

Abstract
A single episode of status epilepticus (SE) causes numerous structural and functional changes in the brain that can lead to the development of a chronic epileptic condition. Most studies of this plasticity have focused on changes in excitatory and inhibitory synaptic properties. However, the intrinsic firing properties that shape the output of the neuron to a given synaptic input may also be persistently affected by SE. Thus, 54% of CA1 pyramidal cells, which normally fire in a regular mode, are persistently converted to a bursting mode after an episode of SE induced by the convulsant pilocarpine. In this model, intrinsic bursts evoked by threshold-straddling depolarizations, and their underlying spike afterdepolarizations (ADPs), were resistant to antagonists of N-, P/Q-, or L-type Ca2+ channels but were readily suppressed by low (30–100 μm) concentrations of Ni2+ known to block T- and R-type Ca2+ channels. The density of T-type Ca2+ currents, but not of other pharmacologically isolated Ca2+ current types, was upregulated in CA1 pyramidal neurons after SE. The augmented T-type currents were sensitive to Ni2+ in the same concentration range that blocked the novel intrinsic bursting in these neurons (IC50 = 27 μm). These data suggest that SE may persistently convert regular firing cells to intrinsic bursters by selectively increasing the density of a Ni2+-sensitive T-type Ca2+current. This nonsynaptic plasticity considerably amplifies the output of CA1 pyramidal neurons to synaptic inputs and most probably contributes to the development and expression of an epileptic condition after SE.
Keywords: T-type Ca2+ channel, status epilepticus, intrinsic burst discharge, plasticity, CA1, hippocampus
Both in humans and in experimental animals, continuous seizure activity as occurs during status epilepticus (SE) results in a large number of plastic changes within the hippocampus and adjacent brain areas (Coulter and DeLorenzo, 1999;Sloviter, 1999; Ben Ari, 2001). The SE-induced modifications in neuronal function may consist either of altered synaptic function or of changes in the intrinsic membrane properties of neurons. Hitherto, a multitude of structural and functional synaptic changes have been shown to follow SE, such as reorganization of excitatory axons (Sutula et al., 1989), altered function of excitatory neurotransmitter receptors (Turner and Wheal, 1991; Lothman et al., 1995; Chen et al., 1999), or changes in GABAA receptor-mediated inhibition (Mangan et al., 1995; Gibbs et al., 1997; Brooks-Kayal et al., 1998; Cossart et al., 2001). Many of these synaptic changes have been suggested to contribute to the development of temporal lobe epilepsy (TLE) that is frequently the outcome of a single or multiple episodes of SE.
Much less is known about SE-induced changes in intrinsic neuronal properties. However, a recent study in the pilocarpine model, in which an episode of SE induced by the convulsant pilocarpine progresses to a chronic epileptic condition resembling human TLE (Turski et al., 1983), suggests that such alterations may be highly significant. In this model, Sanabria et al. (2001) found a marked upregulation of intrinsic neuronal bursting associated with the development of TLE. Although almost all pyramidal neurons in the hippocampal CA1 area are regular firing cells in normal conditions (Jensen et al., 1994), ∼50% of CA1 pyramidal cells in hippocampal tissue removed from rats that experienced SE were found to be intrinsically burst-firing cells. Moreover, the upregulation of intrinsic bursting seemed to result from the de novoappearance of Ca2+-dependent bursting that is not ordinarily seen in this class of principal hippocampal neurons (Azouz et al., 1996). It was also found that a subset of the newly formed bursters, namely, the spontaneously bursting pyramidal cells, acted as pacemakers of spontaneous interictal-like epileptiform bursts, recruiting the entire CA1 population of pyramidal cells, nonbursters and bursters alike, into synchronized discharge (Sanabria et al., 2001).
Because the switch in neuronal firing mode after SE may be important for the development and expression of TLE, and because it may represent a novel mechanism of activity-induced neuronal plasticity, we have used a combination of electrophysiological and pharmacological techniques to characterize in detail the ionic mechanisms underlying the increased intrinsic bursting. Here we report that the dramatic change in firing mode of CA1 neurons after SE is most probably caused by the increased density of a Ni2+-sensitive T-type Ca2+ current. Our findings demonstrate that epilepsy-related changes in intrinsic neuronal properties may result from activity-dependent differential regulation of specific ion channel subtypes and suggest that subunit-selective T-type channel antagonists may be promising future targets for rational anti-epileptic drug design.
MATERIALS AND METHODS
Induction of SE. Experimental protocols were approved by the local animal care and use committees. Male Sabra or Wistar rats (150–200 gm) were injected with a single high dose of the muscarinic agonist pilocarpine (300–380 mg/kg, i.p.), which induced SE in most (∼80%) animals. Peripheral muscarinic effects were reduced by previous administration of methyl-scopolamine (1 mg/kg, s.c.; 30 min before injection of pilocarpine). Diazepam (0.1 mg/kg, s.c.) was administered to all animals 2 hr after the pilocarpine injection. It terminated the convulsions in the responsive rats and sedated all animals. Experiments were performed on hippocampal slices from rats that experienced SE after pilocarpine injection (SE-experienced rats) and from those that did not (sham-control rats). A third group of untreated, age-matched rats served as an additional control (naive-control rats). Previous work has found no differences in electrophysiological properties between sham- and naive-control rats (Sanabria et al., 2001).
Preparation of hippocampal slices and dissociated neurons. Animals were decapitated under ether anesthesia 6–40 d after pilocarpine treatment, and transverse hippocampal slices (250 or 400 μm) were prepared with a vibrating microslicer (Campden Instruments or Leica) and transferred to a storage chamber perfused with oxygenated (95% CO2–5% O2) artificial CSF (ACSF) containing (in mm): NaCl 125, KCl 3, NaH2PO41.25, MgCl2 1, CaCl2 2, NaHCO3 25, and glucose 20, pH 7.4, osmolarity 305 mOsm, where they were maintained at room temperature. For intracellular recordings, 400-μm-thick hippocampal slices were placed in an interface chamber (33.5°C) and perfused with oxygenated ACSF containing (in mm): NaCl 124, KCl 3.5, NaH2PO4 1.25, MgSO4 2, CaCl2 2, NaHCO3 26, and d-glucose 10. In experiments in which Ni2+ was used, NaH2PO4 was omitted. Ca2+ channel blockers, or the glutamate receptor antagonists 6-cyano-7-nitro-quinoxaline-2,3-dione (CNQX) and 2-amino-5-phosphono-valeric acid (APV), were added to the ACSF as indicated. For patch-clamp recordings in the slice preparation, 250 μm hippocampal slices were placed in a submerged recording chamber (21–24°C) and perfused continuously (2.5 ml/min) with oxygenated ACSF containing (in mm): NaCl 125, KCl 2.5, NaHCO3 26.7, CaCl2 2.5, MgCl2 1, HEPES 13, glucose 12.5, pH 7.3, osmolarity 300 mOsm. Pyramidal cells in the CA1 field were visualized at 400× magnification with Dodt-Gradient-Contrast optics (Luigs and Neumann) using a ZeissAxioskop microscope and an infra-red video camera (Hamamatsu, Shizuoka, Japan).
