged (data not shown). The reductions in striatal DA and TH activity levels were accompanied by a significant increase in DA turnover, measured as the DOPAC/DA ratio (Fig. 6C). This increase was only transient in the rAAV-GFP-treated animals, but was maintained at a level of ∼160% of normal at 8 and 27 weeks after injection in the α-synuclein-treated animals. No change in DA turnover was observed in either nucleus accumbens or prefrontal cortex.
Fig. 6.

Reductions in striatal DA and DOPAC and TH activity levels over time. A, B, Striatal DA content (A) and TH activity (B) were reduced by ∼40–50% at all time points; C, striatal DA turnover, as assessed by the DOPAC/DA ratio, was increased by ∼60% at all time points in the α-synuclein transduced animals (filled circles) but only transiently increased in the GFP group (open circles). † p < 0.05 (effect of group, two-way repeated measures ANOVA).
The long-lasting reductions in striatal DA and TH activity levels in the α-synuclein transduced animals are consistent with the observed cell loss in the SNc. However, the fact that striatal DA levels and TH activity was already maximally reduced at 3 weeks, i.e., at a time when the loss of TH-positive cells in the SNc was only partial (∼20%), suggests that elevated cytoplasmic levels of α-synuclein (both wt and mutant) may exert an inhibitory effect on DA synthesis and storage, independent of its neurodegenerative action. Such an inhibitory effect on neurotransmitter function may also explain why striatal DA and TH enzyme activity remained low (−40 to −50%) at 6 months, despite that TH-positive fiber density in the striatum had recovered to ∼75% of normal at this time point.
Impairments in motor behavior
Motor behavior, as monitored in a battery of drug-induced and spontaneous behavioral tests, was overall only marginally affected in the α-synuclein-treated animals at all time points. This suggests that the observed magnitude of DA neuron cell loss, and striatal DA depletion, was insufficient to induce easily detectable behavioral impairments. This is in agreement with previous studies, which have shown that significant impairments in drug-induced and spontaneous motor behavior will appear only in animals in which the nigral DA neurons and striatal DA levels are reduced by >50–60% (Lee et al., 1996; Kirik et al., 1998). Closer inspection of the individual animals, however, revealed a marked variability in the behavioral performance among the animals in the α-synuclein-treated group (Fig.7A,B). Thus, in both the apomorphine rotation and the paw-reaching tests, ∼25% of the α-synuclein transduced animals were significantly impaired compared with the GFP controls (Fig. 7A,B, filled circles). This level of impairment (10–20 pellets retrieved in the paw reaching test, compared with 30 ± 4 pellets in the rAAV-GFP control group) is compatible with a loss of 60–80% of the nigral DA neurons (Lee et al., 1996; Kirik et al., 1998).
Fig. 7.

Changes in motor behavior. In all three tests the overall performance of the α-synuclein transduced animals did not differ from the GFP group (p > 0.05 for all contrasts). However, in both the apomorphine rotation test (A; data from the 8 week test) and the paw reaching test (B; performed at 24 weeks) 25% of the α-synuclein transduced animals were clearly impaired in that they scored lower than any animal in the GFP control group (filled circles). Bars in A and B give means ± 1 SD. C, A single low dose of the DA synthesis inhibitor α-methyl-dl-p-tyrosine, given on the fourth day of testing, produced a significant impairment in the stepping test. † Different from both predrug test and GFP group atp < 0.0125.
These data suggest that behavioral impairment developed in those α-synuclein-treated rats where TH-positive nigral cell loss exceeded a critical threshold of ∼60%. It is known that partial lesions of the nigrostriatal DA projection may be partly compensated for by an increased DA turnover in the residual afferents (Zigmond et al., 1990), which explains why normal striatal function can be maintained in animals where 50–60% of the striatal DA innervation is lost. Such compensated animals, however, are sensitive to blockers of DA synthesis, which will induce motor impairments at dose levels that have no effect in intact animals (Heffner et al., 1977; Marshall, 1979). This approach, therefore, can be used to reveal the underlying functional deficit in neurochemically compensated partially lesioned animals.
The effect of low-dose DA synthesis blockade was investigated 8 weeks after injection in rats given wt rAAV-α-synuclein (n = 10), mutant rAAV-α-synuclein (n= 10), or rAAV-GFP (n = 10), using the forelimb stepping test (Fig. 7C). In the 3 d predrug test, all groups performed at normal levels (∼11 steps). On the fourth day, the rats were given a single low dose of the TH enzyme inhibitor, α-methyl-p-tyrosine. Six hours later, when TH inhibition was maximal, the α-synuclein transduced animals were significantly impaired both relative to the rAAV-GFP-treated controls and their own predrug baseline (p < 0.01). The effect was similar in both α-synuclein groups (p > 0.05) (combined in Fig. 7C). Gradual recovery of normal motor performance was seen over the subsequent 2 d.
