

Abstract
Endogenous neurosteroids have rapid actions on ion channels, particularly GABAA receptors, which are potentiated by nanomolar concentrations of 3α-hydroxypregnane neurosteroids. Previous evidence suggests that 3β-hydroxypregnane steroids may competitively antagonize potentiation induced by their 3α diastereomers. Because of the potential importance of antagonists as experimental and clinical tools, we characterized the functional effect of 3β-hydroxysteroids. Although 3β-hydroxysteroids reduced the potentiation induced by 3α-hydroxysteroids, 3β-hydroxysteroids acted noncompetitively with respect to potentiating steroids and inhibited the largest degrees of potentiation most effectively. Potentiation by high concentrations of barbiturates was also reduced by 3β-hydroxysteroids. 3β-Hydroxysteroids are also direct, noncompetitive GABAA receptor antagonists. 3β-Hydroxysteroids coapplied with GABA significantly inhibited responses to ≥15 μm GABA. The profile of block was similar to that exhibited by sulfated steroids, known blockers of GABAA receptors. This direct, noncompetitive effect of 3β-hydroxysteroids was sufficient to account for the apparent antagonism of potentiating steroids. Mutated receptors exhibiting decreased sensitivity to sulfated steroid block were insensitive to both the direct effects of 3β-hydroxysteroids on GABAAresponses and the reduction of potentiating steroid effects. At concentrations that had little effect on GABAergic synaptic currents, 3β-hydroxysteroids and low concentrations of sulfated steroids significantly reversed the potentiation of synaptic currents induced by 3α-hydroxysteroids. We conclude that 3β-hydroxypregnane steroids are not direct antagonists of potentiating steroids but rather are noncompetitive, likely state-dependent, blockers of GABAAreceptors. Nevertheless, these steroids may be useful functional blockers of potentiating steroids when used at concentrations that do not affect baseline neurotransmission.
Keywords: neurosteroids, inhibitory postsynaptic current, GABAA receptors, pregnenolone sulfate, anesthetic, hippocampal culture
GABAAreceptors mediate most fast inhibitory neurotransmission in the CNS. Many important neuroactive compounds, including benzodiazepines, barbiturates, neuroactive steroids, and other general anesthetics, allosterically interact with GABAAreceptors and thereby influence the balance between neuronal excitation and inhibition (Macdonald and Olsen, 1994). These drugs enhance the activity produced by low concentrations of GABA and/or directly gate GABAA receptor channels in the absence of GABA (Majewska, 1992).
Neuroactive steroids are of particular interest, because they are synthesized in the CNS and periphery and are present in the CNS at concentrations that may endogenously modulate GABAA receptor function (Robel and Baulieu, 1994). (3α,5α)-3-Hydroxypregnan-20-one (3α5αP), (3α,5β)-3-hydroxypregnan-20-one (3α5βP), (3α,5β)-3,21-dihydroxypregnan-20-one (3α5βTHDOC), and (3α,5α)-3,21-dihydroxypregnan-20-one (3α5αTHDOC) act as potent, efficacious positive modulators of GABAAreceptors (Gasior et al., 1999; Lambert et al., 2001). Other endogenous neuroactive steroids, such as pregnenolone sulfate (PS) and (3β,5α)-3-hydroxypregnan-20-one sulfate (3β5αPS), inhibit GABA responses at high nanomolar to micromolar concentrations (for review, see Lambert et al., 2001). The role of endogenous neuroactive steroids in modulating GABAA receptor function remains unclear because of the lack of specific antagonists at the steroid-modulating sites. Precise sites of action of potentiating steroids on the GABAA receptor have also remained elusive (Lambert et al., 2001). Identification of antagonists may help to clarify interactions between potentiating steroids and the GABAA receptor.
Several previous studies explored potential steroid antagonists against potentiation by neuroactive steroids. In studies of [3H]flunitrazepam binding, which is a validated measure of GABAA receptor potentiation (Turner et al., 1989), both (3β,5β)-3-hydroxypregnan-20-one (3β5βP) and (3β,5α)-3-hydroxypregnan-20-one (3β5αP) produced insignificant changes in [3H]flunitrazepam binding when administered alone. However, both competitively antagonized the potentiation of [3H]flunitrazepam binding by 3α5βP and 3α5αP (Prince and Simmonds, 1992, 1993). In electrophysiological studies, 3β5βP antagonized the 3α5βP-induced enhancement of GABA current (Garrett and Gan, 1998;Maitra and Reynolds, 1998). Similarly, 3β5αP and 3β5βP diminished the inhibitory effects of 3α5αP and 3α5βP on population spikes evoked in rat hippocampal CA1 stratum pyramidale (Wang et al., 2000).
In the present study, we examined the effects of a series of endogenous and synthetic 3β-hydroxypregnane and 3β-hydroxyandrostane steroids on GABA-induced currents in Xenopus oocytes expressing recombinant GABAA receptors containing α1, β2, and γ2L subunits and on GABA-mediated synaptic responses in cultured rat hippocampal neurons. We report here that 3β-hydroxypregnane steroids exhibit block of GABAA receptor function that is dependent on GABA concentration, similar to block by sulfated steroids. The direct effect on GABAA receptors explains the apparent ability of 3β-hydroxysteroids to antagonize the effects of positive modulators.
MATERIALS AND METHODS
Chemicals. (3β,5β)-3,21-dihydroxypregnan-20-one, (3α,5β)-3-hydroxypregnan-20-one sulfate (3α5βPS), (3α,5α)-3-hydroxypregnan-20-one sulfate (3α5αPS), and 3β5αPS were obtained from Steraloids Inc. (Newport, RI), and flumazenil was from Roche (Basel, Switzerland). (3β,5β)-3-Hydroxypregnan-20-one sulfate (3β5βPS) was synthesized as described previously (Park-Chung et al., 1997) and was a generous gift of Dr. Robert H. Purdy (Scripps Research Institute, La Jolla, CA). (3β,5β,17β)-3-hydroxyandrostane-17-carbonitrile (3β5βACN) was synthesized as described previously (Han et al., 1996). Synthesis of (3β,5β,7α,17β)-3-hydroxy-7-methylpregnan-20-one [(7αMe)3β5βP] was also described previously (Zeng et al., 2001). All of the remaining chemicals were from RBI/Sigma (St. Louis, MO). The steroids were dissolved in DMSO. Pentobarbital was dissolved in 0.1% NaOH. All chemicals were then diluted in saline for experiments. The concentration of DMSO in experimental solutions was ≤0.1%.
Hippocampal microcultures. Primary microisland cultures of hippocampal cells were prepared from 1- to 3-d-old postnatal Sprague Dawley rats using established methods (Mennerick et al., 1995). Under halothane anesthesia, rats were decapitated, and the hippocampi were dissected and cut into 500-μm-thick transverse slices. The slices were dissociated with 1 mg/ml papain in oxygenated Leibovitz L-15 medium and mechanical trituration in modified Eagle's medium containing 5% horse serum, 5% fetal calf serum, 17 mmd-glucose, 400 μm glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. Isolated cells were plated onto 35 mm plastic culture dishes at a density of 75 cells/mm2. Before plating, culture dishes were coated with a layer of 0.15% agarose, dried overnight, and sprayed with small droplets of rat tail collagen using a microatomizer (Thomas Scientific, Swedeboro, NJ). The agarose layer serves as a nonpermissive background for cell adhesion. Cultures were treated with cytosine arabinoside (5–10 μm) after 3 din vitro to halt glial proliferation. Electrophysiological recordings were performed 8–15 d after plating.
Culture electrophysiology. Whole-cell recordings were performed on solitary, inhibitory, hippocampal microculture neurons, using an Axopatch 1D amplifier (Axon Instruments, Foster City, CA) interfaced to a Pentium III-based computer via a Digidata 1200 acquisition board (Axon Instruments). Recordings were at room temperature. Electrodes had resistances of 1.5–4 MΩ for whole-cell recordings. Access resistance was electronically compensated 90–100%. Autaptic release of neurotransmitter was stimulated in voltage-clamped solitary neurons with a 2 msec voltage pulse to 0 mV from a holding potential of −70 mV. This stimulation protocol elicits an escaped action potential in the partially clamped axons that triggers transmitter release (Bekkers and Stevens, 1991; Mennerick et al., 1995). Whenever possible, at least three traces in each experimental condition were acquired for analysis. For all experiments, the interval between data sweeps was 25 sec for synaptic responses. Control conditions were interleaved with experimental conditions to counterbalance any time-dependent changes. Data sampling frequency was 5–10 kHz.
