

Abstract
Several signal transduction pathways have been implicated in the induction of long-term potentiation (LTP), yet the signal transduction mechanisms behind the maintenance–expression phase of LTP are still poorly understood. We investigated the role of phosphatidylinositol 3-kinase (PI3-kinase) in LTP at Schaffer collateral/commissural fiber–CA1 synapses in rat hippocampal slices using biochemical approaches and extracellular electrophysiological recordings. We observed that PI3-kinase activity was induced in the CA1 region during LTP of field EPSPs (fEPSPs) and that two structurally unrelated PI3-kinase inhibitors, LY294002 and wortmannin, abated established LTP, suggesting that PI3-kinase is involved in the maintenance–expression phase of LTP. However, LTP recovered after washout of the reversible PI3-kinase inhibitor LY294002, confirming that LTP maintenance and expression are distinct events and indicating that PI3-kinase activity is required for LTP expression rather than for its maintenance. Interestingly, preincubation with LY294002 did not prevent LTP induction. In fact, if LY294002 was withdrawn 5 min after high-frequency stimulation, an LTP of fEPSP was seen. Last, a voltage-dependent calcium channel-dependent form of LTP in the CA1 could also be reversibly abated by LY294002, raising the possibility that PI3-kinase could be required for the expression of multiple forms of synaptic potentiation.
Keywords: long-term potentiation, synaptic plasticity, hippocampus, PI3-kinase, signal transduction, NMDA, voltage-dependent calcium channels, AMPA
Activity-dependent synaptic changes are believed to be crucial in learning and memory. Long-term potentiation (LTP) of EPSPs is a long-lasting increase of synaptic strength that can be induced with tetanic stimulation of afferent fibers (Malenka and Nicoll, 1999). At Schaffer collateral/commissural fiber–CA1 synapses, LTP is characterized by a persistent enhancement of the responses of AMPA-sensitive glutamatergic receptors (Malenka and Nicoll, 1999; Malinow et al., 2000). Such an enhancement is currently believed to be primarily attributable to increased postsynaptic density of AMPA receptors (Malenka and Nicoll, 1999;Luscher et al., 2000; Malinow et al., 2000). Induction of LTP is triggered by an initial elevation of cytosolic calcium to which both NMDA receptors and L-type voltage-dependent calcium channels (VDCC) can contribute (Teyler et al., 1994; Malenka and Nicoll, 1999). Multiple signal transduction pathways are then recruited to translate the Ca2+ signal into increased synaptic strength, including α-calcium/calmodulin-dependent protein kinase II (CaMKII), which is believed to play a pivotal role (Malenka and Nicoll, 1999). However, although prevailing views implicate CaMKII also in the maintenance–expression of LTP, specific inhibitors of CaMKII do not abate LTP when applied after its induction (Malinow et al., 1989; Ito et al., 1991; Bortolotto and Collingridge, 1998; Chen et al., 2001) nor do inhibitors of other kinases implicated in LTP, such as mitogen-activated protein kinase (MAPK) and Src (English and Sweatt, 1997; Salter, 1998). These observations suggest that distinct signal transduction events could be involved in the induction and in the maintenance–expression phases of LTP. A distinction has also been proposed between the maintenance and the expression of LTP (Malinow et al., 1988). In fact, it has been observed that the wide-spectrum kinase inhibitor H-7 can abate established LTP in a reversible manner, suggesting that the molecular mechanisms underlying the maintenance of LTP are not affected by this inhibitor, despite the inhibition of LTP expression (Malinow et al., 1988). A late phase of LTP has also been identified that is protein kinase A (PKA) dependent and requiresde novo gene expression (Frey et al., 1993; Huang, 1998).
In the present study, we investigated the role of phosphatidylinositol 3-kinase (PI3-kinase) (Whitman et al., 1988; Wymann and Pirola, 1998;Leevers et al., 1999) in the early phase of LTP at Schaffer collateral/commissural fiber–CA1 synapses using biochemical and electrophysiological approaches. PI3-kinase phosphorylates the D-3 position of the inositol ring of phosphoinositides (PtdIns) (Whitman et al., 1988), which are also the precursors for the second messengers phosphoinositols and diacylglycerol (Clapham, 1995). Current views suggest that D-3-phosphorylated PtdIns act as membrane-embedded second messengers in the regulation of a broad array of biological functions (Whitman et al., 1988; Wymann and Pirola, 1998; Leevers et al., 1999). Among PI3-kinase downstream effectors are phosphoinositide-dependent kinases 1 (PDK1) and the members of the AGC subfamily of protein kinases Akt (also known as protein kinase B), p70S6k, and atypical protein kinase C isozymes, such as PKCζ (Whitman et al., 1988; Wymann and Pirola, 1998; Leevers et al., 1999). PDK1 is involved in the activation of other PI3-kinase targets, including Akt and p70S6k, which are believed to mediate most of the PI3-kinase effects (Franke et al., 1997; Chou et al., 1998; Chan et al., 1999; Romanelli et al., 1999).