DISCUSSION
These results show that overexpression of wt or mutant α-synuclein can induce a progressive neurodegenerative pathology in the nigrostriatal DA neurons, characterized by α-synuclein-positive cytoplasmic and axonal inclusions, dystrophic and fragmented neurites, and cell death. These degenerative changes were specific for the nigral DA neurons, and were not seen in any of the three mesencephalic nondopaminergic neuron systems that were efficiently transduced by the rAAV-α-synuclein vectors. The impact of α-synuclein expression appeared to be twofold: first, a suppression of TH enzyme activity and striatal DA levels (by ∼40–50%) at a stage when α-synuclein had a diffuse cytoplasmic distribution (3 weeks after injection), followed by a loss of 30–80% of the nigral DA neurons, coincident with the appearance of cytoplasmic inclusions and dystrophic neurites, and the development of significant motor impairment in those animals in which DA neuron cell loss exceeded a critical threshold of 50–60%. More than 90% of the TH-positive neurons in the SNc were transduced by the rAAV vector. Consistent with previous studies using this vector (Mandel et al., 1998; Bjorklund et al., 2000; Kirik et al., 2000) the α-synuclein and GFP transgenes showed stable, long-term expression in the nigrostriatal neurons and high expression was maintained 27 weeks after vector injection. Because rAAV is a single-stranded DNA virus, it is possible that two virions with the cDNA in complementary directions must infect a cell for successful transduction (Ferrari et al., 1996). This opens the possibility that widely varying numbers of genome copies are inserted into a given cell. The final expression level, therefore, is likely to vary among the transduced cells. This may readily explain why only some of the transduced SNc neurons degenerated and why cell loss was variable from animal to animal.
Previous in vitro studies suggest that overexpression of α-synuclein can be toxic to cells and that this protein can interact with oxidative stress and mitochondrial damage to exert its cytotoxic actions (Ostrerova et al., 1999; Hsu et al., 2000; Kanda et al., 2000;Tabrizi et al., 2000). Thus, oxidative stress, as well as increased cellular α-synuclein levels, may promote α-synuclein aggregation (Giasson et al., 2000b; Hsu et al., 2000), which in turn may lead to mitochondrial damage and increased production of toxic free radicals (Ostrerova et al., 1999; Hsu et al., 2000). Moreover, Giasson et al. (2000b) have reported that α-synuclein is a target for oxidative damage and that aggregation of α-synuclein into toxic inclusions may be caused by nitration of the protein by reactive oxygen and nitrogen species. Our results, as well as those of a recent study inDrosophila (Feany and Bender, 2000), indicate that DA neurons may be particularly vulnerable to α-synuclein overexpression. This may be explained by the fact that free radical production is high in these types of cells, because of their content of both DA and iron. Similarly, oxidative stress in combination with reduced levels of mitochondrial complex I and α-synuclein aggregation have been proposed to underlie the selective death of nigral DA neurons in human PD (Hsu et al., 2000; Tabrizi et al., 2000).
This “double-hit” model may account for the fact that α-synuclein overexpression in the present study induced pathological changes and cell death in some, but not all nigral DA neurons. The signs of ongoing pathology, including α-synuclein-positive inclusions and dystrophic neurites, subsided over time, despite the fact that the cellular expression of wt or mutant α-synuclein protein was maintained at a high level. This suggests that those neurons that survived the initial impact of α-synuclein overexpression could survive and function even in the presence of a maintained, increased cellular level of the transgene product. α-Synuclein per se, therefore, may not be toxic to the cells as long as they are not exposed to excessive oxidative stress or other events that may impair mitochondrial function. The neurochemical data, however, suggest that α-synuclein may have a negative effect on DA neuron function, even in the absence of any overt pathological signs. Thus, in the long-term surviving animals, 6 months after vector injection, striatal DA levels and striatal TH enzyme activity was reduced by an average of 40–50%, whereas the striatal TH-positive innervation showed a partial recovery at this time point (to ∼75% of normal). Consistent with the observations in the short-term surviving animals, i.e., 3 weeks after vector injection, these data suggest that increased intracellular levels of α-synuclein may have a direct inhibitory effect on DA synthesis and storage.