At the time of experiments, culture medium was replaced with an extracellular recording solution consisting of (in mm): 138 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, and 0.025d-APV, pH 7.25. Solutions were exchanged via a local multibarrel perfusion pipette, with a common delivery port placed 0.5 mm from the cell under study. The standard pipette solution for autaptic responses contained (in mm): 140 KCl, 4 NaCl, 0.5 CaCl2, 5 EGTA, and 10 HEPES, pH 7.25. IPSCs were easily distinguished from AMPA receptor-mediated EPSCs under these conditions by the >10-fold slower 10–90% decay times of IPSCs. There was no effect of either 3α-hydroxy (500 nm 3α5αP) or 3β-hydroxy (10 μm 3β5βTHDOC) neurosteroids on the amplitude or decay of AMPA receptor-mediated EPSCs (n = 4). IPSCs exhibited decays that were fitted by two or three exponential components, and potentiating steroid had most prominent effects on the slow components of decay (Zorumski et al., 1998). For ease of comparing effects of several drugs among cells with heterogeneous kinetics, we used the model-independent 10–90% decay time of IPSCs as our primary measure of IPSC duration (see Fig. 10).
Fig. 10.

Similar synaptic effect of 3β5βTHDOC and 3β5αPS. A, Autaptic IPSCs were elicited from solitary GABAergic hippocampal neurons in microcultures with 2 msec voltage pulse to 0 mV from a holding potential of −70 mV. Thetraces represent responses obtained in the absence and presence of 10 μm 3β5βTHDOC. Note that the drug alone only slightly affected the IPSC. B, Thetraces represent responses obtained in the absence and presence of 0.5 μm 3α5αP and 0.5 μm3α5αP plus 10 μm 3β5βTHDOC. Note that 3α5αP significantly prolonged the decay time course of the IPSC. 3β5βTHDOC inhibited the prolongation induced by potentiating steroid. C, Summary of the effect of 10 μm3β5βTHDOC and 2 μm 3β5αPS on IPSCs in the absence and presence of 0.5 μm 3α5αP (n = 6 neurons). For this analysis, we used 10–90% decay times, which averaged 315 ± 71 msec in the control situation. We also fit IPSC decays with multiple exponential components (Zorumski et al., 1998). Weighted time constants (∑aiτI, whereai is the fractional amplitude and τI is the time constant of each exponential component) were also used to quantify the data (Jones and Westbrook, 1997). Raw values from the weighted time constant analysis were 137 ± 30 msec (control), 119 ± 20 msec (3β5βTHDOC alone), 101 ± 18 msec (3β5αPS alone), 486 ± 63 msec (3α5αP alone), 176 ± 20 msec (3α5αP plus 3β5βTHDOC), and 266 ± 48 msec (3α5αP plus 3β5αPS).
Expression in Xenopus oocytes. Stage V–VI oocytes were harvested from sexually mature female Xenopus laevis (Xenopus One, Northland, MI) under 0.1% tricaine (3-aminobenzoic acid ethyl ester) anesthesia. Oocytes were defolliculated by shaking for 20 min at 37°C in collagenase (2 mg/ml) dissolved in calcium-free solution containing (in mm): 96 NaCl, 2 KCl, 1 MgCl2, and 5 HEPES, pH 7.4. Capped mRNA, encoding rat GABAA receptor α1, β2, and γ2L subunits, was transcribed in vitro using the mMESSAGE mMachine Kit (Ambion, Austin, TX) from linearized pBluescript vectors containing receptor coding regions. Subunit transcripts were injected in equal parts (20–40 ng of total RNA) 8–24 hr after defolliculation. Oocytes were incubated up to 5 d at 18°C in ND96 medium containing 96 mm NaCl, 1 mmKCl, 1 mm MgCl2, 2 mm CaCl2 and 5 mm HEPES at pH 7.4, supplemented with 5 mm pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μg/ml gentamycin. The cDNAs for the rat GABAA receptor subunits were provided by A. Tobin (α1) (University of California, Los Angels, CA), P. Malherbe (β2) (Hoffman-La Roche, Basal, Switzerland), and C. Fraser (γ2L) (National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD). The V256S mutation, a point mutation in the α1 subunit at the second residue N terminal to the beginning of transmembrane domain 2 (position 2′), was characterized previously and shown to dramatically reduce pregnenolone sulfate block of GABAAreceptors (Akk et al., 2001).
Oocyte electrophysiology. Two-electrode voltage-clamp experiments were performed with an Axoclamp 2B amplifier or Warner Instruments (Hamden, CT) OC725 amplifier 2–5 d after RNA injection. The extracellular recording solution was ND96 medium with no supplements. Intracellular recording pipettes were filled with 3m KCl and had open tip resistances of ∼1 MΩ. Drugs were applied from a common tip via a gravity-driven multibarrel drug-delivery system. Drugs were always coapplied with GABA and were not preapplied in the absence of GABA. Cells were clamped at −70 mV for all experiments, and current at the end of 20–30 sec drug applications was measured for quantification of current amplitudes. In three oocytes, we pretreated cells with a low concentration of potentiating steroid (3α5αP, 100 nm for 40 sec) before coapplying GABA. This protocol resulted in little difference in response size compared with the standard (coapplication only) protocol used for all figures (101 ± 11% potentiation with pretreatment, followed by coapplication; 85 ± 12% with coapplication alone). Likewise, pretreatment with a moderate concentration of blocking steroid (3β5βTHDOC, 6 μm for 40 sec) yielded similar block at the end of 20 μm GABA coapplication as with coapplication alone (36 ± 4% depression with pretreatment vs 36 ± 2% depression with coapplication alone) (see Fig. 6).
Fig. 6.

3β-Hydroxypregnane steroids block responses to high concentrations of GABA. A, Sample traces showing lack of effect of 10 μm 3β5βTHDOC on responses to 2 μm GABA but inhibition of responses to 20 μm GABA. Drugs were coapplied for 20 sec. Note the change in vertical calibration bars between theleft and right panels. B, GABA concentration–response curves in the absence (filled circles) and presence (open squares) of 10 μm 3β5βTHDOC. Eachpoint is calculated relative to the normalizing response (IN) activated by 2 μmGABA. The solid line through GABA concentration–response values (circles) is the best fit of the Hill equation, with an EC50 of 12.6 μmand a Hill coefficient of 2.6 (n = 8). Thesolid line through GABA plus 3β5βTHDOC interaction values (squares) is the best fit of the Hill equation, with an EC50 of 9.4 μm and a Hill coefficient of 2.1 (n = 8). C. The graph shows the concentration–response curve of 3β5βTHDOC against 20 μm GABA. The normalizing current (IN) was the response to 20 μm GABA. The solid lines are the fit of the Hill equation, with an IC50 of 6.8 μm and a Hill coefficient of 0.8, with maximum inhibition of −1.0 (n = 6).
Data analysis. Data acquisition and analysis were performed with pClamp software (Axon Instruments). Data plotting and curve fitting were done with Sigma Plot software (SPSS, Chicago, IL). Data are presented in the text and figures as mean ± SE. Statistical differences were determined using a two-tailed Student's ttest or a one-way ANOVA. The percentage of modulation of GABA-activated current was calculated as (IM −IN)/IN, where IN (normalizing current) andIM (measured current) are the amplitudes of the GABA-activated current in the absence and presence of the test substance, respectively. Fitting of the dose–response relationships were performed using the Hill equation as follows:I = Imax ×Cn/(EC50n+ Cn), where C is the concentration of steroid (or GABA) (see Fig. 6),Imax is the maximum current amplitude, EC50 is the concentration of steroid (or GABA) that produces 50% of Imax, andn is the Hill coefficient.
RESULTS
Noncompetitive antagonism of steroid and barbiturate potentiation by 3β-hydroxypregnane steroids
To characterize the action of 3β-hydroxypregnane steroids at GABAA receptors, we examined their putative antagonist profile against GABA-potentiating neurosteroids. Figure1 shows the pregnane–androstane steroid ring system, and the chiral centers at carbon 3 (C3) and C5 are highlighted. The steroid shown is a 3α,5α steroid. The steroids used in the present study varied in the side chain group at C17 (Fig.1), but these side groups all conformed to the general rule that the side chain contained a hydrogen bond acceptor (Covey et al., 2001). This side chain group at C17 in the β configuration is necessary (but not sufficient) for GABAA receptor potentiation (Lambert et al., 2001). Potentiation by neuroactive steroids is also critically dependent on a hydrogen bond donor in the α configuration at C3 (Lambert et al., 2001). For naturally occurring steroids, such as 3α5αP, 3α5βP, and 3α5βTHDOC, this hydrogen bond donor is a hydroxyl group. We tested the hypothesis in the current work that the 3β diastereomers of these potentiating neurosteroids are competitive antagonists of the potentiators (Prince and Simmonds, 1992, 1993).
Fig. 1.