Dissociated hippocampal neurons were prepared from 400-μm-thick slices primarily as described previously (Beck et al., 1999). After an equilibration period (>60 min), enzymatic digestion was performed for 10 min at 37°C and 5 min at room temperature in 5 ml of incubation medium containing (in mm): sodium methanesulfonate 145, KCl 3, CaCl2 0.5, MgCl2 1, HEPES 10, glucose 15, and 2 mg/ml pronase (protease type XIV; Sigma, St. Louis, MO) (pH 7.4, osmolarity 310 mOsm, 100% O2). After washing with enzyme-free incubation medium, the CA1 region was dissected and triturated with fire-polished glass pipettes. The cell suspension was placed in a Petri dish and superfused with modified ACSF containing (in mm): sodium methanesulfonate 125, tetraethylammonium chloride (TEA) 20, 4-aminopyridine 4, BaCl2 5, MgCl2 1, KCl 3, glucose 10, HEPES 10, tetrodotoxin (TTX) 0.5 μm, pH 7.4, osmolarity 315 mOsm. Cells were visualized on a inverted microscope (Zeiss Axiovert).
Sharp microelectrode recordings in slices. Intracellular recordings were obtained using sharp glass microelectrodes containing 1m K+-acetate (70–90 MΩ). An active bridge circuit in the amplifier (Axoclamp 2B, Axon Instruments) allowed simultaneous injection of current and measurement of membrane potential. The signals were filtered on-line at 5 kHz, digitized at a sampling rate of 10 kHz, and stored on hard disk (TL-1 DMA and pClamp, Axon Instruments). In some experiments, bipolar platinum (50 μm) electrodes connected to a stimulator by an isolation unit were used for focal stimulation of afferent fibers in stratum radiatum near the CA2/CA3 border (orthodromic stimulation).
Patch-clamp recordings in slices and dissociated neurons. Recording patch pipettes (2–4 MΩ) were pulled from borosilicate glass on a vertical puller (List-Medical or Narishige, Tokyo, Japan). Pipettes were coated with Sylgard resin for recordings in the slice preparation (Dow Corning Chemical) and filled with an intracellular solution containing (in mm): Cs-methanesulfonate 100, EGTA 5, HEPES 10, TEA 20, MgCl2 2, CaCl2 0.1, pH 7.35. For recordings from dissociated CA1 neurons, pipettes were filled with (in mm): Cs-methanesulfonate 87.5, TEA 20, CaCl2 0.5, MgCl2 5, BAPTA 5, HEPES 10, glucose 10, Na+2-ATP 10, and GTP 0.5, pH 7.2 (NaOH), osmolarity adjusted to 300 mOsm with sucrose. Tight-seal whole-cell recordings were obtained using a patch-clamp amplifier (Axopatch 200A, Axon Instruments, or EPC9, HEKA Instruments). The signals were collected on-line as described above. Series resistance compensation was used to improve the voltage-clamp control (60–85%) so that the maximal residual voltage error did not exceed 5 mV. A liquid junction potential of ∼10 mV was measured between the intracellular and extracellular solutions and corrected on-line.
Chemicals and drugs. In slice experiments the drugs were applied via the perfusing ACSF. Application of drugs to dissociated neurons was performed via a pipette placed at a distance of 30–50 μm from the cell body. All drugs were obtained from Sigma, except the Ca2+ channel toxins (Bachem or Alomone Labs) and CNQX (Tocris Neuroamin). Stock solutions of the toxins (0.2–1 mm) were prepared in deoxygenated solution containing 0.1% BSA, 100 mm NaCl, 10 mm Trizma, 1 mm EDTA, pH 7.5. In experiments using the toxins, all solutions contained 0.1 mg/ml cytochrome c to prevent unspecific peptide binding to tubing. Stock solutions (10 mm) of nifedipine and nicardipine were prepared in DMSO.
Analysis. A measure for the size of the spike afterdepolarization was provided by the area delimited by the ADP waveform and resting membrane potential. The concentration-dependent reduction of Ca2+ current amplitude was fit with a modified Hill function of the form: E =Emax ∗cn/(cn+ IC50n), whereE is the fraction of current blocked,Emax corresponds to the maximal block,c is the concentration of antagonist, n is the Hill coefficient, and the IC50 is the concentration at which half-maximal blocking effects were observed.
The decay of tail currents after mock action potentials was fit with a biexponential equation of the form: Ι(t) =A1 ∗ (1 − exp(−t/τf)) +A2 ∗ (1 − exp(−t/τs)), whereI(t) is the current amplitude at the time pointt after onset of the voltage command,τf andτs are the fast and slow decay time constants, respectively, and A is the amplitude contribution of the different time constants. Data were fit using a Levenberg-Marquard nonlinear curve-fitting procedure.
Significant differences between groups were tested with a two-tailed Student's t test with the significance level set top < 0.05.
RESULTS
Altered intrinsic bursting after pilocarpine-induced SE
First, we have replicated the recent findings of Sanabria et al. (2001), demonstrating enhanced bursting behavior in SE-experienced rats compared with naive-control and sham-control animals, in a new set of experiments. Sharp microelectrodes were used to impale CA1 pyramidal cells and to stimulate them with threshold-straddling, long (200 msec) and brief (4 msec) depolarizing current pulses (Azouz et al., 1996). In both the naive-control (n = 15 cells; data not shown) and sham-control (n = 42 cells) (Fig.1A) groups, all except one neuron fired a series of independent spikes in response to the long pulse and a single spike in response to the brief pulse (Fig.1Aa,B). The remaining neuron in each group burst-fired in response to the long pulse. In marked contrast, more than half of the CA1 neurons in the SE-experienced group (54%;n = 97) generated a burst of action potentials as a minimal response to threshold-straddling long current injections and in many cases burst-fired also in response to brief stimulation (Fig.1Ab,B). As expected (Sanabria et al., 2001), the burst responses were unaffected by blocking synaptic transmission with CNQX (15 μm), APV (50 μm), and bicuculline methiodide (10 μm; n = 26). Thus, as described previously, pilocarpine-induced SE triggers a long-lasting switch in firing mode from a nonbursting to a bursting behavior in a large fraction of CA1 pyramidal cells.
Fig. 1.
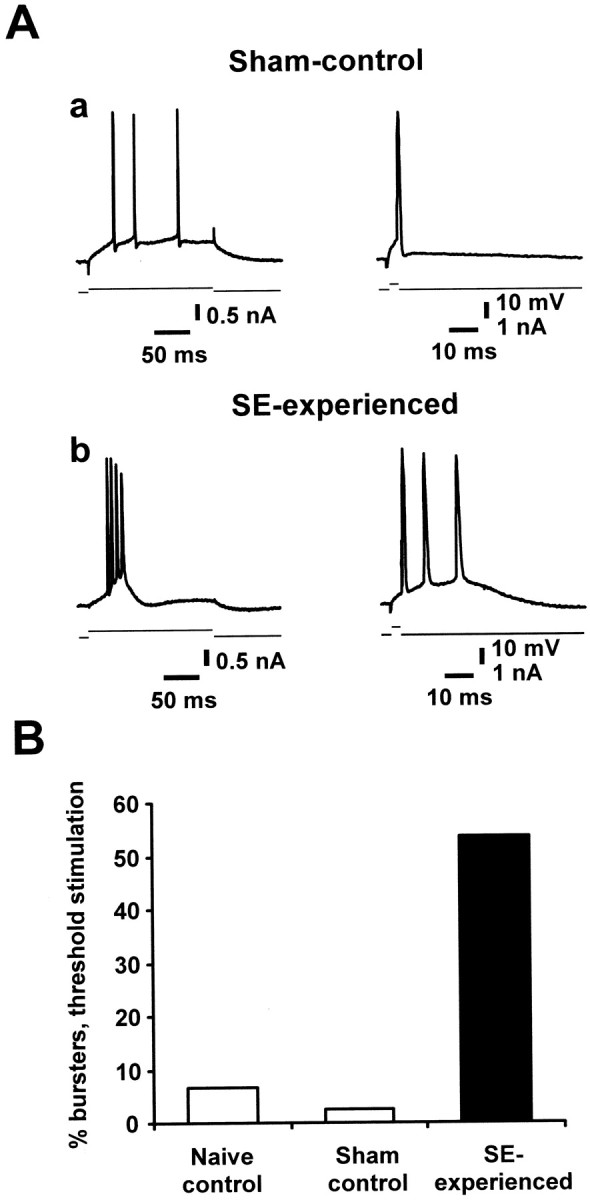
Upregulation of intrinsic burst-firing in CA1 pyramidal cells from SE-experienced versus control animals.A, Representative responses of CA1 pyramidal cells from sham-control (a) and SE-experienced animals (b) 30 d after pilocarpine treatment. In each panel, the responses of CA1 pyramidal cells to long (leftmost traces) and brief (rightmost traces) depolarizing current pulses (injected through the recording microelectrode) are shown. Top andbottom traces depict the neuronal response and the current stimulus, respectively. B, Most of the CA1 pyramidal cells in the sham-control (n = 42) as well as in the naive-control group (n = 15) were regular firing cells, with only a small fraction displaying burst discharges to threshold stimulation (white bars). In contrast, CA1 pyramidal cells in the SE-experienced group showed a high incidence of intrinsic bursting (54%, n = 97), with bursting neurons displaying all-or-none bursts of three to four clustered spikes in response to either brief or long current injection (Ab).