MATERIALS AND METHODS
Hippocampal slice preparations and electrophysiological techniques. Hippocampal slices were prepared as described previously (Sanna et al., 2000). Briefly, we killed Wistar rats (28–35 d of age) by decapitation under halothane (3% in air) anesthesia. The brains were then rapidly removed and transferred into ice-cold artificial CSF (ACSF) [in mm: 130 NaCl, 3.5 KCl, 24 NaHCO3, 1.25 NaH2PO4, 2.2 CaCl2, 10 glucose, and 2 MgSO4, pH 7.4 (oxygenated by bubbling a mixture of 95% O2–5%CO2)] and sliced with a Leica (Wetzlar, Germany) VT1000E automatic vibratome slicer to obtain transverse hippocampal slices (400 μm). Hippocampal slices were collected in oxygenated ACSF and preincubated for at least 1 hr at room temperature. For recording, slices were transferred to a submerged recording chamber, perfused with oxygenated ACSF, and maintained at 31 ± 1°C. They were further incubated for at least 40 min before the recording session. The flow was maintained at 1.2 ml/min in a chamber volume of 1.5 ml. Drug applications were performed by changing the chamber infusion to ACSF containing the agent. Microelectrodes, pulled from 1.5-mm-outer diameter glass tubing with the use of a Flaming/Brown micropipette puller (Sutter Instruments, Novato, CA), were filled with ACSF (resistance of 3–5 MΩ). Bipolar stimulating electrodes were placed in the stratum radiatum to activate the Schaffer collateral/commissural fibers (see below, LTP paradigms). Recordings of field EPSPs (fEPSPs) were made in the middle of the stratum radiatum with an Axoclamp 2B (Axon Instruments, Foster City, CA). Test intensities were set to obtain fEPSP slopes of ∼50% of those at which population spikes were detectable based on input–output curves obtained in each slice. An average of five stimulus-evoked responses was collected every 3 min. As an indicator of synaptic efficacy, we measured the initial slope of the rising phase of the fEPSP because it is not affected by population spikes or altered by EPSP–spike (E–S) potentiation (Abraham et al., 1987). The initial slope of fEPSP was measured near its onset for an interval of 1.2 msec. The acquisition and analysis were performed with the LABVIEW software package (National Instruments, Austin, TX). Intracellular recordings were made using glass micropipettes filled with 2 m K-acetate, pH 7.3 (80–120 MΩ). Cells with stable resting membrane potentials (r.m.p.) during the recording section were selected (r.m.p., −67.4 ± 0.75 mV; input resistance, 36 ± 5.1 MΩ; n = 5). Stimuli of 0.08 msec duration and 0.9 ± 0.05 mA intensity were applied once every 20 sec to the Schaffer collateral/commissural fibers to evoke EPSPs NMDA. Before each synaptic activation, the input resistance was measured injecting into the cell a pulse of current (200 msec, −0.2 nA). The bridge balance and r.m.p. were carefully adjusted as needed. All of the data presented were obtained in slices from multiple animals
LTP paradigms. fEPSPs were recorded for at least 20 min before high-frequency stimulation (HFS) to ensure stability of excitability. fEPSP slopes were normalized as the percentage of mean fEPSP over such period of time. fEPSPs at the time point preceding HFS were used as the basal fEPSP level. A two-pathway paradigm was used in which two stimulating electrodes were placed in the stratum radiatum on opposite sides of the recording electrode to stimulate two separate groups of Schaffer collateral/commissural fibers. The pathway stimulated by the electrode on the medial side of the stratum radiatum was designated as S1, and the pathway stimulated by the electrode on the lateral (fimbrial) side was called S2. The independence of the two pathways was demonstrated by the absence of paired-pulse facilitation of fEPSP when two sets of orthodromic stimuli (0.08 msec duration) were delivered to the two pathways at 40 msec interval (in either order, S1–S2 or S2–S1); however, paired-pulse facilitation of fEPSP was observed when the stimuli were applied to the same electrode. Independence of the two pathways was also demonstrated by the lack of potentiation in the untetanized pathway (S1) after induction of LTP to the tetanized one (S2). Test stimuli were delivered at one per 15 sec. For LTP induction, two trains of 500 msec duration each at 100 Hz with an interval of 10 sec at the test intensity were delivered to one of the pathways. Results obtained with the two-pathway paradigm were confirmed in a single pathway paradigm and by inverting the order of stimulation of the two pathways. Some experiments (see Fig. 2) were performed in the single-pathway paradigm in the same manner. An L-type VDCC-dependent LTP was obtained in the CA1 with 10 trains of 200 Hz, each 200 msec long, delivered at 5 sec intervals in the presence ofd-AP-5 (50 μm) and bicuculline (10 μm), as described by Grover and Yan (1999). LY294002, wortmannin, nifedipine, andd-AP-5 were obtained from Calbiochem (San Diego, CA). We operationally define as interfering with LTP induction treatments that can prevent LTP only when applied during tetanic stimulation but that do not abate established LTP. Conversely, we define as interfering with LTP expression treatments capable of reversibly abating established LTP after tetanic stimulation, whereas only a treatment with a reversible mechanism of action that would permanently abate LTP could be seen as an inhibitor of LTP maintenance.
Fig. 2.
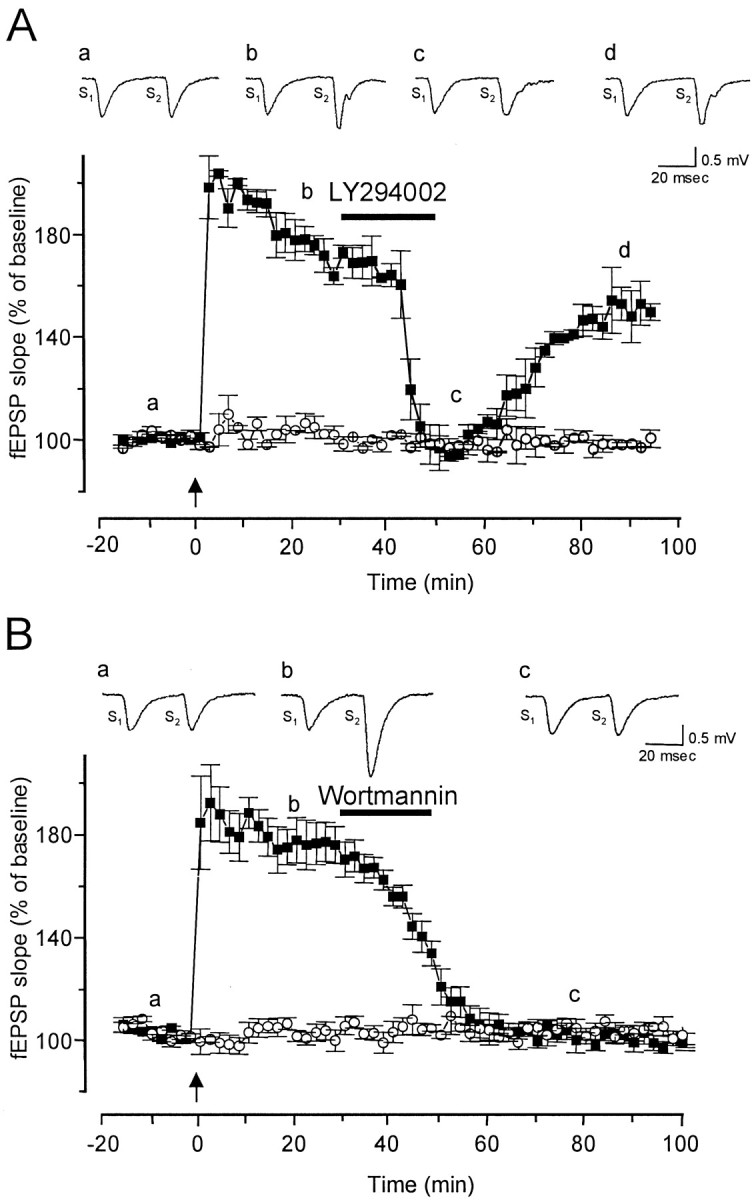
Inhibitors of PI3-kinase abate established LTP of fEPSP in the CA1 region. Synaptic potentials were simultaneously monitored in two independent pathways [white circles, stimulus 1 (S1); black squares, stimulus 2 (S2)]. The structurally unrelated inhibitors of PI3-kinase LY294002 (100 μm) and wortmannin (5 μm) were applied 30 min after delivery of HFS to one of the two pathways (S2).Insets are representative traces of extracellular fEPSPs recorded at the times marked by lowercase letters. Each representative trace is an average of five responses. Graphs represent the mean normalized fEPSP slopes plotted against time.Arrows indicate when tetanic stimulation to one pathway (black squares) was given at time 0. A, A transient 20 min application of LY294002 (100 μm) 30 min after LTP induction abated LTP in the potentiated pathway (n = 7) (black squares), but no change was seen in the untetanized pathway (white circles). B, Similar results were obtained with wortmannin, a structurally unrelated PI3-kinase inhibitor (5 μm) (n = 5). As expected, inhibition by wortmannin was irreversible.