Steroid structure, with emphasis (arrows) on the chiral centers at C3 and C5. The structure shown is 3α,5α,17β. 3β-Hydroxypregnane steroids have been suggested to antagonize the potentiating actions of steroids with a 3α-hydroxy configuration. For the steroids tested in this work, the R group was CN, COCH3, or COCH2OH.
Figure 2A shows raw traces from a Xenopus oocyte expressing a combination of α1β2γ2L GABAA receptor subunits. We applied a low concentration of GABA (2 μm) alone, or we coapplied (with GABA) varied concentrations of 3α5αP plus or minus 10 μm 3β5αP. Note that 3β5αP significantly inhibited only the response to the high concentration of potentiating steroid. Figure 2B shows a similar experiment using another 3β-hydroxypregnane steroid, 3β5βTHDOC, which exhibited a similar profile of apparent antagonism. Figure 2C–E shows summaries of the effect of three different 3β-hydroxypregnane steroids (3β5αP, 3β5βTHDOC, and 3β5βP) on potentiation by 3α5αP. Table1 gives values from fits to the Hill equation of the concentration–response curves shown in Figure2C–E. Note that the EC50 values for potentiation were not substantially affected by the presence of the 3β-hydroxysteroid, suggesting a noncompetitive mechanism of reduced potentiation. The similarity of the effects of the three different steroids (Fig. 2C–E) suggests that neither the configuration of the hydrogen atom at C5 nor the structure of the hydrogen bond acceptor group at C17 is critical to the blocking effect.
Fig. 2.
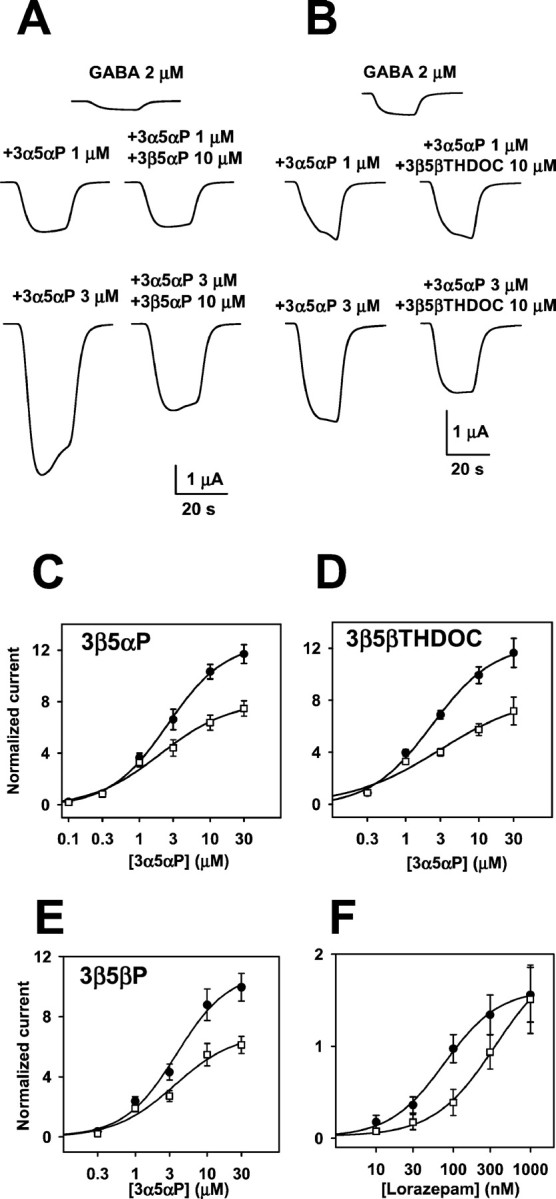
3β5αP and 3β5βTHDOC reversed the effect of high concentrations of GABA potentiating steroids. A, Sample traces showing that 10 μm 3β5αP inhibited potentiation by high but not low concentrations of 3α5αP.B, Similar traces for 10 μm 3β5βTHDOC.C–E, Summary of effect of 3β5αP (C), 3β5βTHDOC (D), and 3β5βP (E) from oocytes tested with a range of 3α5αP concentrations. C, Summary of the effect of 10 μm 3β5αP against increasing concentrations of 3α5αP. Normalized responses in this and subsequent figures were calculated as follows: (IM −IN)/IN, where IM is the amplitude of the measured current in a given experimental condition, andIN is the normalizing current. For these data and data in D and E,IN was the response to 2 μmGABA in the absence of modulator. The solid linesrepresent least-squares fits of the data to the Hill equation as follows: I = Imax×Cn/(EC50n+ Cn), where C is the concentration of potentiator, EC50 is the concentration of potentiator that produced half-maximum potentiation, andn is the Hill coefficient. Parameters of the fit for potentiators in the absence (filled circles) and presence (open squares) of 3β5αP are given in Table 1. D, Summary of the effect of 10 μm 3β5βTHDOC under similar experimental conditions to those in C. E, Concentration–response relationship for 3α5αP in the presence and absence of 3β5βP (10 μm). F, Concentration–response relationship of the benzodiazepine agonist lorazepam in the presence and absence of the benzodiazepine antagonist flumazenil. The solid line through lorazepam concentration–response values (circles) is the best fit of the Hill equation, with an EC50 of 76.7 nm and a Hill coefficient of 1.2 (n = 7). The solid line through lorazepam–flumazenil interaction values (squares) is the best fit of the Hill equation, with an EC50 of 348.5 nm and a Hill coefficient of 1.1 (n = 7).
Table 1.
Fit parameters for potentiation in the presence and absence of 3β-hydroxysteroids
| EC50(μm) | Hill n | % Potentiation | % Reduction | # Cells | |||
|---|---|---|---|---|---|---|---|
| Potentiator alone | + 3β steroid | Potentiator alone | + 3β steroid | 10 μm potentiator alone ± SEM | 10 μmpotentiator + 10 μm 3β steroid* | ||
| 3α5αP versus 3β5αP | 2.5 | 2.0 | 1.1 | 0.9 | 1033 ± 56 | −38.3 | 11 |
| 3α5αP versus 3β5βTHDOC | 2.3 | 3.0 | 1.0 | 0.7 | 992 ± 64 | −42.1 | 9 |
| 3α5αP versus 3β5βP | 3.7 | 3.5 | 1.2 | 1.0 | 779 ± 205 | −42.6 | 6 |
| 3α5βP versus 3β5βTHDOC | 2.5 | 1.7 | 1.0 | 1.1 | 1021 ± 61 | −41.9 | 8 |
| 3α5βTHDOC versus 3β5βTHDOC | 3.0 | 2.1 | 1.3 | 1.3 | 629 ± 22 | −29.4 | 8 |
| 3α5αTHDOC versus 3β5βTHDOC | 3.2 | 2.6 | 1.6 | 1.2 | 1003 ± 49 | −43.4 | 10 |
| 3α5αP versus 3β5αPS | 3.0 | 2.2 | 1.2 | 1.2 | 854 ± 117 | −38.5* | 6 |
In experiments in which data were fit to the Hill equation, GABA was present at 2 μm, and 3β-hydroxysteroids, when present, were used at 10 μm. Percentage of potentiation was calculated by comparing the current in response to 2 μm GABA alone with the current in the presence of 2 μm GABA plus 10 μm 3α-hydroxysteroid. Reduction was calculated by comparing the size of the current in the presence of 10 μm potentiator alone with the current amplitude in the presence of equimolar concentrations (10 μm) of potentiator and 3β-hydroxysteroid.
indicates that 3β5αPS was used at 300 nm instead of 10 μm because of its greater potency compared with 3β-hydroxysteroids.
We were surprised by the apparently noncompetitive nature of the interaction between the 3β-hydroxysteroids and 3α5αP (Fig.2C–E). To be sure that our protocol was capable of detecting a competitive interaction between potentiator and antagonist, we examined the effect of the benzodiazepine antagonist flumazenil on the effect of a potentiating benzodiazepine lorazepam. Flumazenil (100 nm) competitively inhibited the GABA-enhancing effect of lorazepam by shifting lorazepam concentration–response curves to the right (n = 7) (Fig.2F). The EC50 of lorazepam increased from 76.7 to 348.5 nm in the presence of 100 nm flumazenil (Fig. 2F). The Hill coefficients (Fig. 2F, see legend) and maximal response were quite close between two lorazepam concentration–response curves. This control experiment suggests that our experimental protocol should have detected a competitive interaction between a steroid potentiator and antagonist if present.
We explored the actions of several other 3β-hydroxysteroids at 10 μm, and, consistent with the results of Figure 2, we found little structural specificity in the action against the potentiating neurosteroid 3α5αP (Fig.3A). Addition of an α methyl group at C7 did not affect block of potentiation, nor did substitution of a carbonitrile group for the acetyl side group at C17 (Fig.3A).
