

Abstract
The mechanisms by which neurons and synapses are lost in Alzheimer's disease (AD) are not completely understood. To characterize potential signaling events linked to AD pathogenesis, activation-specific antibodies were used to examine mitogen-activated protein kinase (MAPK) kinase pathways at various ages in mice transgenic for human amyloid precursor protein-695 with the Swedish familial AD mutations (Tg2576) and homozygous for a P264L familial AD mutation introduced by targeting of the presenilin-1 gene (PS1P264L). Although the c-Jun N-terminal kinase (JNK) and p38 pathways were significantly activated in the cortex at both 7 and 12 months of age, there was no significant activation of the extracellular signal-regulated kinase pathway. MAPK kinase-4, an upstream activator of JNK, was also significantly activated at 7 and 12 months, whereas c-Jun, a downstream effector of JNK-associated apoptotic signaling, was not induced. The lack of c-Jun activation is consistent with the absence of neuronal loss in both cortex and hippocampal CA1 at 12 months. The JNK activation was localized to amyloid deposits, within neurites containing phosphorylated tau. Synaptophysin was quantified biochemically as a measure of synaptic integrity and was significantly reduced in an age-dependent manner in the Tg2576/PS1P264L cortex but not in either PS1P264L or Tg2576 cortex. Stress-responsive MAP kinase pathways were activated in the brain of the Tg2576/PS1P264L AD model, and this activation was coincident with the age-dependent increase in amyloid deposition, tau phosphorylation, and loss of synaptophysin.
Keywords: JNK, p38, ERK, tau, presenilin, synaptophysin, amyloid precursor protein
The memory impairments in Alzheimer's disease (AD) are attributable to the loss of both neurons and synapses (West et al., 1994; DeKosky et al., 1996;Gomez-Isla et al., 1997). Toxic proteins that accumulate in the AD brain are thought to precipitate this loss. Amyloid β protein (Aβ) is a cleavage product of the amyloid precursor protein (APP) and accumulates as extracellular plaque, a diagnostic lesion of the AD brain. Elevated levels of soluble Aβ precede disease pathology in familial forms of AD (FAD) (Scheuner et al., 1996) and in Down's syndrome (Tokuda et al., 1997).
Hyperphosphorylated tau protein accumulates as insoluble paired helical filaments (PHF) within neuronal cell bodies [neurofibrillary tangles (NFTs)] and amyloid-associated processes (dystrophic neurites). Hyperphosphorylated tau binds poorly to microtubules (Iqbal and Grundke-Iqbal, 1996; Mandelkow et al., 1996), likely reducing the efficiency of axonal transport and contributing to neuronal dysfunction. Although tau mutations cause NFTs in frontotemporal dementia with parkinsonism (Hutton et al., 1998), tau mutations resulting in AD have not been demonstrated.
Accompanying increased brain levels of Aβ and tau are elevations in the inflammation-related proteins of the complement cascade, as well as interleukin-1β and tumor necrosis factor-α (Akiyama et al., 2000). Any or all of the above mentioned proteins are potential triggers for the neuronal death and synaptic loss; however, the mechanism(s) by which these losses occur is unknown.
Mitogen-activated protein kinase (MAPK) signaling pathways regulate many cellular processes, including gene expression, differentiation, and cell death (Chang and Karin, 2001). In AD brain, activation of three MAPK pathways has been demonstrated in neurons and dystrophic neurites: c-Jun N-terminal kinase (JNK) (Shoji et al., 2000; Zhu et al., 2001a), p38 (Hensley et al., 1999; Zhu et al., 2000), and extracellular signal-regulated protein (ERK) (Perry et al., 1999;Ferrer et al., 2001). Furthermore, ERK activation was reported in hippocampal slices after treatment with soluble Aβ1–42 (Dineley et al., 2001), whereas JNK pathway activation occurred in both cortical neurons treated with Aβ25–35 and differentiated PC12 cells after exposure to aggregated Aβ1–42 (Bozyzcko-Coyne et al., 2001;Morishima et al., 2001; Troy et al., 2001). Inhibition of the JNK pathway significantly reduced the toxicity attributable to Aβ in all three studies. Increased p38 activity has been reported after Aβ treatment of microglia (McDonald et al., 1998; Pyo et al., 1998).