Kinase assay and Western blotting. The PI3-kinase assay was performed as described previously (Macara et al., 1984; Alessi et al., 1996), with minor modifications. We obtained tissue extract by sonication of CA1 hippocampal regions in 50 μl of lysis buffer containing 10 mm Tris-HCl, pH 7.6, 50 mm NaCl, 30 mmNa4P2O7, 1% NP-40, and protease (10 μg/ml leupeptin, 2.5 μl/ml aprotinin, and 1 mm PMSF) and phosphatase inhibitors (20 mm NaF and 1 mm activated Na3VO4). Homogenized samples were clarified by centrifugation for 10 min at 15,000 rpm at 4°C. PI3-kinase activity was analyzed in immunoprecipitates obtained with 5 μl of an anti-phosphotyrosine antibody (Sigma, St. Louis, MO) performed at 4°C with gentle shaking, using an agarose-conjugated goat anti-mouse antiserum (Sigma). The PI3-kinase reaction mixture contained 20 mm HEPES, 10 mm MgCl2, 100 μmNa3VO4, 40 μm cold ATP, 3.5 μl of [γ-32P]ATP (1 mCi, 3000 Ci/mmol; NEN, Boston, MA), and 0.3 μg of PI (Avanti, Alabaster, AL) from a stock prepared in 50 mm HEPES, 1 mm EDTA, pH 7.0, and 0.5% w/v cholic acid, after drying under a stream of N2. After incubation at room temperature for 20 min, reactions were stopped with 114 μl of 2.4 m HCl in 50% MeOH. Phospholipids were then extracted in 50 μl of chloroform separated by thin-layer chromatography (TLC) on plates (Whatman, Clifton, NJ) prerun for 3 hr with 1.2% potassium oxalate (C2O4K2) w/v in 50% MeOH in H2O. TLC running buffer consisted of H20/MeOH/glacial acetic acid/acetone/chloroform (7:13:12:15:40). After running for 1 hr, TLC plates were dried and exposed on Kodak Biomax MS film (Eastman Kodak, Rochester, NY). We performed Western blots as described previously (Sanna et al., 2000) and exposed them on Kodak Biomax MS film. Autoradiograms were scanned and the signal quantified with the NIH Image 1.61 software package. Statistical analyses were performed by ANOVA with Statview software (Abacus Concepts, Calabasas, CA). Antibodies to phosphorylated Akt (Thr308) and phosphorylated p70S6K (Thr389) were obtained from Cell Signaling Technology (Beverly, MA). These residues are phosphorylated in a PI3-kinase-dependent manner and are predictors of the activation of the two kinases (Alessi et al., 1996; Balendran et al., 1999). Antibodies to total Akt (Santa Cruz Biotechnology, Santa Cruz, CA) and total p70S6K (Cell Signaling Technology) served as controls.
RESULTS
PI3-kinase and its downstream effectors are persistently activated during LTP
We induced LTP of fEPSP at Schaffer collateral/commissural fiber–CA1 synapses in hippocampal slices with an HFS paradigm consisting of two trains of electrical stimuli of 100 Hz with a duration of 500 msec each at an interval of 10 sec. The CA1 region was rapidly dissected and immediately frozen for biochemical analyses at different time points before and after delivery of LTP-inducing HFS. All slices used for biochemistry were monitored electrophysiologically. By use of a PI3-kinase assay, we observed that the activity of PI3-kinase in CA1 regions was significantly (p< 0.05) elevated in tetanized slices during both the post-tetanic potentiation (PTP) and LTP, 30 min after HFS (Fig.1A). Consistently, phosphorylation of the PI3-kinase downstream effectors Akt and p70S6K was also significantly (p < 0.05) induced during PTP and LTP (Fig.1B–D). Inhibition of PI3-kinase with the specific inhibitor of PI3-kinase LY294002 (100 μm) (Vlahos et al., 1994) prevented increased phosphorylation of Akt and p70S6K after HFS (Fig.1E,F).
Fig. 1.

PI3-kinase and its downstream effectors Akt and p70S6K are activated in LTP. A, PI3-kinase activity was increased after LTP-inducing HFS. PI3-kinase activity was tested in CA1 protein extracts using phosphatidylinositol as a substrate in the presence of [γ-32P]ATP. Accumulation of radioactive phosphatidylinositol-3P (PIP) was increased shortly after HFS (PTP) and 30 min after HFS (LTP) when compared with untetanized control slices (C). B–D, Phosphorylation of PI3-kinase downstream effectors Akt and p70S6K was also significantly increased (p < 0.05 for both), as revealed by Western blotting with specific phosphorylation state-specific antibodies. C, D, Mean phosphorylation of Akt and p70S6K, respectively; error bars indicate SEM. *p < 0.05 indicates different from control. E, Incubation of hippocampal slices with the PI3-kinase-specific inhibitor LY294002 (100 μm) prevented increased phosphorylation of Akt and p70S6K 30 min after HFS; representative blots.F, Cumulative results from these experiments. *p < 0.05 indicates different from basal; ‡p < 0.05 indicates different from potentiated slices.
