

Abstract
Stress affects seizure susceptibility in animals and humans, but the underlying mechanisms are obscure. Here, we provide evidence that GABAA receptor-modulating neurosteroids derived from deoxycorticosterone (DOC) play a role in stress-related changes in seizure control. DOC, an adrenal steroid whose synthesis is enhanced during stress, undergoes sequential metabolic reduction by 5α-reductase and 3α-hydroxysteroid oxidoreductase to form 5α-dihydrodeoxycorticosterone (DHDOC) and allotetrahydrodeoxycorticosterone (THDOC), a GABAAreceptor-modulating neurosteroid with anticonvulsant properties. Acute swim stress in rats significantly elevated plasma THDOC concentrations and raised the pentylenetetrazol (PTZ) seizure threshold. Small systemic doses of DOC produced comparable increases in THDOC and PTZ seizure threshold. Pretreatment with finasteride, a 5α-reductase inhibitor that blocks the conversion of DOC to DHDOC, reversed the antiseizure effects of stress. DOC also elevated plasma THDOC levels and protected mice against PTZ, methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate, picrotoxin, and amygdala-kindled seizures in mice (ED50 values, 84–97 mg/kg). Finasteride reversed the antiseizure activity of DOC (ED50, 7.2 mg/kg); partial antagonism was also obtained with indomethacin (100 mg/kg), an inhibitor of 3α-hydroxysteroid oxidoreductase. Finasteride had no effect on seizure protection by DHDOC and THDOC, whereas indomethacin partially reversed DHDOC but not THDOC. DHDOC, like THDOC, potentiated GABA-activated Cl− currents in cultured hippocampal neurons (≤1 μm) and directly activated GABAAreceptor currents (≥1 μm), compatible with a role for DHDOC in the antiseizure activity of DOC. DOC is a mediator of the physiological effects of acute stress that could contribute to stress-induced changes in seizure susceptibility through its conversion to neurosteroids with modulatory actions on GABAA receptors including THDOC and possibly also DHDOC.
Keywords: stress; seizure; pentylenetetrazol; kindling; neurosteroid; deoxycorticosterone; 5α-dihydrodeoxycorticosterone; 5α,3αtetrahydrodeoxycorticosterone (3α,21-dihydroxy-5α-pregnan-20-one); finasteride; indomethacin; GABAA receptor
Neurosteroids such as the progesterone metabolite allopregnanolone (3α-hydroxy-5α-pregnane-20-one) and the deoxycorticosterone (DOC) metabolite allotetrahydrodeoxycorticosterone (THDOC; 3α,21-dihydroxy-5α-pregnan-20-one) are powerful endogenous positive modulators of GABAA receptors with anticonvulsant, anxiolytic, and sedative properties (Crawley et al., 1986; Bitran et al., 1991; Kokate et al., 1994; Lambert et al., 1995;Reddy and Kulkarni, 2000). Although there is emerging evidence that allopregnanolone has a role in the pathophysiology of menstrual cycle-related disorders such as catamenial epilepsy (Reddy and Rogawski, 2000; Reddy et al., 2001) and premenstrual syndrome (Smith et al., 1998), the clinical importance of THDOC remains unclear. THDOC is synthesized from DOC, an adrenal steroid, by two sequential A-ring reductions. 5α-Reductase isoenzymes first convert DOC to the intermediate 5α-dihydrodeoxycorticosterone (DHDOC), which is then further reduced by 3α-hydroxysteroid oxidoreductase to form THDOC (Fig. 1). In contrast to allopregnanolone, which is present in the brain even after adrenalectomy and gonadectomy, THDOC appears to be derived nearly exclusively from adrenal sources (Purdy et al., 1991).
Fig. 1.

Sequential enzymatic conversion of DOC to DHDOC and THDOC by 5α-reductase and 3α-hydroxysteroid oxidoreductase. Finasteride is a specific irreversible inhibitor of 5α-reductase; indomethacin competitively blocks 3α-hydroxysteroid oxidoreductase.
Adrenocorticotrophic hormone, released by the activation of the hypothalamic–pituitary–adrenal axis, is a key coordinator of neuroendocrine and behavioral responses to stress. Although DOC is an intermediate in aldosterone synthesis in the adrenal zona glomerulosa and itself has weak mineralocorticoid activity, much more DOC is produced in the zona fasciculata, where its synthesis is under the control of adrenocorticotrophic hormone, and its secretion correlates with that of glucocorticoids and not aldosterone (Tan and Mulrow, 1975;Kater et al., 1989). Thus, in addition to its well recognized role as a mineralocorticoid precursor, there is substantial evidence that DOC participates in the hypothalamic–pituitary–adrenal axis response to acute stress. Presumably because of enhanced DOC availability, acute stressors such as swimming, foot shock, or carbon dioxide exposure elicit an increase in THDOC concentrations in plasma and brain (Purdy et al., 1991; Barbaccia et al., 1996, 1997; Serra et al., 2000; Vallee et al., 2000). It has therefore been proposed that THDOC, through its actions on GABAA receptors, could play a physiological role in mediating the effects of stress on CNS function.
Although the underlying mechanisms are poorly understood, it is well recognized that emotional stress can be a factor affecting seizure control in temporal lobe epilepsy and other seizure syndromes (Feldman and Paul, 1976; Minter, 1979; Temkin and Davis, 1984; Frucht et al., 2000). Moreover, experimental stress, including swim stress, has anticonvulsant effects in animals (Goldberg and Salama, 1969; Soubrie et al., 1980; Abel and Berman, 1993; Peric̆ićet al., 2000, 2001). THDOC exhibits anticonvulsant activity in a variety of animal seizure models (Kokate et al., 1994, 1996), and it is attractive to speculate that DOC-derived THDOC could play a role in the effects of stress on seizure susceptibility. This would imply that DOC itself should have anticonvulsant properties. In fact, shortly after its isolation and chemical synthesis (von Steiger and Reichstein, 1937), DOC was reported to protect against pentylenetetrazol (PTZ) seizures in rats (Selye, 1942; Craig, 1966). However, the mechanism of this anticonvulsant action has remained obscure. In this study, we investigate the role of DOC and its neurosteroid metabolite THDOC in the regulation of seizure susceptibility by stress. Our results demonstrate that the previously observed anticonvulsant activity of DOC in the PTZ seizure model is attributable to its conversion to neurosteroids that promote GABAergic synaptic inhibition. Moreover, this pathway is likely to be physiologically relevant in mediating the effects of stress on seizure susceptibility.
MATERIALS AND METHODS
Animal testing and plasma THDOC determinations
Animals. Male Sprague Dawley rats (250–300 gm) were purchased from Taconic (Germantown, NY). Male National Institutes of Health (NIH) Swiss mice (25–30 gm) were obtained from the NIH animal program. Male adrenalectomized Swiss mice were purchased from Taconic. Animals were allowed to acclimatize with access to food and waterad libitum for at least 24 hr before use and were group-housed under a 12 hr light/dark cycle in an environmentally controlled animal facility. All procedures were performed in strict compliance with the NIH Guide for the Care and Use of Laboratory Animals under a protocol approved by the National Institutes of Neurological Disorders and Stroke Animal Care and Use Committee.
Swim stress and PTZ seizure threshold determination. Rats were subjected to swim stress (10 min) in water at ambient temperature (22°C) in an acrylic cylindrical container (36 × 54 inches) filled to 75% capacity. After swimming, the rats were gently dried with towels and warmed by a heating lamp. After 15 min, the tail vein was cannulated with a 25 gauge butterfly needle and the animals were infused with 20 mg/ml PTZ solution in 0.9% sterile saline at a rate of 0.5 ml/min using a Harvard Apparatus (Holliston, MA) syringe infusion pump. The times between the start of infusion and the onset of myoclonic forelimb clonus were recorded in seconds, and the threshold convulsant dose in milligrams of PTZ per kilogram was calculated.
Estimation of THDOC. Mice were anesthetized with CO2 gas, and 2 ml of carotid blood was collected in heparinized tubes. The plasma was separated by centrifugation at 12,000 × g for 10 min and stored at −20°C in 10 ml glass tubes containing 7.5% EDTA solution (68 μl). The concentration of THDOC was quantified by liquid chromatography–mass spectrometry using a Hewlett-Packard (Palo Alto, CA) liquid chromatograph (analytical column: Genesis C18, 4 μm, 3 × 30 mm; Jones Chromatography, Lakewood, CO) and a Micromass Quattro II mass spectrometer (Reddy and Rogawski, 2000). Briefly, a 200 μl plasma sample was added to a tube containing evaporated internal standard (5β,3α-pregnanolone). The steroid and internal standard were extracted with 4 ml of hexane. Each sample was analyzed using the atmospheric pressure chemical ionization technique under acidic conditions. A standard curve was plotted using pure THDOC in methanol mixed with 0.2 ml of blank plasma. Plasma samples that had levels of THDOC below the detection limit (5 ng/ml) were spiked with 20 ng of THDOC, which was subtracted from the final readings.
Chemoconvulsant seizure tests. DOC, DHDOC, and THDOC were evaluated for their ability to protect against subcutaneous PTZ (85 mg/kg)-induced clonic seizures in mice (White et al., 1995). DOC was also evaluated against seizures induced by the GABAA receptor antagonists picrotoxin (3.2 mg/kg, s.c.) and methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate (DMCM; 7.5 mg/kg, s.c.). In brief, mice were injected intraperitoneally with the test compound and 15 min (DHDOC and THDOC) or 30 min (DOC) later, or at the indicated intervals in the time course studies, received an injection of PTZ, picrotoxin, or DMCM. Animals were then observed for a 30 min period. Mice failing to show clonic spasms lasting >5 sec were scored as protected.