MAPK pathways have also been implicated in tau hyperphosphorylation. Cell-free assays have demonstrated that JNK, p38, and ERK phosphorylate tau at sites found in AD (Goedert et al., 1997; Reynolds et al., 2000), yet cell-associated phosphorylation of tau by these kinases has not been reported.
Here, MAPK activation was studied using a mouse model incorporating two FAD mutations: (1) a human APP695 transgene with the Swedish FAD mutations (Tg2576) (Hsiao et al., 1996) and (2) a presenilin 1 (PS1)-P264L FAD knock-in mutation (Flood et al., 2001). This model exhibits an aggressive rate of amyloid deposition and associated pathology, achieving 24% cortical amyloid burden at 12 months (Flood et al., 2001). We now report JNK activation in Tg2576/PS1P264L cortex that colocalizes with phospho-tau staining in the abnormal neurites that surround amyloid deposits. Notably, this activation is absent in neuronal cell bodies. p38 is also activated in the cortex of the AD model, whereas ERK is not. Accompanying kinase activation and tau phosphorylation is a reduction in synaptophysin, indicating synaptic abnormalities.
MATERIALS AND METHODS
Transgenic and gene-targeted mouse lines. Mice overexpressing human APP695 with the Swedish FAD mutations (SwAPP695) have been described previously (Tg2576; Hsiao et al., 1996). These transgenic animals were crossed with mice expressing a PS1 FAD knock-in mutation at P264L (Campion et al., 1995; Wasco et al., 1995). Details of PS1 cloning, targeting vector construction, homologous recombination, and chimera production have been reported previously (Siman et al., 2000; Flood et al., 2001). Tg2576/PS1P264L/+ heterozygous males were crossed with nontransgenic PS1P264L/+heterozygous females to generate the four genotypes used in this study. The following littermate-controlled genotypes were analyzed: (1) Tg2576/PS1P264L/P264L(Tg2576/PS1P264L or TgPS1, Tg and homozygous for PS1 mutation), (2) PS1P264L/P264L(PS1P264L or PS1, homozygous for PS1 mutation and non-Tg), (3) Tg2576/PS1+/+(Tg2576, Tg and carrying the normal mouse PS1), and (4) non-Tg/PS1+/+ (wild type or WT). The shortened designations of the genotypes are used hereafter. The use of animals in this study was in accordance with Cephalon's Institutional Animal Care and Use Committee and followed National Institutes of Health guidelines.
Antibodies. The following antibodies from Cell Signaling Technology (Beverly, MA) were used for either immunohistochemistry (IHC) or Western blotting (WB): phospho-JNK (9255; WB), phospho-MAPK kinase-4 (MKK4) (9151; IHC and WB), phospho-ser63-c-Jun (9261; WB), c-Jun (9162; WB), p38 (9212; WB), phospho-ERK (9106; WB), and ERK (9102; WB). Additional antibodies were received from Santa Cruz Biotechnology (Santa Cruz, CA): phospho-JNK (sc6254; IHC), MKK4 (sc837; WB), tau (sc5587; WB), and actin (sc1616; WB). The following antibodies were from Innogenetics (Alpharetta, GA): phospho-tau-ser202 (AT8; IHC and WB) and phospho-tau-thr231 (AT180; IHC and WB). JNK (15691A; WB) was from PharMingen (San Diego, CA), and phospho-p38 (M8177; WB) and synaptophysin (S5768; WB) were from Sigma (St. Louis, MO). Antibody 1153, recognizing human Aβ sequences 1–28, was generated as described previously (IHC; Savage et al., 1994).