Although oxidative stress has long been hypothesized to be involved in the pathogenesis of PD, dysfunction of protein degradation by the ubiquitin-proteosome processing system has only recently been implicated as a major pathogenic mechanism. The interest in this protein degradation mechanism is attributable to the identification of multiple forms of familial PD in which the mutated genes: α-synuclein, parkin, and ubiquitin C-terminal hydrolase L1 (UCHL1) are all related to the proteosome degradation system (Shimura et al., 2000). Thus, overexpression of α-synuclein in nigral DA neurons, at the levels achieved in this study, may overwhelm the ability of the proteosomal pathway to successfully process and remove excess α-synuclein from the cell. Moreover, α-synuclein overexpression may lead to increased levels of the 22 kDa form of α-synuclein in the cell (Shimura et al., 2001). Whereas this glycosylated α-synuclein isoform has not been well characterized, it may be toxic to some neurons but not others (Shimura et al., 2000). According to this model, all forms of α-synuclein, wt or mutant, may be toxic if they are expressed at sufficiently high levels, or if they are subjected to oxidative damage (e.g., by oxidative metabolism of DA in nigral neurons), or because the cellular handling of the α-synuclein protein is impaired (as may be the case in patients with mutations in the parkin gene; Shimura et al. 2001). This contention would further suggest that nondopaminergic neurons, outside the substantia nigra, or the resistant DA neurons within the substantia nigra, may be protected by more efficient handling of excess α-synuclein. The fact that the α-synuclein-positive inclusions seen here did not stain for ubiquitin would be consistent with the fact that α-synuclein itself may not be a substrate for ubiquitination (Tofaris et al., 2001). The speed by which neurodegeneration occurs (3–8 weeks in the overexpression model studied here, in contrast to months or years in PD patients), furthermore, may be explained, at least in part, by the level of overexpression of α-synuclein within the cells and the ability of the cells to handle degradation of this protein.
The technique for targeted α-synuclein overexpression in the nigrostriatal system using rAAV vectors provides a new model of PD that reproduces some of the cardinal pathological, neurochemical, and behavioral features of the human disease, and may, therefore provide an interesting tool to elucidate the pathogenetic mechanisms underlying neurodegeneration in familial or idiopathic PD. The use of viral vectors to overexpress putative toxic proteins, such as α-synuclein, may offer distinct advantages over standard transgenic mice. First, viral vectors may be administered at any time during the life span of the animal to any specific anatomical location in the brain. Second, viral vectors can be administered in one hemisphere, allowing the other hemisphere to serve as a control, and in the model system used here, allow the use of functional tests that rely on behavioral asymmetries. Third, viral vector transduction has the advantage that it can be used in rats, i.e., in a species that is more useful for behavioral studies than mice. Finally, the use of the viral vector strategy is also applicable to other species and may thus also allow studies on the effects of α-synuclein overexpression in primates. Indeed, α-synuclein overexpression using the viral vector strategy resulted in more pronounced neuropathology and cell death in the nigrostriatal DA system than has been achieved so far using transgenic technology in mice. The viral vector strategy will thus add an important research tool to complement other, more well known, in vivo gene modification strategies.
Footnotes
This work was supported by Swedish Medical Research Council Grants 04X-3874 and 99-XG-13285, NsGene A/S (Denmark), and National Institutes of Health Grant PO1 NS36302 (N.M.). We thank Kerstin Fogelström and Ulla Jarl for expert technical assistance, the Powell Gene Therapy Center Vector Core Laboratory for production of the vectors, and Dr. Virginia M. Lee for generous supply of the α-synuclein antibody.
Correspondence should be addressed to Anders Björklund, Wallenberg Neuroscience Center, BMC A11, S-221 84, Lund, Sweden. E-mail: Anders.Bjorklund@mphy.lu.se.