PI3-kinase is required for LTP expression but not for the maintenance or the induction of LTP
LTP of fEPSP was induced at Schaffer collateral/commissural fiber–CA1 synapses in hippocampal slices with the HFS paradigm described above. A 20 min bath application of the PI3-kinase inhibitor LY294002 (100 μm) 30 min after delivery of HFS abated established LTP of fEPSP in the tetanized pathway without affecting fEPSPs in the untetanized one of a two-pathway paradigm (Fig.2A). In these slices (n = 7), mean ± SE normalized fEPSP slopes of the tetanized pathway were 171.3 ± 2.6% of baseline before application of LY294002. During application of the drug, fEPSP slope of the tetanized pathway decreased to baseline levels (94.5 ± 1.0%). However, fEPSPs recovered to a potentiated level (154.9 ± 1.0%) ∼40 min after LY294002 washout (Fig. 2A). This result suggests that PI3-kinase activity is required for the expression rather than the maintenance of LTP. The concentration of LY294002 used here is in the high range of doses used in tissue culture (Vogelbaum et al., 1998; Akasaki et al., 1999; Cox et al., 1999; Sajan et al., 1999) in which drug-effective doses are usually lower than in slice preparations. When tested on a battery of purified ATP-requiring enzymes, including PKC, PKA, MAPK, Src, epidermal growth factor receptor, and PI4-kinase, LY294002 had no effect on the activity of any of them at a concentration of 50 μm, whereas its IC50 on purified PI3-kinase was 1.4 μm (Vlahos et al., 1994). At 100 μm, LY294002 did not cause cellular toxicity as measured by MTT oxidation (Vlahos et al., 1994).
To validate the specificity of the effect of LY294002 on established LTP, we tested the structurally unrelated PI3-kinase inhibitor wortmannin (Powis et al., 1994; Norman et al., 1996). Like LY294002, wortmannin (5 μm) could abate established LTP in the tetanized pathway when applied 30 min after tetanization (Fig.2B). In these slices, as seen with LY294002, a 20 min application of wortmannin reversed established LTP (n = 5) to baseline levels (fEPSP slope, 98.8 ± 3.0%). Treatment with wortmannin did not affect fEPSP slopes in the untetanized pathway (Fig.2B). However, whereas LY294002 is a reversible inhibitor of PI3-kinase (Vlahos et al., 1994), wortmannin is an irreversible one (Powis et al., 1994; Norman et al., 1996). Predictably, therefore, LTP expression did not recover after wortmannin withdrawal (Fig. 2B).
In hippocampal slices continuously incubated in LY294002 (100 μm), no LTP of fEPSP was observed after delivery of HFS (Fig. 3A). However, if LY294002 was withdrawn 5 min after HFS, an LTP of fEPSP was seen (Fig.3B). In fact, in these slices (n = 6), mean ± SE normalized fEPSP slope was 157.1 ± 5.1% at 60 min after HFS (Fig. 3B). This result indicates that PI3-kinase activity is not required for LTP induction. As expected, pretreatment with wortmannin prevented LTP induction in the CA1, and no LTP was seen during washout (data not shown). PTP was not significantly affected by bath application of LY294002 (100 μm), nor was paired-pulse facilitation, which, like PTP, is a presynaptic form of short-term plasticity (Kamiya and Zucker, 1994; Fisher et al., 1997). Intracellular recording from CA1 pyramidal neurons (n = 5) also demonstrated that a 20 min application of LY294002 (100 μm) did not modify the amplitudes of NMDA glutamate receptor-mediated EPSPs pharmacologically isolated with the AMPA receptor antagonist CNQX (10 μm), the GABAA receptor antagonist bicuculline (30 μm), and the GABAB receptor antagonist CGP55845A (1 μm) (Fig. 3C). Resting membrane potentials and input resistance were also unaffected by bath application of LY294002 (100 μm) (n = 5) (data not shown).
Fig. 3.

PI3-kinase is not required for LTP induction.A, Hippocampal slices continuously incubated in LY294002 (100 μm) did not display LTP of fEPSP after delivery of HFS (n = 6). B, If LY294002 was withdrawn 5 min after HFS, an LTP of fEPSP was seen (n = 6). C, Pharmacologically isolated NMDA receptor-mediated EPSP amplitudes were not modified by a 20 min application of LY294002 (100 μm) (102.2 ± 2.5%) as revealed by intracellular recordings from CA1 pyramidal neurons (n = 5).
PI3-kinase is required for the expression of VDCC-dependent LTP
Activation of NMDA receptors is the primary trigger for LTP with the HFS protocol used above. In fact, although induction of LTP in this manner is prevented by the NMDA receptor antagonist d-AP-5 (50 μm), it is not prevented by the L-type VDCC blocker nifedipine (30 μm) (Fig.4). Even when such a tetanization protocol was delivered in the presence of nifedipine (30 μm), inhibition of PI3-kinase with LY294002 (100 μm) abated established LTP of fEPSP without affecting the untetanized pathway (n = 6) (Fig. 4). As in the experiments described above, the effect of LY294002 was reversible, and potentiation recovered during washout (Fig. 4). However, with a stronger tetanization protocol (see Materials and Methods) (Teyler et al., 1994; Grover and Yan, 1999), an L-type VDCC-dependent form of LTP can also be induced at Schaffer collateral/commissural fibers–CA1 synapses in the presence of the NMDA blockerd-AP-5 (50 μm) (Teyler et al., 1994; Grover and Yan, 1999). The dependence of this LTP on VDCC is demonstrated by its sensitivity to nifedipine (30 μm) (data not shown) (Teyler et al., 1994;Grover and Yan, 1999). Thus, we investigated whether inhibition of PI3-kinase could also abate such VDCC-dependent LTP. We observed that, when applied 30 min after induction of VDCC-dependent LTP, LY294002 (100 μm) abated established potentiation of fEPSP in the tetanized pathway without affecting fEPSP slopes in the untetanized one (Fig. 4). In these slices (n = 6), mean ± SE normalized fEPSP slopes of the tetanized pathway were 160.7 ± 0.5% of baseline before application of LY294002 (100 μm). A 20 min bath application of LY294002 30 min after delivery of HFS abated established LTP of fEPSP to baseline levels (109.5 ± 1.2%). The fEPSPs recovered to a potentiated level (138.5 ± 0.7%) 40–50 min after LY294002 washout (Fig. 4). These results suggest that PI3-kinase is required for the expression of both a strictly NMDA-dependent and a VDCC-dependent form of LTP at Schaffer collateral/commissural fiber–CA1 synapses.