Immunohistochemistry. Mice were perfused with 10 ml of Ringer's solution after deep anesthesia with Avertin (1.25% 2,2,2-tribromoethanol and 2.5% 2-methyl-2-butanol). Brains were removed, post-fixed in 70% ethanol and 0.15 mNaCl for 48 hr, and then paraffin embedded. Sections were cut at 10 μm, deparaffinized, and rinsed with 0.01 mPBS. For detection of phospho-JNK, sections were microwaved at 50% power for 10 min in 0.01 m citrate buffer, pH 4.5, and rinsed with water. Detection of phospho-tau, phospho-MKK4, or Aβ deposition required no pretreatment. Endogenous peroxidase was quenched with 0.3% methanol/water (50:50) for 30 min, and nonspecific binding was blocked with 5% goat serum in 0.1 mPBS. After overnight incubation at 4°C with primary antibody in 2% goat serum, horseradish peroxidase (HRP) immunohistochemistry was performed using detection kits from Biogenex (San Ramon, CA) and nickel-intensified 3, 3′-diaminobenzidine. To demonstrate specific staining, primary antibody was eliminated.
Tissue processing and Western blots. Mouse brains were removed after deep anesthesia with Avertin. Cortex was dissected and frozen at −70°C. Tissue was homogenized in 4°C lysis buffer (10 mm Tris, pH 7.6, 50 mmNaCl, 0.03 μm sodium pyrophosphate, 50 mm sodium fluoride, and 1% Triton X-100) containing 1 mm phenylmethylsulfonyl fluoride, 20 μg/ml aprotinin, and 1 mm sodium vanadate. Samples were then centrifuged at 14,000 × g for 10 min at 4°C, and supernatant was assayed for total protein concentration using a BCA assay kit (Pierce, Rockford, IL). Samples of 3, 10, or 100 μg of protein per well were added in NuPage sample buffer (Invitrogen, Carlsbad, CA) to visualize synaptophysin/actin, JNK, and all other proteins, respectively. Proteins were resolved using SDS-PAGE with 10% NuPage gels and morpholinopropanesulfonate buffer (both Invitrogen) and transferred to polyvinylidene difluoride membranes. Membranes were blocked for nonspecific binding with 5% nonfat dry milk in Tris-buffered saline and 0.05% Tween 20 detergent. Primary antibodies were used in this same blocking buffer and incubated with membranes overnight at 4°C. After incubation with HRP-linked secondary antibodies (Southern Biotechnology Associates, Birmingham, AL), proteins were detected using ECL detection reagent (Amersham Biosciences, Piscataway, NJ). Linear ranges were determined for each protein. For quantification of both phosphorylated and nonphosphorylated proteins, RFLPscan software (Scanalytics, Billerica, MA) was used. Phosphoprotein levels were normalized, calculating a density ratio of phosphoprotein/phospho-independent protein (or synaptophysin/actin) within the same sample and gel, first visualizing phosphoprotein (phospho-JNK) and then stripping the blot for detection of specific protein (JNK). This ratio is termed the active fraction for the kinase pathway members. To control for inter-gel variability, the average density ratios for wild-type samples on each gel were set to 100%. Statistical differences were determined using Kruskal–Wallis one-way ANOVA on ranks (SigmaStat software; SPSS Science, Chicago, IL) with Dunnett's method for comparison with wild-type control. Significance was indicated on each graph with anasterisk at p < 0.05.