Critical role of Ni2+-sensitive Ca2+ channels in SE-induced intrinsic bursting
Previous work has suggested that Ca2+currents play a critical role in the SE-induced intrinsic bursting of CA1 pyramidal cells (Sanabria et al., 2001). In normal tissue, CA1 pyramidal cells express multiple types of voltage-gated Ca2+ channels, namely, L-, N-, P-/Q-, R-, and T-types (Yaari et al., 1987; Takahashi et al., 1989; Fisher et al., 1990; Thompson and Schwindt, 1991; Christie et al., 1995). To identify the Ca2+ channel types that mediate intrinsic bursting induced by SE, we have compared the effects of selective organic Ca2+ channel blockers: namely, nicardipine (10 μm; n = 6) or nifedipine (30 μm;n = 5) to block L-type, ω-conotoxin GVIA (1 μm;n = 7) or neomycin (0.5 mm;n = 6) to block N-type, ω-conotoxin MVIIC (5 μm; n = 5) to block N-/P-/Q-type, and ω-agatoxin TK (300 nm;n = 3) to block P-type Ca2+ channels. Additionally, the divalent cation Ni2+ was applied at low concentrations (50–100 μm;n = 32) that predominantly block T-/R-type Ca2+ channels (Soong et al., 1993;Williams et al., 1994; Lee et al., 1999b). The blockers were added to the ACSF perfusing the slices from SE-experienced animals for at least 45 min. The dihydropyridines (Fig.2A, comparea, b) did not affect bursting in any of the neurons tested, and neither did neomycin (data not shown) or the toxin blockers of Ca2+ channels (Fig.2B–D, compare a,b).
Fig. 2.

Critical role of Ni2+-sensitive Ca2+ channels in intrinsic burst firing in SE-experienced animals. Recordings were performed on intrinsically bursting CA1 neurons from SE-experienced rats (Aa,Ba, Ca, and Da).A–D, No change in intrinsic bursting behavior could be observed after 40 min of perfusion with 10 μm nicardipine (Ab) or 1 μmω-conotoxin GVIA (Bb; 30 min). Likewise, application of 5 μm ω-conotoxin MVIIC (Cb; 40–80 min) or 300 nm ω-agatoxin TK (Db; 40–60 min) did not affect intrinsic burst generation. In all cases, additional perfusion of 50 μmNi2+ blocked bursting entirely (Ac,Bc, Cc, and Dc).
To test whether intrinsic bursting is generated synergistically by several types of Ca2+ channels, we have added the organic Ca2+ channel blockers sequentially to the ACSF (without washing them out) while recording from a single neuron. In these experiments (n = 5), intrinsic bursting was unaffected in all cases (Fig.3Aa1–d1). In contrast, when 50 or 100 μmNi2+ was added to the ACSF containing one or more of the organic Ca2+ channel blockers, intrinsic bursting was entirely suppressed in 26 of 32 (81%) neurons (Figs. 2Ac–Dc, 3Ae1). To confirm that the organic Ca2+ channel blockers are pharmacologically active when applied to the slice, we monitored their effects on the orthodromically evoked EPSPs. In all experiments, ω-conotoxin MVIIC, ω-conotoxin GVIA, ω-agatoxin TK, and nicardipine produced a partial reduction of the EPSPs within 30 min (Fig.3Aa2–d2), presumably consequent to blocking a subset of presynaptic voltage-gated Ca2+ channels (Wu and Saggau, 1994; Qian and Noebels, 2001).
Fig. 3.

Combined application of organic Ca2+ channel antagonists fails to block bursting.A, Ca2+ channel blockers were sequentially added to the ACSF, and the efficacy of Ca2+ channel blockade was assessed by the progressive block of the EPSP evoked by orthodromic stimulation of the Schaffer collaterals (Aa2–Ae2). Combined application of organic Ca2+antagonists did not block bursting (Aa1–Ad1) but additional application of 50 μmNi2+ did (Ae1). Blockers were applied for at least 30 min (ω-conotoxin GVIA 1 μm, 62 min; ω-agatoxin TK 300 nm, 40 min; nicardipine 10 μm, 30 min; Ni2+ 50 μm, 50 min).
We also examined the consequences of blocking only T-/R-type Ca2+ channels with low concentrations of Ni2+. Addition of 10 μmNi2+ to the ACSF had no effect on intrinsic bursting (n = 4), but 30 μm Ni2+ shortened the bursts in two of four neurons. Addition of 100 μm Ni2+ to the ACSF reversibly suppressed intrinsic bursts in 35 of 42 (83%) bursters examined (Fig.4Aa1–c1). Raising the concentration of Ni2+ further (up to 1 mm) did not block intrinsic bursts that were insensitive to 100 μmNi2+ (n = 5; data not shown), indicating that Ni2+-insensitive bursting is not mediated by any type of voltage-gated Ca2+ channel. Rather, it may involve activation of persistent Na+ channels (Azouz et al., 1996; Su et al., 2001), but this mechanism was not investigated further in this study.
Fig. 4.

Low concentrations of Ni2+suppress burst firing by reducing the spike ADP. A, Intrinsically bursting CA1 pyramidal neuron from an SE-experienced animal in normal ACSF (a1), after block of the burst discharge by perfusion of 50 μm Ni2+(b1), and after washout (c1). The slow depolarization underlying the burst was unmasked by delivering a brief (4 msec) hyperpolarizing current pulse immediately after the first spike (a2–c2). The time course of the ADP was similar ina1 anda2 (for comparison at larger magnification, see d). Adding 50 μmNi2+ to the ACSF reversibly suppressed the ADP (b2,c2; for comparison at larger magnification, see e). B, Regular firing CA1 pyramidal cell in a sham-control animal before (a) and after (b) application of 100 μm Ni2+. The ADP was not affected by Ni2+ (for comparison at larger magnification, see c).