Maximal electroshock seizure test. Thirty min after intraperitoneal injection of DOC or 15 min after DHDOC and THDOC, mice were subjected to a 50 mA, 0.2 sec, 60 Hz electrical stimulus via corneal electrodes (5-mm-diameter stainless steel balls) wetted with normal saline. Animals failing to show tonic hindlimb extension were scored as protected.
Amygdala kindling. Electrode implantation and stimulation procedures for mouse amygdala kindling were as described previously (Rogawski et al., 2001). Briefly, mice were anesthetized by intraperitoneal injection of a mixture of ketamine (100 mg/kg) and xylazine (20 mg/kg). A twisted bipolar stainless steel wire electrode (model MS303/1; Plastic One, Roanoke, VA) was stereotaxically implanted in the right amygdala complex (1.3 mm posterior and 3.0 mm lateral to bregma and 4.6 mm below the dorsal surface of the skull) and anchored with dental acrylic to three jeweler's screws placed in the skull. A period of 7–10 d was allowed for recovery. The stimulation paradigm consisted of 1-msec-duration, bipolar, square current pulses delivered at 60 Hz for 1 sec. The afterdischarge threshold was determined by stimulating at 5 min intervals beginning with an intensity of 75 μA and increasing in steps of 50 μA until an afterdischarge of at least 5 sec was obtained. Stimulation on subsequent days used a stimulation intensity of 125% of the threshold value. Seizure activity after each stimulation was rated according to the criterion of Racine (1972) as modified for the mouse: stage 0, no response or behavior arrest; stage 1, chewing or head nodding; stage 2, chewing and head nodding; stage 3, forelimb clonus; stage 4, bilateral forelimb clonus and rearing; stage 5, falling. The afterdischarge was recorded from the amygdala electrode with a Grass CP511 AC electroencephalogram preamplifier (Astro-Med, West Warwick, RI) and stored in digital form using Axotape 2.02 (Axon Instruments, Foster City, CA). Kindling stimulation was delivered daily until stage 5 seizures were elicited on 3 consecutive days. To examine the ability of neurosteroids to suppress the expression of kindled seizures, the kindled mice underwent a 5 d test protocol. On the first day, they were verified to exhibit a stimulation-induced stage 5 seizure. They were then tested on days 2 and 4 after having received an intraperitoneal injection of DOC or THDOC, respectively, 30 or 15 min before stimulation. The animals received control stimulations on days 3 and 5. The two seizure scores after drug administration and the three control seizure scores were averaged.
Motor toxicity test. Motor toxicity was evaluated using a modification of the horizontal screen test (Coughenour et al., 1977) that determines an animal's ability to support its own body weight by grasping a grid. Mice were placed on a horizontally oriented grid (consisting of parallel 1.5-mm-diameter rods situated 1 cm apart), and the grid was inverted. Animals that fell from the grid within 1 min were scored as positive. Drug-free mice never fell from the grid.
Drugs. Stock solutions of steroids for injection were made in 30% hydroxypropyl-β-cyclodextrin (β-cyclodextrin) in water, and additional dilutions were made using normal saline. By itself, β-cyclodextrin at concentrations as high as 45% failed to affect PTZ-, maximal electroshock (MES)-, or kindling-induced seizures. Indomethacin was suspended in 0.1% methylcellulose solution, which per se had no significant effect on PTZ-induced seizures. Drug solutions were administered in a volume equaling 1% of the animal's body weight. All drugs and chemicals were obtained from Sigma (St. Louis, MO).
Data analysis. Plasma THDOC concentration data are expressed as the mean ± SEM. The significance of differences in the mean plasma concentrations and mean PTZ doses in the PTZ threshold test was assessed by one-way ANOVA, followed by Dunnett's t test. ED50 values (the doses at which 50% of tested animals were protected from seizures) and CD50 values (the doses at which 50% of tested animals exhibited seizures) with 95% confidence limits were determined by log-probit analysis using the Litchfield and Wilcoxon procedure. In the construction of dose–response curves, at least seven animals were tested at each dose. The significance of differences between steroid dose–response curves in the PTZ seizure test was assessed with the Litchfield and Wilcoxon χ2 test. Kindling group data are expressed as the mean ± SEM; the means were compared by the Mann–Whitney U test. In all tests, the criterion for statistical significance was p < 0.05. Statistical analyses were performed with PHARM/PCS version 4.2 (Microcomputer Specialists, Philadelphia, PA).
Cellular electrophysiology
The effect of neurosteroids on the GABAAreceptor Cl− currents was evaluated in cultured hippocampal neurons using whole-cell patch-clamp recording. Cultures of hippocampal neurons were prepared from 19-d-old rat embryos according to previously published methods (Segal, 1983; Donevan et al., 1992). Hippocampi were dissected and triturated in modified minimal essential medium with Earle's salt (Advanced Biotechnologies Inc., Columbia, MD) by repeated passage through a 10 ml pipette. The cell suspension was then plated onto 35 mm polystyrene Petri dishes (Falcon 3001; Becton Dickinson Labware, Oxnard, CA) precoated with Matrigel at a density corresponding to 1–1.5 hippocampus per dish. The plating medium was supplemented with horse serum (Invitrogen, San Diego, CA), fetal calf serum, and N3 (composed of transferrin, putrescine, sodium selenite, triiodothyronine, insulin, progesterone, and corticosterone). Cell cultures were placed in a humidified atmosphere containing 10% CO2 at 37°C for 6–12 d before use. Fresh growth medium (without fetal calf serum and N3) was added after 6 d in culture.
Electrophysiological recordings from neurons were performed in 35 mm tissue culture dishes on the stage of an inverted phase-contrast microscope (Nikon Diaphot; Nikon, Tokyo, Japan) at room temperature (23–25°C). Before each experiment, the culture medium was replaced with filtered extracellular solution composed of 142 mmNaCl, 8.1 mm CsCl, 1 mmCaCl2, 6 mmMgCl2, 10 mm HEPES, and 1 μm tetrodotoxin. The pH of the solution was adjusted to 7.3 using NaOH and to an osmolality of 325–330 mOsm/kg H2O by adding sucrose. Patch electrodes (prepared from 1.5 mm borosilicate glass capillaries with tip openings of ∼2 μm and resistances of 3–8 MΩ) were filled with an intracellular solution composed of (in mm): 153 CsCl, 1 MgCl2, 5 EGTA, and 10 HEPES. The pH of the pipette solution was adjusted to 7.3 using CsOH and to an osmolality of 315–320 mOsm/kg H2O using sucrose. Medium-sized neurons with two or three large processes were selected for study. Recordings were made using an Axopatch 200A amplifier (Axon Instruments, Burlingame, CA). Membrane currents were filtered at 1 kHz (−3 dB, four-pole, low-pass Bessel filter), and the corresponding voltages were acquired in digitized form using an on-line data acquisition system (pClamp, Axon Instruments).
Drug solutions were applied using a microprocessor-controlled, gravity-fed, multibarrel microperfusion system that allowed rapid (1 msec) switching between bathing medium and solutions of various test substances alone or in combination. The perfusion pipette tip (diameter ∼500 μm) was positioned 400–500 μm from the cell surface. To examine modulation of GABA responses, a 10 sec preapplication of either external buffer or steroid solution was followed by a 10 sec application of GABA plus steroid, followed by a 10 sec wash with external buffer solution. To examine the direct actions of the steroids, the steroids were continuously perfused for 20 sec. Stock solutions of steroids were prepared in dimethylsulfoxide (DMSO) and diluted to final concentration in extracellular solution (DMSO concentration < 0.01–0.1%). To avoid artifacts caused by effects of DMSO on the steroid-induced currents, all other solutions, including GABA, also contained 0.05% DMSO. Applications of GABA and steroids either alone or in combination were separated by a time interval of at least 1 min to minimize desensitization.
Fractional potentiation produced by the steroids was calculated as (IS −IGABA)/IGABA, where IGABA is the peak amplitude of the control GABA response and IS is the peak amplitude of the response produced during coapplication of GABA and the test steroid. The estimated concentration producing a doubling in the amplitude of the GABA response (EC100) was determined by interpolation from the concentration-response data. Concentration-response data for direct activation of Cl− currents were fitted using the Levenberg–Marquardt nonlinear least-squares method to the logistic equation F =Fmax/{1 + (EC50/[S])nH}, where F represents the ratio of potentiated peak current and the peak current evoked by 10 μm GABA (IGABA),Fmax is the maximum ratio, [S] is the steroid (or GABA) concentration, andnH is an empirical parameter describing the steepness of fit (equivalent to the Hill coefficient). Data are presented as the mean ± SEM.
RESULTS
Swim stress elevation of PTZ seizure threshold and plasma THDOC levels in rats: reversal by finasteride
Acute swim stress (10 min) resulted in a significant increase (22%; p < 0.05) in the threshold for PTZ-induced clonic seizures (Fig. 2). To examine whether the effect of stress on seizure susceptibility is related to neurosteroids, male rats were pretreated by intraperitoneal injection 90 min before the start of swimming (115 min before PTZ infusion) with 100 mg/kg finasteride, an irreversible inhibitor of types I and II 5α-reductase in rodents (Normington and Russell, 1992; Thigpen and Russell, 1992; Azzolina et al., 1997), which has been shown previously to inhibit neurosteroid synthesis (Kokate et al., 1999). Finasteride did not affect the seizure threshold of nonstressed (naive) animals. It did, however, completely prevent the stress-induced elevation of seizure threshold. Moreover, in finasteride-treated stressed rats, the seizure threshold was reduced significantly below the threshold in nonstressed animals.