Fig. 3.
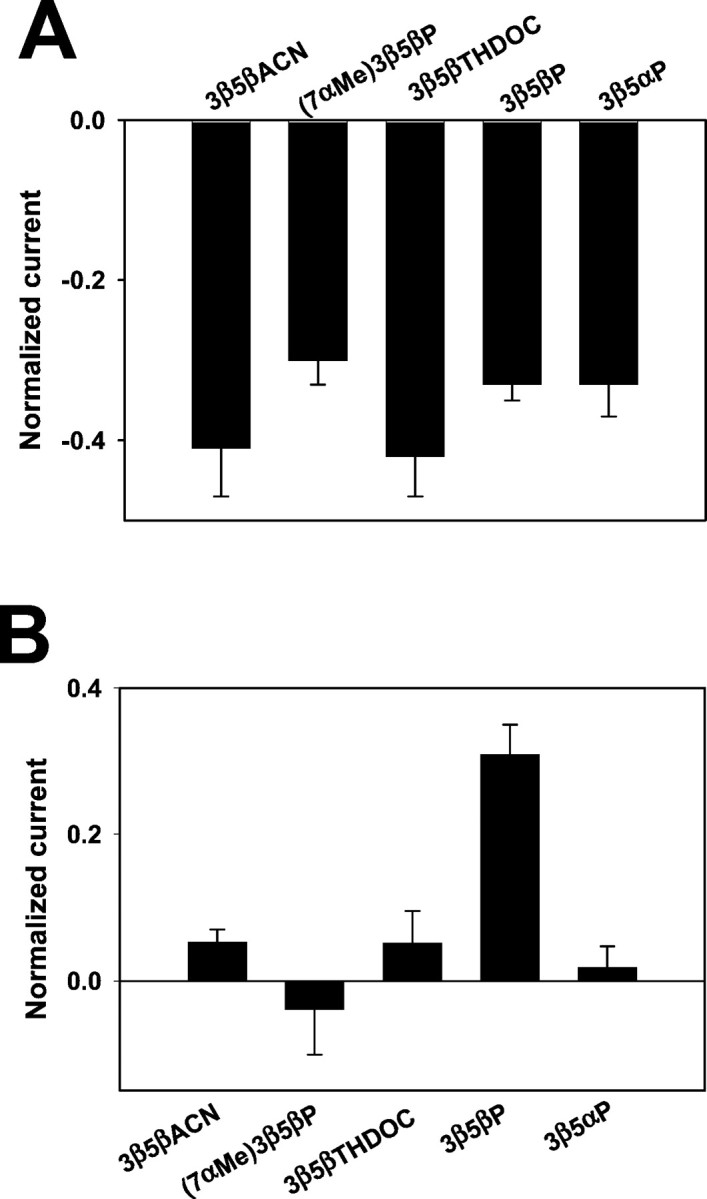
Effects of a series 3β-hydroxysteroids on 2 μm GABA responses in the presence and absence of 3α5αP. A, Effects of several 3β-hydroxysteroids (10 μm) against potentiation induced by 3α5αP (3 μm) in the presence of 2 μm GABA. For these data, the normalizing response (IN, representing 0 on the y-axis) was the current in the combined presence of 2 μm GABA plus 3 μm3α5αP (n = 6). B, Effects of several 3β-hydroxysteroids on responses to 2 μm GABA alone. Most compounds were effectively inert at 10 μm, except for 3β5βP, which slightly potentiated responses to 2 μm GABA. IN represented the response to 2 μm GABA alone (n = 5).
We also examined this panel of 3β-hydroxysteroids (using a fixed concentration of 10 μm) against the response to 2 μm GABA alone (Fig. 3B). We found that most 3β steroids were inert, but, as reported previously (Puia et al., 1990; Woodward et al., 1992; Kokate et al., 1994), 10 μm 3β5βP mildly potentiated GABA responses. For many of the remaining experiments, we used 3β5βTHDOC as our standard 3β-hydroxysteroid because of its relative inertness against GABA alone (Fig. 3B), the slightly better blocking activity than some other 3β-hydroxysteroids (Fig. 3A), and its commercial availability.
Given that the interaction between 3β-hydroxysteroids and 3α5αP was noncompetitive, we examined whether this noncompetitive interaction held true for other potentiating neurosteroids. Indeed, Figure4A–C shows that 3β5βTHDOC similarly inhibited potentiation by 5α- and 5β-reduced neuroactive steroids. A noncompetitive profile was evident for all steroid potentiators. When we tested other classes of GABA potentiators, we were surprised to find that barbiturate (Fig.5B,C) but not benzodiazepine (Fig. 5A,C) potentiation was inhibited by 3β-hydroxysteroids. The potentiation induced by 150 μm pentobarbital was significantly reduced by 10 μm3β5βTHDOC (Fig. 5B,C). As with steroid potentiation, the effect on pentobarbital potentiation was dependent on potentiator concentration. The potentiation by 50 μm pentobarbital (121 ± 25%) was not affected by 3β5βTHDOC (130 ± 28%; n = 13) (data not shown). Also, we found that maximum lorazepam potentiation (1 μm; 142 ± 8% potentiation) was unaffected by 3β5βTHDOC (138 ± 14% potentiation) (Fig.5C).
Fig. 4.

Effect of 3β-hydroxypregnane steroids on other 5α-reduced and 5β-reduced steroid potentiators of GABAAresponses. A, Concentration dependence of 3α5αTHDOC potentiation in the absence (filled circles) and presence (open squares) of 10 μm3β5βTHDOC. GABA (2 μm) was coapplied with steroids, and the response to 2 μm GABA was used as the normalizing response (IN). B,C, Similar analyses using the 5β-reduced steroids 3α5βP and 3α5βTHDOC as potentiators. In allpanels, the solid line through the concentration–response values in the absence and presence of 3β5βTHDOC is the best fit of the Hill equation. Parameters of the fits and n values are given in Table 1.
Fig. 5.

Effect of 3β-hydroxysteroids on two other classes of GABAA receptor potentiators. A, Effect of 3β5βTHDOC on potentiation by the benzodiazepine lorazepam in an oocyte. B, Effect of 3β5βTHDOC on potentiation by the barbiturate pentobarbital in another oocyte. C, Summary of the effects of 10 μm 3β5βTHDOC on pentobarbital (150 μm; n = 13) and lorazepam (1 μm; n = 5) potentiation. Note that the more robust potentiation by the barbiturate was more effectively antagonized. *p < 0.01 indicates significant inhibition.
At high concentrations, 3α-hydroxysteroids can weakly gate the GABAA receptor (Barker et al., 1987). We investigated whether this direct gating was antagonized by 3β-hydroxy steroids. We found that 3α5αP (30 μm) in the absence of GABA gated a current with an amplitude 34 ± 4% of response to 2 μm GABA in the same cell. Consistent with a lack of direct antagonism between 3β-hydroxysteroids and effects of 3α-hydroxysteroids, we observed no significant reduction in this steroid-gated current when 3β5βTHDOC was coapplied with 30 μm 3α5αP (30 ± 6% of the response to 2 μm GABA; n = 4).
Direct 3β-hydroxysteroid inhibition of GABA responses mimics the action of sulfated steroids and explains steroid antagonism
The pattern of noncompetitive inhibition of potentiation, the lack of structural specificity to the blocking effects, and the promiscuity of the effects of 3β-hydroxysteroids against at least two classes of potentiators suggest that the mechanism of 3β-hydroxysteroids does not result from a direct interaction with potentiating steroids sites on the GABAA receptor. Among all three classes of potentiators, a pattern emerged in which only strong potentiation was inhibited by 3β-hydroxysteroids (effects on only high concentrations of steroids and barbiturates, no effect on the modest benzodiazepine potentiation). This pattern suggested the hypothesis that block by 3β-hydroxysteroids may correlate with opening of GABAA receptors.
To test this hypothesis, we examined the effect of 3β-hydroxysteroids on responses to GABA alone over a range of GABA concentrations. We found that responses to low concentrations (<15 μm) of GABA were unaffected by 10 μm 3β5βTHDOC (Fig.6A,B). However, responses to ≥15 μm GABA were significantly depressed by 10 μm 3β5βTHDOC. The profile of 3β5βTHDOC effects against GABA was similar to that observed against potentiation by 3α-hydroxysteroids (Fig.6B) (compare Figs. 2D, 4). The effect of 3β5βTHDOC was concentration dependent, with an IC50 against 20 μm GABA responses of 6.8 μm (n = 6) (Fig. 6C). We again tested the panel of 3β-hydroxysteroids shown in Figure 3 and found that, at a fixed concentration of 10 μm, all similarly blocked currents in response to 20 μm GABA (n = 6) (data not shown). IC50 values for other 3β-hydroxysteroids against 20 μm GABA were as follows: 3β5βP, 12.8 μm (n= 5); 3β5αP, 14.4 μm (n = 5); and 3β5βACN, 9.5 μm (n= 6).