Low concentrations of Ni2+ suppress intrinsic bursting by reducing the spike afterdepolarization
In CA1 pyramidal cells, the fast spike activates a slow somatodendritic inward current that generates a slow spike ADP in the soma. Although in regular firing cells the ADP is subthreshold, in bursters it redepolarizes the neuron sufficiently to generate additional spikes (Azouz et al., 1996). We tested in four bursters whether low Ni2+ concentrations suppress the spike ADP underlying the burst discharge. This slow potential was unmasked by delivering a brief (4 msec) hyperpolarizing current pulse immediately after the first spike. The intensity of this pulse was adjusted to the minimum required to inhibit further discharge (Wong and Prince, 1981; Jensen et al., 1996). This procedure disclosed a large ADP with a duration similar to that of the depolarizing burst envelope (Fig.4Aa1,a2; overlaid and enlarged in d). Adding 50 μm Ni2+ to the ACSF suppressed the burst discharge (Fig.4Aa1,b1) and concurrently attenuated the size of the ADP in these cells (Fig.4Aa2,b2; overlaid and enlarged in e). We also examined the effect of low Ni2+ concentrations on the spike ADP in six regular firing CA1 pyramidal cells in sham-control hippocampal slices. The spike ADPs in these neurons were significantly smaller than in neurons from SE-experienced animals (108.0 ± 5.1 vs 250.8 ± 7.1 mV · msec, respectively) and were insensitive to 100 μm Ni2+ (Fig.4Ba,b; overlaid and enlarged in c). The Ni2+-insensitive ADP components most likely are generated by persistent Na+current (Azouz et al., 1996; Su et al., 2001).
Depolarization from resting membrane potential abolishes intrinsic bursting
These data so far have suggested that in CA1 pyramidal cells from SE-experienced animals, Ni2+-sensitive, T-/R-type Ca2+ current furnishes the main depolarizing drive for the spike ADP leading to burst generation. Both T- and R-type Ca2+ channels are steeply voltage-dependent and manifest inactivation after depolarization from the resting membrane potential (Randall and Tsien, 1997; Kozlov et al., 1999). To further explore their role in bursting, we tested how depolarization with constant current injection affects this activity. In five of seven neurons examined, the burst responses were suppressed by depolarizing the neurons 3–5 mV from their resting membrane potential (Fig. 5, compare Aa,Ba). Depolarizing these neurons by >5 mV suppressed the burst responses in all cases and induced the appearance of spontaneous single spike firing. Although there may be other explanations, these results are consistent with the notion that bursting is generated by T-/R-type Ca2+ currents. Further supporting this notion was the finding that although Ni2+ suppressed bursting at resting membrane potential presumably by reducing the spike ADP (Fig.5Aa,b; overlaid and enlarged in Ac), it had no effect on the spike ADP at the slightly more depolarized potentials (Fig. 5Ba,b; overlaid and enlarged inBc).
Fig. 5.

Modest depolarization from resting potential suppresses burst discharges. A, Pyramidal neuron from an SE-experienced animal showing Ni2+-sensitive bursting (compare a, b; for comparison at larger magnification, see c). B, Depolarization of the neuron from a resting potential of −61 mV to a potential of −58 mV (compare Aa, Ba) converts the burst discharge into a single spike. Under these conditions, Ni2+ did not affect the time course of the spike ADP (compare Ba, Bb; for comparison at larger magnification, see c).
Increase in T-type Ca2+ current density in SE-experienced animals
The de novo appearance of Ni2+-sensitive intrinsic bursting in CA1 pyramidal cells described above might be caused by a genuine increase in the density of functional T-/R-type Ca2+ channels, a more effective recruitment of existing channels by the first somatodendritic spike, or both. We have therefore directly measured the densities of T- and R-type Ca2+ currents in CA1 pyramidal cells in naive-control and SE-experienced rats using the whole-cell patch-clamp technique.
We first compared the densities of these currents between the two experimental groups in visually identified CA1 pyramidal cells in hippocampal slices. Voltage steps from −90 to −60 and −50 mV evoked transient Ca2+ currents (Fig.6Aa) (example shown for −60 mV voltage steps). The densities of these currents were significantly larger (2.2-fold and 1.9-fold increase for command pulses to −60 and −50 mV, respectively; p < 0.01) in SE-experienced animals (n = 10 cells; 6–12 d after pilocarpine-induced SE) compared with the naive-control animals (n = 12 cells) (Fig. 6Ac).
Fig. 6.

Increased density of T-type Ca2+ currents in SE-experienced animals.A, Recordings of T-type Ca2+ current in a CA1 pyramidal cell in the slice preparation using voltage steps from −90 to −60 or −50 mV (a) or in a dissociated CA1 neuron perfused with ACSF containing 2 μmω-conotoxin GVIA, 3 μm ω-conotoxin MVIIC, 200 nm ω-agatoxin GIVA, and 10 μm nifedipine to block most of the high-voltage-activated Ca2+channel types (b; see also Fig.8Ab). In this preparation, Ca2+ currents were evoked with voltage steps to −50 mV from a holding potential of −80 mV after a conditioning prepulse to −100 mV (5 sec). Average T-type amplitudes measured in SE-experienced versus naive-control animals in either CA1 neurons in slices (n = 10 and 12, respectively) or dissociated CA1 neurons (n = 14 and 15, respectively) are depicted in c. The current amplitudes were normalized to the cell capacitance in all cases. The current density showed a significant increase in SE-experienced animals (black bars) compared with naive-control animals (white bars) for recordings from both dissociated and intact CA1 neurons (*p < 0.05). B, Ni2+ sensitivity of the T-type current in dissociated CA1 neurons. The T-type current was markedly and reversibly reduced by micromolar concentrations of Ni2+, as shown in an exemplary recording from an SE-experienced animal (a). Concentration-dependence of the T-type Ca2+ current block by Ni2+ in both SE-experienced (●) and naive-control groups (○) (SE-experienced group: n= 3, 6, 6, and 3 cells; naïve-control group:n = 4, 2, 7, and 5 cells for 10, 30, 100, and 300 μm Ni2+; b). Data were fit by a Hill equation using a Levenberg-Marquard nonlinear curve-fitting procedure. The fitting curve is shown superimposed on the data points.
Similar experiments were performed in acutely dissociated CA1 pyramidal cells using depolarizing voltage steps to −50 mV preceded by a 5 sec prepulse to −100 mV (Fig. 6Ab). In these experiments we also superfused the neurons with ACSF containing organic Ca2+ channel blockers (2 μm ω-conotoxin GVIA, 3 μm ω-conotoxin MVIIC, 200 nm ω-agatoxin GIVA, and 10 μm nifedipine) to avoid even a small contribution of N-, P/Q-, or L-type Ca2+channels (Avery and Johnston, 1996). As found in the slice preparation, the densities of T-type Ca2+ currents in dissociated CA1 pyramidal cells were significantly larger (3.3-fold increase; p = 0.0001) in SE-experienced (n = 14 cells; 30–40 d after SE) compared with naive-control animals (n = 15 cells) (Fig.6Ac).
Ni2+ sensitivity of T-Type Ca2+ current in SE-experienced animals
Recent studies of voltage-gated Ca2+channels have identified three pore-forming α1subunits, namely, α1G, α1H, and α1I, that give rise to T-type Ca2+ channels (Cribbs et al., 1998; Perez-Reyes et al., 1998; Lee et al., 1999a). All three subunits are expressed in hippocampal CA1 pyramidal cells (Talley et al., 1999). They differ, however, in their sensitivity to blockage by Ni2+, with α1Hbeing an order of magnitude more sensitive to Ni2+ than α1G and α1I (Lee et al., 1999b). We have therefore examined the Ni2+ sensitivity of the T-type Ca2+ current in these neurons in SE-experienced as well as in the naive-control animals. In both groups, 100 μm Ni2+ almost completely and reversibly blocked the T-type Ca2+ current (Fig. 6Ba). Dose–response curves for Ni2+ (10–300 μm) (Fig. 6Bb) yielded similar IC50 values for SE-experienced (24.6 ± 2.6 μm; maximal block 84.5 ± 4.1%) and naive-control animals (27.9 ± 1.6 μm;maximal block 79.6 ± 2.0%).