Fig. 2.

Acute swim stress elevates clonic seizure threshold and plasma THDOC levels: inhibition by finasteride. Rats were subjected to swimming for 10 min in water at ambient temperature, and the seizure threshold was estimated 15 min later by intravenous infusion of PTZ. Immediately after seizure occurrence, plasma samples were collected for THDOC determination by liquid chromatography–mass spectrometry. Pretreatment with finasteride (100 mg/kg, i.p.) 90 min before swim stress significantly decreased the stress-induced increase of THDOC level and seizure threshold. Data represent mean ± SEM (n = 8 per group). *p < 0.05 versus naive controls (Dunnett's t test); others are not significantly different from naive controls.
Swim stress was associated with an increase in THDOC plasma levels at the time of seizure testing (Fig. 2). In naive rats, THDOC plasma levels were only slightly and nonsignificantly decreased by pretreatment with finasteride. However, finasteride completely prevented the stress-induced increase in plasma THDOC (p < 0.01). The ability of finasteride to prevent the stress-induced increase in seizure threshold and rise in THDOC suggested that acute stress modulates seizure susceptibility by enhancing 5α-pregnane-derived neurosteroids such as THDOC. Because THDOC is derived from DOC, these results raised the possibility that the anticonvulsant effects of acute stress could be mediated by increased DOC synthesis or availability.
DOC-induced elevation of PTZ seizure threshold in rats: correlation with plasma THDOC levels
To evaluate the possibility that the anticonvulsant effects of swim stress are related to enhanced DOC synthesis and conversion to THDOC, we sought to confirm that DOC itself has anticonvulsant activity that is correlated with THDOC levels. Intraperitoneal administration of DOC (1.9–30 mg/kg) in rats caused a dose-dependent elevation of the PTZ seizure threshold (Fig. 3). There was a corresponding dose-dependent increase in plasma THDOC levels. The lowest DOC dose tested (1.9 mg/kg) caused a 12% elevation in seizure threshold (p < 0.05) and was associated with an increase in mean plasma THDOC from 3.8 to 27 ng/ml (p < 0.001).
Fig. 3.

Systemic DOC administration elevates clonic seizure threshold and plasma THDOC levels. Rats were injected intraperitoneally with vehicle (control) or various doses of DOC. Thirty minutes later, the seizure threshold was estimated by intravenous infusion of PTZ. Immediately after seizure occurrence, plasma samples were collected for THDOC determination by liquid chromatography–mass spectrometry. Data represent mean ± SEM (n = 7–8 per group). *p < 0.05 versus control; **p < 0.01 versus control (Dunnett's t test). Plasma THDOC values are all significantly different from control (p < 0.001).
Anticonvulsant actions of DOC in mice
Additional anticonvulsant testing was performed using an all-or-none mouse model with an ED97 dose of PTZ (85 mg/kg, s.c.). This model provides greater reproducibility and allows quantification of anticonvulsant ED50values but is less sensitive to the anticonvulsant effects of neurosteroids. As in the rat PTZ threshold model, acute administration of DOC protected male mice against PTZ-induced clonic seizures in a dose-dependent manner (Fig. 4); the ED50 value determined by log-probit analysis is presented in Table 1. THDOC plasma levels were determined in a separate group of animals at the same time as the initiation of the seizure testing. For increasing doses of DOC, there was a corresponding dose-dependent increase in plasma THDOC levels that was highly correlated with the dose–response relationship for seizure protection in the PTZ test (r = 0.91). The interpolated plasma THDOC level at the ED50 dose of DOC is 595 ng/ml.
Fig. 4.

Protective activity of DOC in the PTZ seizure test correlates with plasma THDOC levels. DOC (25–200 mg/kg, i.p.) was administered to male mice 30 min before injection of PTZ (85 mg/kg, s.c.) or plasma collection. Each point in the seizure test curve represents eight mice; each point in the plasma THDOC curve represents the mean ± SEM of THDOC determinations in a separate group of six mice. There is a high correlation (r = 0.91;p < 0.001) between the dose–response relationship for seizure protection and the mean plasma THDOC levels.
Table 1.
ED50 values of DOC, DHDOC, and THDOC in seizure and motor toxicity tests in mice
| DOC (mg/kg) | DHDOC (mg/kg) | THDOC (mg/kg) | |
|---|---|---|---|
| Seizure tests | |||
| PTZ | 84 (68–104) | 26 (21–32) | 19 (12–28) |
| Picrotoxin | 97 (77–122) | ND | ND |
| DMCM | 88 (62–125) | ND | ND |
| MES | 108 (35–166) | 58 (48–71) | 48 (35–66) |
| Screen test | 73 (57–94) | 27 (20–36) | 57 (37–90) |
DOC was administered 30 min before testing; DHDOC and THDOC were administered 15 min before testing. In the seizure tests, mice that failed to show clonic spasms during a 30 min period after injection of the chemoconvulsant were scored as protected. ED50 values were determined according to the Litchfield and Wilcoxon procedure. Numbers in parentheses are 95% confidence intervals. ND, Not determined. DOC (≥300 mg/kg, i.p.) had no protective activity against seizures induced by strychnine (1.3 mg/kg, s.c.), kainic acid (32 mg/kg, s.c.), 4-aminopyridine (13.3 mg/kg, s.c.), and NMDA (257 mg/kg, s.c.).
Activity of DOC in other chemoconvulsant models, MES test, and screen test
In addition to characterizing its actions in the PTZ seizure test, we examined DOC for protective activity against seizures induced by various other chemoconvulsants in mice. DOC was also examined for activity in the MES test and for motor toxicity in the screen test. The results of these studies are summarized in Table 1. DOC protected in a dose-dependent manner against seizures induced by picrotoxin and DMCM. At higher doses, it was protective in the MES test. At doses as high as 300 mg/kg, it did not protect against seizures induced by strychnine (1.3 mg/kg, s.c.), kainic acid (32 mg/kg, s.c.), 4-aminopyridine (13.3 mg/kg, s.c.), and NMDA (257 mg/kg, s.c.). At doses comparable with those that were effective in the PTZ test, DOC induced motor impairment as detected by the screen test.
Effects of finasteride and indomethacin on anticonvulsant activity of DOC in PTZ test
If the anticonvulsant effects of DOC are caused by its conversion to THDOC, then inhibitors of the two reduction steps through which THDOC is synthesized from DOC should interfere with its protective activity. We used finasteride as in the swim stress experiments to block the initial 5α-reductase step, and we also studied indomethacin, which has been reported to act as a competitive inhibitor of 3α-hydroxysteroid oxidoreductase (Penning et al., 1985). Finasteride (50 mg/kg, i.p.) caused an 84% reduction in the conversion of DOC to THDOC as assessed by plasma THDOC determinations 30 min after a 150 mg/kg intraperitoneal injection of DOC administered 60 min after finasteride (control THDOC, 1081 ± 91 ng/ml; finasteride, 176 ± 11 ng/ml).
As illustrated in Figure 5A, pretreatment with finasteride (10 mg/kg, i.p.) caused a marked rightward shift in the dose–response relationship for DOC protection against PTZ seizures. However, the highest DOC dose tested (300 mg/kg) almost completely overcame the reversal. Finasteride significantly increased the anticonvulsant ED50 values of DOC derived from these data from a control value of 84 mg/kg [95% confidence limits (CL), 35–145 mg/kg] to 180 mg/kg (95% CL, 139–233 mg/kg). Similarly, indomethacin also inhibited the antiseizure activity of DOC [ED50 after indomethacin treatment, 129 mg/kg (95% CL, 107–154 mg/kg)]; this inhibitory action could also be completely overcome at high DOC doses. Figure 5B shows the dose–response relationships for finasteride and indomethacin inhibition of seizure protection by a 2× ED50 (168 mg/kg) dose of DOC. The ED50 value for finasteride inhibition of the antiseizure activity of DOC was 7.2 mg/kg (95% CL, 5–30 mg/kg). Similarly, high doses of indomethacin significantly but only partially (39% at 300 mg/kg) inhibited the antiseizure activity of these high doses of DOC (Fig. 5B). The expected inhibition determined by interpolation for 100 mg/kg indomethicin from the experiment shown in Figure 5A is 11%, which compares favorably with the expected 10% inhibition by the same dose in the experiment shown in Figure 5B.
Fig. 5.