REFERENCES
- 1.Arima K, Ueda K, Sunohara N, Hirai S, Izumiyama Y, Tonozuka-Uehara H, Kawai M. Immunoelectron-microscopic demonstration of NACP/alpha-synuclein- epitopes on the filamentous component of Lewy bodies in Parkinson's disease and in dementia with Lewy bodies. Brain Res. 1998;808:93–100. doi: 10.1016/s0006-8993(98)00734-3. [DOI] [PubMed] [Google Scholar]
- 2.Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 3.Bjorklund A, Kirik D, Rosenblad C, Georgievska B, Lundberg C, Mandel RJ. Towards a neuroprotective gene therapy for Parkinson's disease: use of adenovirus, AAV and lentivirus vectors for gene transfer of GDNF to the nigrostriatal system in the rat Parkinson model. Brain Res. 2000;886:82–98. doi: 10.1016/s0006-8993(00)02915-2. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Braak E. Pathoanatomy of Parkinson's disease. J Neurol [Suppl 2] 2000;247:3–10. doi: 10.1007/PL00007758. [DOI] [PubMed] [Google Scholar]
- 5.Conway JE, Zolotukhin S, Muzyczka N, Hayward GS, Byrne BJ. Recombinant adeno-associated virus type 2 replication and packaging is entirely supported by a herpes simplex virus type 1 amplicon expressing Rep and Cap. J Virol. 1997;71:8780–8789. doi: 10.1128/jvi.71.11.8780-8789.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duda JE, Lee VM, Trojanowski JQ. Neuropathology of synuclein aggregates. J Neurosci Res. 2000;61:121–127. doi: 10.1002/1097-4547(20000715)61:2<121::AID-JNR1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 7.Feany MB, Bender WW. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 8.Ferrari FK, Samulski T, Shenk T, Samulski RJ. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virol. 1996;70:3227–3234. doi: 10.1128/jvi.70.5.3227-3234.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giasson BI, Jakes R, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VM. A panel of epitope-specific antibodies detects protein domains distributed throughout human alpha-synuclein in Lewy bodies of Parkinson's disease. J Neurosci Res. 2000a;59:528–533. doi: 10.1002/(SICI)1097-4547(20000215)59:4<528::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 10.Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000b;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 11.Hayashida K, Oyanagi S, Mizutani Y, Yokochi M. An early cytoplasmic change before Lewy body maturation: an ultrastructural study of the substantia nigra from an autopsy case of juvenile parkinsonism. Acta Neuropathol. 1993;85:445–448. doi: 10.1007/BF00334457. [DOI] [PubMed] [Google Scholar]
- 12.Heffner TG, Zigmond MJ, Stricker EM. Effects of dopaminergic agonists and antagonists of feeding in intact and 6-hydroxydopamine-treated rats. J Pharmacol Exp Ther. 1977;201:386–399. [PubMed] [Google Scholar]
- 13.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. alpha-synuclein promotes mitochondrial deficit, oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irizarry MC, Growdon W, Gomez-Isla T, Newell K, George JM, Clayton DF, Hyman BT. Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson's disease and cortical Lewy body disease contain alpha-synuclein immunoreactivity. J Neuropathol Exp Neurol. 1998;57:334–337. doi: 10.1097/00005072-199804000-00005. [DOI] [PubMed] [Google Scholar]
- 15.Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van Der Putten H, Probst A, Kremmer E, Kretzschmar HA, Haass C. Subcellular localization of wild-type and Parkinson's disease-associated mutant alpha-synuclein in human and transgenic mouse brain. J Neurosci. 2000;20:6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanda S, Bishop JF, Eglitis MA, Yang Y, Mouradian MM. Enhanced vulnerability to oxidative stress by alpha-synuclein mutations and C-terminal truncation. Neuroscience. 2000;97:279–284. doi: 10.1016/s0306-4522(00)00077-4. [DOI] [PubMed] [Google Scholar]
- 17.Kirik D, Rosenblad C, Bjorklund A. Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Exp Neurol. 1998;152:259–277. doi: 10.1006/exnr.1998.6848. [DOI] [PubMed] [Google Scholar]
- 18.Kirik D, Rosenblad C, Bjorklund A, Mandel RJ. Long-term rAAV-mediated gene transfer of GDNF in the rat Parkinson's model: intrastriatal but not intranigral transduction promotes functional regeneration in the lesioned nigrostriatal system. J Neurosci. 2000;20:4686–4700. doi: 10.1523/JNEUROSCI.20-12-04686.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klein RL, Meyer EM, Peel AL, Zolotukhin S, Meyers C, Muzyczka N, King MA. Neuron-specific transduction in the rat septohippocampal or nigrostriatal pathway by recombinant adeno-associated virus vectors. Exp Neurol. 1998;150:183–194. doi: 10.1006/exnr.1997.6736. [DOI] [PubMed] [Google Scholar]
- 20.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 21.Lee CS, Sauer H, Bjorklund A. Dopaminergic neuronal degeneration and motor impairments following axon terminal lesion by intrastriatal 6-hydroxydopamine in the rat. Neuroscience. 1996;72:641–653. doi: 10.1016/0306-4522(95)00571-4. [DOI] [PubMed] [Google Scholar]
