

Abstract
We have previously shown (Otani et al., 1999b) that bath application of (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG IV), the agonist of group II metabotropic glutamate receptors (mGluRs), induces postsynaptic Ca2+-dependent long-term depression (LTD) of layer I-II to layer V pyramidal neuron glutamatergic synapses of rat medial prefrontal cortex. In the present study, we examined detailed mechanisms of this DCG IV-induced LTD. First, the group II mGluR antagonist (RS)-α-methylserine-O-phosphate monophenyl ester blocked DCG IV-induced LTD, and another group II agonist (2S,3S,4S)-CCG/(2S,1′S,2′S)-2-(carboxycyclopropyl)glycine-induced LTD, suggesting that LTD is indeed mediated by the activation of group II mGluRs. Second, DCG IV-induced LTD was blocked by the NMDA receptor antagonist AP-5, whereas DCG IV did not potentiate NMDA receptor-mediated synaptic responses. Interruption of single test stimuli during DCG IV application blocked DCG IV-induced LTD. These results suggest that small NMDA receptor-mediated responses evoked by single synaptic stimuli contribute to DCG IV-induced LTD. Third, DCG IV-induced LTD was blocked or reduced by the following drugs: phospholipase C inhibitor U-73122 (bath-applied or postsynaptically injected), postsynaptically injected IP3receptor blocker heparin, phospholipase D-linked mGluR blocker PCCG-13, PKC inhibitor RO318220, postsynaptically injected PKC inhibitor PKC(19–36), and PKA inhibitor KT-5720. Fourth, fluorescent Ca2+ analysis techniques revealed that DCG IV increases Ca2+ concentration in prefrontal layer V pyramidal neurons. These Ca2+ rises and the LTD were both blocked by postsynaptic heparin in the same cells. Taken together, these results suggest that postsynaptic group II mGluRs, linked to phospholipase C and probably also phospholipase D, induce LTD through postsynaptic PKC activation and IP3receptor-mediated postsynaptic increases of Ca2+concentration.
Keywords: long-term depression, synaptic plasticity, prefrontal cortex, group II metabotropic glutamate receptors, IP3receptors, calcium release, phospholipase C, phospholipase D, protein kinase C, protein kinase A
It is well known that primate dorsolateral prefrontal cortex (PFC) plays important role in attention- and memory-guided behavior, such as spatial working memory (Goldman-Rakic, 1995), selection and remembering of relevant stimuli (Rainer et al., 1998), and remembering of contextually relevant, cross-modal stimulus association (Fuster et al., 2000). In rats, these functions may be performed by prelimbic area (Zhart et al., 1997;Birrell and Brown, 2000), i.e., the medial aspect of frontal cortex, which may be anatomically analogous to human–primate PFC (Kolb, 1984).
Several examples of plasticity of glutamatergic synapses in rat prelimbic area (hereafter called rat PFC) have been reported (Hirsch and Crepel, 1990; Law-Tho et al., 1995; Vickery et al., 1997; Gurden et al., 1999; Morris et al., 1999; Takita et al., 1999; Blond et al., 2002). We have recently shown that the induction of long-term depression (LTD) in rat PFC is facilitated by dopamine, and that this LTD induction depends on concurrent synaptic activation of groups I and II metabotropic glutamate receptors (mGluRs) (Otani et al., 1998b,1999b). We proposed that a mechanism for the cooperativity between dopamine receptors (DA-Rs) and the mGluRs for LTD induction is postsynaptic converging activation of mitogen-activated protein kinases (MAPKs) by these receptors. Somewhat surprisingly, in this study (Otani et al., 1999b), we also found that LTD can be induced solely by bath-application (10–15 min) of the potent group II mGluR agonist (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG IV) (Ishida et al., 1993; Brabet et al., 1998). DCG IV-induced LTD was blocked by postsynaptic injection of the Ca2+ chelator bis-(o-aminophenoxy)-N,N,N′,N′-tetra-acetic acid (BAPTA). The source of critical Ca2+ for the DCG IV-induced LTD is not a flow through voltage-activated Ca2+ channels, because DCG IV did not depolarize the postsynaptic membrane or increase neuronal excitability or synaptic responses (Otani et al., 1999b).
These DCG IV data are inconsistent with the view that group II mGluRs are presynaptically located and inhibitory in the forebrain neurons (Baskys and Malenka, 1991; Lovinger, 1991). However, an immunohistological study suggests that group II mGluRs exist in postsynaptic sites in the cortex (Petralia et al., 1996). Moreover, biochemical data show that DCG IV increases the activity of phospholipase C (PLC) and phospholipase D (PLD) in hippocampal slices (Klein et al., 1997), or enhances the turnover of phosphoinositides in neonatal rat cerebral cortex slices (Mistry et al., 1998). Indeed, the brain-abundant PLC-β can be activated by βγ subunits of Gi/o-type G-protein (Rebecchi and Pentyala, 2000). Gi/o-type G-protein is associated with group II mGluRs (Conn and Pin, 1997), and its α subunit inhibits adenylate cyclase (Nestler and Duman, 1994). Thus, the available data point to the possibility that activation of group II mGluRs can lead to postsynaptic Ca2+ concentration ([Ca2+]) rises through release from internal Ca2+ stores. In the present study, therefore, we tested whether the group II mGluR agonist DCG IV induces LTD through its postsynaptic action on PLC and IP3 receptors, and through consequential postsynaptic protein kinase activation and Ca2+ rises.
Some of the results have appeared in abstract form (Otani et al., 1998a, 1999a).
MATERIALS AND METHODS
Slice preparation and intracellular recording. Male Sprague Dawley rats (23- to 30-d-old) were decapitated. The brain was rapidly removed, and coronal slices (300 μm; 2.2–3.7 mm from bregma) were sectioned by the use of a Campden vibratome in chilled (≈0°C) oxygenated (95% O2 and 5% CO2) artificial CSF (ACSF). The composition of ACSF was as follows (mm): NaCl 124, KCl 2, NaHCO326, KH2PO4 1.15, MgCl2 1, CaCl22, andd-glucose 11. The slices were allowed to recover for at least 2 hr at room temperature (≈20°C) in a container filled with continuously oxygenated ACSF. For the experiments, a slice was transferred to a submerged-type recording chamber where it was perfused with warmed ACSF (28°C) at the rate of 1 ml/min.
The soma of layer V pyramidal neurons in the prelimbic area of the medial frontal cortex was penetrated by sharp, glass micropipettes filled with 3 m K-acetate (80–120 MΩ tip resistance). Negative currents were initially injected by the use of an Axoclamp 2A amplifier (Axon Instruments, Foster City, CA), but after stabilization of the cells, most or all currents were removed. The cells had mean resting membrane potential of −72 ± 0.4 mV (SEM) with initial input resistance of 48 ± 1.5 MΩ. Mean membrane potential held during the experiments was −73 ± 0.4 mV. Cells that had stable membrane potential more negative than −65 mV, a spike height approximately ≥70 mV, and initial input resistance of at least 30 MΩ were considered healthy, and the experiments were started. When cells showed a sign of degradation (a constant decline in any of these parameters reaching more than ∼10% changes of the initial values), the experiments were stopped. The mode of spike discharge was routinely examined before the experiment by the application of a depolarizing current step (500 msec) from resting membrane potential. The intensity of the depolarizing step was set so that a 30 msec application at that intensity caused the cell to discharge one action potential. Of 137 cells tested, 51 cells (37%) were classified as regular spiking cell, and 33 cells (24%) as bursting cell. Twenty-two cells (16%) showed initial bursting followed by regular spiking typically with adaptation. The remaining 31 cells (23%) showed initial repetitive, or rather sporadic, spikes that abruptly ceased by a strong adaptation. As in our previous studies (Otani et al., 1998b, 1999b), there was no correlation between a discharge mode and synaptic plasticity induction.
A bipolar, Teflon-coated tungsten stimulating electrode (external diameter, 125 μm) was placed on layer I-II of the prelimbic area (immediately interior to pial surface). The EPSP of ∼10 mV amplitude was evoked at 0.033 Hz by the application of monophasic square voltage pulses (100 μsec duration; Digitimer isolated stimulator). During a typical experiment, at least a 20 min period of baseline collection was followed by a drug application phase. Then, the EPSP was continuously monitored until 45 min after the beginning of the drug washout. All evoked responses were fed to an Axoclamp 2A amplifier at current-clamp mode and were digitized at 5–10 kHz with a Labmaster interface and stored in an on-line IBM computer for later analyses (ACQUIS1 program; developed by Dr. G. Sadoc, Institut Alfred Fessard, Centre National de la Recherche Scientifique, Gif-sur-Yvette, France). All experiments were performed in the presence of GABA-A antagonist bicuculline methiodide (1 μm) in the bathing medium.
For the analysis of the EPSP, initial rising slope (1 msec period from its onset; in millivolts per millisecond), which contains only a monosynaptic component (Hirsch and Crepel, 1990), was measured. To express changes in the EPSP slope, we typically averaged responses from the 10 min period just before a drug application (baseline), the final 2–3 min period of the drug application phase (to assess acute effects of drugs), and the 35–45 min period after the beginning of drug washout (to assess long-term effects of the drugs). We then calculated percentage of changes of the initial slope from the baseline value. In the text, the 35–45 min period will be referred to as “40 min after drug washout.” These percentage changes were compared between different drug groups. Statistical analyses (two-tailed Student'st test) were performed with p < 0.05 considered as significant. All values were expressed as mean ± SEM.
For morphological identification of the neurons, biocytin (1.5%; Sigma, St. Louis, MO) was routinely included in recording electrodes and allowed to diffuse to postsynaptic cells. Immediately after the termination of the experiments, the slices were fixed in 4% paraformaldehyde dissolved in potassium PBS (0.01 m) at least overnight. They were then washed in the PBS solution three times (10 min each) and placed in 1 ml of 0.1% PBS–Triton X-100 solution containing 25 μl of solution A (Avadin) and solution B (biotinylated horseradish peroxidase H), as provided by Vectastain Standard ABC kit (Vector Laboratories, Burlingame, CA), for up to 48 hr. The slices were washed again in PBS solution. They were then placed in diaminobenzidine tetrahydrochloride (DAB) solution (Peroxidase Substrate Kit, SK-4100; Vector Laboratories) for 10 min. The slices were washed three times in PBS solution before being mounted on microscope slides.
Ca2+ analysis with fluorescent indicators and patch-clamp recording. For global analysis of Ca2+ concentration ([Ca2+]) in prefrontal areas (cf. Takita et al., 1997), cortical slices were first stained with fura-2 AM (10 μm with 0.001% Cremophore EL, a nontoxic surface-active reagent) for 30 min, then incubated in normal solution for at least 1 hr at room temperature. A stained slice was mounted on a thin glass (0.15 mm) chamber (300 μl bath volume) located above a raised silicon-intensified target video camera attached to an emission filter (510 nm), and was perfused with ACSF at the rate of 2 ml/min at 32°C. Two types of excitation light (340 and 360 nm) were applied alternately by an automatic filter changer. The angles for the excitation lights were each at 45°-oblique arranged by a diagonally set dual light fiber. Camera images of 340 and 360 nm were accumulated consecutively from 64 video frames and recorded every 30 sec by a calcium image processor (Argus-50/CA; Hamamatsu, Tokyo, Japan). The analysis was performed on the basis of 340:360 nm ratio images.