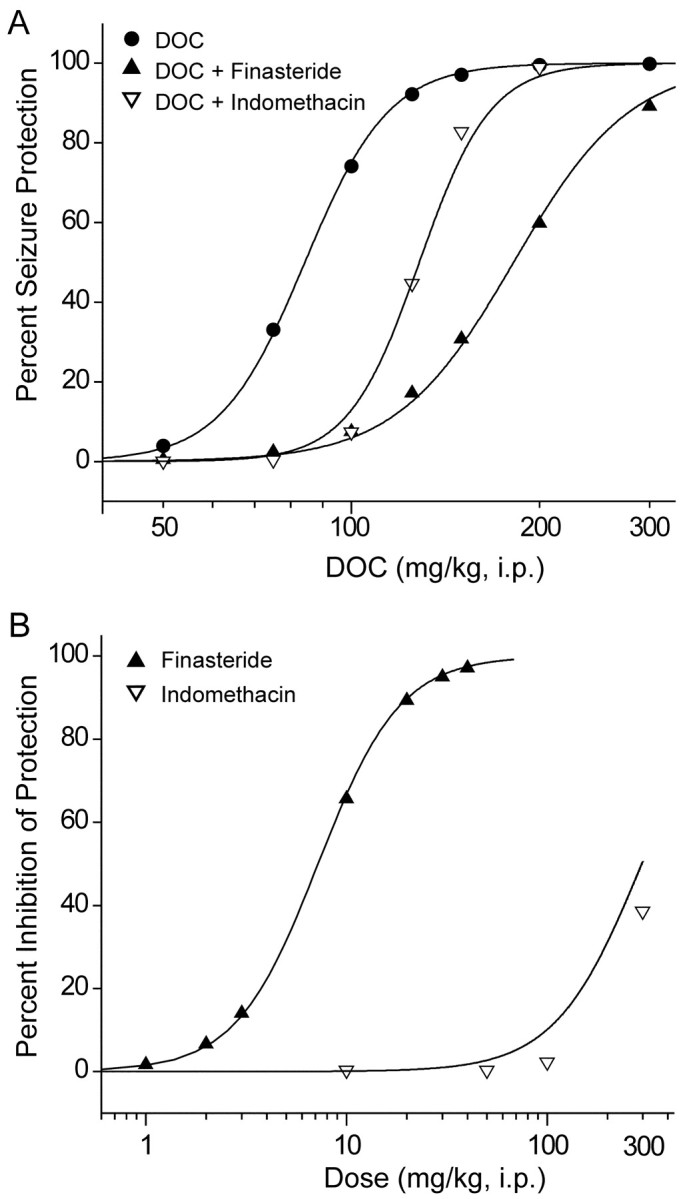
Finasteride and indomethacin inhibit the protective activity of DOC in the PTZ seizure test. A, Dose–response relationship for DOC (50–300 mg/kg, i.p.) protection against PTZ seizures is shifted to the right by pretreatment with finasteride (10 mg/kg, i.p.) or indomethacin (100 mg/kg, i.p.).B, Dose–response relationship for finasteride (1–50 mg/kg, i.p.) or indomethacin (10–300 mg/kg, i.p.) inhibition of the anticonvulsant activity of DOC (168 mg/kg) in the PTZ test.A and B, Finasteride or indomethacin was administered 60 or 30 min, respectively, before the DOC injection. Each point represents data from eight mice.
We have shown previously that finasteride is neither proconvulsant nor has any nonspecific anticonvulsant actions in the PTZ test in mice (Kokate et al., 1999). To ascertain whether indomethacin affects the PTZ seizure threshold, we compared the dose–response relationships for PTZ (30–85 mg/kg) 30 min after animals were injected with 100 mg/kg indomethacin or vehicle. The CD50 values for the vehicle control [48 mg/kg (95% CL, 42–54 mg/kg)] and indomethacin pretreatment groups [46 mg/kg (95% CL, 41–52 mg/kg)] did not differ significantly, indicating that indomethacin did not affect the PTZ seizure test.
These data provide strong evidence that the antiseizure activity of DOC in the PTZ model is mediated by its A-ring reduced metabolites. Moreover, the dramatic activity of finasteride in conjunction with the limited efficacy of indomethacin suggested that DHDOC as well as THDOC could participate in the anticonvulsant activity of DOC.
Anticonvulsant activity of DHDOC: comparison with DOC and THDOC
To investigate the possibility that the intermediate DHDOC as well as THDOC contributes to the anticonvulsant activity of DOC, we administered the intermediate directly. As predicted, DHDOC exhibited a dose-dependent protective activity in the mouse PTZ test and was only slightly less potent than THDOC (Fig.6A). At higher doses, both DHDOC and THDOC also protected in a dose-dependent manner against MES-induced seizures and impaired motor function as assessed with the screen test (ED50 values in Table 1).
Fig. 6.

Anticonvulsant activity of DHDOC and THDOC in the PTZ seizure test: effects of finasteride (Finast.) and indomethacin (Indomet.). A, Dose–response relationships for protection by DOC, DHDOC, and THDOC in the PTZ seizure test. B, Time courses for protection against PTZ-induced seizures by DOC (168 mg/kg, i.p.), DHDOC (52 mg/kg, i.p.), and THDOC (38 mg/kg, i.p.) administered at time 0. Thepoints at time 0 represent the simultaneous administration of PTZ and a neurosteroid. The linesrepresent arbitrary fits to the data points. C, Dose–response relationship for DHDOC protection against PTZ seizures is shifted to the right by indomethacin (100 mg/kg, i.p.) pretreatment but not by finasteride (10 mg/kg, i.p.) pretreatment. D, Lack of effect of finasteride and indomethacin pretreatment on the dose–response relationship for THDOC protection against PTZ seizures. Finasteride or indomethacin was administered 60 or 30 min, respectively, before the DHDOC or THDOC injections. Each point represents data from eight mice.
Time courses for seizure protection
The time courses for seizure protection after a 2× ED50 dose of DOC, DHDOC, and THDOC are shown in Figure 6B. DOC exhibited a slow onset to peak effect (30 min) and a sustained duration of action (up to 180 min). In contrast, for DHDOC and THDOC, protection was maximal at 15 min and diminished during the 60 min period after the injection. These results are compatible with the possibility that DOC is activated by conversion to DHDOC and THDOC.
Effects of finasteride and indomethacin on anticonvulsant activity of DHDOC and THDOC
The anticonvulsant activity of DHDOC could be either attributable to its own direct action or mediated via conversion to THDOC. If DHDOC is activated by conversion to THDOC, its anticonvulsant activity should be sensitive to indomethacin. As shown in Figure 6C, indomethacin (100 mg/kg), but not finasteride (10 mg/kg), significantly reduced the antiseizure activity of DHDOC [control ED50, 25.5 mg/kg (95% CL, 23–53 mg/kg), indomethacin treatment, 48 mg/kg (95% CL, 34–68 mg/kg);p < 0.05] (Fig. 6C). Neither finasteride nor indomethacin affected THDOC protection against PTZ-induced seizures (Fig. 6D).
Effects of pharmacological antagonists on anticonvulsant activity of DOC in MES test
In contrast to its effects in the PTZ test, finasteride (up to 200 mg/kg) did not block the protective effects of high doses of DOC (214 mg/kg) on tonic hindlimb extension in the MES seizure test (only one of nine mice showed tonic hindlimb extension with 200 mg/kg finasteride pretreatment). In addition, mifepristone (3–30 mg/kg, s.c.), a progesterone receptor antagonist, and spironolactone (5–30 mg/kg, s.c.), a mineralocorticoid receptor antagonist, did not inhibit the antiseizure activity of DOC (168 mg/kg) against PTZ-induced seizures, indicating that it does not act through classic steroid nuclear hormone receptors (data not shown).
Activity of DOC in adrenalectomized mice
To exclude the possibility that the anticonvulsant activity of DOC is related to adrenal conversion to neurosteroids or to the modification of adrenal steroid synthesis, we examined its activity in adrenalectomized mice. The ED50 for DOC protection against PTZ-induced seizures was 67 mg/kg (95% CL, 51–90 mg/kg), which is not significantly different from the value in naive controls (Table 1), indicating that adrenal cortical 5α-reductase (Yokoi et al., 1998) is not required for the anticonvulsant activity of DOC. In adrenalectomized mice, finasteride (30 mg/kg) reversed the protective effect of a 2× ED50 dose of DOC (134 mg/kg) in five of six animals. This dose of DOC protected six of six control animals. These results indicate that extra-adrenal conversion of DOC to anticonvulsant neurosteroids requires 5α-reductase.
Antiseizure activity of DOC and THDOC in kindling model of epilepsy
DOC and THDOC were also examined for protective activity against fully kindled seizures in the mouse amygdala-kindling model of epilepsy. Mice with a bipolar stimulating electrode implanted unilaterally into the right amygdala were stimulated once daily at 125% of their afterdischarge threshold until they exhibited stage 5 kindled seizures on 3 successive days (Rogawski et al., 2001). There was a progressive increase in seizure stage, with all animals achieving stage 5 after 17 stimulations. The mean afterdischarge duration did not vary significantly during the course of stimulation in saline-treated animals. Administration of DOC (50–100 mg/kg, i.p.) 30 min before electrical stimulation in fully kindled animals caused a dose-dependent suppression of the behavioral seizure scores (Fig.7A) but did not affect the afterdischarge duration (data not shown). Pretreatment with finasteride (10 and 30 mg/kg, i.p.) produced a dose-dependent reversal of the protective effect of 100 mg/kg DOC. Neither the seizure score nor the afterdischarge duration was significantly affected by finasteride alone. Similarly, administration of THDOC (3–20 mg/kg, i.p.) 15 min before stimulation also caused a dose-dependent reduction in the seizure score in fully kindled animals (Fig. 7B). However, finasteride (10 and 30 mg/kg) failed to produce a significant reversal of the inhibition of kindled seizures produced by 20 mg/kg THDOC. These results indicate that the anticonvulsant activity of DOC against kindled seizures is mediated via its 5α-reduced metabolites.
Fig. 7.

Inhibition of fully kindled seizures by DOC and THDOC: reversal of DOC but not THDOC by finasteride. A, Mice that exhibited stage 5 seizures on 3 consecutive days received an intraperitoneal injection of vehicle or DOC (50–100 mg/kg) 30 min before stimulation on days 1 and 3 of stimulation. Some animals received finasteride (10 or 30 mg/kg) 1 hr before DOC.B, Similar experiment to A except that THDOC (3, 10, or 20 mg/kg 15 min before stimulation) was substituted for DOC. Each bar represents the mean ± SEM of data from six to eight animals. *p < 0.05, **p < 0.01 versus vehicle (Veh);#p < 0.05 versus DOC (100 mg/kg) alone group (Mann–Whitney U test).