It is well known that another class of endogenous neurosteroids, steroids sulfated at C3, block GABAAreceptor-mediated responses (Majewska et al., 1988). Block is not dependent on the stereochemistry of the sulfate group at C3, with 3β-sulfated steroids blocking GABA responses with similar potency as 3α-sulfated steroids (Park-Chung et al., 1999). Block is not dependent on the presence of a sulfate group, because steroids containing other anionic groups also block GABA currents (Park-Chung et al., 1999). This class of blockers exhibits additional structural nonspecificity, because the charged group can be placed at C24 rather than C3 (Mennerick et al., 2001). Previous work has also shown that charge is not essential for blocking activity, because dehydroepiandrosterone sulfate and dehydroepiandrosterone both block GABAA receptors (Demirgoren et al., 1991; Le Foll et al., 1997). We addressed the possibility that 3β-hydroxysteroids are GABAA receptor blockers, acting similarly to sulfated steroids. As a first test of this hypothesis, we repeated the experiments shown in Figure 4, substituting 300 nm 3β5αPS for 10 μm 3β5βTHDOC. The 3β-sulfated steroid produced inhibition of steroid potentiation that was essentially indistinguishable from the inhibition produced by the 3β-hydroxysteroids (Fig.7A1,A2). In addition, 3β5αPS clearly inhibited responses to high GABA concentrations more effectively than it inhibited responses to low concentrations of GABA (Fig. 7B). When examined against a fixed GABA concentration of 20 μm, the IC50 for 3β5αPS block was ∼190 nm (n = 6) (Fig. 7C), much lower than the IC50 for 3β5βTHDOC under the same conditions (Fig. 6C). Pregnenolone sulfate was also quite potent, with an IC50 against 20 μm GABA responses of 385 nm (data not shown).
Fig. 7.

The action of 3β-hydroxysteroids is similar to that of sulfated steroid block. A, Inhibition of potentiation by a sulfated steroid. A1, Sample traces showing the very small effect of 300 nm 3β5αPS against potentiation by 1 μm 3α5αP but stronger inhibition against GABA responses potentiated by 10 μm 3α5αP.A2, Summary of experiments like that shown inA1, in which multiple concentrations of potentiator were examined. GABA (2 μm) was coapplied with varied concentrations of 3α5αP either without (filled circles) or with (open squares) 300 nm 3β5αPS. Parameters of fits to the Hill equation and experimental n are given in Table 1. B, Sulfated steroid block, like 3β-hydroxysteroid block, is dependent on GABA concentration. Shown are concentration– response curves for GABA in the absence (filled circles) and presence (open squares) of 300 nm 3β5αPS. In the absence of steroid, the EC50 for GABA was 12.5 μm, with a Hill coefficient of 2.8 (n= 7). In the presence of steroid, the GABA EC50 was 11.4 μm, with a Hill coefficient of 3.0 (n= 7). The normalized maximum response was decreased from 13.9 to 7.8 in the presence of steroid. C, Against a fixed GABA concentration of 20 μm, 3β5αPS produced a concentration-dependent inhibition of responses. The solid line is a fit of the Hill equation, with an IC50 of 189 nm and a Hill coefficient of 0.7 (n= 6).
To further explore whether the action of 3β-hydroxypregnane steroids is similar to that of sulfated steroids, we used a recently characterized point mutation causing resistance to sulfated steroid block (Akk et al., 2001). The mutant carries a valine to serine substitution at position 256 of the α1 subunit, located on the cytoplasmic side of transmembrane domain 2. Because we observed that the degree of block by both 3β-hydroxysteroids and 3β-sulfated steroids was dependent on GABA concentration, we examined the GABA concentration–response profile for wild-type and mutated receptors. We found that the mutation caused a large (∼20-fold) leftward shift of the GABA concentration–response relationship. To ensure that we examined the effect of 3β-hydroxysteroids and sulfated steroids under conditions that should promote block, we examined the effect of the steroids against 5 μm GABA in mutated receptors, a concentration that produced nearly maximum responses (Fig.8A, arrow). Comparison was made with wild-type responses at 20 μm, a concentration somewhat higher than the EC50 (Fig. 8A,arrow).
Fig. 8.
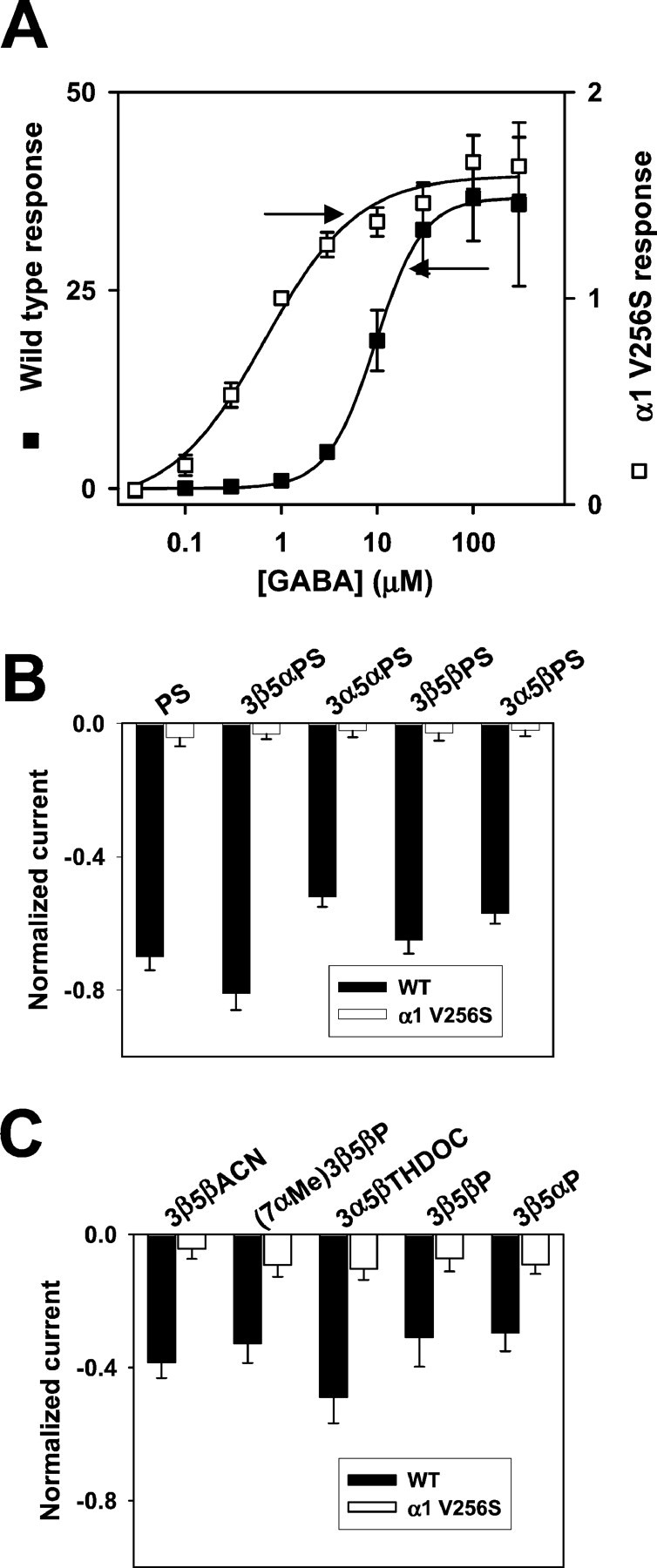
A point mutation in the α1 subunit (V256S) reduces sulfate and 3β-hydroxysteroid block of GABAAreceptors. A, GABA concentration–response curves for wild-type (filled squares; n= 5) and mutated (open squares; n = 6) GABAA receptors. Responses at 1 μm were set to 1 for both wild-type and mutated receptors, and other responses in the same oocyte were normalized to this response. Note that thelefty-axis corresponds to the wild-type normalized responses, and the righty-axis corresponds to the normalized responses from mutated receptors. Absolute amplitudes of maximum responses did not notably differ between wild-type and mutated receptors, but the mutation resulted in an ∼20-fold leftward shift in the GABA EC50, from 9.6 to 0.53 μm, resulting in the apparently larger normalized maximum responses for wild-type receptors. Arrows indicate concentrations of GABA used in subsequent studies of steroid block of GABAA receptors (B, C). B, Sulfated steroid effects (1 μm) in wild-type (WT,filled bars; n = 5) and α1V256S mutated (open bars; n = 5) GABAA receptors. C, 3β-Hydroxysteroid effects (10 μm) in wild-type (WT,filled bars; n = 6) and α1V256S (open bars; n = 9) mutated GABAA receptors.