For single-neuron [Ca2+] analysis and patch-clamp recording, layer V pyramidal neurons were selected by visual guidance with Nomarski optic through a 40× water immersion objective. All experiments were performed with whole-cell configuration achieved at somatic level in the voltage-clamp mode, using an AXOPATCH 1D amplifier (Axon Instruments). The patch pipettes (2–3.5 MΩ tip resistance) were filled with internal solution containing (mm): KCl or potassium gluconate 140, NaCl 8, HEPES 10, ATP-Mg 2, and 100 μm Fluo-3 (final pH 7.3 with KOH, 300 mOsm/l). Before starting a Ca2+fluorometric recording session, or a fluorometric session combined with electrophysiological recording, the neurons were initially recorded in current-clamp mode, and neuronal discharge was tested by applying a depolarizing current step. Only neurons exhibiting at least a spike height of ∼70 mV from resting membrane potential were retained to continue experiments. Throughout the experiments, the cells were maintained at a holding potential of −80 mV, and the passive electrical properties as well as the stability of access resistances were continuously monitored by the application of −10 mV hyperpolarizing voltage steps. As previously described (Daniel et al., 1999), fluorescence measurements were performed with the high-affinity Ca2+-sensitive indicator fluo-3 loaded to postsynaptic cells via patch pipettes. The fluorometric recording session started 20–30 min after whole-cell “break in.” This period was required to allow dye diffusion and to washout background fluorescence in the surrounding tissue that occurs by dye leakage before sealing. The Ca2+-sensitive dye was excited by light from a 100 W Xenon lamp, and epifluorescence excitation wavelength was at 485 ± 22 nm. Emitted light was collected by a photometer through a barrier filter at 530 ± 30 nm, from the area (40 × 40 μm) centered on the soma. Because this single wavelength method does not determine absolute free Ca2+ levels, fluorescence changes of fluo-3 were expressed as a ratio to the initial background-corrected resting fluorescence. The fluorescence data were analyzed both on-line and off-line by the use of the ACQUIS1 program. For the sessions combining Ca2+ fluorometric and electrophysiological measurements, the EPSCs were evoked at 0.067 Hz by the application of monophasic square voltage pulses (20 μsec; Digitimer isolated stimulator) through a monopolar stimulating electrode placed on layer I-II. As for intracellular experiments, all electrical recordings achieved in the patch-clamp experiments were analyzed both on-line and off-line by the use of the ACQUIS1 program.
Drugs used were: dl-2-amino-5-phosphonopentanoic acid (dl-AP-5; Tocris Cookson, Bristol, UK), (RS)-1-aminoindan-1,5-dicarboxylic acid (AIDA; Tocris Cookson), bicuculline methiodide (Tocris Cookson), bisindolylmaleimide IX methanesulfonate (RO 31–8220; Alexis Biochemicals, Paris, France), (2R,1′S,2′R,3′S)-2-(2′-carboxy-3′-phenylcyclopropyl)glycine (PCCG-13; Alexis Biochemicals), (2S,3S,4S)-CCG/(2S,1′S,2′S)-2-(carboxycyclopropyl)glycine (l-CCG-I; Tocris Cookson), (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG IV; Tocris Cookson), heparin (Prolabo), KT5720 (Alexis Biochemicals), 1-[6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)-amino)hexyl]-1H-pyrrole-2,5-dione (U-73122; Calbiochem, La Jolla, CA), (RS)-α-methylserine-O-phosphate monophenyl ester (MSOPPE; Tocris Cookson), and protein kinase C (19–36) (Alexis Biochemicals).
RESULTS
DCG IV acts on group II mGluRs to induce LTD
As we have previously shown (Otani et al., 1999b), bath application of specific group II mGluR agonist DCG IV (50–100 nm, 15 min) acutely depressed the EPSP of layer V pyramidal neurons evoked by the stimulation of layer I-II fibers (−38 ± 5.7% at the end of the drug application; n = 9), which was followed by a partial recovery and a subsequent plateau, i.e., LTD (Fig. 1A). The postsynaptic membrane potential and input resistance were stable throughout the course of the experiments (Fig. 1A). Mean percentage decrease of the slope of the EPSP at 40 min after DCG IV washout was −20 ± 2.8% (n = 9). We have already concluded (Otani et al., 1999b) that this DCG IV-induced LTD is not a result of an insufficient washout of DCG IV, because a late application of specific group II mGluR antagonist MSOPPE did not reverse the DCG IV-induced LTD. Also, postsynaptic presence of BAPTA spared acute depression and selectively blocked LTD phase. In this study, first, we verified that DCG IV indeed acts on group II mGluRs to initiate LTD by the use of MSOPPE (200–300 μm). MSOPPE was applied in the bath 10 min before DCG IV application until the end of the DCG IV application. As shown in Figure 1B, in the presence of MSOPPE (n = 5), the acute depression induced by DCG IV was smaller, by ∼50%, than that seen in the absence of MSOPPE (−22 ± 4.2 vs −38 ± 5.7%; p< 0.05). Furthermore, LTD induction by DCG IV was completely blocked in the presence of MSOPPE (9.2 ± 4.9% 40 min after drug washout; n = 5;p < 0.0002 vs DCG IV alone group). Membrane properties did not change significantly during the experiments (Fig.1B). However, the acute effect of DCG IV, albeit much smaller than normal, was still present under the MSOPPE condition (see also the result of Huang et al., 1999). Therefore, to further confirm that the LTD is induced by group II mGluR stimulation, we tested the effect of another group II agonist l-CCG-I (10–20 μm; Brabet et al., 1998) on the synaptic transmission. l-CCG-I was applied in the bath for 15 min. As shown in Figure 1C,l-CCG-I caused a large acute synaptic depression (−70 ± 10% at the end of the application. n = 6), which was not accompanied by membrane depolarization, and induced LTD (−24 ± 7.6% at 40 min after washout; n = 6). The amplitude of l-CCG-I-induced LTD was comparable to that of DCG IV-induced LTD (−20 ± 2.8%).
Fig. 1.
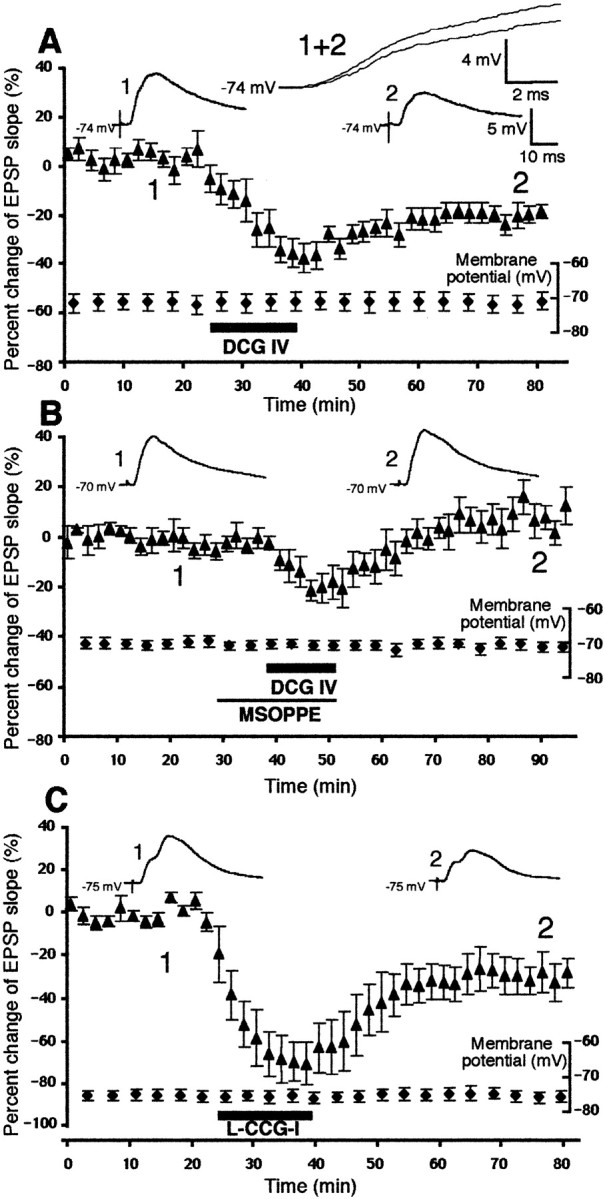
Agonist stimulation of group II mGluRs induces LTD in rat prefrontal neurons. A, Bath-application of the potent group II mGluR agonist DCG IV (50–100 nm) acutely depressed the slope of the monosynaptic EPSP (−38 ± 5.7% decrease at the end of drug application; n = 9) and induced LTD (−20 ± 2.8% decrease 40 min after drug washout;n = 9). These changes were not accompanied by changes in postsynaptic membrane potential. Membrane input resistance was also stable (initial value 57 ± 7.3 MΩ, and the end value 58 ± 8.5 MΩ). The large waveforms shown at thetop are superimposed averaged responses taken from the periods just before DCG IV application (1) and 35–45 min after DCG IV washout (2). The smaller waveforms are the same responses plotted over a longer time scale.B, The presence of the group II mGluR antagonist MSOPPE (200–300 μm) in the bath blocked DCG IV-induced LTD. The EPSP slope change 40 min after washout of the drugs was 9.2 ± 4.9% (n = 5; p < 0.0002 vs DCG IV alone group, depicted in A). Averaged responses taken just before MSOPPE application (1) and 35–45 min after drug washout (2) are also shown. Membrane potential (plotted) and membrane input resistance (initial value 48 ± 8.5 MΩ, and the value 45 ± 8.9 MΩ) were stable during the recording. C, Bath-application of another group II mGluR agonist l-CCG-I (10–20 μm) also induced LTD (−24 ± 7.6% 40 min after washout; n = 6). Membrane potential (plotted) and input resistance (initial value 48 ± 8.7 MΩ, end value 48 ± 9.1 MΩ) were stable during the experiments.
DCG IV-induced LTD requires synaptic activation of NMDA receptors but not group I mGluRs
The concentrations of DCG IV used in the present study (50–100 nm) are much lower than the concentrations at which the drug was shown to act as a NMDA receptor agonist (>10 μm; Ishida et al., 1993; Wilsch et al., 1994). The above results with MSOPPE and l-CCG-I also indicate that DCG IV did not act on NMDA receptors to induce LTD. However, provided that certain forms of LTD are NMDA-dependent (Dudek and Bear, 1992; Mulkey and Malenka, 1992), it is possible that synaptically activated, small NMDA receptor-mediated component contributes to DCG IV-induced LTD. To test this possibility, the NMDA receptors were blocked by the antagonist dl-AP-5 (100 μm) during DCG IV application. As shown in Figure2A, althoughdl-AP-5 did not affect DCG IV-induced acute depression of the EPSP (−49 ± 5.7%; n = 6;p > 0.05 compared with DCG IV alone group), it blocked DCG IV-induced LTD (3.6 ± 6.5% 40 min after drug washout,p < 0.02 compared with DCG IV alone group).