Fig. 4.

PI3-kinase is required for the expression of both NMDA- and VDCC-dependent forms of CA1 LTP.A, L-type VDCC-dependent form of LTP of fEPSP was induced at Schaffer collateral/commissural fiber–CA1 synapses in the presence of d-AP-5 (50 μm). A 20 min application of LY294002 (100 μm) 30 min after the induction of VDCC-dependent LTP abated LTP in the tetanized pathway (n = 6) (black squares), but no change was seen in the fEPSP in the untetanized pathway (white circles). B, Inhibition of PI3-kinase with LY294002 (100 μm) also abated NMDA-dependent LTP induced in the presence of nifedipine (30 μm).
DISCUSSION
We observed that PI3-kinase activity was induced during LTP of fEPSP at Schaffer collateral/commissural fiber–CA1 synapses. Two structurally unrelated PI3-kinase inhibitors, LY294002 and wortmannin, abated established LTP when applied after delivery of HFS, suggesting that PI3-kinase is involved in the maintenance–expression phase of LTP. After withdrawal of LY294002, a reversible inhibitor of PI3-kinase, fEPSPs returned to a potentiated level. Additionally, PI3-kinase activity was dispensable for LTP induction, because delivery of HFS during perfusion with LY294002 did not prevent LTP expression after washout of this inhibitor, suggesting that different signal transduction mechanisms are involved in the induction and in the expression–maintenance phases of LTP. Last, a VDCC-dependent form of LTP was also reversibly abated by LY294002.
Several signal transduction pathways have been implicated in the induction of LTP in the CA1 region, including CaMKII, Src, PKC, and MAPK (Salter, 1998; Malenka and Nicoll, 1999; Winder and Sweatt, 2001). The cAMP–PKA– cAMP response element-binding protein pathway has been shown to be required for the late phase of LTP in the CA1 region (Frey et al., 1993; Bourtchuladze et al., 1994), although a role in early events can also be demonstrated in certain paradigms (Blitzer et al., 1995). However, the signal transduction events behind the maintenance–expression phase of LTP are still poorly understood (Malenka and Nicoll, 1999). In fact, although established LTP can be abated by a broad-spectrum Ser/Thr kinase inhibitor at high doses (Malinow et al., 1988), specific inhibitors of CaMKII, Src, PKC, or the ERK–MAPK pathway, although they can prevent LTP induction when applied during HFS, do not affect established LTP (Malinow et al., 1989; Ito et al., 1991; English and Sweatt, 1997; Bortolotto and Collingridge, 1998;Salter, 1998; Chen et al., 2001). Established LTP was found to be selectively abated by two inhibitors of PI3-kinase, LY294002 and wortmannin. The reversibility of the action of LY294002 on established LTP confirms that LTP maintenance and expression are distinct events, as proposed by Malinow et al. (1988), and indicates that PI3-kinase is required for the expression rather than the maintenance of LTP (Fig.5). PI3-kinase activity was also dispensable for LTP induction. In fact, if HFS was delivered during incubation of hippocampal slices with LY294002, an LTP of fEPSP was seen during withdrawal of the drug. A necessary role for PI3-kinase in the maintenance–expression phase of LTP is corroborated by the fact that wortmannin, a PI3-kinase inhibitor structurally unrelated to LY294002, could also selectively abate established LTP. However, unlike LY294002, wortmannin blocks PI3-kinase irreversibly by covalent modification of the kinase, and, thus, PI3-kinase activity cannot recover without new protein synthesis (Powis et al., 1994; Norman et al., 1996). Therefore, wortmannin did not allow us to differentiate between LTP maintenance and expression because LTP did not recover after withdrawal of the inhibitor. Additionally, when we pretreated hippocampal slices with wortmannin, LTP in the CA1 was prevented, and the irreversible action of the drug did not allow it to recover during washout. Consistently, LTP was not seen in the dentate gyrus in animals pretreated with wortmannin (Kelly and Lynch, 2000). Although the term “expression” was used in that report (Kelly and Lynch, 2000), pretreatment with an irreversible inhibitor like wortmannin does not allow one to distinguish between inhibition of LTP induction and of the maintenance–expression phase of LTP. Recently, preincubation with inhibitors of PI3-kinase was also shown to prevent LTP of fEPSP in the basolateral amygdala (Lin et al., 2001).
Fig. 5.
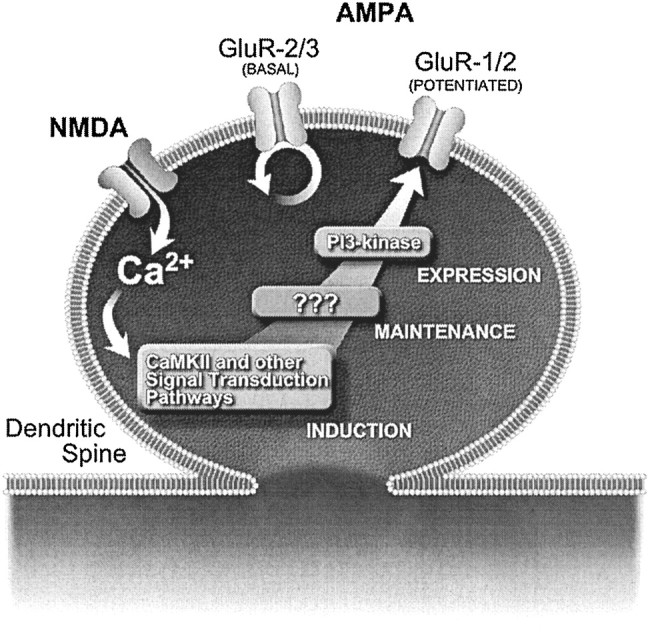
A model for the role of PI3-kinase in LTP. AMPA receptors composed of subunits 2 and 3 (GluR2/3) are believed to be responsible for basal AMPA responses. They are present at variable levels at postsynaptic sites in which they constitutively cycle between intracellular compartments and presynaptic membranes (Passafaro et al., 2001; Shi et al., 2001). Addition of GluR1 AMPA receptors in complex with GluR2 (GluR1/2) to postsynaptic membranes is believed to be the basis for LTP (Shi et al., 2001). We showed here that PI3-kinase is required for LTP but not for basal AMPA transmission. This observation is consistent with a role for PI3-kinase in the insertion of AMPA receptors during LTP but not in regulating basal AMPA receptor density. Consistent with this view, it has been shown recently that NMDA-dependent exocytosis of GluR1-containing AMPA receptor in primary hippocampal neurons is PI3-kinase dependent, unlike the exocytosis of GluR2 subunits not associated with GluR1 (Passafaro et al., 2001). Interestingly, delivery of HFS during perfusion with the reversible PI3-kinase inhibitor LY294002 did not prevent expression of LTP after washout of this inhibitor, and addition of LY294002 to established LTP caused a reversible inhibition of LTP. The former observation suggests that PI3-kinase is not responsible for the induction of LTP. The latter observation confirms that LTP maintenance and expression are distinct events (Malinow et al., 1988) and indicates that PI3-kinase activity is required for LTP expression rather than for its maintenance.