T-type Ca2+ currents invariably show characteristically slow deactivation rates that distinguish them from the much more rapidly deactivating R-type channels (Randall and Tsien, 1997; Nakashima et al., 1998; Kozlov et al., 1999). Therefore, tail currents after brief depolarizing steps that activate both T- and R-type Ca2+ currents may be used to analyze these two current components separately. As illustrated in Figure 7Aa, activation of pharmacologically isolated T-/R-type Ca2+currents by mock action potentials as voltage commands was followed by biphasic tail currents in both experimental groups of neurons. These currents were best fitted by a biexponential curve (Fig.7Ab). The fast (τf) and slow (τs) decay time constants of these tail currents were, respectively, 478 ± 275 μsec and 4.1 ± 2.5 msec in naive-control animals (n = 7 cells), and 712 ± 122 μsec and 4.7 ± 1.6 msec in SE-experienced animals (n = 5 cells). The rapidly deactivating component most probably corresponds to an R-type current, whereas the slowly deactivating component is caused by T-type Ca2+ currents (Kozlov et al., 1999). Indeed, the amplitude of the slowly deactivating component of the tail current derived from biexponential fitting was tightly correlated with the amplitude of the T-type Ca2+ current measured with rectangular voltage steps (correlation coefficient 0.88;p = 0.0005; n = 12 cells). The amplitude of the slow (presumably T-type) tail Ca2+ current component was considerably larger in the SE-experienced group than in the control group (3.9-fold increase; p = 0.005), whereas that of the rapid (presumably R-type) component was not different (Fig. 7B). These results support our conclusion of a selective upregulation of T-type Ca2+ current in CA1 pyramidal cells in SE-experienced animals.
Fig. 7.

Increased T-type Ca2+ currents after SE are associated with an increase in slowly deactivating Ca2+ tail currents after mock action potentials.A, Application of mock action potentials (rising phase 1 msec; decaying phase 2 msec; peak +30 mV) as a voltage command in dissociated CA1 pyramidal neurons from naive-control animals (a1) and SE-experienced animals (a2). Current traces were derived by subtraction of traces obtained in the presence of organic Ca2+ blockers and 300 μmNi2+ from traces obtained only in the presence of organic Ca2+ channel blockers and thus reflect Ca2+ influx through T-/R-type channels. The decay of the inward tail current immediately after termination of the mock action potential was fit by a biexponential equation superimposed on the data points (b). B, Amplitudes of the fast and slowly decaying component of the tail currents normalized to the cell capacitance. A significant increase was observed only for the slower decaying component (*p < 0.005).
Changes in densities of high voltage-activated Ca2+ currents in SE-experienced animals
In the slice preparation, voltage steps to potentials above −40 mV evoked large Ca2+ currents that escaped voltage-clamp control in neurons from both experimental groups. Therefore, changes in the densities of high voltage-activated Ca2+ currents were investigated only in acutely dissociated CA1 pyramidal cells, in which these currents could be adequately voltage clamped (Beck et al., 1999). Stepping the membrane voltage from −100 to 0 mV evoked large Ca2+ currents (69.5 ± 4.0 pA/pF in naïve control animals; 69.0 ± 5.7 pA/pF in SE-experienced animals), which were reduced to approximately one-third by perfusing the neurons with ACSF containing the organic Ca2+ channel blockers (Fig.8Aa,b). The residual current evoked with test pulses to 0 mV comprises mostly R-type Ca2+ currents, with a small contribution of T-type currents (Thompson and Schwindt, 1991). Despite the increase in T-type current amplitude, the density of the compound T-/R-type current was not different between SE-experienced (n = 21) and naive-control neurons (n = 20) (Fig. 8Ac), indicating that the R-type current is either unaltered or slightly reduced. This conclusion is supported also by the lack of change in amplitude of the rapidly decaying tail Ca2+ current component after mock action potentials (Fig. 7B).
Fig. 8.

Functional analysis of high-threshold current components. A, Recordings were obtained in a dissociated CA1 pyramidal neuron from a naive-control animal. Ca2+ currents were evoked with the voltage paradigm shown in the inset. To isolate the T-/R-type Ca2+ current, the organic blockers ω-conotoxin GVIA (2 μm), ω-conotoxin MVIIC (3 μm), ω-agatoxin GIVA (200 nm), and nifedipine (10 μm) were co-applied, unmasking a rapidly inactivating residual component (a). The current traces in a were recorded at the time points indicated by the lowercase letters inb. Pharmacologically isolated T-/R-type Ca2+ currents were compared in dissociated CA1 pyramidal neurons from SE-experienced (n = 21 cells) and naive-control animals (n = 20 cells) (c). Current densities were obtained by normalizing current amplitudes to cell capacitance. B, In another dissociated neuron from a naive-control animal, the organic Ca2+ channel blockers ω-conotoxin GVIA (2 μm), ω-agatoxin IVa (200 nm), and nifedipine (10 μm) were added sequentially (a; duration of application is indicated byhorizontal bars, b). This allowed assessment of the individual contributions of N-, P/Q-, and L-type Ca2+ current components to the whole-cell current (indicated at right margin of b). Thecurrent traces in a were recorded at thetime points indicated by the lowercase letters in b. High-voltage-activated Ca2+ current densities in naïve control (n = 9–10) versus SE-experienced rats (n = 10) are shown in c. The N-type current density was significantly reduced (*p < 0.05), whereas the density of P/Q- and L-type currents was unaltered.
We also compared the densities of Ni2+-insensitive, high-threshold Ca2+ currents in acutely dissociated CA1 pyramidal cells from SE-experienced versus naive-control animals. The amplitudes of N-, P/Q-, and L-type Ca2+currents were determined by sequentially applying the selective antagonists 2 μm ω-conotoxin GVIA (n = 10 for both groups), 200 nm ω-agatoxin IVa (n = 10 for both groups), and 10 μm nifedipine (naive-control, n= 9 cells; SE-experienced, n = 10 cells), respectively (Fig. 8Ba,b). The effects of ω-conotoxin GVIA and ω-agatoxin IVa were primarily irreversible (Fig.8Bb). The N-type Ca2+current density was significantly reduced in the SE-experienced compared with the naive-control group, whereas the densities of P/Q- and L-type currents were not significantly altered (Fig.8Bc). Thus, of all the different types of Ca2+ currents expressed in CA1 pyramidal neurons, only T-type Ca2+ channel density is upregulated in SE-experienced animals.
Impact of altered firing mode on neuronal responses to synaptic excitation
The conversion of CA1 pyramidal cells from regular firing to burst firing after pilocarpine-induced SE would be expected to amplify their spike output in response to excitatory synaptic inputs. To examine this notion, we compared the synaptic activation of regular firing neurons in sham-control animals (Fig.9Aa) (n = 8 cells) with that of bursting neurons in SE-experienced animals (Fig.9Ba) (n = 4 cells). Single-shock stimuli were applied to afferent fibers in stratum radiatum, and their intensity was adjusted to evoke threshold-straddling EPSPs (Fig.9Ab,Bb). As expected, the minimal response in sham-control animals was a single spike (Fig. 9Ab). In contrast, the minimal response in SE-experienced animals was a spike burst similar in pattern to that evoked by direct current injection (Fig. 9Bb). To confirm that in the latter group the threshold EPSPs recruit the intrinsic burst mechanism rather than produce multiple spiking by virtue of their longer duration (Fig. 9, compare top panels in Ab, Bb), we exposed them to Ni2+. Because a small blocking effect of Ni2+ was observed on EPSPs, the stimulus strength was readjusted to yield threshold EPSPs. Adding 100 μm Ni2+to the ACSF suppressed the intrinsic burst response (Fig.9Bc) and reduced the burst responses to threshold EPSPs to single spikes (Fig. 9Bd) without affecting the duration of these EPSPs (Fig. 9Ba,c, top traces). These data clearly demonstrate that SE, by modifying intrinsic neuronal properties of CA1 pyramidal cells, markedly and persistently amplifies the neuronal output to a given synaptic input.