Fig. 2.
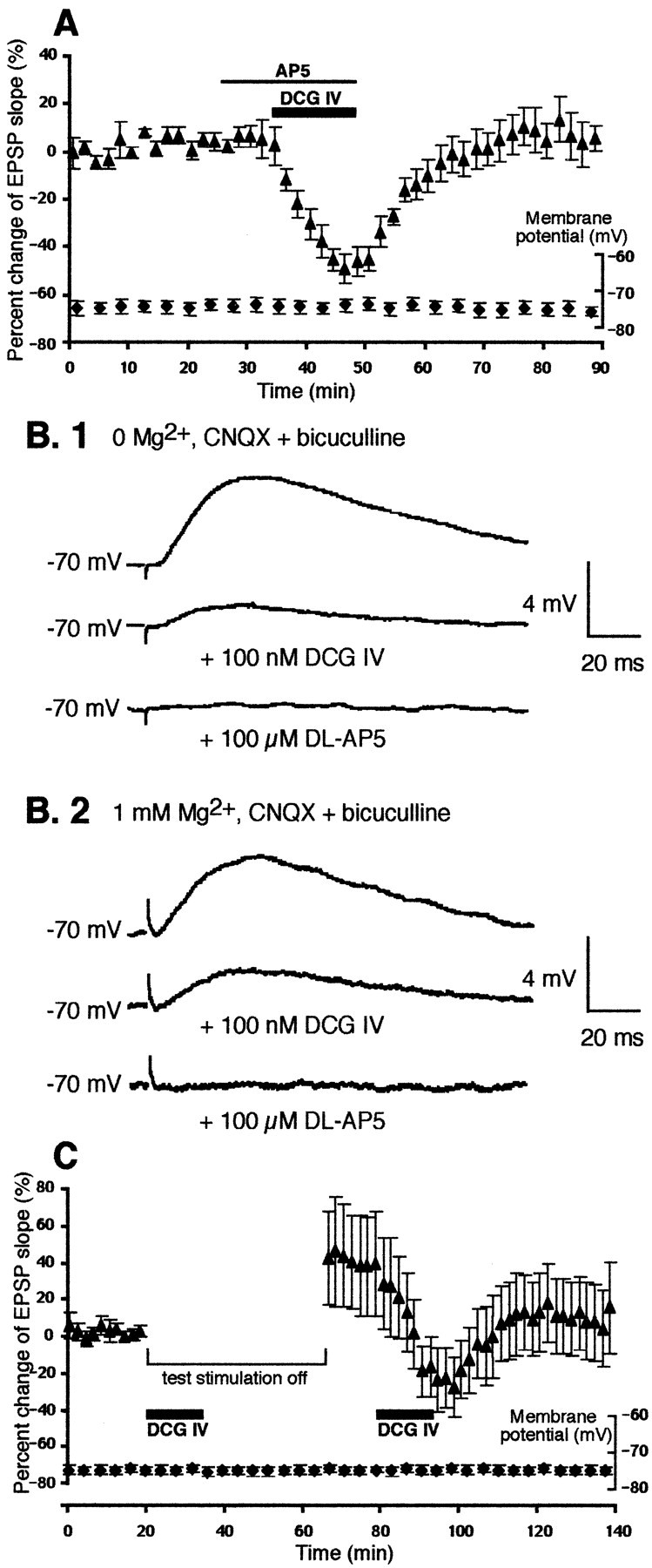
DCG IV-induced LTD requires synaptic activation of NMDA receptors, but DCG IV does not potentiate NMDA responses.A, The presence of NMDA receptor antagonistdl-AP-5 (100 μm) blocked DCG IV-induced LTD. The change of the EPSP slope 40 min after drug washout was 3.6 ± 6.5% (p < 0.02 compared with DCG IV alone group depicted in Fig. 1A). The membrane potentials during the course of the experiments are also plotted. Membrane input resistance: initial value 47 ± 4.9 MΩ, end value 49 ± 8.6 MΩ.B, However, the manner of NMDA receptor involvement in DCG IV-induced LTD is not such that DCG IV potentiates NMDA component of synaptic responses. The NMDA component was isolated by the addition of CNQX (5 μm) and bicuculline (1 μm, routine application) in 0 Mg2+ solution (B.1;n = 3) or in normal solution (B.2;n = 4). Stimulus intensity was increased to visualize NMDA component clearly, particularly in the latter case. In both conditions, DCG IV (100 nm) depressed NMDA component of synaptic transmission and never potentiated it. The traces are averaged responses from representative experiments.C, Absence of the small NMDA-mediated responses during DCG IV application by the interruption of the 0.033 Hz single test stimuli blocked DCG IV-induced LTD. The test stimuli were stopped just before DCG IV application until 30 min after the beginning of DCG IV washout (n = 12). Under this condition, only two cells showed LTD, and 10 cells showed no change or potentiation (36 ± 26% mean change 40 min after washout;n = 12). If LTD is defined as a ≥15% decrease at 40 min after DCG IV washout, the frequency of the LTD occurrence (2 of 12 cases) is significantly lower than that in DCG IV alone group (7 of 9 cases) (p < 0.01; Fisher's exact probability test). A second application of DCG IV with test pulses delivered induced LTD (−18 ± 4.9%, taking the EPSP level before the second DCG IV application as a new baseline; p> 0.7 compared with DCG IV alone group depicted in Fig.1A). Membrane potential was stable during the experiments. Membrane input resistance: initial value 42 ± 3.4 MΩ, end value 42 ± 3.2 MΩ.
We then decided to verify that DCG IV does not potentiate NMDA receptor-mediated synaptic responses. First, the NMDA responses were isolated in nominally 0 Mg2+ medium by the addition of 5 μm CNQX and 1 μm bicuculline (routine application) to the bathing medium (n = 3). As shown in Figure 2B.1, DCG IV depressed the NMDA receptor-mediated synaptic transmission and did not potentiate it. It was still possible, however, that DCG IV alters voltage dependence of Mg2+ block on the NMDA receptors so that net transmission through the receptors at resting membrane potential increases in the presence of DCG IV. This was also found not to be the case. The synaptic NMDA component was isolated in the presence of Mg2+ by the addition of CNQX and bicuculline (Fig. 2B.2). DCG IV still depressed the NMDA responses under this condition (n = 4). Taken together, these results show that DCG IV does not potentiate NMDA component of the synaptic transmission: the drug generally depresses the synaptic responses irrespective of whether the response is mediated by AMPA or NMDA receptors, perhaps through presynaptic mechanisms (Huang et al., 1999).
The above results with AP-5 and with the isolated NMDA component indicate that the small NMDA receptor-mediated synaptic transmission at resting membrane potential, evoked by the 0.033 Hz single test stimuli, contributes to LTD. If this is the case, the interruption of test pulses during DCG IV application should block the induction of LTD. We therefore performed another set of experiments in which the 0.033 Hz test stimuli were stopped just before a 15 min DCG IV application until 30 min after the beginning of DCG IV washout. Of 12 neurons tested, only two of them showed LTD when test stimuli were resumed (−50% and −17% from baseline 40 min after DCG IV washout). Among the other 10 neurons, five showed no lasting changes (−4.2 ± 1.9%), and the other five showed variable degrees of potentiation (104 ± 49%). In Figure 2C, we plotted mean changes calculated from all 12 neurons (36 ± 26% mean change 40 min after DCG IV washout;n = 12). Because of the large variation, the mean value was only marginally different (p = 0.08) when compared with LTD in the DCG IV alone group (i.e., −20 ± 2.8% as depicted in Fig. 1A). However, if LTD is defined as a 15% decrease or more from the baseline at 40 min after DCG IV washout, the frequency of LTD induction in the test pulse-interruption condition (2 of 12 cases) is significantly lower than that in the DCG IV alone group (seven of nine cases, p < 0.01, Fisher's exact probability test; Ichihara, 1990). Test pulse interruption itself does not affect the amplitude of the synaptic responses (Otani et al., 1999b). In these 12 neurons, we successively applied DCG IV with the 0.033 Hz test pulses continuously delivered. In this case, DCG IV induced LTD (Fig. 2C). The LTD value is −18 ± 4.9% from the period just before the second DCG IV application (no difference from DCG IV-induced LTD depicted in Fig.1A; p > 0.7). These results support the possibility that synaptically activated NMDA receptors play role in the induction of LTD by DCG IV and indicate that the DCG IV-induced LTD is a homosynaptic type of LTD.
Another possibility was that synaptically activated group I mGluRs contribute to DCG IV-induced LTD. This possibility was necessary to consider, because (1) dopamine-facilitated LTD requires synaptic activation of both group I and group II mGluRs (Otani et al., 1999b), (2) DCG IV potentiates the effect of group I agonist (S)-3,5-dihydroxyphenylglycine (DHPG) on phosphoinositide turnover in neonatal cortex slices (Mistry et al., 1998), and (3) DCG IV potentiates the effects of DHPG on membrane inward currents and postsynaptic [Ca2+] augmentation in perirhinal cortex slices (Cho et al., 2000). To test whether group I mGluR blockade blocks DCG IV-induced LTD, we bath-applied specific group I mGluR antagonist AIDA (200 μm;Otani et al., 1999b) 10 min before DCG IV application until the end of the 15 min application (n= 7; data not depicted). AIDA blocked neither the acute synaptic depression by DCG IV (−45 ± 4.5%; n = 7) nor DCG IV-induced LTD (−18 ± 7.5% 40 min after drug washout;n = 5; p > 0.8; intracellular recording of two cells was lost during experiment). Application of AIDA alone for 25 min did not change baseline synaptic responses during the 40 min post-drug period (−3.0 ± 6.3% 40 min after AIDA;n = 5). We conclude that synaptic activation of group I mGluRs is not required for the induction of LTD by DCG IV.