Neuronal counting. Mice were perfused with Ringer's solution, followed by 4% paraformaldehyde in 0.1m phosphate buffer, pH 7.4. Frozen sections were cut at 30 μm in the coronal plane, slide mounted, and stained with cresyl violet to visualize neuronal nuclei. Using a 100× objective lens, all neurons with a visible nucleolus were counted. Every 12th section was counted, yielding an average of seven sections counted per brain for CA1 and 18 sections for cortex. Neurons were counted with the CASTGrid System (Olympus Optical, Denmark, Copenhagen) using the optical dissector technique (West and Gundersen, 1990). Total neuronal number was estimated by calculating the volume of the CA1 pyramidal layer or the cortex of both hemispheres (excluding piriform and entorhinal cortex) according to the method of Cavalieri. Neuronal number was estimated using ∼100 dissectors for CA1 and 200 dissectors for the cortex. Dissectors were 8 μm high (in an average actual section thickness of 13 μm, measured with an optical encoder attached to the microscope), and the frame area was 880 μm2 for the cortex and 440 μm2 for CA1. The estimation of total neurons was calculated by multiplying the neuronal density by the volume of CA1 or cortex. A single examiner (M.J.S.) performed the counting and was blinded as to genotype. Coefficients of error were <5% with the parameters used to estimate counts in both regions.
RESULTS
Activation of the JNK pathway in AD model cortex
The active, phosphorylated fractions of JNK, MKK4, and c-Jun in the brains of Tg2576/PS1P264L mice at various ages and levels of amyloid deposition were measured by immunoblot. At 3, 7, and 12 months, amyloid deposition was ∼2, 12, and 24% of the cortical area, respectively (Flood et al., 2001). Wild-type controls were examined at 12 months and had no deposited amyloid. Levels of both phospho-JNK (Fig.1A, top) and total JNK (Fig. 1A, bottom) migrating at two molecular weights, p54 and p46, are demonstrated from the same samples. There was an age-dependent increase in phosphorylation of p54 JNK, whereas steady-state levels of the protein decreased. p46 demonstrated a relatively constant steady-state phosphorylation with decreased total protein. When the densities of the phospho-JNK bands were standardized to the total JNK bands for each sample and wild-type ratios were set to 100% to control for inter-gel variability, p54 demonstrated an age-dependent increase at 2.7-fold and 5.8-fold over the wild-type ratio at 7 and 12 months, respectively (Fig.1B). There was a twofold increase in the active fraction of the p46 form at 12 months, with no significant elevation at 7 months.
Fig. 1.

JNK activation is age dependent in Tg2576/PS1P264L cortex. A, Thetop demonstrates 10 μg of cortical protein per sample stained with an antibody against phospho-JNK (phospho-thr183/phospho-thr185). These sites can be phosphorylated by MKK4 and indicate the presence of activated JNK in older Tg2576/PS1P264L cortex. The bottomdemonstrates total JNK protein from the same blot after stripping of the phospho-JNK antibodies. JNK protein levels decrease in the Tg2576/PS1P264L cortex with age. Brepresents the active fraction of JNK from a density ratio of phospho-JNK to total JNK for each form. Six samples per group were assayed in triplicate on immunoblot. *p ≤ 0.05 indicates significant differences. WT 12, Wild-type mice at 12 months of age.
To confirm that JNK activation was attributable to amyloid deposition and not an effect of either APP overexpression or the PS1-P264L mutation alone, the following littermate-controlled cortical samples at 12 months were compared with wild-type cortex for JNK activation: (1) PS1P264L homozygous knock-in mutation, (2) Tg2576 with human SwAPP695 overexpression, or (3) the combined Tg2576/PS1P264L genotypes. Relatively constant levels of phospho-JNK were present in all groups, except Tg2576/PS1P264L, in which levels of phospho-JNK were increased substantially (Fig.2A, middle). When the active fraction of JNK was again represented as a percentage of the wild-type samples on each gel, the JNK activity continued to be significantly upregulated in the Tg2576/PS1P264L cortex at 12 months (Fig.2B). In this group, the p54 JNK was increased 5.5-fold, whereas the p46 JNK was increased ∼2.5-fold. Interestingly, the level of phospho-ser202-tau, detected with the AT8 antibody (Fig.2A, top), mirrored the JNK activation. In Tg2576 cortex, with 0.5% amyloid burden (Flood et al., 2001), the increased JNK activity was not statistically significant compared with wild-type samples.