Effects of DHDOC and THDOC on GABAAreceptor function
The ability of DHDOC and THDOC to modulate GABAA receptor function was investigated in whole-cell voltage-clamp recordings of GABAAreceptor Cl− currents in cultured hippocampal neurons (Fig. 8). Perfusion with 10 μm GABA evoked inward currents that were blocked by 100 μm bicuculline methiodide and 100 μmpicrotoxin, indicating that they are mediated by GABAA receptors (Fig.9B). As demonstrated previously (Kokate et al., 1994), preapplication (10 sec) and then coapplication of THDOC (10–1000 nm) with 10 μm GABA resulted in enhancement of the current response in comparison with the magnitude of the response obtained with GABA alone. Sample currents obtained with 10 μmGABA alone or in combination with THDOC are illustrated in Figure8A. As shown in Figure 8B, there was a concentration-dependent increase in the degree of potentiation, with concentrations as low as 10 nm producing a significant increase. The concentration that produced a doubling of the control GABA current (EC100) estimated by interpolation was 150 nm. Similarly, DHDOC (10–1000 nm) also produced a concentration-dependent enhancement of GABA-activated Cl− currents that was evident at concentrations as low as 30 nm but was quantitatively smaller than the potentiation produced by THDOC; the interpolated EC100 was ∼500 nm.
Fig. 8.

Modulatory and direct actions of THDOC and DHDOC on GABAA receptor Cl− currents in cultured hippocampal neurons. A, Sampletraces showing Cl− currents evoked during perfusion with 10 μm GABA alone and coapplication of GABA and 1 μm THDOC or 1 μm DHDOC. The steroids were preapplied for 10 sec before the onset of the GABA coapplication. Dotted lines indicate baseline current. Time scale applies to both traces. B, Concentration–response curves for THDOC and DHDOC derived from experiments similar to those shown in A. Peak amplitude values during coapplication of GABA and neurosteroid were compared with the amplitude of 10 μm GABA responses in the same cells (see Materials and Methods). Each point represents the mean ± SEM of fractional increase values from five to eight cells. The curves show arbitrary logistic fits to the mean values; parameters could not be derived because plateau responses were not achieved. Approximate EC100 values determined by interpolation for THDOC and DHDOC are 150 and 500 nm, respectively. C, Inward currents evoked by 10 μm GABA and 1 and 100 μm DHDOC alone. Thethick trace in the middle panel shows the trace expanded 10-fold. D, Concentration dependence of peak GABA-activated Cl− current expressed as a fraction of the current evoked by 100 μm GABA. EC50, 19.6 μm;nH, 1.1; mean current for 100 μm GABA, 6.1 ± 1.1 nA. E, Concentration–response curves for direct activation of inward current by DHDOC or THDOC alone. Currents are expressed as a fraction of the peak current evoked by 10 μm GABA in the same cell. Curves are fitted as described in Materials and Methods. EC50 and Fmax values for THDOC and DHDOC are 19.0 ± 2.3 μm and 0.58 ± 0.03 (nH = 2.1), and 30.2 ± 6.0 μm and 0.41 ± 0.06 (nH = 2.9), respectively.D and E, Each point represents the mean ± SEM of data from 5 to 12 cells. Holding potential in all experiments, −60 mV.
Fig. 9.

Currents evoked by THDOC are blocked by the GABAA receptor antagonists picrotoxin (PTX) and bicuculline (BIC).A, Representative current traces showing block of currents activated by 30 μm THDOC in two separate cells by 100 μm PTX (left) and 100 μm BIC (right). B, Fractional block of currents evoked by 30 μm THDOC (left) and 10 μm GABA (right) by PTX and BIC in experiments similar to those shown in A. Peak current amplitudes in the presence of PTX and BIC are normalized to the peak control current amplitudes in the absence of antagonist. Each bar represents the mean ± SEM of data from three to five cells. Mean amplitudes of THDOC- and GABA-evoked currents are 1030 ± 170 pA (n = 5) and 1920 ± 380 pA (n = 5), respectively. Holding potential, −60 mV.
At high concentrations, neurosteroids, in the absence of GABA, directly activate GABAA receptor Cl− currents (Kokate et al., 1994). We confirmed this effect for THDOC and observed a similar direct effect of DHDOC (Fig. 8C,E). As illustrated in Figure 8C, perfusion of hippocampal neurons with 1 or 100 μm DHDOC (in the absence of GABA) resulted in activation of inward current responses that were similar to the response evoked by GABA. The maximum amplitudes of the THDOC or DHDOC responses were 58 and 41% of the 10 μmGABA response amplitude, which was 32% of the maximal GABA response amplitude (Fig. 8D). Thus, even the maximal steroid-evoked currents were only a fraction (<20%) of the maximal GABA-activated Cl− current. Like GABA-evoked responses, the currents evoked directly by THDOC and DHDOC were blocked by coapplication of picrotoxin and bicuculline methiodide, as illustrated for THDOC in Figure 9, indicating that they are attributable to activation of GABAAreceptors.
DISCUSSION
Swim stress elevation in seizure threshold is mediated by neurosteroids
Our observation that swim stress increases the threshold for clonic seizures in rats induced by intravenous PTZ is consistent with several previous studies demonstrating that acute swim stress is associated with anticonvulsant effects against seizures induced by PTZ and other GABAA receptor antagonists (Soubrie et al., 1980; Abel and Berman, 1993; Peric̆ić et al., 2000,2001). At the time of seizure protection, swim stress was associated with a 2.9-fold elevation in plasma THDOC levels. Similar increases in plasma THDOC levels have been observed previously in response to swim stress and other stressors, including footshock (Purdy et al., 1991;Barbaccia et al., 1996). Moreover, we observed that both the elevation in seizure threshold and the rise in THDOC were eliminated by pretreatment with finasteride, consistent with the possibility that the anticonvulsant effect is mediated by 5α-reduced neurosteroids. [Our results differ from those of Peric̆ić et al. (2000), possibly because of differences in the seizure models.] Finasteride did not affect seizure threshold or THDOC levels in nonstressed animals, indicating that THDOC turnover under basal conditions is substantially longer than the 90 min pretreatment interval and that the stress-induced increase in THDOC is related to de novosynthesis. In rodents, finasteride blocks the type I 5α-reductase isoenzyme, which is the predominant form in the brain, as well as the type II form of the prostate and gonads (Russell and Wilson, 1994;Mensah-Nyagan et al., 1999; Poletti et al., 1999). Consequently, the neurosteroids responsible for stress-induced seizure protection could be synthesized in peripheral tissues and then transported by the circulation to the brain, or they could be produced locally in the brain. In fact, because of local biosynthesis, brain THDOC levels may be substantially higher than plasma levels after stressful events (Purdy et al., 1991). Because the synthesis of DOC, the precursor of THDOC, is increased by stress (Barbaccia et al., 1996) and THDOC is well recognized to have anticonvulsant activity (Kokate et al., 1994), THDOC is a prime candidate for the neurosteroid responsible for the swim stress effects on seizures. In this regard, it is noteworthy that 3α-hydroxysteroid oxidoreductase, the enzyme responsible for the final step in the synthesis of THDOC, is also present in the brain (Li et al., 1997; Stoffel-Wagner et al., 2000). In addition to blocking THDOC biosynthesis, finasteride also inhibits the conversion of progesterone to allopregnanolone (Azzolina et al., 1997). It is therefore possible that allopregnanolone or other anticonvulsant 5α-reduced neurosteroids might also have contributed to the antiseizure effects of swim stress. In fact, there is evidence that levels of allopregnanolone, like those of THDOC, are increased during stress (Purdy et al., 1991; Barbaccia et al., 1996). However, glucocorticoids, which reach higher levels than DOC during stress (Kater et al., 1989), are not likely to be involved in the protective effects of swim stress on seizures, because they either decrease or do not affect seizure thresholds in animals (Heuser and Eidelberg, 1961;Conforti and Feldman, 1975; Roberts and Keith, 1994;Peric̆ić et al., 2000). We unexpectedly observed that the seizure threshold of finasteride-treated stressed rats was significantly reduced below the baseline control value. This suggests the existence of a proconvulsant factor released during stress whose activity is masked by the concomitant release of anticonvulsant neurosteroids.
GABAA receptor-modulating neurosteroids are responsible for the antiseizure activity of DOC
To obtain support for the hypothesis that DOC contributes to stress-related reductions in seizure susceptibility, we examined DOC as an anticonvulsant. We confirmed early studies showing that DOC protects against PTZ-induced seizures (Selye, 1942; Craig, 1966) and extended this work by demonstrating an effect of the steroid in several other chemoconvulsant models and against fully kindled seizures. We found that seizure protection in the rat PTZ threshold test occurred with low doses of DOC that were associated with levels of plasma THDOC comparable with those in the stressed rats. However, the magnitude of the seizure threshold elevation after swim stress was greater than that occurring with the lowest dose of DOC (22% vs 12%), although the elevation in plasma THDOC was less after stress than with this dose of DOC (threefold vs sevenfold). This supports the possibility that additional factors (such as allopregnanolone) may contribute to the anticonvulsant activity of swim stress.