Figure 8B (filled bars) shows a summary of the effect of various sulfated steroids at a fixed concentration of 1 μm against responses to 20 μm GABA in wild-type receptors. All sulfated steroids blocked more than half the current under these conditions. In contrast, none of the sulfated steroids significantly inhibited responses of mutated receptors, despite the use of a functionally higher GABA concentration than used for the wild-type receptors (Fig.8B, open bars). We found a similar pattern when a panel of 3β-hydroxysteroids were examined against wild-type and mutated receptors (Fig. 8C). In contrast, we found no difference in the ability of other GABAA receptor antagonists (gabazine, bicuculline, and picrotoxin) to inhibit mutated receptors (n = 8) (data not shown). These results are consistent with the idea that 3β-hydroxysteroids and sulfated steroids block GABAA receptors similarly, albeit with different potencies.
To address whether the direct effects on GABAAreceptors are sufficient to explain the apparent antagonism of potentiating steroids, in the same oocytes, we matched the size of GABA currents produced by a moderate concentration of GABA alone to that produced by 2 μm GABA plus potentiating steroid (Fig.9A,C). We found that 20 μm GABA produced responses of similar amplitude to those produced by 3 μm3α5αP plus 2 μm GABA. Responses to 20 μm GABA alone or the combination of 2 μm GABA plus 3α5αP were inhibited to a similar degree by 10 μm 3β5βTHDOC (Fig.9A,C). These results are consistent with the idea that the direct effect on GABAAreceptors can account for the apparent antagonism of potentiating steroid effects.
Fig. 9.
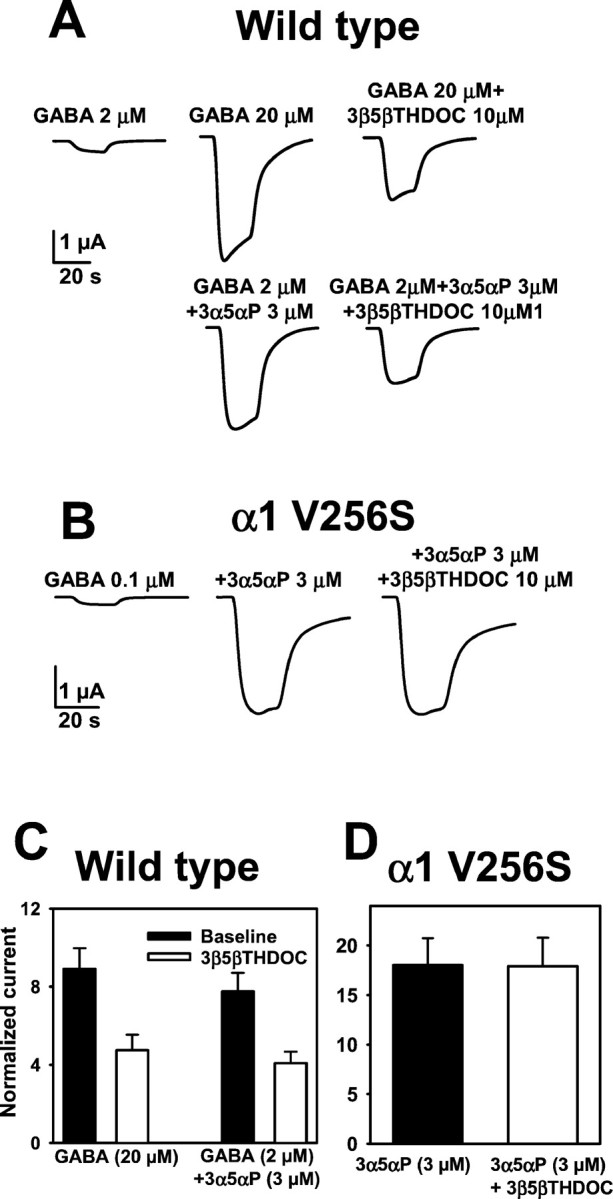
Direct GABAA receptor block explains 3β-hydroxysteroid inhibition of potentiation. A, Comparison of responses “potentiated” by increasing GABA concentration or by adding 3 μm 3α5αP. Thetraces show that 20 μm GABA and 3 μm 3α5αP cause a similar increase in current relative to 2 μm GABA. 3β5βTHDOC (10 μm) inhibited both increased currents to a similar degree.B, Mutated receptors are potentiated by 3α-hydroxysteroids, but steroid potentiation is not inhibited by 3β-hydroxysteroids. C, Summary of the blocking actions of 3β5βTHDOC on 20 μm GABA-activated current and potentiation of 2 μm GABA-activated current by 3 μm 3α5αP (n = 7) in oocytes expressing wild-type receptors. The bar graph represents current amplitudes normalized to the 2 μm GABA response.D, The bar graph represents a summary of the lack of effect of 3β5βTHDOC on potentiation by 3α5αP in mutated receptors. Normalizing current was the response activated by 0.1 μm GABA alone (n = 8).
As another test of whether direct block of GABAAreceptor function can account for the apparent antagonism of potentiating steroids, we examined steroid potentiation of α1 V256S mutated receptors and the effect of 3β-hydroxypregnane steroids on potentiation. For studies of potentiation at wild-type receptors, we typically used 2 μm GABA, approximately fivefold below the GABA EC50. For studies of potentiation at mutated receptors, we used 100 nm GABA to functionally match the GABA concentration with that used on wild-type receptors (Fig. 8A). We found that 3 μm 3α5αP robustly potentiated currents generated by mutated GABAA receptors. No additional potentiation was observed at 10 μm3α5αP (n = 8) (data not shown). In contrast to wild-type receptors, we observed no significant antagonism of this potentiation by 3β5βTHDOC (Fig. 9, compareB,D with C).
Actions of 3β-hydroxysteroids and sulfated steroids at GABA synapses
Although these experiments show that 3β-hydroxypregnane steroids are not true antagonists of potentiating steroids and act through an action similar to sulfated steroids, it is possible that either class of steroid could be a useful functional antagonist against postsynaptic potentiation at synapses. It is thought that a high GABA concentration is present at the synapse but only briefly (∼1 msec) (Maconochie et al., 1994; Jones and Westbrook, 1995). If binding and action of 3β-hydroxysteroids or sulfated steroids is dependent on GABA concentration and time, it is possible that blocking steroids are relatively inert against normal GABA transmission but more effective against responses potentiated by 3α-hydroxysteroids or other postsynaptic potentiators. In pilot experiments on cultured hippocampal neurons, we found that 2 μm 3β5αPS produced inhibition of responses to exogenous GABA that was equivalent to 10 μm 3β5βTHDOC (data not shown). This suggests that, in hippocampal cells, there is only approximately fivefold difference in potency between the 3β-hydroxysteroid and the sulfated steroid compared with the 35-fold difference observed in oocytes (Figs.6C, 7C). We therefore examined the effect of 10 μm 3β5βTHDOC and 2 μm 3β5αPS on IPSCs generated by recurrent (autaptic) synapses formed in culture (Mennerick et al., 1995). We found that 3β5βTHDOC and 3β5αPS at these concentrations had little effect on IPSCs generated in the absence of postsynaptic modulator (13 ± 12% decrease in 10–90% decay time for 3β5βTHDOC; 20 ± 9% decrease for 3β5αPS;n = 6) (Fig.10A,C). Despite the relative inertness of the steroids on baseline IPSCs, the same concentrations significantly inhibited the ability of a potentiating steroid (3α5αP, 0.5 μm) to prolong IPSCs (Fig. 10B,C). Also, consistent with a direct effect on GABAAreceptors rather than a competitive inhibition of the potentiating steroid, 30 μm 3β5βTHDOC alone significantly speeded the 10–90% decay time of baseline IPSCs (38 ± 14%; n = 15). Neither 10 nor 30 μm 3β5βTHDOC had any appreciable effect on the peak IPSC amplitude (4 ± 3 and 8 ± 4% increase, respectively). In summary, these data are consistent with the idea that 3β-hydroxysteroids and sulfated steroids share a similar mechanism and suggest that these blockers might be useful under some circumstances as functional antagonists of postsynaptic potentiation at GABAergic synapses.