We then investigated whether the expression of LTP induced with a different triggering mechanism at Schaffer collateral/commissural fiber–CA1 synapses would also display the same PI3-kinase dependence. As mentioned above, we observed that activation of PI3-kinase with our standard HFS paradigm was dependent on NMDA receptors rather than VDCC. Consistently, we observed that a strictly NMDA-dependent LTP induced in the presence of nifedipine was reversibly abated by the PI3-kinase inhibitor LY294002. We then investigated whether LY294002 could also reversibly abate a form of VDCC-dependent LTP at Schaffer collateral/commissural fiber–CA1 synapses. In fact, activation of VDCC can induce an LTP that, perhaps because of the extrasynaptic localization of VDCC, appears to recruit a different set of signal transduction pathways than those required for NMDA-dependent LTP (Cavus and Teyler, 1996). However, we observed that VDCC-dependent LTP induced by strong tetanization in the presence of d-AP-5 was reversibly abated by LY294002, like NMDA-dependent LTP. This result raises the possibility that PI3-kinase could be a final common pathway for the expression of multiple forms of synaptic potentiation.
It is believed currently that central to synaptic plasticity are dynamic modifications of dendritic spines leading to changes in the density of AMPA receptors on postsynaptic membranes (Malenka and Nicoll, 1999; Luscher et al., 2000; Malinow et al., 2000) (but seeGrosshans et al., 2002). Cytoskeletal elements such as actin have been implicated in regulating spine morphology (Halpain, 2000; Matus, 2000) and AMPA redistribution (Allison et al., 1998; Shen et al., 2000). Mounting evidence supports that PI3-kinase and its effectors are part of the actin polymerizing system in several cell types, including neurons (Wymann and Arcaro, 1994; Higaki et al., 1996; Burnett et al., 1998). PI3-kinase has also been shown to be involved in multiple aspects of membrane trafficking in different cell types. Among them is the surface translocation of glucose transporters (Corvera and Czech, 1998), Na+/H+exchanger (Kurashima et al., 1998; Janecki et al., 2000; Yudowski et al., 2000), and transferrin receptor (Shepherd et al., 1995), all of which cycle between the plasmalemma and intracellular membrane compartments. Thus, PI3-kinase could play a role in the cytoskeletal and signaling events responsible for regulating AMPA receptor density at postsynaptic sites. AMPA receptors of different subunit composition appear to be responsible for basal AMPA responses and for the increased AMPA-mediated transmission seen in LTP (Shi et al., 1999; Luscher et al., 2000; Malinow et al., 2000; Shi et al., 2001). AMPA receptors in the hippocampus are mostly hetero-oligomers composed of GluR1/GluR2 or GluR2/GluR3 subunits (Wenthold et al., 1996). GluR2/3 AMPA receptors are present at variable levels at postsynaptic sites, and they constitutively cycle between intracellular compartments and presynaptic membranes through the interaction of GluR2 withN-ethylmaleimide-sensitive factor and group II PDZ (postsynaptic density-95/Discs large/zona occludens-1) domain proteins (Sheng and Pak, 2000; Passafaro et al., 2001; Shi et al., 2001). AMPA receptors containing subunits 1 and 2 (GluR1/2) are delivered to postsynaptic sites after LTP induction through the interaction of GluR1 and group I PDZ domain proteins (Shi et al., 2001). Some synapses are believed to lack AMPA receptors, the so-called “silent synapses,” and to be converted to functional synapses during LTP (Malenka and Nicoll, 1999; Luscher et al., 2000). We observed that PI3-kinase inhibitors can abate LTP but do not affect basal AMPA transmission. This suggests a requirement of PI3-kinase for the insertion and/or continued surface expression of additional AMPA receptors during LTP but not for regulating basal AMPA receptor density (Fig. 5). Consistent with this view, it has been shown recently that exocytosis of GluR1-containing AMPA receptor can be induced in primary hippocampal neurons by NMDA or insulin in a PI3-kinase-dependent manner (Passafaro et al., 2001). However, exocytosis of GluR2 subunits not associated with GluR1 in primary hippocampal neurons is unaffected by the PI3-kinase inhibitor wortmannin (Passafaro et al., 2001).
The present data demonstrate that PI3-kinase is required for the expression but not for the induction or maintenance of LTP at Schaffer collateral/commissural fiber–CA1 synapses. Although several signal transduction pathways are known to be involved in the induction of LTP, PI3-kinase is the first one to be found to be required for its expression. Both NMDA-dependent and VDCC-dependent forms of LTP at Schaffer collateral/commissural fiber–CA1 synapses were found to require PI3-kinase activity for their expression, suggesting that PI3-kinase could be a final common pathway in the expression of multiple forms of synaptic potentiation.
Footnotes
This study was supported in part by National Institutes of Health Grants MH62140 and MH64376 (P.P.S. and W.F.) and P50AA06420 (F.E.B. and P.P.S.). We are grateful to Drs. George Siggins, Paul Schweitzer, and Steven Henriksen (from The Scripps Research Institute) for critical review of this manuscript and Dr. George Koob (also from The Scripps Research Institute) for his support and encouragement.