In the mouse all-or-none PTZ test, we found that the anticonvulsant activity of DOC was well correlated with plasma THDOC levels, although the absolute concentrations required for seizure protection were substantially greater than in the rat PTZ threshold model. Finasteride completely blocked the antiseizure activity of DOC and markedly blunted the increase in plasma THDOC obtained after administration of the precursor. We also found that indomethacin, an inhibitor of 3α-hydroxysteroid oxidoreductase, significantly reduced but did not completely block the anticonvulsant activity of DOC. Together, these results indicate that DOC itself is not anticonvulsant and must be activated by A-ring reduction. The relatively delayed onset and more prolonged duration of seizure protection conferred by DOC compared with DHDOC and THDOC (Fig. 6B) is compatible with the possibility that DOC is an inactive precursor that must be metabolically activated. Nevertheless, it is apparent that DOC is rapidly converted to its active metabolites, inasmuch as some seizure protection was apparent even at times as brief as 7 min after its parenteral administration.
The observation that indomethacin is not fully effective at eliminating the anticonvulsant activity of DOC is consistent with the possibility that DHDOC could contribute to the antiseizure activity. The spectrum of activity exhibited by DOC in animal seizure models was similar to that of anticonvulsants that modulate GABAAreceptors, providing additional support for the concept that the protection is ultimately mediated through GABAAreceptors. Thus, like GABAA receptor modulators (Rogawski and Porter, 1990), DOC exhibited protective activity against seizures induced by the GABAA receptor antagonists PTZ, picrotoxin, and DMCM but did not confer protection against seizures induced by the glutamate receptor agonists NMDA and kainate and the glycine receptor antagonist strychnine. In addition, DOC was highly active against fully kindled seizures, as are drugs that facilitate GABAergic neurotransmission (Albertson et al., 1980;Morimoto et al., 1993, 1997). Neuroactive steroids have been reported to protect against corneal and PTZ-kindled seizures (Carter et al., 1997; Gasior et al., 2000). We now extend these observations by showing that THDOC and also DOC are protective against seizures in the amygdala-kindling model. There are several early reports that DOC is effective against electroshock seizures (Woodbury and Davenport, 1949;Craig, 1966; Ferngren, 1969). We also found that high doses of DOC are protective in the MES test, as we reported previously for high doses of progesterone and its A-ring reduced metabolite allopregnanolone (Kokate et al., 1999). However, the anticonvulsant activity of DOC in the MES test, like that of progesterone in our previous study, was unaffected by finasteride, indicating that it occurs through mechanisms unrelated to conversion to GABAA receptor-modulating neurosteroids.
Identity of the neurosteroids that mediate the anticonvulsant activity of DOC
Finasteride and indomethacin inhibited the anticonvulsant activity of DOC without themselves affecting seizure threshold (Kokate et al., 1999; Reddy and Rogawski, 2000), providing strong evidence that A-ring reduced neurosteroid metabolites contribute to the anticonvulsant activity of the adrenal steroid. Several considerations indicate that DHDOC and THDOC represent these activated metabolites. The observation that finasteride can completely block the anticonvulsant activity of DOC indicates that the 5β-reduced isomers of DHDOC and THDOC, which could be produced by 3-ketosteroid 5β-reductase in the liver (Charbonneau and The, 2001) and brain (Kawahara et al., 1975), do not play a role in the anticonvulsant activity, although the 5β-isomers have anticonvulsant activity that is only modestly less potent than their 5α-congeners (Kokate et al., 1994). However, indomethacin did not fully block the anticonvulsant activity of DOC, suggesting that the intermediate DHDOC may have anticonvulsant activity itself. Alternatively, metabolites of DHDOC other than THDOC could have anticonvulsant activity. In this regard, it is noteworthy that 3β-reduced analogs have low GABAAreceptor modulatory activity and are correspondingly weak anticonvulsants (Kokate et al., 1994). Moreover, steroid 3β-reducing activity has not been reported. Therefore, 3β-reduced DHDOC metabolites are unlikely to contribute to the anticonvulsant activity of DOC. The conclusion that DHDOC is an active DOC metabolite relies on the effectiveness of indomethacin as an inhibitor of 3α-hydroxysteroid oxidoreductase. Indomethacin is a highly potent and effective inhibitor of the enzyme in vitro that is activein vivo even at doses that are substantially lower than those required to affect the anticonvulsant activity of DOC (Penning and Talalay, 1983; Penning et al., 1985; Smithgall and Penning, 1985;Gallo and Smith, 1993; Beyer et al., 1999). The fact that DHDOC can potentiate GABAA receptor responses, albeit with less potency than THDOC, suggests that DHDOC could contribute to the anticonvulsant activity of DOC, particularly in the presence of 3α-hydroxysteroid oxidoreductase inhibition, when DHDOC levels would be expected to become significant.
Antiseizure activity of DOC occurs via neurosteroid potentiation of GABAA receptors
Our electrophysiological studies confirm that THDOC acts as a positive modulator of GABAA receptors (Majewska et al., 1986; Harrison et al., 1987; Lambert et al., 1990; Purdy et al., 1990; Wetzel et al., 1999), and we now show that the intermediate DHDOC acts in a similar manner. Both of the steroids directly activate GABAA receptor Cl−currents and potentiate the action of GABA, as does the progesterone-derived neurosteroid allopregnanolone (Kokate et al., 1994; Lambert et al., 1995; Park-Chung et al., 1999). The mean plasma concentration of THDOC determined after swim stress (6.2 ng/ml = 19 nm) is within the range of concentrations found to enhance GABA-activated Cl− currents. Comparable concentrations of THDOC (27 ng/ml) were associated with a statistically significant elevation in the rat PTZ seizure threshold (Fig. 3). However, these concentrations do not appear to be adequate for seizure protection in the less sensitive mouse PTZ test (Fig. 4). In any case, plasma concentrations might not fully reflect brain levels because of local synthesis. The direct agonist actions of THDOC and DHDOC were antagonized by bicuculline and picrotoxin to an extent similar to GABA, demonstrating that the currents obtained were caused by activation of GABAA receptors. The micromolar concentrations at which these direct effects occur were not achieved in the stress paradigm used in this study but were attained with exogenous administration of doses of DOC active in the PTZ test (plasma concentration after ED50 DOC dose, 1.8 μm).
Implications
It has been recognized for centuries that emotional factors can affect seizure control (Minter, 1979). Although most systematic studies have observed that high stress levels and stressful events are associated with more frequent epileptiform electroencephalographic abnormalities and seizures (Temkin and Davis, 1984; Frucht et al., 2000), there are situations in which stress or alerting has been demonstrated to reduce epileptiform electrographic abnormalities (Minter, 1979). Nevertheless, it is generally accepted that stress triggers seizures. How can the present observations be reconciled with this view? There are undoubtedly many neural and endocrine pathways through which stress can alter brain function and thereby affect seizure susceptibility. The likelihood of seizures represents a balance between these pathways, some of which promote seizures (e.g., glucocorticoids, CRF, hypercarbia associated with hyperventilation) and others (e.g., neurosteroids) that protect against seizures. Stress-induced seizures would therefore occur when the balance is shifted to favor the proconvulsant factors, outweighing the anticonvulsant action of endogenous GABAAreceptor-modulating neurosteroids. We have shown previously that withdrawal from allopregnanolone dramatically enhances seizure susceptibility (Reddy and Rogawski, 2000, 2001). Similarly, withdrawal of adrenal-derived neurosteroids in association with variations in the level of stress during a stressful episode could be another factor that predisposes to stress-induced seizures.
Footnotes
The expert assistance of Nassim Tabatabai, Karen Wayns, and Dr. Shun-ichi Yamaguchi is gratefully acknowledged. We thank Dr. Gregory L. Holmes for bringing to our attention early work on the anticonvulsant activity of DOC.
Correspondence should be addressed to Dr. Michael A. Rogawski at the above address. E-mail: michael.rogawski@nih.gov.
D. S. Reddy's present address: Department of Anatomy, Physiological Sciences, and Radiology, North Carolina State University College of Veterinary Medicine, Raleigh, NC 27606.