DISCUSSION
We characterized the actions of 3β-hydroxypregnane steroids, which previous reports suggested are competitive antagonists against 3α-hydroxypregnane steroid potentiators. Although the 3β-hydroxysteroids behave as functional inhibitors of GABAA receptor potentiation, we find that the antagonism is noncompetitive with respect to 3α-hydroxypregnane steroids. In fact, 3β-hydroxysteroids are also noncompetitive antagonists of GABAA receptors themselves by actions similar to that of sulfated steroids. Both sulfated steroids and 3β-hydroxysteroids block GABAA receptors more effectively under conditions that promote channel opening, suggesting that the direct antagonism of GABA responsiveness may represent ligand-dependent or state-dependent block. This direct action at GABAA receptors accounts for the apparent antagonism of potentiating steroid effects.
Our results contrast with those reported by others using different methods. Using [3H]flunitrazepam binding, it was shown previously that 3β5βP competitively reduced the increase in binding produced by potentiating steroids (Prince and Simmonds, 1992, 1993). Other studies suggest that 3β5βP acts as a partial agonist at the potentiating steroid site (Pignataro and Fiszer de Plazas, 1997). Our results do not support the competitive interaction, observed in binding studies, between 3β-hydroxypregnane steroids and 3α-hydroxypregnane steroids.
Antagonism of potentiation by 3β-hydroxysteroids has also been reported in previous electrophysiological studies (Garrett and Gan, 1998; Maitra and Reynolds, 1998), but the nature of the interaction between 3α- and 3β-hydroxysteroids was not explored in any of these studies. In previous studies, 3β5βP marginally potentiated or had no effect on responses to GABA alone at GABA concentrations less than EC50, similar to our results (Puia et al., 1990;Kokate et al., 1994; Le Foll et al., 1997; Poisbeau et al., 1997;Garrett and Gan, 1998). These studies either did not explore the effect of 3β5βP on GABA concentrations higher than the EC50 or did not explore the effects of 3β5βP at concentrations near 10 μm, conditions under which we find that the direct inhibitory effects of 3β5βP become apparent. An exception is one study that observed significant inhibition of GABA responses at 100 μm GABA but not at 1 or 10 μm, similar to our results (Woodward et al., 1992).
Our results also suggest that 3β-hydroxypregnane (and 3β-hydroxyandrostane) steroids share similar actions with sulfated steroids, including the endogenous steroids pregnenolone sulfate and 3β5αPS. 3β5αPS, at an appropriate concentration, produces a very similar profile of GABAA receptor block as 3β-hydroxysteroids (Figs. 7, 10). A mutation that inhibits sulfated steroid block of GABAA receptors also inhibits direct block of receptors by 3β-hydroxysteroids and the interaction of 3β-hydroxysteroids with potentiators (Figs. 8, 9).
The direct effect of 3β-hydroxysteroids on GABAA receptors is compatible with previous suggestions regarding the structural requirements for steroid block of GABAA receptors. Several previous studies have shown that block and potentiation occur through different sites on the GABAA receptor. Our work with the α1 V256S mutation, which exhibits intact potentiation (present study) but interferes with block by sulfated steroids (Akk et al., 2001; present study), further confirms that block and potentiation are likely to be independent phenomena. Block is not sensitive to the stereochemistry of the sulfate at C3, and other charged groups can substitute for the sulfate group (Park-Chung et al., 1999; Mennerick et al., 2001). We also recently presented evidence that steroids with a carboxylate group attached to C24 (at the opposite end of the steroid nucleus from C3) are effective, albeit less potent, blockers of GABAA receptor function than steroids with a C3 sulfate group. Block by carboxylated steroids could be prevented by lowering the pH of the extracellular solution, suggesting that charge is important for block (Mennerick et al., 2001). However, the present work suggests that, for some steroids (i.e., 3β-hydroxysteroids), charge is not absolutely critical for blocking function. It has been similarly proposed that charge is not necessary for blocking action of dehydroepiandrosterone sulfate, because dehydroepiandrosterone also blocks GABAA receptors (Demirgoren et al., 1991;Le Foll et al., 1997).
We propose that sulfated and 3β-hydroxysteroids block GABAA receptors more effectively under conditions that promote agonist binding or channel opening. This conclusion apparently conflicts with some studies, which have suggested no use dependence to pregnenolone sulfate block (Zaman et al., 1992; Akk et al., 2001). A recent analysis of single-channel behavior in the presence of GABA and pregnenolone sulfate suggested no difference in the ability of pregnenolone sulfate to block liganded closed versus liganded open receptors (Akk et al., 2001). These results leave open the possibility that pregnenolone sulfate may prefer liganded over unliganded receptors, consistent with our proposal of some state dependence to pregnenolone sulfate actions. Other previous studies have noted that pregnenolone sulfate (Woodward et al., 1992) or carboxylated steroids (Mennerick et al., 2001) are more effective against GABA responses gated by high concentrations of GABA. Also, it has been proposed that pregnenolone sulfate may block receptors by promoting fast desensitization (Shen et al., 2000), a process correlated with channel opening. Part of the reconciliation of these disparate conclusions regarding the dependence of steroid block on receptor opening (use) may lie in the observation that sulfated steroid block is not a classical use-dependent block (Woodward et al., 1992). The inhibition does not involve open channel block, because single-channel studies have suggested no evidence of changes in channel open times with pregnenolone sulfate block (Mienville and Vicini, 1989; Akk et al., 2001). Several studies have noted recovery from block in the absence of agonist (Woodward et al., 1992; Shen et al., 2000) and little or no voltage dependence (Majewska et al., 1988; Akk et al., 2001), both inconsistent with a channel block mechanism. Additional work will be needed to clarify the mechanism by which steroids more effectively block larger macroscopic GABA currents.
Our findings suggest that baseline GABAergic transmission was only slightly affected by 10 μm 3β5βTHDOC, but this same concentration of steroid reversed the prolongation of IPSCs caused by potentiating steroids. These data are consistent with a use-dependent mechanism of block by 3β-hydroxysteroids and suggest that, during normal transmission, GABA concentration is not sufficiently high and/or receptors are not open sufficiently long for 10 μm3β5βTHDOC to block. Postsynaptic potentiation of receptor by 3α-hydroxysteroid or by barbiturate allows sufficient potentiation of channel opening to permit block by 3β-hydroxysteroid. Thus, under some conditions, 3β-hydroxypregnane steroids or sulfated steroids may be useful as functional antagonists of endogenous or exogenous postsynaptic potentiators, with the understanding that the mechanism is not one of competitive or direct antagonism.
Footnotes
This work was supported by National Institutes of Health Grants MH45493 (C.F.Z.) and GM47969 (D.F.C., J.H.S., and C.F.Z.), the Klingenstein Foundation, and National Institutes of Health Grants NS40488 and AA12952 (S.M.). M.W. was supported by a postdoctoral fellowship from Umeå University–Washington University Network and European Union regional funds. C.F. was supported by a Howard Hughes Medical Institute Undergraduate Fellowship. L.N.E. was supported by National Institutes of Health Training Grant T32-NS07205-20. We thank Bob Cormier for discussion.
Correspondence should be addressed to Dr. Steven Mennerick, Department of Psychiatry, Washington University School of Medicine, 660 South Euclid Avenue, Campus Box 8134, St. Louis, MO 63110. E-mail:menneris@psychiatry.wustl.edu.
M. Wang's present address: Department of Clinical Science, Section of Obstetrics and Gynecology, Umeå University, S-901 87 Umeå, Sweden.