DCG IV-induced LTD requires activation of postsynaptic PLC, postsynaptic IP3 receptors, and PLD
DCG IV-induced LTD requires postsynaptic increases of [Ca2+] (Otani et al., 1999b), and the evidence shows that group II mGluRs stimulate PLC (Klein et al., 1997;Mistry et al., 1998). Therefore, we tested whether DCG IV-induced LTD requires postsynaptic PLC activation. We used a PLC inhibitor U-73122, which inhibits muscarinic acetylcholine receptor agonist-stimulated phosphoinositide hydrolysis in human neuroblastoma cells (Thompson et al., 1991) and in rat pancreatic acinar cells (Yule and Williams, 1992), with an IC50 value being 3.7 μm in one measure (Thompson et al., 1991). U-73122 has no effect on secretin-stimulated cAMP formation in rat pancreatic acinar cells (Yule and Williams, 1992). In the first series of experiment, U-73122 (4 μm) was bath-applied 10 min before the application of DCG IV until the end of the 15 min application. As shown in Figure 3A, U-73122 did not affect the acute depression of the EPSP by DCG IV (−41 ± 7.3% at the end of DCG IV application; n = 7; p > 0.05 compared with DCG IV alone group depicted in Fig.1A) but blocked DCG IV-induced LTD. Percentage of change of the EPSP slope 40 min after washout of the drugs (3.4 ± 6.8% from baseline; n = 7) is significantly different from the same measure taken from DCG IV alone group (p < 0.02). Next, we tested whether U-73122 itself has any effects on the EPSP. As shown in Figure 3B, a 25 min application of U-73122 (4 μm) only slightly increased the slope of the EPSP (8.0 ± 5.3% at the end of the application and 5.5 ± 5.6% 40 min after drug washout; n = 7). These results show that PLC activation is required for DCG IV-induced LTD. In the next series of experiment, we injected U-73122 in postsynaptic neurons to test the locus of PLC activation. We included in recording electrodes 20 μm U-73122 (1% DMSO) and allowed it to diffuse into the cells. As shown in Figure3C, the postsynaptic injection of U-73122 totally blocked DCG IV-induced LTD (6.8 ± 7.4% 40 min after DCG IV;n = 5; p < 0.02 compared with DMSO control, see below) without affecting acute depression by DCG IV (−39 ± 8.9%; n = 5; p > 0.05). Identical postsynaptic injection of 1% DMSO alone did not change the degree of DCG IV-induced LTD (−19 ± 3.5% at 40 min;n = 5; p > 0.05; data not shown). In addition, neurons injected with U-73122 and 1% DMSO did not show any signs of deterioration of the baseline EPSP during the equivalent period of experiment (4.6 ± 9.7% change; n = 4). We conclude that critical PLC activation for LTD occurs in postsynaptic sites.
Fig. 3.
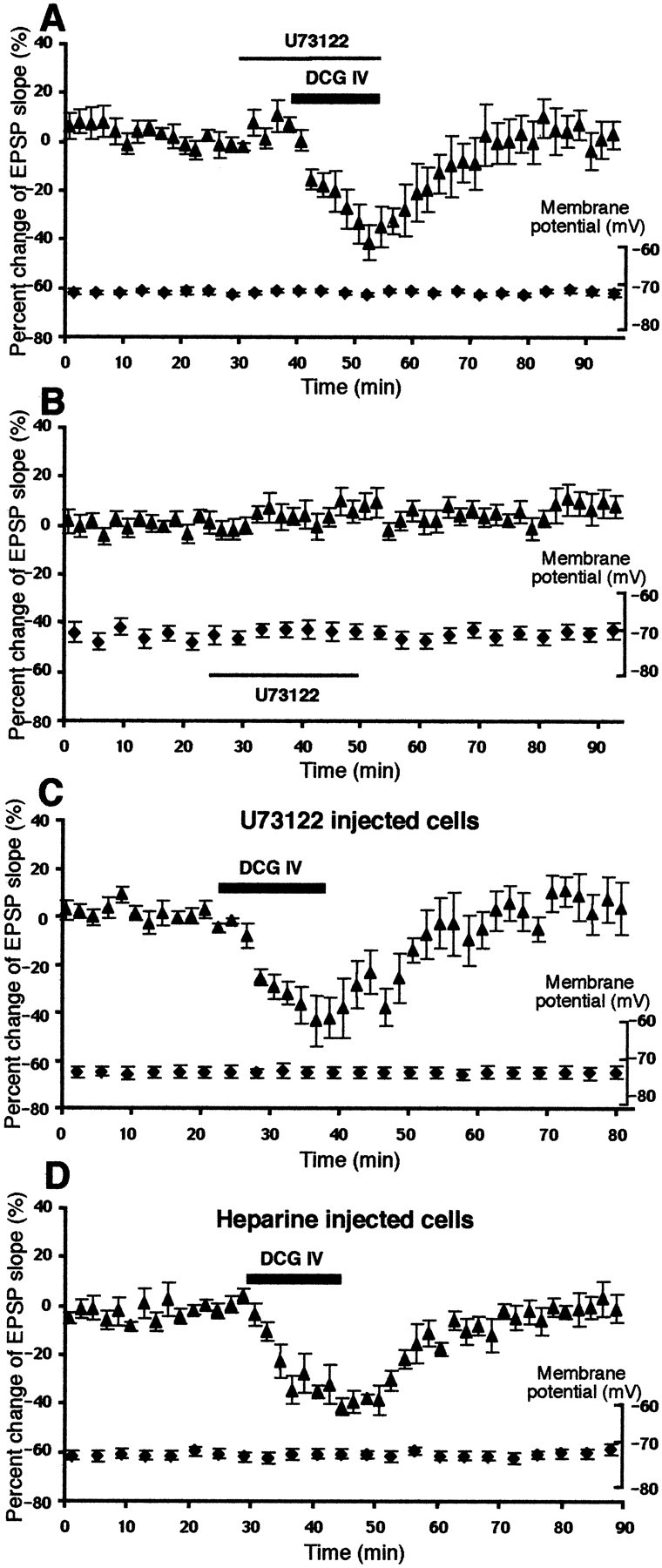
DCG IV-induced LTD requires the activation of postsynaptic PLC and postsynaptic IP3 receptors.A, Bath-application of PLC inhibitor U-73122 (4 μm) blocked DCG IV-induced LTD. The percentage change of the EPSP slope 40 min after drug washout was 3.4 ± 6.8% from baseline (n = 7; p < 0.02 compared with DCG IV alone group depicted in Fig.1A). During the course of the experiments, membrane potential was stable. Membrane input resistance: initial value 44 ± 3.8 MΩ, end value 46 ± 5.5 MΩ. B,Bath-application of U-73122 (4 μm) alone did not primarily change the synaptic responses (8.0 ± 5.3% at the end of the 25 min application and 5.5 ± 5.6% 40 min after drug washout; n = 7). The membrane potential was also stable. Membrane input resistance: initial value 48 ± 7.5 MΩ, end value 50 ± 6.3 MΩ. C, Postsynaptic injection of U-73122 (20 μm in recording electrodes) also completely blocked DCG IV-induced LTD (6.8 ± 7.4% change at 40 min; n = 5; p < 0.02 compared with DMSO control, see Results). D, Postsynaptic injection of the inositol trisphosphate IP3 receptor blocker heparin (4 mg/ml in recording electrodes) also blocked DCG IV-induced LTD. Percentage change of the EPSP slope 40 min after DCG IV washout in this group was −0.5 ± 4.8% (n = 5; p < 0.025 compared with DCG IV alone group). Membrane potential (plotted) and membrane input resistance (initial value 44 ± 6.0 MΩ, end value 44 ± 5.1 MΩ) were stable.
A key messenger generated by PLC activation is inositol trisphosphate (IP3), which stimulates IP3receptors on endoplasmic reticulum and triggers Ca2+ release from the internal stores (Agranoff and Fisher, 1994). To test whether postsynaptic IP3 receptor activation is important in DCG IV-induced LTD, we postsynaptically injected IP3receptor blocker heparin through recording electrodes (4 mg/ml). As shown in Figure 3D, in the heparin-injected neurons, baseline synaptic responses and the acute depression by DCG IV (−35 ± 5.3%; p > 0.05) were unaffected, but DCG IV-induced LTD was blocked (−0.5 ± 4.8% 40 min after DCG IV washout; n = 5; p < 0.025). Thus, postsynaptic IP3 receptors are required for DCG IV-induced LTD.
DCG IV has been shown to stimulate PLD (Klein et al., 1997). Therefore, we also used PCCG-13, a selective blocker against PLD activation by certain mGluR agonists, with an IC50 value being 100 nm (Pellegrini-Giampietro et al., 1996). As in the case of U-73122, PCCG-13 (2 μm) was present in the bath from 10 min before DCG IV application until the end of the application. PCCG-13 did not affect acute EPSP depression by DCG IV (−31 ± 5.2%; n = 6; p > 0.05) but blocked DCG IV-induced LTD (Fig.4A) (−0.8 ± 5.7% 40 min after drug washout; n = 5;p < 0.025 compared with DCG IV alone group). Intracellular recording of one cell was lost 25 min after the beginning of the drug washout, but the depression remaining in this cell was only −5.2%. PCCG-13 alone slightly increased the EPSP slope during application (6.5 ± 5.0% at the end of 25 min application;n = 6) (Fig. 4B), but this increase returned to the baseline level within 40 min after washout (−0.1 ± 4.7%; n = 6).
Fig. 4.

DCG IV-induced LTD requires the activation of PLD.A, PCCG-13 (2 μm), the inhibitor of PLD activation by certain mGluR agonists, blocked DCG IV-induced LTD (−0.8 ± 5.7% change 40 min after drug washout;n = 5; p < 0.025 compared with DCG IV alone group depicted in Fig. 1A). Membrane potential during the course of the experiments is also plotted. Membrane input resistance: initial value 47 ± 6.2 MΩ, end value 44 ± 6.6 MΩ. B, PCCG-13 alone did not change the slope of the EPSP (−0.1 ± 4.7% from baseline 40 min after drug washout; n = 6). Membrane potential (plotted) and membrane input resistance (initial value 44 ± 4.2 MΩ, end value 45 ± 4.3 MΩ) were also stable.
DCG IV-induced LTD requires activation of postsynaptic PKC
Another key second messenger activated after PLC and PLD stimulation is PKC (Tanaka and Nishizuka, 1994; Klein et al., 1995). We therefore tested PKC involvement in DCG IV-induced LTD. First, we used the cell-permeable, specific and potent PKC inhibitor RO318220 (Davis et al., 1989; Harris et al., 1996), which has already been shown to block cerebellar LTD (Linden and Connor, 1991). RO318220 (0.2 μm) was bath-applied 10 min before DCG IV application until the end of the 15 min application. As shown in Figure5A, RO318220 did not affect the acute EPSP depression by DCG IV (−25 ± 7.1% at the end of DCG IV application; n = 8; p > 0.05) but blocked DCG IV-induced LTD (−3.6 ± 5.1% 40 min after drug washout; n = 8; p < 0.015). The effect of RO318220 alone is depicted in Figure 5B. A 25 min application of RO318220 had no large effects on the EPSP slope (1.0 ± 6.3% at the end of application and −0.8 ± 4.5% 40 min after drug washout; n = 7).
Fig. 5.