REFERENCES
- 1.Abel EL, Berman RF. Effects of water immersion stress on convulsions induced by pentylenetetrazol. Pharmacol Biochem Behav. 1993;45:823–825. doi: 10.1016/0091-3057(93)90127-f. [DOI] [PubMed] [Google Scholar]
- 2.Albertson TE, Peterson SL, Stark LG. Anticonvulsant drugs and their antagonism of kindled amygdaloid seizures in rats. Neuropharmacology. 1980;19:643–652. doi: 10.1016/0028-3908(80)90038-6. [DOI] [PubMed] [Google Scholar]
- 3.Azzolina B, Ellsworth K, Andersson S, Geissler W, Bull HG, Harris GS. Inhibition of rat 5α-reductases by finasteride: evidence for isozyme differences in the mechanism of inhibition. J Steroid Biochem Mol Biol. 1997;61:55–64. doi: 10.1016/s0960-0760(97)00002-2. [DOI] [PubMed] [Google Scholar]
- 4.Barbaccia ML, Roscetti G, Trabucchi M, Mostallino MC, Concas A, Purdy RH, Biggio G. Time-dependent changes in rat brain neuroactive steroid concentrations and GABAA receptor function after acute stress. Neuroendocrinology. 1996;63:166–172. doi: 10.1159/000126953. [DOI] [PubMed] [Google Scholar]
- 5.Barbaccia ML, Roscetti G, Trabucchi M, Purdy RH, Mostallino MC, Concas A, Biggio G. The effects of inhibitors of GABAergic transmission and stress on brain and plasma allopregnanolone concentrations. Br J Pharmacol. 1997;120:1582–1588. doi: 10.1038/sj.bjp.0701046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beyer C, Gonzalez-Flores O, Ramirez-Orduna JM, Gonzalez-Mariscal G. Indomethacin inhibits lordosis induced by ring A-reduced progestins: possible role of 3α-oxidoreduction in progestin-facilitated lordosis. Horm Behav. 1999;35:1–8. doi: 10.1006/hbeh.1998.1457. [DOI] [PubMed] [Google Scholar]
- 7.Bitran D, Hilvers RJ, Kellogg CK. Anxiolytic effects of 3α-hydroxy-5α[β]-pregnan-20-one: endogenous metabolites of progesterone that are active at the GABAA receptor. Brain Res. 1991;561:157–161. doi: 10.1016/0006-8993(91)90761-j. [DOI] [PubMed] [Google Scholar]
- 8.Carter RB, Wood PL, Wieland S, Hawkinson JE, Belelli D, Lambert JJ, White HS, Wolf HH, Mirsadeghi S, Tahir SH, Bolger MB, Lan NC, Gee KW. Characterization of the anticonvulsant properties of ganaxolone (CCD 1042; 3α-hydroxy-3β-methyl-5α-pregnan-20-one), a selective, high-affinity, steroid modulator of the γ-aminobutyric acidA receptor. J Pharmacol Exp Ther. 1997;280:1284–1295. [PubMed] [Google Scholar]
- 9.Charbonneau A, The VL. Genomic organization of a human 5β-reductase and its pseudogene and substrate selectivity of the expressed enzyme. Biochim Biophys Acta. 2001;1517:228–235. doi: 10.1016/s0167-4781(00)00278-5. [DOI] [PubMed] [Google Scholar]
- 10.Conforti N, Feldman S. Effect of cortisol on the excitability of limbic structures of the brain in freely moving rats. J Neurol Sci. 1975;26:29–38. doi: 10.1016/0022-510x(75)90111-2. [DOI] [PubMed] [Google Scholar]
- 11.Coughenour AG, McLean JR, Parker RB. A new device for the rapid measurement of impaired motor function in mice. Pharmacol Biochem Behav. 1977;6:351–353. doi: 10.1016/0091-3057(77)90036-3. [DOI] [PubMed] [Google Scholar]
- 12.Craig CR. Anticonvulsant activity of steroids: separability of anticonvulsant from hormonal effects. J Pharmacol Exp Ther. 1966;153:337–343. [Google Scholar]
- 13.Crawley JN, Glowa JR, Majewska MD, Paul SM. Anxiolytic activity of an endogenous adrenal steroid. Brain Res. 1986;398:382–385. doi: 10.1016/0006-8993(86)91500-3. [DOI] [PubMed] [Google Scholar]
- 14.Donevan SD, Jones SM, Rogawski MA. Arcaine blocks N-methyl-d-aspartate receptor responses by an open channel mechanism: whole-cell and single-channel recording studies in cultured hippocampal neurons. Mol Pharmacol. 1992;41:727–735. [PubMed] [Google Scholar]
- 15.Feldman RG, Paul NL. Identity of emotional triggers in epilepsy. J Nerv Ment Dis. 1976;162:345–353. doi: 10.1097/00005053-197605000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Ferngren H. High frequency electro-shock seizures and their antagonism in developing mice. III. Effects of cortisol, corticosterone, deoxycorticosterone, dexamethasone and a synthetic corticotropine. Acta Pharm Suec. 1969;6:339–348. [PubMed] [Google Scholar]
- 17.Frucht MM, Quigg M, Schwaner C, Fountain NB. Distribution of seizure precipitants among epilepsy syndromes. Epilepsia. 2000;41:1534–1539. doi: 10.1111/j.1499-1654.2000.001534.x. [DOI] [PubMed] [Google Scholar]
- 18.Gallo MA, Smith SS. Progesterone withdrawal decreases latency to and increases duration of electrified prod burial: a possible rat model of PMS anxiety. Pharmacol Biochem Behav. 1993;46:897–904. doi: 10.1016/0091-3057(93)90219-j. [DOI] [PubMed] [Google Scholar]
- 19.Gasior M, Ungard JT, Beekman M, Carter RB, Witkin JM. Acute and chronic effects of the synthetic neuroactive steroid, ganaxolone, against the convulsive and lethal effects of pentylenetetrazol in seizure-kindled mice: comparison with diazepam and valproate. Neuropharmacology. 2000;39:1184–1196. doi: 10.1016/s0028-3908(99)00190-2. [DOI] [PubMed] [Google Scholar]
- 20.Goldberg ME, Salama AI. Effect of drum stress on maximal electroconvulsive seizure latency in mice. Int J Neuropharmacol. 1969;8:161–167. doi: 10.1016/0028-3908(69)90009-4. [DOI] [PubMed] [Google Scholar]
- 21.Harrison NL, Majewska MD, Harrington JW, Barker JL. Structure-activity relationships for steroid interactions with the γ-aminobutyric acid-A receptor complex. J Pharmacol Exp Ther. 1987;241:346–353. [PubMed] [Google Scholar]
- 22.Heuser E, Eidelberg E. Steroid-induced convulsions in experimental animals. Endocrinology. 1961;69:915–924. doi: 10.1210/endo-69-5-915. [DOI] [PubMed] [Google Scholar]
- 23.Kater CE, Biglieri EG, Brust N, Chang B, Hirai J, Irony I. Stimulation and suppression of the mineralocorticoid hormones in normal subjects and adrenocortical disorder. Endocr Rev. 1989;10:149–164. doi: 10.1210/edrv-10-2-149. [DOI] [PubMed] [Google Scholar]
- 24.Kawahara FS, Berman ML, Green OC. Conversion of progesterone-1,2–3-H to 5β-pregnane-3,20-dione by brain tissue. Steroids. 1975;25:459–463. doi: 10.1016/0039-128x(75)90023-9. [DOI] [PubMed] [Google Scholar]
- 25.Kokate TG, Svensson BE, Rogawski MA. Anticonvulsant activity of neurosteroids: correlation with γ-aminobutyric acid-evoked chloride current potentiation. J Pharmacol Exp Ther. 1994;270:1223–1229. [PubMed] [Google Scholar]
- 26.Kokate TG, Cohen AL, Karp E, Rogawski MA. Neuroactive steroids protect against pilocarpine- and kainic acid-induced limbic seizures and status epilepticus in mice. Neuropharmacology. 1996;35:1049–1056. doi: 10.1016/s0028-3908(96)00021-4. [DOI] [PubMed] [Google Scholar]
- 27.Kokate TG, Banks MK, Magee T, Yamaguchi S, Rogawski MA. Finasteride, a 5α-reductase inhibitor, blocks the anticonvulsant activity of progesterone in mice. J Pharmacol Exp Ther. 1999;288:679–684. [PubMed] [Google Scholar]
- 28.Lambert JJ, Peters JA, Sturgess NC, Hales TG. Steroid modulation of the GABAA receptor complex: electrophysiological studies. Ciba Found Symp. 1990;153:56–71. doi: 10.1002/9780470513989.ch4. [DOI] [PubMed] [Google Scholar]
- 29.Lambert JJ, Belelli D, Hill-Venning C, Peters JA. Neurosteroids and GABAA receptor function. Trends Pharmacol Sci. 1995;16:295–303. doi: 10.1016/s0165-6147(00)89058-6. [DOI] [PubMed] [Google Scholar]
- 30.Li X, Bertics PJ, Karavolas HJ. Regional distribution of cytosolic and particulate 5α-dihydroprogesterone 3α-hydroxysteroid oxidoreductases in female rat brain. J Steroid Biochem Mol Biol. 1997;60:311–318. doi: 10.1016/s0960-0760(96)00195-1. [DOI] [PubMed] [Google Scholar]
- 31.Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM. Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science. 1986;232:1004–1007. doi: 10.1126/science.2422758. [DOI] [PubMed] [Google Scholar]
- 32.Mensah-Nyagan AG, Do-Rego JL, Beaujean D, Luu-The V, Pelletier G, Vaudry H. Neurosteroids: expression of steroidogenic enzymes and regulation of steroid biosynthesis in the central nervous system. Pharmacol Rev. 1999;51:63–81. [PubMed] [Google Scholar]
- 33.Minter RE. Can emotions precipitate seizures: a review of the question. J Fam Pract. 1979;8:55–59. [PubMed] [Google Scholar]
- 34.Morimoto K, Sanei T, Sato K. Comparative study of the anticonvulsant effect of γ-aminobutyric acid agonists in the feline kindling model of epilepsy. Epilepsia. 1993;34:1123–1129. doi: 10.1111/j.1528-1157.1993.tb02144.x. [DOI] [PubMed] [Google Scholar]