Correspondence should be addressed to Pietro Paolo Sanna or Walter Francesconi, Department of Neuropharmacology, The Scripps Research Institute, 10550 N. Torrey Pines Road, La Jolla, CA 92037. E-mails:psanna@scripps.edu and wfranc@scripps.edu.
REFERENCES
- 1.Abraham WC, Gustafsson B, Wigstrom H. Long-term potentiation involves enhanced synaptic excitation relative to synaptic inhibition in guinea-pig hippocampus. J Physiol (Lond) 1987;394:367–380. doi: 10.1113/jphysiol.1987.sp016875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akasaki T, Koga H, Sumimoto H. Phosphoinositide 3-kinase-dependent and -independent activation of the small GTPase Rac2 in human neutrophils. J Biol Chem. 1999;274:18055–18059. doi: 10.1074/jbc.274.25.18055. [DOI] [PubMed] [Google Scholar]
- 3.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 4.Allison DW, Gelfand VI, Spector I, Craig AM. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18:2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balendran A, Currie R, Armstrong CG, Avruch J, Alessi DR. Evidence that 3-phosphoinositide-dependent protein kinase-1 mediates phosphorylation of p70 S6 kinase in vivo at Thr-412 as well as Thr-252. J Biol Chem. 1999;274:37400–37406. doi: 10.1074/jbc.274.52.37400. [DOI] [PubMed] [Google Scholar]
- 6.Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 7.Bortolotto ZA, Collingridge GL. Involvement of calcium/calmodulin-dependent protein kinases in the setting of a molecular switch involved in hippocampal LTP. Neuropharmacology. 1998;37:535–544. doi: 10.1016/s0028-3908(98)00058-6. [DOI] [PubMed] [Google Scholar]
- 8.Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 9.Burnett PE, Blackshaw S, Lai MM, Qureshi IA, Burnett AF, Sabatini DM, Snyder SH. Neurabin is a synaptic protein linking p70 S6 kinase and the neuronal cytoskeleton. Proc Natl Acad Sci USA. 1998;95:8351–8356. doi: 10.1073/pnas.95.14.8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cavus I, Teyler T. Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. J Neurophysiol. 1996;76:3038–3047. doi: 10.1152/jn.1996.76.5.3038. [DOI] [PubMed] [Google Scholar]
- 11.Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 12.Chen HX, Otmakhov N, Strack S, Colbran RJ, Lisman JE. Is persistent activity of calcium/calmodulin-dependent kinase required for the maintenance of LTP? J Neurophysiol. 2001;85:1368–1376. doi: 10.1152/jn.2001.85.4.1368. [DOI] [PubMed] [Google Scholar]
- 13.Chou MM, Hou W, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- 14.Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- 15.Corvera S, Czech MP. Direct targets of phosphoinositide 3-kinase products in membrane traffic and signal transduction. Trends Cell Biol. 1998;8:442–446. doi: 10.1016/s0962-8924(98)01366-x. [DOI] [PubMed] [Google Scholar]
- 16.Cox D, Tseng CC, Bjekic G, Greenberg S. A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J Biol Chem. 1999;274:1240–1247. doi: 10.1074/jbc.274.3.1240. [DOI] [PubMed] [Google Scholar]
- 17.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 18.Fisher SA, Fischer TM, Carew TJ. Multiple overlapping processes underlying short-term synaptic enhancement. Trends Neurosci. 1997;20:170–177. doi: 10.1016/s0166-2236(96)01001-6. [DOI] [PubMed] [Google Scholar]
- 19.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 20.Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- 21.Grosshans DR, Clayton DA, Coultrap SJ, Browning MD. LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat Neurosci. 2002;5:27–33. doi: 10.1038/nn779. [DOI] [PubMed] [Google Scholar]
- 22.Grover LM, Yan C. Blockade of GABAA receptors facilitates induction of NMDA receptor-independent long-term potentiation. J Neurophysiol. 1999;81:2814–2822. doi: 10.1152/jn.1999.81.6.2814. [DOI] [PubMed] [Google Scholar]
- 23.Halpain S. Actin and the agile spine: how and why do dendritic spines dance? Trends Neurosci. 2000;23:141–146. doi: 10.1016/s0166-2236(00)01576-9. [DOI] [PubMed] [Google Scholar]
- 24.Higaki M, Sakaue H, Ogawa W, Kasuga M, Shimokado K. Phosphatidylinositol 3-kinase-independent signal transduction pathway for platelet-derived growth factor-induced chemotaxis. J Biol Chem. 1996;271:29342–29346. doi: 10.1074/jbc.271.46.29342. [DOI] [PubMed] [Google Scholar]
- 25.Huang EP. Synaptic plasticity: going through phases with LTP. Curr Biol. 1998;8:R350–R352. doi: 10.1016/s0960-9822(98)70219-2. [DOI] [PubMed] [Google Scholar]
- 26.Ito I, Hidaka H, Sugiyama H. Effects of KN-62, a specific inhibitor of calcium/calmodulin-dependent protein kinase II, on long-term potentiation in the rat hippocampus. Neurosci Lett. 1991;121:119–121. doi: 10.1016/0304-3940(91)90663-e. [DOI] [PubMed] [Google Scholar]
- 27.Janecki AJ, Janecki M, Akhter S, Donowitz M. Basic fibroblast growth factor stimulates surface expression and activity of Na+/H+ exchanger NHE3 via mechanism involving phosphatidylinositol 3-kinase. J Biol Chem. 2000;275:8133–8142. doi: 10.1074/jbc.275.11.8133. [DOI] [PubMed] [Google Scholar]
- 28.Kamiya H, Zucker RS. Residual Ca2+ and short-term synaptic plasticity. Nature. 1994;371:603–606. doi: 10.1038/371603a0. [DOI] [PubMed] [Google Scholar]
- 29.Kelly A, Lynch MA. Long-term potentiation in dentate gyrus of the rat is inhibited by the phosphoinositide 3-kinase inhibitor, wortmannin. Neuropharmacology. 2000;39:643–651. doi: 10.1016/s0028-3908(99)00169-0. [DOI] [PubMed] [Google Scholar]
- 30.Kurashima K, Szabo EZ, Lukacs G, Orlowski J, Grinstein S. Endosomal recycling of the Na+/H+ exchanger NHE3 isoform is regulated by the phosphatidylinositol 3-kinase pathway. J Biol Chem. 1998;273:20828–20836. doi: 10.1074/jbc.273.33.20828. [DOI] [PubMed] [Google Scholar]