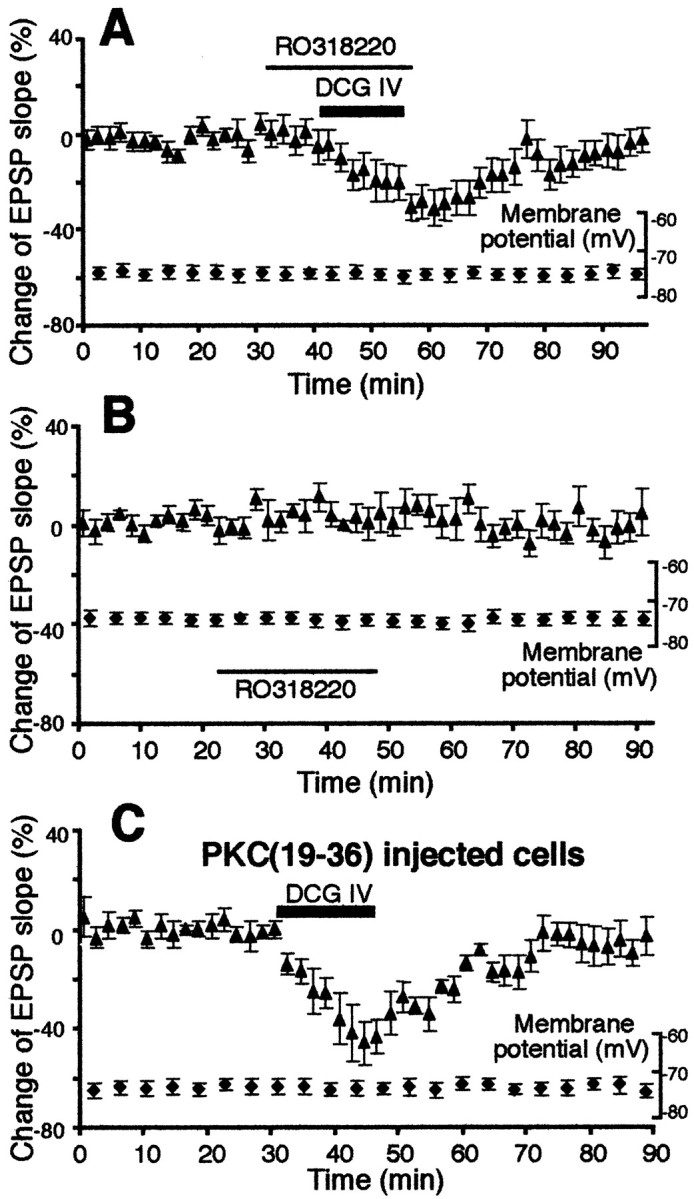
DCG IV-induced LTD requires postsynaptic activation of PKC. A, The cell-permeable, specific, and potent PKC inhibitor RO318220 (0.2 μm) blocked DCG IV-induced LTD (−3.6 ± 5.1% change 40 min after drug washout;n = 8; p < 0.015 compared with DCG IV alone group depicted in Fig. 1A). Membrane potential (plotted) and membrane input resistance (initial value 38 ± 2.7 MΩ, end value 41 ± 4.6 MΩ) were stable.B, RO318220 (0.2 μm) alone did not largely change synaptic responses (−0.8 ± 4.5% change 40 min after drug washout; n = 7). Membrane potential (plotted) and membrane input resistance (initial value 57 ± 8.0 MΩ, end value 55 ± 7.2 MΩ) were also stable.C, Postsynaptic presence of PKC inhibitor pseudosubstrate peptide PKC(19–36) (1–2 mm in recording electrodes) significantly reduced DCG IV-induced LTD. The EPSP slope change 40 min after DCG IV washout was −6.3 ± 5.0% from baseline (n = 6) and significantly different from DCG IV-induced LTD depicted in Figure 1A(p < 0.05). Membrane potential during the course of the experiments is also plotted. Membrane input resistance: initial value 48 ± 3.6 MΩ, end value 51 ± 6.4 MΩ.
To show postsynaptic locus of PKC activation for DCG IV-induced LTD, we injected pseudosubstrate peptide PKC(19–36) to postsynaptic neurons (Fig. 5C). PKC(19–36) (1–2 mm in recording electrodes) was allowed to diffuse to postsynaptic sites for at least 1 hr before DCG IV application (cf. Otani and Connor, 1998). There was no significant shift in baseline responses during the diffusion period. Postsynaptic PKC(19–36) did not affect acute EPSP depression induced by DCG IV (−45 ± 8.7%; n = 6; p > 0.05) but significantly reduced DCG IV-induced LTD (Fig. 5C). Percentage change of the EPSP slope 40 min after DCG IV washout in these cells is only −6.3 ± 5.0% from baseline (n = 6), and this value is significantly different from DCG IV-induced LTD depicted in Figure1A (p < 0.05).
Reid et al. (1996), Schoepp et al. (1996), and Sortino et al. (1996)suggested that group II mGluRs can increase cAMP levels. Therefore finally, in additional cells, we tested the effect of selective PKA inhibitor KT5720. KT5720 shows a strong specificity to PKA over PKC and PKG (Ki = 60 nmfor PKA, and Ki > 2000 nm for both PKC and PKG; Kase et al., 1987). KT5720 (0.2 μm) was present in the bath 10 min before DCG IV application until the end of the application. As shown in Figure 6A, KT5720 significantly reduced DCG IV-induced LTD (−6.4 ± 4.5%;n = 6; p < 0.04 compared with DCG IV alone group depicted in Fig. 1A). A 25 min application of KT5720 alone slightly decreased the slope of the EPSP (−17 ± 8.6% at the end of the application; n = 6), but this decrease recovered to baseline level within 40 min (−1.3 ± 3.7%; n = 6) (Fig.6B).
Fig. 6.

DCG IV-induced LTD requires PKA activation.A, The potent and selective PKA inhibitor KT5720 (0.2 μm) reduced DCG IV-induced LTD. The percentage change of the EPSP slope 40 min after drug washout was −6.4 ± 4.5% from baseline (n = 6), and this value is significantly different from DCG IV-induced LTD depicted in Figure 1A(p < 0.04). Membrane potential (plotted) and membrane input resistance (initial value 47 ± 7.2 MΩ, end value 48 ± 6.5 MΩ) were stable. B, KT5720 (0.2 μm) application alone did not largely affect synaptic responses (−1.3 ± 3.7% from baseline 40 min after washout;n = 6). During the course of the experiments, membrane potential did not fluctuate significantly. Membrane input resistance: initial value 56 ± 8.5 MΩ, end value 55 ± 12 MΩ.
DCG IV augments Ca2+ concentration in layer V pyramidal neurons by stimulating IP3 receptors
Together with our previous study (Otani et al., 1999b), the results so far suggest that DCG IV acts on MSOPPE-sensitive mGluRs and stimulates PLC and PLD, which leads to IP3-mediated postsynaptic [Ca2+] increases and postsynaptic PKC activation. To further show postsynaptic metabotropic action of DCG IV, we analyzed [Ca2+] increases induced by DCG IV in PFC neurons. As a pilot study, we first used the cell-permeable fluorescent Ca2+ indicator fura-2 AM to ratio-image areas in coronal brain slices (n = 5; data not shown). DCG IV application (100 nm, 10 min) rapidly increased [Ca2+] in both deep and superficial layers of prelimbic area (i.e., the area of our interest). Often (n = 3), the rapid [Ca2+] increases during DCG IV application were followed by slower, oscillatory [Ca2+] fluctuations during washout periods.
We then performed fluorescent Ca2+analysis in single postsynaptic layer V pyramidal neurons. The cells were filled with high-affinity Ca2+indicator fluo-3 via patch pipettes attached to their soma and were maintained in voltage-clamp mode at −80 mV. In the first series of experiment (not depicted), the synaptic responses were not simultaneously recorded. Nevertheless, DCG IV application (100 nm) induced transient increases in [Ca2+] in postsynaptic cells, in the absence and in the presence of 100 μm AP-5. Mean amplitudes of relative fluorescence variation (ΔF/F) were 6.5 ± 1.0% in the absence of dl-AP-5 (n = 4) and 8.0 ± 2.9% in the presence of dl-AP-5 (n = 4). We never observed inward depolarizing currents associated with the [Ca2+] increases by DCG IV, consistent with our observation using the sharp electrode intracellular recording (Fig. 1A). In three separate cells, the effect of another group II mGluR agonistl-CCG-I (20 μm) on [Ca2+] was tested.l-CCG-I also transiently increased postsynaptic [Ca2+]. Mean amplitude of fluorescence changes with l-CCG-I was 8.2 ± 2.7% (ΔF/F) (n = 3). Across the different drug groups, we noted that in 5 of these 11 cells, immediate transient [Ca2+] changes were followed by some oscillatory [Ca2+] waves during washout periods of the drugs, reminiscent of those observed in the whole slice imaging study described above.
Based on these pilot studies, we conducted a new series of experiments to correlate DCG IV-induced [Ca2+] signals with LTD. Thus, the Ca2+ indicator fluo-3 was identically injected to postsynaptic layer V neurons, and simultaneously, layer I-II to layer V neuron synaptic responses were collected under voltage-clamp mode (at −80 mV). First, as shown in the top graph of Figure 7A, DCG IV rapidly augmented postsynaptic [Ca2+] (n = 6). There were no inward depolarizing currents during these [Ca2+] rises. Time course of the [Ca2+] rises differed among cells, creating the large data variations during the increase phase, as seen in the top graph. However, when peak [Ca2+] amplitude from each cell was taken, mean peak percentage of [Ca2+] increase associated with the DCG IV application was 6.7 ± 0.3% (n = 6). Importantly, as shown in the bottom graph, the [Ca2+] rises were accompanied by acute DCG IV action on the synaptic responses followed by LTD (−27 ± 3.8% 40 min after DCG IV washout; n = 6). We detected no significant difference in peak [Ca2+] increases between this synaptic input condition and the above no-stimulation condition (6.7 ± 0.3% vs 6.5 ± 1.0%). This observation may indicate that synaptically activated NMDA receptors do not potentiate DCG IV-induced Ca2+ signals to induce LTD. However, this cannot be concluded until local Ca2+ signals at stimulated distal dendritic sites and spines are analyzed under the synaptic stimulation condition.
Fig. 7.