- 22.Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson's disease. Nature. 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- 23.Mandel RJ, Rendahl KG, Spratt SK, Snyder RO, Cohen LK, Leff SE. Characterization of intrastriatal recombinant adeno-associated virus-mediated gene transfer of human tyrosine hydroxylase and human GTP- cyclohydrolase I in a rat model of Parkinson's disease. J Neurosci. 1998;18:4271–4284. doi: 10.1523/JNEUROSCI.18-11-04271.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marshall JF. Somatosensory inattention after dopamine-depleting intracerebral 6-OHDA injections: spontaneous recovery and pharmacological control. Brain Res. 1979;177:311–324. doi: 10.1016/0006-8993(79)90782-0. [DOI] [PubMed] [Google Scholar]
- 25.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 26.Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, Farrer M, Hardy J, Duff K, Przedborski S, Di Monte DA. Lack of nigral pathology in transgenic mice expressing human alpha- synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–539. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- 27.McLaughlin SK, Collis P, Hermonat PL, Muzyczka N. Adeno-associated virus general transduction vectors: analysis of proviral structures. J Virol. 1988;62:1963–1973. doi: 10.1128/jvi.62.6.1963-1973.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P. Failure of the ubiquitin-proteasome system in Parkinson's disease. Nat Rev Neurosci. 2001;2:589–594. doi: 10.1038/35086067. [DOI] [PubMed] [Google Scholar]
- 29.Muzyczka N. Use of adeno-associated virus as a general transduction vector for mammalian cells. Curr Top Microbiol Immunol. 1992;158:97–129. doi: 10.1007/978-3-642-75608-5_5. [DOI] [PubMed] [Google Scholar]
- 30.Ostrerova N, Petrucelli L, Farrer M, Mehta N, Choi P, Hardy J, Wolozin B. alpha-Synuclein shares physical and functional homology with 14–3-3 proteins. J Neurosci. 1999;19:5782–5791. doi: 10.1523/JNEUROSCI.19-14-05782.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 32.Rathke-Hartlieb S, Kahle PJ, Neumann M, Ozmen L, Haid S, Okochi M, Haass C, Schulz JB. Sensitivity to MPTP is not increased in Parkinson's disease-associated mutant alpha-synuclein transgenic mice. J Neurochem. 2001;77:1181–1184. doi: 10.1046/j.1471-4159.2001.00366.x. [DOI] [PubMed] [Google Scholar]
- 33.Reinhard JF, Jr, Smith GK, Nichol CA. A rapid and sensitive assay for tyrosine-3-monooxygenase based upon the release of 3H2O and adsorption of [3H]-tyrosine by charcoal. Life Sci. 1986;39:2185–2189. doi: 10.1016/0024-3205(86)90395-4. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt RH, Ingvar M, Lindvall O, Stenevi U, Björklund A. Functional activity of substantia nigra grafts reinnervating the striatum: neurotransmitter metabolism and [14C]2-deoxy-d-glucose autoradiography. J Neurochem. 1982;38:737–748. doi: 10.1111/j.1471-4159.1982.tb08693.x. [DOI] [PubMed] [Google Scholar]
- 35.Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 36.Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science. 2001;293:263–269. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- 37.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 38.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tabrizi SJ, Orth M, Wilkinson JM, Taanman JW, Warner TT, Cooper JM, Schapira AH. Expression of mutant alpha-synuclein causes increased susceptibility to dopamine toxicity. Hum Mol Genet. 2000;9:2683–2689. doi: 10.1093/hmg/9.18.2683. [DOI] [PubMed] [Google Scholar]
- 40.Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E. Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am J Pathol. 1998;152:367–372. [PMC free article] [PubMed] [Google Scholar]
- 41.Tofaris GK, Layfield R, Spillantini MG. alpha-synuclein metabolism, aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001;509:22–26. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- 42.van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren WP, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M, Bilbe G. Neuropathology in mice expressing human alpha-synuclein. J Neurosci. 2000;20:6021–6029. doi: 10.1523/JNEUROSCI.20-16-06021.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu L, Daly T, Gao C, Flotte TR, Song S, Byrne BJ, Sands MS, Ponder KP. CMV-beta-actin promoter directs higher expression from an adeno-associated viral vector in the liver than the cytomegalovirus or elongation factor 1alpha promoter and results in therapeutic levels of human factor X in mice. Hum Gene Ther. 2001;12:563–573. doi: 10.1089/104303401300042500. [DOI] [PubMed] [Google Scholar]
- 44.Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends Neurosci. 1990;13:290–296. doi: 10.1016/0166-2236(90)90112-n. [DOI] [PubMed] [Google Scholar]
- 45.Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]