- 31.Leevers SJ, Vanhaesebroeck B, Waterfield MD. Signalling through phosphoinositide 3-kinases: the lipids take centre stage. Curr Opin Cell Biol. 1999;11:219–225. doi: 10.1016/s0955-0674(99)80029-5. [DOI] [PubMed] [Google Scholar]
- 32.Lin C, Yeh S, Lu K, Leu T, Chang W, Gean P. A role for the pi-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron. 2001;31:841–851. doi: 10.1016/s0896-6273(01)00433-0. [DOI] [PubMed] [Google Scholar]
- 33.Luscher C, Nicoll RA, Malenka RC, Muller D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci. 2000;3:545–550. doi: 10.1038/75714. [DOI] [PubMed] [Google Scholar]
- 34.Macara IG, Marinetti GV, Balduzzi PC. Transforming protein of avian sarcoma virus UR2 is associated with phosphatidylinositol kinase activity: possible role in tumorigenesis. Proc Natl Acad Sci USA. 1984;81:2728–2732. doi: 10.1073/pnas.81.9.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 36.Malinow R, Madison DV, Tsien RW. Persistent protein kinase activity underlying long-term potentiation. Nature. 1988;335:820–824. doi: 10.1038/335820a0. [DOI] [PubMed] [Google Scholar]
- 37.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 38.Malinow R, Mainen ZF, Hayashi Y. LTP mechanisms: from silence to four-lane traffic. Curr Opin Neurobiol. 2000;10:352–357. doi: 10.1016/s0959-4388(00)00099-4. [DOI] [PubMed] [Google Scholar]
- 39.Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–758. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- 40.Norman BH, Shih C, Toth JE, Ray JE, Dodge JA, Johnson DW, Rutherford PG, Schultz RM, Worzalla JF, Vlahos CJ. Studies on the mechanism of phosphatidylinositol 3-kinase inhibition by wortmannin and related analogs. J Med Chem. 1996;39:1106–1111. doi: 10.1021/jm950619p. [DOI] [PubMed] [Google Scholar]
- 41.Passafaro M, Piëch V, Sheng M. Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci. 2001;4:917–926. doi: 10.1038/nn0901-917. [DOI] [PubMed] [Google Scholar]
- 42.Powis G, Bonjouklian R, Berggren MM, Gallegos A, Abraham R, Ashendel C, Zalkow L, Matter WF, Dodge J, Grindey G, Vlahos CJ. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994;54:2419–2423. [PubMed] [Google Scholar]
- 43.Romanelli A, Martin KA, Toker A, Blenis J. p70 S6 kinase is regulated by protein kinase Czeta and participates in a phosphoinositide 3-kinase-regulated signalling complex. Mol Cell Biol. 1999;19:2921–2928. doi: 10.1128/mcb.19.4.2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sajan MP, Standaert ML, Bandyopadhyay G, Quon MJ, Burke TR, Jr, Farese RV. Protein kinase C-zeta and phosphoinositide-dependent protein kinase-1 are required for insulin-induced activation of ERK in rat adipocytes. J Biol Chem. 1999;274:30495–30500. doi: 10.1074/jbc.274.43.30495. [DOI] [PubMed] [Google Scholar]
- 45.Salter MW. Src, N-methyl-d-aspartate (NMDA) receptors, and synaptic plasticity. Biochem Pharmacol. 1998;56:789–798. doi: 10.1016/s0006-2952(98)00124-5. [DOI] [PubMed] [Google Scholar]
- 46.Sanna PP, Berton F, Cammalleri M, Tallent MK, Siggins GF, Bloom FE, Francesconi W. A role for Src kinase in spontaneous epileptiform activity in the CA3 region of the hippocampus. Proc Natl Acad Sci USA. 2000;97:8653–8657. doi: 10.1073/pnas.140219097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen L, Liang F, Walensky LD, Huganir RL. Regulation of AMPA receptor GluR1 subunit surface expression by a 4.1 N-linked actin cytoskeletal association. J Neurosci. 2000;20:7932–7940. doi: 10.1523/JNEUROSCI.20-21-07932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheng M, Pak DT. Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu Rev Physiol. 2000;62:755–778. doi: 10.1146/annurev.physiol.62.1.755. [DOI] [PubMed] [Google Scholar]
- 49.Shepherd PR, Nave BT, Siddle K. Involvement of PI 3-kinase in stimulation of glucose transport and recruitment of transferrin receptors in 3T3–L1 adipocytes. Biochem Soc Trans. 1995;23:201S. doi: 10.1042/bst023201s. [DOI] [PubMed] [Google Scholar]
- 50.Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- 51.Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- 52.Teyler TJ, Cavus I, Coussens C, DiScenna P, Grover L, Lee YP, Little Z. Multideterminant role of calcium in hippocampal synaptic plasticity. Hippocampus. 1994;4:623–634. doi: 10.1002/hipo.450040602. [DOI] [PubMed] [Google Scholar]
- 53.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 54.Vogelbaum MA, Tong JX, Rich KM. Developmental regulation of apoptosis in dorsal root ganglion neurons. J Neurosci. 1998;18:8928–8935. doi: 10.1523/JNEUROSCI.18-21-08928.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whitman M, Downes CP, Keeler M, Keller T, Cantley L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature. 1988;332:644–646. doi: 10.1038/332644a0. [DOI] [PubMed] [Google Scholar]
- 57.Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- 58.Wymann M, Arcaro A. Platelet-derived growth factor-induced phosphatidylinositol 3-kinase activation mediates actin rearrangements in fibroblasts. Biochem J. 1994;298:517–520. doi: 10.1042/bj2980517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta. 1998;1436:127–150. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- 60.Yudowski GA, Efendiev R, Pedemonte CH, Katz AI, Berggren PO, Bertorello AM. Phosphoinositide-3 kinase binds to a proline-rich motif in the Na+, K+-ATPase alpha subunit and regulates its trafficking. Proc Natl Acad Sci USA. 2000;97:6556–6561. doi: 10.1073/pnas.100128297. [DOI] [PMC free article] [PubMed] [Google Scholar]