REFERENCES
- 1.Akk G, Bracamontes J, Steinbach JH. Pregnenolone sulfate block of GABAA receptors: mechanism and involvement of a residue in the M2 region of the α subunit. J Physiol (Lond) 2001;532:673–684. doi: 10.1111/j.1469-7793.2001.0673e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barker JL, Harrison NL, Lange GD, Owen DG. Potentiation of gamma-aminobutyric-acid-activated chloride conductance by a steroid anaesthetic in cultured rat spinal neurones. J Physiol (Lond) 1987;386:485–501. doi: 10.1113/jphysiol.1987.sp016547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci USA. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Covey DF, Evers AS, Mennerick S, Zorumski CF, Purdy RH. Recent developments in structure-activity relationships for steroid modulators of GABAA receptors. Brain Res Brain Res Rev. 2001;37:91–97. doi: 10.1016/s0165-0173(01)00126-6. [DOI] [PubMed] [Google Scholar]
- 5.Demirgoren S, Majewska MD, Spivak CE, London ED. Receptor binding and electrophysiological effects of dehydroepiandrosterone sulfate, an antagonist of the GABAA receptor. Neuroscience. 1991;45:127–135. doi: 10.1016/0306-4522(91)90109-2. [DOI] [PubMed] [Google Scholar]
- 6.Garrett KM, Gan J. Enhancement of gamma-aminobutyric acidA receptor activity by alpha-chloralose. J Pharmacol Exp Ther. 1998;285:680–686. [PubMed] [Google Scholar]
- 7.Gasior M, Carter RB, Witkin JM. Neuroactive steroids: potential therapeutic use in neurological and psychiatric disorders. Trends Pharmacol Sci. 1999;20:107–112. doi: 10.1016/s0165-6147(99)01318-8. [DOI] [PubMed] [Google Scholar]
- 8.Han M, Zorumski CF, Covey DF. Neurosteroid analogues. IV. The effect of methyl substitution at the C-5 and C-10 positions of neurosteroids on electrophysiological activity at GABAA receptors. J Med Chem. 1996;39:4218–4232. doi: 10.1021/jm960304p. [DOI] [PubMed] [Google Scholar]
- 9.Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 10.Jones MV, Westbrook GL. Shaping of IPSCs by endogenous calcineurin activity. J Neurosci. 1997;17:7626–7633. doi: 10.1523/JNEUROSCI.17-20-07626.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kokate TG, Svensson BE, Rogawski MA. Anticonvulsant activity of neurosteroids: correlation with gamma-aminobutyric acid-evoked chloride current potentiation. J Pharmacol Exp Ther. 1994;270:1223–1229. [PubMed] [Google Scholar]
- 12.Lambert JJ, Peters JA, Harney S, Belelli D. Steroid modulation of GABAA receptors. In: Mohler H, editor. Handbook of experimental pharmacology. Springer; Berlin: 2001. pp. 117–140. [Google Scholar]
- 13.Le Foll F, Louiset E, Castel H, Vaudry H, Cazin L. Electrophysiological effects of various neuroactive steroids on the GABAA receptor in pituitary melanotrope cells. Eur J Pharmacol. 1997;331:303–311. doi: 10.1016/s0014-2999(97)01042-x. [DOI] [PubMed] [Google Scholar]
- 14.Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- 15.Maconochie DJ, Zempel JM, Steinbach JH. How quickly can GABAA receptors open? Neuron. 1994;12:61–71. doi: 10.1016/0896-6273(94)90152-x. [DOI] [PubMed] [Google Scholar]
- 16.Maitra R, Reynolds JN. Modulation of GABAA receptor function by neuroactive steroids: evidence for heterogeneity of steroid sensitivity of recombinant GABAA receptor isoforms. Can J Physiol Pharmacol. 1998;76:909–920. doi: 10.1139/cjpp-76-9-909. [DOI] [PubMed] [Google Scholar]
- 17.Majewska MD. Neurosteroids: endogenous bimodal modulators of the GABAA receptor. Mechanism of action and physiological significance. Prog Neurobiol. 1992;38:379–395. doi: 10.1016/0301-0082(92)90025-a. [DOI] [PubMed] [Google Scholar]
- 18.Majewska MD, Mienville JM, Vicini S. Neurosteroid pregnenolone sulfate antagonizes electrophysiological responses to GABA in neurons. Neurosci Lett. 1988;90:279–284. doi: 10.1016/0304-3940(88)90202-9. [DOI] [PubMed] [Google Scholar]
- 19.Mennerick S, Que J, Benz A, Zorumski CF. Passive and synaptic properties of neurons grown in microcultures and in mass cultures. J Neurophysiol. 1995;73:320–332. doi: 10.1152/jn.1995.73.1.320. [DOI] [PubMed] [Google Scholar]
- 20.Mennerick S, Zeng CM, Benz A, Shen W, Izumi Y, Evers AS, Covey DF, Zorumski CF. Effects on gamma-aminobutyric acid (GABA)A receptors of a neuroactive steroid that negatively modulates glutamate neurotransmission and augments GABA neurotransmission. Mol Pharmacol. 2001;60:732–741. [PubMed] [Google Scholar]
- 21.Mienville JM, Vicini S. Pregnenolone sulfate antagonizes GABAA receptor-mediated currents via a reduction of channel opening frequency. Brain Res. 1989;489:190–194. doi: 10.1016/0006-8993(89)90024-3. [DOI] [PubMed] [Google Scholar]
- 22.Park-Chung M, Wu FS, Purdy RH, Malayev AA, Gibbs TT, Farb DH. Distinct sites for inverse modulation of N-methyl-d-aspartate receptors by sulfated steroids. Mol Pharmacol. 1997;52:1113–1123. doi: 10.1124/mol.52.6.1113. [DOI] [PubMed] [Google Scholar]
- 23.Park-Chung M, Malayev A, Purdy RH, Gibbs TT, Farb DH. Sulfated and unsulfated steroids modulate gamma-aminobutyric acidA receptor function through distinct sites. Brain Res. 1999;830:72–87. doi: 10.1016/s0006-8993(99)01381-5. [DOI] [PubMed] [Google Scholar]
- 24.Pignataro L, Fiszer de Plazas S. Epipregnanolone acts as a partial agonist on a common neurosteroid modulatory site of the GABAA receptor complex in avian CNS. Neurochem Res. 1997;22:221–225. doi: 10.1023/a:1027327910138. [DOI] [PubMed] [Google Scholar]
- 25.Poisbeau P, Feltz P, Schlichter R. Modulation of GABAA receptor-mediated IPSCs by neuroactive steroids in a rat hypothalamo-hypophyseal coculture model. J Physiol (Lond) 1997;500:475–485. doi: 10.1113/jphysiol.1997.sp022034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prince RJ, Simmonds MA. 5 beta-pregnan-3 beta-ol-20-one, a specific antagonist at the neurosteroid site of the GABAA receptor-complex. Neurosci Lett. 1992;135:273–275. doi: 10.1016/0304-3940(92)90454-f. [DOI] [PubMed] [Google Scholar]
- 27.Prince RJ, Simmonds MA. Differential antagonism by epipregnanolone of alphaxalone and pregnanolone potentiation of [3H]flunitrazepam binding suggests more than one class of binding site for steroids at GABAA receptors. Neuropharmacology. 1993;32:59–63. doi: 10.1016/0028-3908(93)90130-u. [DOI] [PubMed] [Google Scholar]
- 28.Puia G, Santi MR, Vicini S, Pritchett DB, Purdy RH, Paul SM, Seeburg PH, Costa E. Neurosteroids act on recombinant human GABAA receptors. Neuron. 1990;4:759–765. doi: 10.1016/0896-6273(90)90202-q. [DOI] [PubMed] [Google Scholar]
- 29.Robel P, Baulieu E-E. Neurosteroids:biosynthesis and function. In: de Kloet R, Sutanto W, editors. Neurobiology of steroids: methods in the neurosciences 22. Academic; San Diego: 1994. pp. 36–50. [Google Scholar]
- 30.Shen W, Mennerick S, Covey DF, Zorumski CF. Pregnenolone sulfate modulates inhibitory synaptic transmission by enhancing GABAA receptor desensitization. J Neurosci. 2000;20:3571–3579. doi: 10.1523/JNEUROSCI.20-10-03571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turner DM, Ransom RW, Yang JS, Olsen RW. Steroid anesthetics and naturally occurring analogs modulate the gamma-aminobutyric acid receptor complex at a site distinct from barbiturates. J Pharmacol Exp Ther. 1989;248:960–966. [PubMed] [Google Scholar]
- 32.Wang MD, Backstrom T, Landgren S. The inhibitory effects of allopregnanolone and pregnanolone on the population spike, evoked in the rat hippocampal CA1 stratum pyramidale in vitro, can be blocked selectively by epiallopregnanolone. Acta Physiol Scand. 2000;169:333–341. doi: 10.1046/j.1365-201x.2000.00744.x. [DOI] [PubMed] [Google Scholar]
- 33.Woodward RM, Polenzani L, Miledi R. Effects of steroids on gamma-aminobutyric acid receptors expressed in Xenopus oocytes by poly(A)+ RNA from mammalian brain and retina. Mol Pharmacol. 1992;41:89–103. [PubMed] [Google Scholar]
- 34.Zaman SH, Shingai R, Harvey RJ, Darlison MG, Barnard EA. Effects of subunit types of the recombinant GABAA receptor on the response to a neurosteroid. Eur J Pharmacol. 1992;225:321–330. doi: 10.1016/0922-4106(92)90106-6. [DOI] [PubMed] [Google Scholar]
- 35.Zeng CM, Benz A, Howell M, Manion B, Evers AS, Zorumski CF, Mennerick S, Covey DF. Effect of C-6 and C-7 methyl substitution on modulatory action of neuroactive steroids at GABAA receptors. Chem Abstr. 2001;2001:640035. [Google Scholar]
- 36.Zorumski CF, Mennerick SJ, Covey DF. Enantioselective modulation of GABAergic synaptic transmission by steroids and benz[e]indenes in hippocampal microcultures. Synapse. 1998;29:162–171. doi: 10.1002/(SICI)1098-2396(199806)29:2<162::AID-SYN7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]