Fig. 9.

Impact of intrinsic plasticity on neuronal responses to synaptic excitation. These experiments were performed in the absence of blockers of synaptic transmission in the control ACSF.A, Recordings from a regular firing CA1 pyramidal cell from a sham-control animal. Firing behavior was determined using long depolarizing current injections through the recording microelectrode (a). Single-shock stimuli were applied to afferent fibers in stratum radiatum and adjusted to evoke threshold-straddling EPSPs (b; different trials with identical stimulation intensity are shown). B, A similar experiment in a bursting neuron (a) from an SE-experienced animal (n = 4 cells). All suprathreshold EPSPs elicited a burst discharge (b; three different trials with identical stimulation intensity; 1.4 V). Application of 100 μm Ni2+ blocked the intrinsic bursts induced by depolarizing current pulses (comparea, c). It also slightly reduced the amplitude of the EPSPs without affecting their duration (data not shown). To elicit threshold EPSPs in the presence of 100 μm Ni2+, stimulation intensity was increased to 1.5 V (d, top trace). Under these conditions, the EPSP-evoked firing responses were converted to single spikes (d; middle andbottom traces).
DISCUSSION
In this study we demonstrate that the intrinsic discharge behavior of hippocampal CA1 neurons is dramatically modified after pilocarpine-induced SE, in congruence with another recent study (Sanabria et al., 2001). This form of plasticity consists of a transition from regular firing to a burst-firing mode, in which the minimal response of >50% of the neurons to threshold depolarization is a burst of several spikes. We also show that this abnormal burst activity is resistant to blockers of L-, N-, and P/Q-types of voltage-sensitive Ca2+ channels but is readily suppressed by low concentrations of Ni2+ that block R- and T-types. Furthermore, we show that the appearance of Ni2+-sensitive intrinsic bursting is associated with a marked increase in the density of T-type, but not R-type, Ca2+ channels. Cumulatively, these data suggest that an increase in T-type Ca2+ current density plays a critical role in the intrinsic neuronal plasticity that is triggered by a single episode of SE.
Role of T-type Ca2+ current in intrinsic neuronal plasticity
It was shown previously that SE-induced intrinsic bursting in pilocarpine-treated animals is suppressed by removal of Ca2+ from, or the addition of 1 mm Ni2+ to, the ACSF, but not by the intracellular application of a Ca2+chelator, suggesting that this activity is driven directly by a Ca2+ current (Sanabria et al., 2001). Our further pharmacological experiments show that none of the organic Ca2+ channel blockers that affect L-, N-, and P-/Q-type Ca2+ channels, applied individually or in combination, blocked intrinsic bursting in this model. In contrast, both intrinsic bursting and its underlying spike ADP were suppressed by low concentrations of Ni2+ (30–100 μm), which block both T- and R-type Ca2+ channels in CA1 pyramidal cells. In addition, moderate depolarization of the neuron from its resting membrane potential, which would augment the steady-state inactivation of both T- and R-type Ca2+ channels (Schneider et al., 1994;Kozlov et al., 1999), abolished intrinsic bursting. These results by themselves are consistent with a role of either T- or R-type Ca2+ channels in intrinsic burst generation.
Two major lines of evidence, however, argue for a critical role of T-type Ca2+ channels in mediating this activity. First, the density of T-type Ca2+ current was considerably (1.9- to 3.3-fold) increased in CA1 pyramidal cells after SE, whereas the density of all other types of Ca2+ current (including the R-type) was either unaltered or decreased. Second, T-type Ca2+ currents display biophysical characteristics that make them particularly suitable to mediate burst discharges (Huguenard, 1996). Thus, T-type, but not R-type, Ca2+ channels display a slow rate of deactivation (time constants in the order of several milliseconds) at potentials close to the neuronal resting potential (Randall and Tsien, 1997; Kozlov et al., 1999). As a consequence of this feature, action potentials induce a slow tail Ca2+ current mediated by T-type Ca2+ currents. We have found that the increase in T-type Ca2+current amplitude after SE is associated with a marked and selective increase of the slowly deactivating tail current component after mock action potentials. Clearly, an increase in slow inward currents after action potentials could initiate a regenerative depolarization that would boost the ADP beyond spike threshold and elicit additional discharge. Additionally, the low threshold for activation and enhanced deinactivation during hyperpolarization of T-type Ca2+ channels would allow them to furnish the initial depolarizing drive for spontaneous burst discharges (Huguenard, 1996).
In normal CA1 pyramidal cells, intrinsic bursting mediated by Ca2+ currents has been demonstrated only after blocking outward K+ currents by millimolar concentrations of 4-aminopyridine (Magee and Carruth, 1999). However, in that condition, bursting was generated by both Ni2+-sensitive and Ni2+-insensitive Ca2+ currents (Magee and Carruth, 1999), reflecting the complement of voltage-gated Ca2+ channels normally expressed in these neurons. It is improbable, therefore, that a persistent reduction in outward K+ currents consequent to pilocarpine-induced SE, by itself, accounts for the change in discharge behavior reported here. However, changes in expression of voltage-gated channels other than Ca2+ channels also may contribute to the intrinsic neuronal plasticity. Indeed, it was found previously (Sanabria et al., 2001), and confirmed in the present study, that some CA1 neurons in SE-experienced animals display a Ca2+-independent burst mechanism. A likely candidate for mediating Ca2+-insensitive bursting in CA1 pyramidal cells is the persistent Na+ current (Azouz et al., 1996; Su et al., 2001), but the contribution of this current to SE-induced intrinsic plasticity has yet to be demonstrated.
Molecular basis of intrinsic neuronal plasticity
Our data allow us to propose that of the three known T-type Ca2+ channel α1subunits (i.e., α1H, α1I, and α1G), α1H most probably underlies the augmented T-type Ca2+ current and associated intrinsic bursting after pilocarpine-induced SE. It was shown that when expressed in human embryonic kidney cells, the Ca2+ currents mediated by the three cloned α1 subunits are differentially blocked by Ni2+, with the α1Hsubunit being ∼20-fold more sensitive (IC50values, 12 μm) than α1G and α1I subunits (IC50values, 250 and 216 μm, respectively) (Lee et al., 1999b). The T-type Ca2+ currents in CA1 pyramidal cells in both control and SE-experienced animals were highly sensitive to Ni2+(IC50 values of 24 and 28 μm, respectively), suggesting that α1H subunits contribute strongly to this current. Likewise, the slow (∼4–5 msec) Ca2+ tail currents that follow mock action potentials suggest the involvement of α1Hand/or α1G subunits, which have a comparably slow deactivation kinetics (Kozlov et al., 1999). Clearly, more experiments will be required to prove unequivocally the role of the α1H Ca2+ channel subunit in SE-induced intrinsic plasticity. Interestingly, it was shown recently that intrinsic bursting in thalamic neurons relies on expression of the α1G subunit (Kim et al., 2001); thus intrinsic bursting in different classes of neurons may be produced by distinct T-type Ca2+ channel subunits.