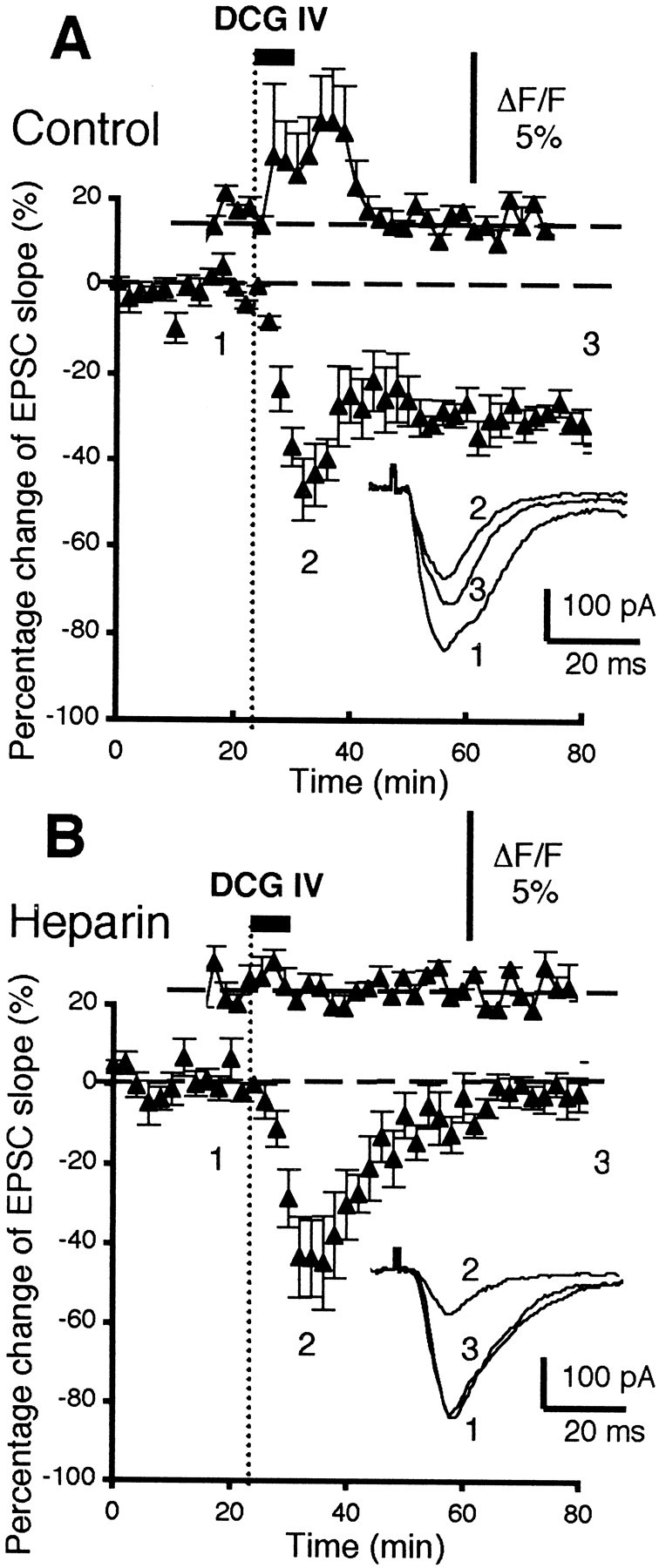
DCG IV-induced LTD is accompanied by postsynaptic increases in [Ca2+], and these increases are dependent on postsynaptic activation of IP3 receptors.A, Control condition: the top graph shows mean percentage [Ca2+] increases in postsynaptic layer V pyramidal neurons induced by DCG IV bath application (100 nm, 5 min, n = 6; see Results for statistics), as revealed by [Ca2+] analysis with fluorescent indicator fluo-3. The bottom graphshows the time course of the changes of the EPSC slope simultaneously recorded from the same six neurons. DCG IV induced acute depression and a subsequent LTD of the synaptic responses (−27 ± 3.8% 40 min after DCG IV washout; n = 6). Theinset shows superimposed averaged EPSCs taken from the indicated time points. B, Heparin-injected neurons: the figure has the same configuration as in A. The postsynaptic presence of the IP3 receptor blocker heparin (1 mg/ml, diffused from the patch pipettes) totally blocked DCG IV-induced [Ca2+] increases (n= 5; top graph; see Results for statistics). Heparin also blocked DCG IV-induced LTD in the same neurons without affecting acute synaptic depression by DCG IV. Mean change of the EPSC slope 40 min after DCG IV washout in these neurons is −0.3 ± 2.8% (n = 5; p < 0.001).
Next, we tested whether a blockade of postsynaptic IP3 receptors blocks DCG IV-induced LTD (Fig.3D) and DCG IV-induced [Ca2+] rises in the same cells (n = 5). The IP3 receptor blocker heparin (1 mg/ml) was added in the fluo-3-loaded patch pipettes and was allowed to diffuse to postsynaptic sites. Under this condition, first, DCG IV-induced [Ca2+] rises were absent (Fig. 7B, top graph). “Peak” amplitude of [Ca2+], i.e., top limit of [Ca2+] fluctuation during DCG IV application, was 1.0 ± 0.01% in the heparin-injected neurons (p < 0.05 compared with the neurons depicted in Fig. 7A). Second, in the same neurons, DCG IV-induced LTD was blocked (Fig. 7B, bottom graph) (−0.3 ± 2.8% EPSP slope change at 40 min; n = 5; p< 0.001 compared with LTD depicted in Fig. 7A). This set of experiments shows that IP3 receptor-mediated postsynaptic [Ca2+] rises are required for DCG IV-induced LTD.
DISCUSSION
Our results show that DCG IV acts on MSOPPE-sensitive postsynaptic mGluRs and induces LTD through a series of biochemical responses. These include PLC and PLD activation, internal [Ca2+] increases via IP3 receptor stimulation, and PKC and PKA activation (Fig.8B).
Fig. 8.

Schematic figures to compare two different LTD induction protocols we found in glutamatergic synapses of rat prefrontal layer V pyramidal neurons. The coexistence of D1-like and D2-like receptors and that of D2-like and glutamate receptors in the same synapse are hypothetical. A shows our “standard” LTD induction protocol. LTD is induced by the application of 50 Hz tetanic stimuli to layer I-II glutamatergic axons in the presence of dopamine (100 μm) in the bath (Otani et al., 1998b). In this LTD, dopamine acts on both D1-like and D2-like receptors. Induction of this type of LTD does not require NMDA receptors, but it requires (1) postsynaptic depolarization during tetanus, which dopamine facilitates, (2) postsynaptic increases of Ca2+ concentration, (3) synaptic activation of groups I and II mGluRs, and (4) activation of MAPKs (ERK1 and ERK2) (Otani et al., 1998b, 1999b). Pharmacologically, this type of LTD is mimicked by combined bath-application of dopamine and 1S-3R-ACPD (the groups I and II mGluR agonist), or even of dopamine and DHPG (specific group I mGluR agonist) (Otani et al., 1999b). Thus, when the mGluRs and DA-Rs are pharmacologically costimulated, tetanic stimuli and the consequential large postsynaptic depolarization can be omitted. In this type of LTD, role of group II mGluRs is relatively small, because in the pharmacological protocol, ACPD can be replaced by DHPG.B, In contrast, strong pharmacological activation of group II mGluRs by bath-application of potent agonist DCG IV (50–100 nm) induces LTD without combined application of dopamine and synaptic stimulation of group I mGluRs (present study). DCG IV-induced LTD is dependent on the activation of postsynaptic PLC, PKC, and IP3 receptors. The major differences of DCG IV-induced LTD from the LTD induced by coactivation of mGluRs and DA-Rs are: (1) DCG IV-induced LTD requires concurrent synaptic activation of NMDA receptors, and (2) DCG IV-induced LTD does not require postsynaptic MAPK activation (−19 ± 2.5% LTD was induced in MAPK pseudosubstrate peptide-injected cells; n = 4), although DCG IV activates MAPKs in prefrontal tissue (Otani et al., 1999b). We do not know at this stage whether group II mGluRs and NMDA receptors convergently activate intracellular factor or factors to induce LTD.
Accumulating evidence suggests that group II mGluRs are positively coupled to the PLC and PLD pathways, in addition to the well known downregulation of adenylate cyclase activity (Tanabe et al., 1992;Mistry et al., 1998). For example, Klein et al. (1997) showed that DCG IV (1–3 μm) increases the activity of PLC and PLD by 30–40% in hippocampal slices prepared from 8-d-old rats (EC50 for PLD activation: 22 nm). The increase of PLD activity was independent of previous stimulation of adenylate cyclase by forskolin, suggesting that the DCG IV effect on PLD is not secondary to inhibition of adenylate cyclase by DCG IV. Second, Mistry et al. (1998) showed that DCG IV stimulates phosphoinositide turnover in neonatal rat cerebral cortex slices. They found also that DCG IV and another group II agonist 2R,4R-4-aminopyrrolidine-2,4-dicarboxylate (2R,4R-APDC) potentiate the effect of group I mGluR agonist DHPG on the phosphoinositide turnover (EC50: 0.28 μm). Third,Schoepp et al. (1996) similarly found a potentiating effect of 2R,4R-APDC on the DHPG action on phosphoinositide turnover in neonatal and adult hippocampal slices. Fourth, in perirhinal cortex slices, DCG IV (0.5 μm) potentiated the effects of DHPG on membrane inward currents and postsynaptic [Ca2+] augmentation (Cho et al., 2000). The results reported in this study are generally consistent with these previous studies. However, to our knowledge, this is the first evidence that DCG IV alone stimulates postsynaptic [Ca2+] mobilization via IP3 receptor stimulation. This is also the first demonstration that DCG IV stimulates postsynaptic PKC. Recently, L. J. Bindman, J. Keelan, and S. H. Morris (personal communication) found that DCG IV increases [Ca2+] in cultured rat PFC neurons maintained in Ca2+-free solution, consistent with our data.
Involvement of group II mGluRs in LTD induction has been shown elsewhere. For example, Manahan-Vaughan (1997) showed that CA1 LTD by 1 Hz stimuli was blocked by group II mGluR antagonists MSOPPE and (2S)-α-ethylglutamic acid. Huang et al. (1997)showed that LTD in the dentate gyrus by 1 Hz stimuli was blocked by another group II antagonist 2S,1S′,2S′-2-methyl-2-(2′-carboxycyclopropyl)glycine. More recently, Huang et al. (1999) reported results clearly consistent to our present data. In the dentate gyrus, bath-application (20 min) of DCG IV (200 nm to 2 μm) or another group II agonist LY354740 (500 nm) induced LTD, and this induction could be inhibited or blocked by MSOPPE, AP-5, RO318220 and H-89, the PKA inhibitor. They did not directly test the loci for DCG IV action and kinase activation. But, at least the expression of the DCG IV-induced LTD was postsynaptic (Kilbride et al., 1998; Huang et al., 1999). Postsynaptic locus of group II mGluR-induced LTD was shown more clearly in lateral amygdala by Heinbockel and Pape (2000). Thus, theta frequency stimulation induced group II mGluR-dependent LTD, and this LTD was blocked by postsynaptic injection of BAPTA. These results taken together, it appears that similar LTD mechanisms involving postsynaptic group II mGluRs exist in the dentate gyrus, the lateral amygdala, and the prefrontal cortex. In some cases, group II mGluRs might induce LTD via a potentiation of group I mGluR actions (Cho et al., 2000). An exception is however striatal LTD; group II mGluR agonists induce LTD in the stratum, but immunostaining indicates only presynaptic localization of group II mGluRs in this structure (Kahn et al., 2001).
Recent results suggest the importance of PLC and PLC-coupled receptors in various forms of LTD. In the hippocampus, Huber et al. (2000, 2001) showed that bath-application of group I mGluR agonist DHPG (50–100 μm, 5 min) induces LTD and that this LTD was absent in mice lacking functional mGluR5 (Huber et al., 2001). Also in the hippocampus, Reyes-Harde and Stanton (1998) showed that LTD induction by 1 Hz stimuli was blocked by postsynaptic injection of the PLC inhibitor U-73122. In the visual cortex, PLC involvement in LTD induction was extended to neuromodulatory systems. Thus, Kirkwood et al. (1999) found that LTD by paired stimuli was facilitated by carbachol, the agonist of the PLC-coupled muscarinic acetylcholine receptors. The LTD was also facilitated by the agonist of α1 adrenergic receptors methoxamine, and the α1Breceptor is a class of PLC-coupled receptors (Weiner and Molinoff, 1994).