- 35.Morimoto K, Sato H, Yamamoto Y, Watanabe T, Suwaki H. Antiepileptic effects of tiagabine, a selective GABA uptake inhibitor, in the rat kindling model of temporal lobe epilepsy. Epilepsia. 1997;38:966–974. doi: 10.1111/j.1528-1157.1997.tb01478.x. [DOI] [PubMed] [Google Scholar]
- 36.Normington K, Russell DW. Tissue distribution and kinetic characteristics of rat steroid 5α-reductase isozymes: evidence for distinct physiological functions. J Biol Chem. 1992;267:19548–19554. [PubMed] [Google Scholar]
- 37.Park-Chung M, Malayev A, Purdy RH, Gibbs TT, Farb DH. Sulfated and unsulfated steroids modulate γ-aminobutyric acidA receptor function through distinct sites. Brain Res. 1999;830:72–87. doi: 10.1016/s0006-8993(99)01381-5. [DOI] [PubMed] [Google Scholar]
- 38.Penning TM, Talalay P. Inhibition of a major NAD(P)-linked oxidoreductase from rat liver cytosol by steroidal and nonsteroidal anti-inflammatory agents and by prostaglandins. Proc Natl Acad Sci USA. 1983;80:4504–4508. doi: 10.1073/pnas.80.14.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Penning TM, Sharp RB, Krieger NR. Purification and properties of 3α-hydroxysteroid dehydrogenase from rat brain cytosol. Inhibition by nonsteroidal anti-inflammatory drugs and progestins. J Biol Chem. 1985;260:15266–15272. [PubMed] [Google Scholar]
- 40.Peric̆ić D, Svob D, Jazvinšćak M, Mirković K. Anticonvulsive effect of swim stress in mice. Pharmacol Biochem Behav. 2000;66:879–886. doi: 10.1016/s0091-3057(00)00267-7. [DOI] [PubMed] [Google Scholar]
- 41.Peric̆ić D, Jazvinšćak M, Svob D, Mirković K. Swim stress alters the behavioural response of mice to GABA-related and some GABA-unrelated convulsants. Epilepsy Res. 2001;43:145–152. doi: 10.1016/s0920-1211(00)00194-7. [DOI] [PubMed] [Google Scholar]
- 42.Poletti A, Celotti F, Maggi R, Melcangi RC, Martini L, Negri-Cesi P. Aspects of hormonal steroid metabolism in the nervous system. In: Baulieu E-E, Robel P, Schumacher M, editors. Contemporary endocrinology: neurosteroids: a new regulatory function in the nervous system. Humana; Totowa, NJ: 1999. pp. 97–123. [Google Scholar]
- 43.Purdy RH, Morrow AL, Blinn JR, Paul SM. Synthesis, metabolism, and pharmacological activity of 3α-hydroxy steroids which potentiate GABA-receptor-mediated chloride ion uptake in rat cerebral cortical synaptoneurosomes. J Med Chem. 1990;33:1572–1581. doi: 10.1021/jm00168a008. [DOI] [PubMed] [Google Scholar]
- 44.Purdy RH, Morrow AL, Moore PH, Jr, Paul SM. Stress-induced elevations of γ-aminobutyric acid type A receptor-active steroids in the rat brain. Proc Natl Acad Sci USA. 1991;88:4553–4557. doi: 10.1073/pnas.88.10.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 46.Reddy DS, Kulkarni SK. Development of neurosteroid-based novel psychotropic drugs. Prog Med Chem. 2000;37:135–175. doi: 10.1016/s0079-6468(08)70059-6. [DOI] [PubMed] [Google Scholar]
- 47.Reddy DS, Rogawski MA. Enhanced anticonvulsant activity of ganaxolone after neurosteroid withdrawal in a rat model of catamenial epilepsy. J Pharmacol Exp Ther. 2000;294:909–915. [PubMed] [Google Scholar]
- 48.Reddy DS, Rogawski MA. Enhanced anticonvulsant activity of neuroactive steroids in a rat model of catamenial epilepsy. Epilepsia. 2001;42:337–344. doi: 10.1046/j.1528-1157.2001.10200.x. [DOI] [PubMed] [Google Scholar]
- 49.Reddy DS, Kim H-Y, Rogawski MA. Neurosteroid withdrawal model of perimenstrual catamenial epilepsy. Epilepsia. 2001;42:328–336. doi: 10.1046/j.1528-1157.2001.10100.x. [DOI] [PubMed] [Google Scholar]
- 50.Roberts AJ, Keith LD. Mineralocorticoid receptors mediate the enhancing effects of corticosterone on convulsion susceptibility in mice. J Pharmacol Exp Ther. 1994;270:505–511. [PubMed] [Google Scholar]
- 51.Rogawski MA, Porter RJ. Antiepileptic drugs: pharmacological mechanisms and clinical efficacy with consideration of promising developmental stage compounds. Pharmacol Rev. 1990;42:223–286. [PubMed] [Google Scholar]
- 52.Rogawski MA, Kurzman PS, Yamaguchi S, Li H. Role of AMPA and GluR5 kainate receptors in the development and expression of amygdala kindling in the mouse. Neuropharmacology. 2001;40:28–35. doi: 10.1016/s0028-3908(00)00112-x. [DOI] [PubMed] [Google Scholar]
- 53.Russell DW, Wilson JD. Steroid 5α-reductase: two genes/two enzymes. Annu Rev Biochem. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- 54.Segal M. Rat hippocampal neurons in culture: response to electrical and chemical stimuli. J Neurophysiol. 1983;50:1249–1264. doi: 10.1152/jn.1983.50.6.1249. [DOI] [PubMed] [Google Scholar]
- 55.Selye H. The antagonism between anesthetic steroid hormones and pentamethylenetetrazol (metrazol). J Lab Clin Med. 1942;27:1051–1053. [Google Scholar]
- 56.Serra M, Pisu MG, Littera M, Papi G, Sanna E, Tuveri F, Usala L, Purdy RH, Biggio G. Social isolation-induced decreases in both the abundance of neuroactive steroids and GABAA receptor function in rat brain. J Neurochem. 2000;75:732–740. doi: 10.1046/j.1471-4159.2000.0750732.x. [DOI] [PubMed] [Google Scholar]
- 57.Smith SS, Gong QH, Li X, Moran MH, Bitran D, Frye CA, Hsu FC. Withdrawal from 3α-OH-5α-pregnan-20-one using a pseudopregnancy model alters the kinetics of hippocampal GABAA-gated current and increases the GABAA receptor α4 subunit in association with increased anxiety. J Neurosci. 1998;18:5275–5284. doi: 10.1523/JNEUROSCI.18-14-05275.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smithgall TE, Penning TM. Indomethacin-sensitive 3-hydroxysteroid dehydrogenase in rat tissues. Biochem Pharmacol. 1985;34:831–835. doi: 10.1016/0006-2952(85)90763-4. [DOI] [PubMed] [Google Scholar]
- 59.Soubrie P, Thiebot MH, Jobert A, Montastruc JL, Hery F, Hamon M. Decreased convulsant potency of picrotoxin and pentetrazol and enhanced [3H]flunitrazepam cortical binding following stressful manipulations in rats. Brain Res. 1980;189:505–517. doi: 10.1016/0006-8993(80)90109-2. [DOI] [PubMed] [Google Scholar]
- 60.Stoffel-Wagner B, Beyenburg S, Watzka MS, Blumcke I, Bauer J, Schramm J, Bidlingmaier F, Elger CE. Expression of 5α-reductase and 3α-hydroxisteroid oxidoreductase in the hippocampus of patients with chronic temporal lobe epilepsy. Epilepsia. 2000;41:140–147. doi: 10.1111/j.1528-1157.2000.tb00133.x. [DOI] [PubMed] [Google Scholar]
- 61.Tan SY, Mulrow PJ. The contribution of the zona fasciculata and glomerulosa to plasma 11-deoxycorticosterone levels in man. J Clin Endocrinol Metab. 1975;41:126–130. doi: 10.1210/jcem-41-1-126. [DOI] [PubMed] [Google Scholar]
- 62.Temkin NR, Davis GR. Stress as a risk factor for seizures among adults with epilepsy. Epilepsia. 1984;25:450–456. doi: 10.1111/j.1528-1157.1984.tb03442.x. [DOI] [PubMed] [Google Scholar]
- 63.Thigpen AE, Russell DW. Four-amino acid segment in steroid 5α-reductase 1 confers sensitivity to finasteride, a competitive inhibitor. J Biol Chem. 1992;267:8577–8583. [PubMed] [Google Scholar]
- 64.Vallee M, Rivera JD, Koob GF, Purdy RH, Fitzgerald RL. Quantification of neurosteroids in rat plasma and brain following swim stress and allopregnanolone administration using negative chemical ionization gas chromatography/mass spectrometry. Anal Biochem. 2000;287:153–166. doi: 10.1006/abio.2000.4841. [DOI] [PubMed] [Google Scholar]
- 65.von Steiger M, Reichstein T. Desoxy-cortico-steron (21-oxyprogesteron) aus Δ5–3-oxy-atio-cholensaure. Helv Chim Acta. 1937;20:1164–1179. [Google Scholar]
- 66.Wetzel CH, Vedder H, Holsboer F, Zieglgansberger W, Deisz RA. Bidirectional effects of the neuroactive steroid tetrahydrodeoxycorticosterone on GABA-activated Cl− currents in cultured rat hypothalamic neurons. Br J Pharmacol. 1999;127:863–868. doi: 10.1038/sj.bjp.0702597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.White HS, Woodhead JH, Franklin MR, Swinyard EA, Wolf HA. General principles: experimental selection, quantification, and evaluation of antiepileptic drugs. In: Levy RH, Mattson RH, Meldrum BS, editors. Antiepileptic drugs, Ed 4. Raven; New York: 1995. pp. 99–110. [Google Scholar]
- 68.Woodbury DM, Davenport VD. Brain and plasma cations and experimental seizures in normal and desoxycorticosterone-treated rats. Am J Physiol. 1949;157:234–240. doi: 10.1152/ajplegacy.1949.157.2.234. [DOI] [PubMed] [Google Scholar]
- 69.Yokoi H, Tsuruo Y, Miyamoto T, Ishimura K. Steroid 5α-reductase type 1 immunolocalized in the adrenal gland of normal, gonadectomized, and sex hormone-supplemented rats. Histochem Cell Biol. 1998;109:127–134. doi: 10.1007/s004180050210. [DOI] [PubMed] [Google Scholar]