Functional consequences of altered intrinsic neuronal properties
Modifications of intrinsic neuronal properties dependent on neuronal activity have been described previously in invertebrate (Turrigiano et al., 1994) and vertebrate (Desai et al., 1999) culture models. In these studies, long-term deprivation of activity causes neurons to fire more rapidly in response to injected current (Desai et al., 1999) or even to switch from a tonic to a burst firing mode (Turrigiano et al., 1994). Such mechanisms are thought to be homeostatic, i.e.,. they serve to stabilize the properties of neural circuits (Stemmler and Koch, 1999; Turrigiano, 1999). The plasticity that we describe here represents the converse: SE produces a long-lasting gain in the input–output properties of CA1 neurons. Increased bursting is observed up to 90 d after status epilepticus (our unpublished data), suggesting that this form of plasticity may be stable over prolonged periods of time.
Computer modeling studies of realistic neuronal networks have previously suggested that intrinsic bursters are pivotal in the generation of epileptiform activity (Traub and Wong, 1982; Miles and Wong, 1983). There is also a large body of experimental evidence in support of this contention. Increased intrinsic bursting is observed in acute models of hippocampal epilepsy, such as the high-K+ model (Jensen et al., 1994, 1996;Azouz et al., 1996, 1997; Su et al., 2001). Moreover, intrinsic bursters were shown to be the forerunners of epileptiform eventsin vitro, suggesting that they may be important in initiating epileptiform discharges (Chagnac-Amitai and Connors, 1989;Jensen and Yaari, 1997; Sanabria et al., 2001). Therefore, identification of the mechanisms responsible for the abnormal intrinsic bursting in epileptic tissue may permit pharmacological inhibition of seizure initiation in experimental and human epilepsy. It should be noted, however, that an episode of SE induced by pilocarpine or other procedures triggers long-term changes in synaptic function that may also play a role in the development of an epileptic condition (McNamara, 1999).
In summary, our observations suggest that the transformation of CA1 pyramidal cells from regular firing to burst-firing after pilocarpine-induced SE involves the persistent augmentation of a T-type Ca2+ current, possibly that mediated by the α1H subunit. This SE-induced intrinsic plasticity leads to a marked change in the input–output properties of hippocampal neurons that, in concert with altered synaptic connectivity, may underlie the hyperexcitability characteristic of TLE. Furthermore, these results suggest that subunit-selective T-type Ca2+ channel blockers may be promising targets for rational anti-epileptic drug design in TLE.
H.S. and D.S. contributed equally to this work.
This research was supported by the German–Israel collaborative research program of the Ministry of Science and the Bundesministerium für Bildung und Forschung, Deutsche Forschungsgemeinschaft EL 122/7, the SFB 6006, the Israel Science Foundation, a University of Bonn Medical Center “BONFOR” grant, the Erna D. and Henry J. Leir Chair for Research in Neurodegenerative Diseases, and a Humboldt Research Award to Y.Y. We thank E. Perez-Reyes, T. Schneider, and A. Konnerth for critical comments.
Correspondence should be addressed to Dr. Heinz Beck, Department of Epileptology, University of Bonn Medical Center, Sigmund-Freud Strasse 25, D-53105 Bonn, Germany. E-mail:heinz.beck@ukb.uni-bonn.de.
REFERENCES
- 1.Avery RB, Johnston D. Multiple channel types contribute to the low-voltage-activated calcium current in hippocampal CA3 pyramidal neurons. J Neurosci. 1996;16:5567–5582. doi: 10.1523/JNEUROSCI.16-18-05567.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azouz R, Jensen MS, Yaari Y. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol (Lond) 1996;492:211–223. doi: 10.1113/jphysiol.1996.sp021302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azouz R, Alroy G, Yaari Y. Modulation of endogenous firing patterns by osmolarity in rat hippocampal neurones. J Physiol (Lond) 1997;502:175–187. doi: 10.1111/j.1469-7793.1997.175bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck H, Steffens R, Heinemann U, Elger CE. Ca2+-dependent inactivation of high-threshold Ca2+ currents in hippocampal granule cells of patients with chronic temporal lobe epilepsy. J Neurophysiol. 1999;82:946–954. doi: 10.1152/jn.1999.82.2.946. [DOI] [PubMed] [Google Scholar]
- 5.Ben Ari Y. Cell death and synaptic reorganizations produced by seizures. Epilepsia. 2001;42[Suppl 3]:5–7. doi: 10.1046/j.1528-1157.2001.042suppl.3005.x. [DOI] [PubMed] [Google Scholar]
- 6.Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- 7.Chagnac-Amitai Y, Connors BW. Synchronized excitation and inhibition driven by intrinsically bursting neurons in neocortex. J Neurophysiol. 1989;62:1149–1162. doi: 10.1152/jn.1989.62.5.1149. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Chad JE, Cannon RC, Wheal HV. Reduced Mg2+ blockade of synaptically activated N-methyl-daspartate receptor-channels in CA1 pyramidal neurons in kainic acid-lesioned rat hippocampus. Neuroscience. 1999;88:727–739. doi: 10.1016/s0306-4522(98)00253-x. [DOI] [PubMed] [Google Scholar]
- 9.Christie BR, Eliot LS, Ito K, Miyakawa H, Johnston D. Different Ca2+ channels in soma and dendrites of hippocampal pyramidal neurons mediate spike-induced Ca2+ influx. J Neurophysiol. 1995;73:2553–2557. doi: 10.1152/jn.1995.73.6.2553. [DOI] [PubMed] [Google Scholar]
- 10.Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Ben Ari Y, Esclapez M, Bernard C. Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci. 2001;4:52–62. doi: 10.1038/82900. [DOI] [PubMed] [Google Scholar]
- 11.Coulter DA, DeLorenzo RJ. Basic mechanisms of status epilepticus. Adv Neurol. 1999;79:725–733. [PubMed] [Google Scholar]
- 12.Cribbs LL, Lee JH, Yang J, Satin J, Zhang Y, Daud A, Barclay J, Williamson MP, Fox M, Rees M, Perez-Reyes E. Cloning and characterization of α1H from human heart, a member of the T-type Ca2+ channel gene family. Circ Res. 1998;83:103–109. doi: 10.1161/01.res.83.1.103. [DOI] [PubMed] [Google Scholar]
- 13.Desai NS, Rutherford LC, Turrigiano GG. Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci. 1999;2:515–520. doi: 10.1038/9165. [DOI] [PubMed] [Google Scholar]
- 14.Fisher RE, Gray R, Johnston D. Properties and distribution of single voltage-gated calcium channels in adult hippocampal neurons. J Neurophysiol. 1990;64:91–104. doi: 10.1152/jn.1990.64.1.91. [DOI] [PubMed] [Google Scholar]
- 15.Gibbs JW, III, Shumate M, Coulter D. Differential epilepsy-associated alterations in postsynaptic GABAA receptor function in dentate granule and CA1 neurons. J Neurophysiol. 1997;77:1924–1938. doi: 10.1152/jn.1997.77.4.1924. [DOI] [PubMed] [Google Scholar]
- 16.Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol. 1996;58:329–348. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- 17.Jensen MS, Yaari Y. Role of intrinsic burst firing, potassium accumulation, and electrical coupling in the elevated potassium model of hippocampal epilepsy. J Neurophysiol. 1997;77:1224–1233. doi: 10.1152/jn.1997.77.3.1224. [DOI] [PubMed] [Google Scholar]