Another interesting observation made in our study was that the NMDA receptor antagonist dl-AP-5 blocked DCG IV-induced LTD (cf.Huang et al., 1999). The NMDA dependency is in contrast to other examples of prefrontal LTD (Hirsch and Crepel, 1991; Otani et al., 1998b), but is consistent with certain forms of LTD in the visual cortex and the hippocampus (Dudek and Bear, 1992; Mulkey and Malenka, 1992; Kirkwood and Bear, 1994). Although DCG IV is a NMDA agonist at high concentrations (>10 μm; Ishida et al., 1993; Wilsch et al., 1994), we suggest for the following reasons that DCG IV does not act on NMDA receptors to induce LTD. First, the DCG IV concentrations used in this study (50–100 nm) are well below the concentrations at which DCG IV acts as a NMDA agonist. Second, another agonist l-CCG-I (20 μm), which has no known action on NMDA receptors, induced LTD. Third, DCG IV did not potentiate isolated synaptic NMDA responses. It is apparent that even at resting membrane potential, the small synaptic transmission through NMDA receptors evoked by single test pulses is sufficient to contribute to LTD. Indeed, interruption of test stimuli during DCG IV application blocked DCG IV-induced LTD. Exact mechanisms underlying the NMDA–group II mGluRs interaction for LTD induction are currently unknown. Mistry et al. (1998) described that their DCG IV effect (<1 μm) on phosphoinositide turnover was blocked by NMDA channel blocker MK801. In our case, test pulse application that evokes NMDA currents in layer V neurons did not augment postsynaptic [Ca2+] increases by DCG IV at least at somatic levels. We, however, may have missed test pulse-induced [Ca2+] potentiation occurring in distal dendritic sites. The already deep locations of healthy neurons within the slices prepared from rats of ≥23 d of age made it difficult to visualize the fine distal dendrites of layer V pyramidal neurons.
In Figure 8, we summarized and compared two different LTD induction protocols we found in rat prefrontal layer V neurons; LTD induced by coactivation of mGluRs and DA-Rs (Otani et al., 1998b, 1999b), and LTD induced by DCG IV (present study). The first type of LTD (Fig.8A) is typically induced by the application of afferent tetanic stimuli in the presence of dopamine in the bath, and the induction is NMDA-independent. This LTD, however, requires synaptic activation of group I and II mGluRs, and LTD was indeed mimicked by a combined application of dopamine and 1-aminocyclopentane-1,3-dicarboxylic acid (1S,3R-ACPD) (the groups I and II mGluR agonist). This pharmacological protocol surpasses the requirement of postsynaptic depolarization in the case of LTD induction by the tetani in the presence of dopamine. Also, in this first type of LTD, role of group II mGluRs is relatively small, because the induction could be mimicked even when ACPD was replaced by DHPG, a specific group I mGluR agonist. We suggested that an induction mechanism underlying this type of LTD is converging postsynaptic activation of MAPKs by DA-Rs (D1 and D2) and the mGluRs. In the second type of LTD (Fig. 8B), strong pharmacological stimulation of postsynaptic group II mGluRs cooperates with NMDA receptors. This LTD is dependent on the activation of group II mGluR-associated PLC, PKC, and IP3receptors. Furthermore, our recent observation suggests that the DCG IV-induced LTD does not require postsynaptic activation of MAPKs: the LTD was still induced (−19 ± 2.5%, n = 4) in the cells injected with MAPK pseudosubstrate peptide (1–2 mm in recording electrodes; see Otani et al., 1999b). Thus, although DCG IV activates MAPKs in rat prefrontal tissue (Otani et al., 1999b), this activation is not necessary for the LTD induction in the presence of DCG IV. At this stage, we do not know whether group II mGluRs and NMDA receptors cooperatively activate other downstream factor(s). Whatever the case, these results clearly suggest that there are different parallel biochemical pathways that converge to an LTD induction in rat prefrontal neurons. In the broad context, the DCG IV-induced LTD adds another variation to the wealth of the examples for receptor cross-talks underlying synaptic plasticity. These include the interactions between group I and group II mGluRs (Schoepp et al., 1997; Mistry et al., 1998; Cho et al., 2000), between groups I and II mGluRs and DA-Rs (Otani et al., 1999b) (Fig. 8A), between dopamine D1 and NMDA receptors (Gurden et al., 2000), between NMDA receptors and MCPG-sensitive mGluRs (Otani and Connor, 1995), and between mGluRs and AMPA–kainate receptors (Kemp and Bashir, 1999). Complex, but important, molecular cooperativity underlies these interactions among different postsynaptic receptors.
Footnotes
This work was financially supported by Biotech Grant CT96-0049. We thank Drs. L. J. Bindman and S. J. Sara for their comments on this manuscript.
Correspondence should be addressed to Dr. Satoru Otani, Neurobiologie des Processus Adaptatifs, Université de Paris VI, Case 8, 7 Quai St. Bernard, 75005 Paris, France. E-mail: satoru.otani@snv.jussieu.fr.
REFERENCES
- 1.Agranoff BW, Fisher SK. Phosphoinositides. In: Siegel SJ, Agranoff BW, Albers RW, Molinoff PB, editors. Basic neurochemistry, Chap 20. Raven; New York: 1994. pp. 417–428. [Google Scholar]
- 2.Baskys A, Malenka RC. Agonists at metabotropic glutamate receptors presynaptically inhibit EPSCs in neonatal rat hippocampus. J Physiol (Lond) 1991;444:687–701. doi: 10.1113/jphysiol.1991.sp018901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Birrell JM, Brown VJ. Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci. 2000;20:4320–4324. doi: 10.1523/JNEUROSCI.20-11-04320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blond O, Crepel F, Otani S. Long-term potentiation in rat prefrontal slices facilitated by phased application of dopamine. Eur J Pharmacol. 2002;438:115–116. doi: 10.1016/s0014-2999(02)01291-8. [DOI] [PubMed] [Google Scholar]
- 5.Brabet I, Parmentier M-L, De Colle C, Bockaert J, Acher F, Pin J-P. Comparative effect of l-CCG-I, DCG-IV and γ-carboxy-l-glutamate on all cloned metabotropic glutamate receptor subtypes. Neuropharmacology. 1998;37:1043–1051. doi: 10.1016/s0028-3908(98)00091-4. [DOI] [PubMed] [Google Scholar]
- 6.Cho K, Kemp N, Noel J, Aggelton JP, Brown MW, Bashir ZI. A new form of long-term depression in the perirhinal cortex. Nat Neurosci. 2000;3:150–156. doi: 10.1038/72093. [DOI] [PubMed] [Google Scholar]
- 7.Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 8.Daniel H, Levenes C, Fagni L, Conquet F, Bockaert J, Crepel F. Inositol-1,4,5-trisphosphate-mediated rescue of cerebellar long-term depression in subtype 1 metabotropic glutamate receptor mutant mouse. Neuroscience. 1999;92:1–6. doi: 10.1016/s0306-4522(99)00136-0. [DOI] [PubMed] [Google Scholar]
- 9.Davis PD, Hill CH, Keech E, Lawton G, Nixon JS, Sedgwick AD, Wadsworth J, Westmacott D, Wilkinson SE. Potent selective inhibitors of protein kinase C. FEBS Lett. 1989;259:61–63. doi: 10.1016/0014-5793(89)81494-2. [DOI] [PubMed] [Google Scholar]
- 10.Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuster JM, Bodner M, Kroger JK. Cross-modal and cross-temporal association in neurons of frontal cortex. Nature. 2000;405:347–351. doi: 10.1038/35012613. [DOI] [PubMed] [Google Scholar]
- 12.Goldman-Rakic P. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 13.Gurden H, Tassin J-P, Jay TM. Integrity of the mesocortical dopaminergic system is necessary for complete expression of in vivo hippocampal-prefrontal cortex long-term potentiation. Neuroscience. 1999;94:1019–1027. doi: 10.1016/s0306-4522(99)00395-4. [DOI] [PubMed] [Google Scholar]
- 14. Gurden H, Takita M, Jay TM. Essential role of D1 but not D2 receptors in the NMDA receptor-dependent long-term potentiation at hippocampal-prefrontal cortex synapses in vivo. J Neurosci 20 2000. RC106(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris T, Persaud SJ, Jones PM. Atypical isoforms of PKC and insulin secretion from pancreatic β-cells: evidence using Gö 6976 and Ro 31–8220 as PKC inhibitors. Biochem Biophys Res Commun. 1996;227:672–676. doi: 10.1006/bbrc.1996.1567. [DOI] [PubMed] [Google Scholar]
- 16. Heinbockel T, Pape HC. Input-specific long-term depression in the lateral amygdala evoked by theta frequency stimulation. J Neurosci 20 2000. RC68(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirsch JC, Crepel F. Use-dependent changes in synaptic efficacy in rat prefrontal neurons in vitro. J Physiol (Lond) 1990;427:31–49. doi: 10.1113/jphysiol.1990.sp018159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirsch JC, Crepel F. Blockade of NMDA receptors unmasks a long-term depression in synaptic efficacy in rat prefrontal neurons in vitro. Exp Brain Res. 1991;85:621–624. doi: 10.1007/BF00231747. [DOI] [PubMed] [Google Scholar]
- 19.Huang L-Q, Rowan MJ, Anwyl R. mGluR II agonist inhibition of LTP induction, and mGluR II antagonist inhibition of LTD induction, in the dentate gyrus in vitro. NeuroReport. 1997;8:687–693. doi: 10.1097/00001756-199702100-00022. [DOI] [PubMed] [Google Scholar]
- 20.Huang L, Killbride J, Rowan MJ, Anwyl R. Activation of mGluRII induces LTD via activation of protein kinase A and protein kinase C in the dentate gyrus of the hippocampus in vitro. Neuropharmacology. 1999;38:73–83. doi: 10.1016/s0028-3908(98)00168-3. [DOI] [PubMed] [Google Scholar]
- 21.Huber KM, Kayser MS, Bear MF. Role of rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1256. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- 22.Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86:321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- 23.Ichihara K. Statistics for bioscience, Chap 4, pp 115–146. Nanko-Do; Tokyo: 1990. [Google Scholar]