- 18.Jensen MS, Azouz R, Yaari Y. Variant firing patterns in rat hippocampal pyramidal cells modulated by extracellular potassium. J Neurophysiol. 1994;71:831–839. doi: 10.1152/jn.1994.71.3.831. [DOI] [PubMed] [Google Scholar]
- 19.Jensen MS, Azouz R, Yaari Y. Spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol (Lond) 1996;492:199–210. doi: 10.1113/jphysiol.1996.sp021301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, McEnery MW, Shin HS. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking α1G T-type Ca2+ channels. Neuron. 2001;31:35–45. doi: 10.1016/s0896-6273(01)00343-9. [DOI] [PubMed] [Google Scholar]
- 21.Kozlov AS, McKenna F, Lee JH, Cribbs LL, Perez-Reyes E, Feltz A, Lambert RC. Distinct kinetics of cloned T-type Ca2+ channels lead to differential Ca2+ entry and frequency-dependence during mock action potentials. Eur J Neurosci. 1999;11:4149–4158. doi: 10.1046/j.1460-9568.1999.00841.x. [DOI] [PubMed] [Google Scholar]
- 22.Lee JH, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, Schneider T, Perez-Reyes E. Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. J Neurosci. 1999a;19:1912–1921. doi: 10.1523/JNEUROSCI.19-06-01912.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E. Nickel block of three cloned T-type calcium channels: low concentrations selectively block α1H. Biophys J. 1999b;77:3034–3042. doi: 10.1016/S0006-3495(99)77134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lothman EW, Rempe DA, Mangan PS. Changes in excitatory neurotransmission in the CA1 region and dentate gyrus in a chronic model of temporal lobe epilepsy. J Neurophysiol. 1995;74:841–848. doi: 10.1152/jn.1995.74.2.841. [DOI] [PubMed] [Google Scholar]
- 25.Magee JC, Carruth M. Dendritic voltage-gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1999;82:1895–1901. doi: 10.1152/jn.1999.82.4.1895. [DOI] [PubMed] [Google Scholar]
- 26.Mangan PS, Rempe DA, Lothman EW. Changes in inhibitory neurotransmission in the CA1 region and dentate gyrus in a chronic model of temporal lobe epilepsy. J Neurophysiol. 1995;74:829–840. doi: 10.1152/jn.1995.74.2.829. [DOI] [PubMed] [Google Scholar]
- 27.McNamara JO. Emerging insights into the genesis of epilepsy. Nature. 1999;399:A15–A22. doi: 10.1038/399a015. [DOI] [PubMed] [Google Scholar]
- 28.Miles R, Wong RK. Single neurones can initiate synchronized population discharge in the hippocampus. Nature. 1983;306:371–373. doi: 10.1038/306371a0. [DOI] [PubMed] [Google Scholar]
- 29.Nakashima YM, Todorovic SM, Pereverzev A, Hescheler J, Schneider T, Lingle CJ. Properties of Ba2+ currents arising from human α1E and α1Eβ3 constructs expressed in HEK293 cells: physiology, pharmacology, and comparison to native T-type Ba2+ currents. Neuropharmacology. 1998;37:957–972. doi: 10.1016/s0028-3908(98)00097-5. [DOI] [PubMed] [Google Scholar]
- 30.Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee JH. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- 31.Qian J, Noebels JL. Presynaptic Ca2+ channels and neurotransmitter release at the terminal of a mouse cortical neuron. J Neurosci. 2001;21:3721–3728. doi: 10.1523/JNEUROSCI.21-11-03721.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Randall AD, Tsien RW. Contrasting biophysical and pharmacological properties of T-type and R- type calcium channels. Neuropharmacology. 1997;36:879–893. doi: 10.1016/s0028-3908(97)00086-5. [DOI] [PubMed] [Google Scholar]
- 33.Sanabria ERG, Su H, Yaari Y (2001) Initiation of network bursts by Ca2+-dependent intrinsic bursting in the rat pilocarpine model of temporal lobe epilepsy. J Physiol (Lond) 205–216. [DOI] [PMC free article] [PubMed]
- 34.Schneider T, Wei X, Olcese R, Costantin JL, Neely A, Palade P, Perez-Reyes E, Qin N, Zhou J, Crawford GD, Smith RG, Appel SH, Stefani E, Birnbaumer L. Molecular analysis and functional expression of the human type E neuronal Ca2+ channel alpha 1 subunit. Receptors Channels. 1994;2:255–270. [PubMed] [Google Scholar]
- 35.Sloviter RS. Status epilepticus-induced neuronal injury and network reorganization. Epilepsia. 1999;40[Suppl 1]:S34–S39. doi: 10.1111/j.1528-1157.1999.tb00876.x. [DOI] [PubMed] [Google Scholar]
- 36.Soong TW, Stea A, Hodson CD, Dubel SJ, Vincent SR, Snutch TP. Structure and functional expression of a member of the low voltage-activated calcium channel family. Science. 1993;260:1133–1136. doi: 10.1126/science.8388125. [DOI] [PubMed] [Google Scholar]
- 37.Stemmler M, Koch C. How voltage-dependent conductances can adapt to maximize the information encoded by neuronal firing rate. Nat Neurosci. 1999;2:521–527. doi: 10.1038/9173. [DOI] [PubMed] [Google Scholar]
- 38.Su H, Alroy G, Kirson ED, Yaari Y. Extracellular calcium modulates persistent sodium current-dependent intrinsic bursting in rat hippocampal neurons. J Neurosci. 2001;21:4173–4182. doi: 10.1523/JNEUROSCI.21-12-04173.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- 40.Takahashi K, Wakamori M, Akaike N. Hippocampal CA1 pyramidal cells of rats have four voltage-dependent calcium conductances. Neurosci Lett. 1989;104:229–234. doi: 10.1016/0304-3940(89)90359-5. [DOI] [PubMed] [Google Scholar]
- 41.Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson SM, Schwindt W. Development of calcium current subtypes in isolated rat hippocampal pyramidal cells. J Physiol (Lond) 1991;439:671–689. doi: 10.1113/jphysiol.1991.sp018687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Traub RD, Wong RK. Cellular mechanism of neuronal synchronization in epilepsy. Science. 1982;216:745–747. doi: 10.1126/science.7079735. [DOI] [PubMed] [Google Scholar]
- 44.Turner DA, Wheal HV. Excitatory synaptic potentials in kainic acid-denervated rat CA1 pyramidal neurons. J Neurosci. 1991;11:2786–2794. doi: 10.1523/JNEUROSCI.11-09-02786.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turrigiano G, Abbott LF, Marder E. Activity-dependent changes in the intrinsic properties of cultured neurons. Science. 1994;264:974–977. doi: 10.1126/science.8178157. [DOI] [PubMed] [Google Scholar]
- 46.Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci. 1999;22:221–227. doi: 10.1016/s0166-2236(98)01341-1. [DOI] [PubMed] [Google Scholar]
- 47.Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res. 1983;9:315–335. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- 48.Williams ME, Marubio LM, Deal CR, Hans M, Brust PF, Philipson LH, Miller RJ, Johnson EC, Harpold MM, Ellis SB. Structure and functional characterization of neuronal alpha 1E calcium channel subtypes. J Biol Chem. 1994;269:22347–22357. [PubMed] [Google Scholar]
- 49.Wong RK, Prince DA. Afterpotential generation in hippocampal pyramidal cells. J Neurophysiol. 1981;45:86–97. doi: 10.1152/jn.1981.45.1.86. [DOI] [PubMed] [Google Scholar]
- 50.Wu G-L, Saggau P. Pharmacological identification of two types of presynaptic voltage-dependent calcium channels at CA3–CA1 synapses of the hippocampus. J Neurosci. 1994;14:5613–5622. doi: 10.1523/JNEUROSCI.14-09-05613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yaari Y, Hamon B, Lux HD. Development of two types of calcium channels in cultured mammalian hippocampal neurons. Science. 1987;235:680–682. doi: 10.1126/science.2433765. [DOI] [PubMed] [Google Scholar]