- 24.Ishida M, Saitoh T, Shimamoto K, Ohfune Y, Shinozaki H. A novel metabotropic glutamate receptor agonist: marked depression of monosynaptic excitation in the newborn rat isolated spinal cord. Br J Pharmacol. 1993;109:1169–1177. doi: 10.1111/j.1476-5381.1993.tb13745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kahn L, Alonso G, Robbe D, Bockaert J, Manzoni OJ. Group 2 metabotropic glutamate receptors induced long term depression in mouse striatal slices. Neurosci Lett. 2001;316:178–182. doi: 10.1016/s0304-3940(01)02397-7. [DOI] [PubMed] [Google Scholar]
- 26.Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun. 1987;142:436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- 27.Kemp N, Bashir ZI. Induction of LTD in the adult hippocampus by the synaptic activation of AMPA/kainate and metabotropic glutamate receptors. Neuropharmacology. 1999;38:495–504. doi: 10.1016/s0028-3908(98)00222-6. [DOI] [PubMed] [Google Scholar]
- 28.Kilbride J, Huang L, Rowan MJ, Anwyl R. Presynaptic inhibitory action of the group II metabotropic glutamate receptor agonists, LY354740 and DCG-IV. Eur J Pharmacol. 1998;356:149–157. doi: 10.1016/s0014-2999(98)00526-3. [DOI] [PubMed] [Google Scholar]
- 29.Kirkwood A, Bear MF. Homosynaptic long-term depression in the visual cortex. J Neurosci. 1994;14:3404–3412. doi: 10.1523/JNEUROSCI.14-05-03404.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF. Modulation of long-term synaptic depression in visual cortex by acetylcholine and norepinephrine. J Neurosci. 1999;19:1599–1609. doi: 10.1523/JNEUROSCI.19-05-01599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein J, Chalifa V, Liscovitch M, Löffelholz K. Role of phospholipase D activation in nervous system physiology and pathophysiology. J Neurochem. 1995;65:1445–1455. doi: 10.1046/j.1471-4159.1995.65041445.x. [DOI] [PubMed] [Google Scholar]
- 32.Klein J, Iovino M, Vakil M, Shinozaki H, Löffelholz K. Ontogenetic and pharmacological studies on metabotropic glutamate receptors coupled to phospholipase D activation. Neuropharmacology. 1997;36:305–311. doi: 10.1016/s0028-3908(97)00024-5. [DOI] [PubMed] [Google Scholar]
- 33.Kolb B. Functions of the frontal cortex of the rat: a comparative review. Brain Res Rev. 1984;8:65–98. doi: 10.1016/0165-0173(84)90018-3. [DOI] [PubMed] [Google Scholar]
- 34.Law-Tho D, Desce JM, Crepel F. Dopamine favours the emergence of long-term depression versus long-term potentiation in slices of rat prefrontal cortex. Neurosci Lett. 1995;188:125–128. doi: 10.1016/0304-3940(95)11414-r. [DOI] [PubMed] [Google Scholar]
- 35.Linden DJ, Connor JA. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science. 1991;254:1656–1659. doi: 10.1126/science.1721243. [DOI] [PubMed] [Google Scholar]
- 36.Lovinger DM. trans-1-Aminocyclopentane-1–3-decarboxylic acid (t-ACPD) decreases synaptic excitation in rat striatal slices through a presynaptic action. Neurosci Lett. 1991;129:17–21. doi: 10.1016/0304-3940(91)90710-b. [DOI] [PubMed] [Google Scholar]
- 37.Manahan-Vaughan D. Group 1 and 2 metabotropic glutamate receptors play differential roles in hippocampal long-term depression and long-term potentiation in freely moving rats. J Neurosci. 1997;17:3303–3311. doi: 10.1523/JNEUROSCI.17-09-03303.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mistry R, Golding N, Challiss RAJ. Regulation of phosphoinositide turnover in neonatal rat cerebral cortex by group I- and II-selective metabotropic glutamate receptor agonists. Br J Pharmacol. 1998;123:581–589. doi: 10.1038/sj.bjp.0701626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris SH, Knevett S, Lerner EG, Bindman LJ. Group I mGluR agonist DHPG facilitates the induction of LTP in rat prelimbic cortex in vitro. J Neurophysiol. 1999;82:1927–1933. doi: 10.1152/jn.1999.82.4.1927. [DOI] [PubMed] [Google Scholar]
- 40.Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- 41.Nestler EJ, Duman RS. G protein and cyclic nucleotides in the nervous system. In: Siegel SJ, Agranoff BW, Albers RW, Molinoff PB, editors. Basic neurochemistry, Chap 21. Raven; New York: 1994. pp. 429–448. [Google Scholar]
- 42.Otani S, Connor JA. Long-term depression of naïve synapses in adult hippocampus induced by asynchronous synaptic activity. J Neurophysiol. 1995;73:2596–2601. doi: 10.1152/jn.1995.73.6.2596. [DOI] [PubMed] [Google Scholar]
- 43.Otani S, Connor JA. Requirement of rapid Ca2+ entry and synaptic activation of metabotropic glutamate receptors for the induction of long-term depression in adult rat hippocampus. J Physiol (Lond) 1998;511:761–770. doi: 10.1111/j.1469-7793.1998.761bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otani S, Auclair N, Desce J-M, Crépel F. Dopaminergic receptors and metabotropic glutamate receptors cooperate to induce LTD in rat prefrontal glutamatergic synapses. Soc Neurosci Abstr. 1998a;24:2024. [Google Scholar]
- 45.Otani S, Blond O, Desce J-M, Crépel F. Dopamine facilitates long-term depression of glutamatergic transmission in rat prefrontal cortex. Neuroscience. 1998b;85:669–676. doi: 10.1016/s0306-4522(97)00677-5. [DOI] [PubMed] [Google Scholar]
- 46.Otani S, Auclair N, Desce J-M, Roisin M-P, Crepel F. LTD induction in rat prefrontal neurons involves converging activation of MAP kinases by dopamine- and mGlu-receptors. Soc Neurosci Abstr. 1999a;25:991. doi: 10.1523/JNEUROSCI.19-22-09788.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Otani S, Auclair N, Desce J-M, Roisin M-P, Crepel F. Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J Neurosci. 1999b;19:9788–9802. doi: 10.1523/JNEUROSCI.19-22-09788.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pellegrini-Giampietro DE, Albani S, Pellicciari R, Moroni F. Metabotropic glutamate receptors coupled to phospholipase D. Neuropharmacology. 1996;35:A23. [Google Scholar]
- 49.Petralia RS, Wang Y-X, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience. 1996;71:949–976. doi: 10.1016/0306-4522(95)00533-1. [DOI] [PubMed] [Google Scholar]
- 50.Rainer G, Asaad WF, Miller EK. Selective representation of relevant information by neurons in the primate prefrontal cortex. Nature. 1998;393:577–579. doi: 10.1038/31235. [DOI] [PubMed] [Google Scholar]
- 51.Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- 52.Reid SNM, Daw NW, Gregory DS, Flavin H. cAMP levels increased by activation of metabotropic glutamate receptors correlate with visual plasticity. J Neurosci. 1996;16:7619–7626. doi: 10.1523/JNEUROSCI.16-23-07619.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reyes-Harde M, Stanton PK. Postsynaptic phospholipase C activity is required for the induction of homosynaptic long-term depression in rat hippocampus. Neurosci Lett. 1998;252:155–158. doi: 10.1016/s0304-3940(98)00496-0. [DOI] [PubMed] [Google Scholar]
- 54.Schoepp DD, Johnson BG, Monn JA. (1S,3R)-1-Aminocyclopentane-1,3-dicarboxylic acid-induced increases in cyclic AMP formation in the neonatal rat hippocampus are mediated by a synergistic interaction between phosphoinositide- and inhibitory cyclic AMP-coupled mGluRs. J Neurochem. 1996;66:1981–1985. doi: 10.1046/j.1471-4159.1996.66051981.x. [DOI] [PubMed] [Google Scholar]
- 55.Schoepp DD, Salhoff CR, Wright RA, Johnson BG, Burnett JP, Mayne NG, Belagaje R, Wu S, Monn JA. The novel metabotropic glutamate receptor agonist 2R,4R-APDC potentiates stimulation of phosphoinositide hydrolysis in the rat hippocampus by 3,5-dihydroxyphenylglycine: evidence for a synergistic interaction between group 1 and group 2 receptors. Neurophamacology. 1997;35:1661–1672. doi: 10.1016/s0028-3908(96)00121-9. [DOI] [PubMed] [Google Scholar]
- 56.Sortino MA, Aleppo G, Copani A, Casabona G, Nicoletti F, Ventra C, Kuhn R, Knöpfel T, Malitschek B, Canonico PL. Immortalized hypothalamic neurons express metabotropic glutamate receptors positively coupled to cyclic AMP formation. Eur J Neurosci. 1996;8:2407–2415. doi: 10.1111/j.1460-9568.1996.tb01204.x. [DOI] [PubMed] [Google Scholar]
- 57.Takita M, Yokoi H, Mizuno T. NMDA receptor clustering in rat prefrontal cortex revealed by in vitro calcium macroimaging. NeuroReport. 1997;8:551–553. doi: 10.1097/00001756-199701200-00034. [DOI] [PubMed] [Google Scholar]
- 58.Takita M, Izaki Y, Jay TM, Kaneko H, Suzuki SS. Induction of stable long-term depression in vivo in the hippocampal-prefrontal cortex pathway. Eur J Neurosci. 1999;11:4145–4148. doi: 10.1046/j.1460-9568.1999.00870.x. [DOI] [PubMed] [Google Scholar]
- 59.Tanabe Y, Masu M, Ishii T, Shigemoto R, Nakanishi S. A family of metabotropic glutamate receptors. Neuron. 1992;8:169–179. doi: 10.1016/0896-6273(92)90118-w. [DOI] [PubMed] [Google Scholar]
- 60.Tanaka C, Nishizuka Y. The protein kinase C family for neuronal signaling. Annu Rev Neurosci. 1994;17:551–567. doi: 10.1146/annurev.ne.17.030194.003003. [DOI] [PubMed] [Google Scholar]
- 61.Thompson AK, Mostafapour SP, Denlinger LC, Bleasdale JE, Fisher S. The aminosteroid U-73122 inhibits muscarinic receptor sequestration and phosphoinositide hydrolysis in SK-N-SH neuroblastoma cells. J Biol Chem. 1991;266:23856–23862. [PubMed] [Google Scholar]
- 62.Vickery RM, Morris SH, Bindman LJ. Metabotropic glutamate receptors are involved in long-term potentiation in isolated slices of rat medial frontal cortex. J Neurophysiol. 1997;78:3039–3046. doi: 10.1152/jn.1997.78.6.3039. [DOI] [PubMed] [Google Scholar]
- 63.Weiner N, Molinoff PB. Catecholamines. In: Siegel SJ, Agranoff BW, Albers RW, Molinoff PB, editors. Basic neurochemistry, Chap 12. Raven; New York: 1994. pp. 261–281. [Google Scholar]
- 64.Wilsch VW, Pidoplichko VI, Opitz T, Shinozaki H, Reymann KG. Metabotropic glutamate receptor agonist DCG-IV as NMDA receptor agonist in immature rat hippocampal neurons. Eur J Pharmacol. 1994;262:287–291. doi: 10.1016/0014-2999(94)90743-9. [DOI] [PubMed] [Google Scholar]
- 65.Yule D, Williams JA. U73122 inhibits Ca2+ oscillations in response to cholecystokinin and carbachol but not to JMV-180 in rat pancreatic acinar cells. J Biol Chem. 1992;267:13830–13835. [PubMed] [Google Scholar]
- 66.Zhart J, Taylor JR, Mathew RG, Arnsten AFT. Supranormal stimulation of D1 dopamine receptors in the rodent prefrontal cortex impairs spatial working memory performance. J Neurosci. 1997;17:8528–8535. doi: 10.1523/JNEUROSCI.17-21-08528.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]