Fig. 2.

JNK activation is genotype dependent.A, The middle demonstrates cortical samples at 12 months stained as in Figure 1 for detection of activated JNK. Increased phosphorylation of p54 JNK in the Tg2576/PS1P264L cortex compared with wild-type, Tg2576, or PS1P264L cortex. Thebottom depicts total JNK as in Figure 1. Thetop shows the same samples stained for a third time for phosphorylated tau at ser202. Note the increase in phospho-tau in the Tg2576/PS1 cortex with the greatest amyloid burden and an intermediate degree of phospho-tau in Tg2576. B depicts a significant increase at p ≤ 0.05 (asterisks) in the active fraction of both p54 and p46 JNK in the Tg2576/PS1P264L cortex compared with wild type. Three samples per group were assayed in triplicate on immunoblot.
In addition to the presence of active JNK, the upstream kinase MKK4 was activated in Tg2576/PS1P264L cortex. Brains were examined for phospho-MKK4 on Western blots and normalized to MKK4 protein within the same samples. There was a significant eightfold and sixfold increase in the ratio of activated MKK4 to total MKK4 protein at 7 and 12 months, respectively (Fig.3B). The increase in phosphorylated MKK4 (Fig. 3A, top) occurred with a subtle decrease in total protein (Fig. 3A,bottom). When brains of Tg2576/PS1P264L mice at 12 months were immunostained for phospho-MKK4, the staining localized to neuronal cell bodies and axons, as seen for total MKK4 in rat brain (Flood et al., 1998). Both amyloid deposits and the surrounding neurites were free of stain. At 12 months, there was no obvious change in staining intensity or location when comparing wild-type and Tg2576/PS1P264L animals.
Fig. 3.

MKK4 is activated in Tg2576/PS1P264L cortex. A, Thetop demonstrates 100 μg of cortical protein per sample stained with an antibody against phosphorylated MKK4 (phospho-thr261). This site is phosphorylated by MAPKK kinases and indicates the presence of more activated MKK4 in 7 and 12 month cortical samples compared with 12-month-old wild-type littermates (WT 12). Thebottom demonstrates total MKK4 using the same blot.B represents the active fraction of MKK4 from a density ratio of phospho-MKK4 to total MKK4. Six samples per group, each assayed in triplicate by immunoblot. *p ≤ 0.05 indicates significant differences.
Unlike JNK and MKK4, there was no demonstrable increase in phospho-c-Jun in the Tg2576/PS1P264Lcortex. Compared with the 12-month-old wild-type animals that had barely detectable phospho-c-Jun, this activity was reduced in the Tg2576/PS1P264L cortex at all ages (Fig.4, top). This phosphorylated species was defined by reaction with a phospho-ser63-c-Jun antibody, molecular weight, and colocalization with total c-Jun after stripping and reprobing with an antibody against c-Jun protein. Like JNK, c-Jun levels were decreased in the cortex of the older Tg2576/PS1P264L animals (Fig. 4,bottom).
Fig. 4.

c-Jun phosphorylation is not increased with age in cortex of Tg2576/PS1P264L mice.Top blot demonstrates cortical protein at 100 μg per well, stained with an antibody against phosphorylated c-Jun (phospho-ser63). This site can be phosphorylated by JNK and induces c-Jun transcription factor activity. Bottom blotdemonstrates a reduction in levels of total c-Jun with age in the Tg2576/PS1P264L cortex using the same blot.WT 12, Wild-type mice at 12 months of age.
Active JNK is found within abnormal neurites
The brains of Tg2576/PS1P264L animals at 2, 4, 6, 9, and 12 months were examined histologically for the location of phospho-JNK immunoreactivity. Serial sections were stained with antibody 1153 (recognizing Aβ sequences 1–28) and antibodies specific for phospho-JNK and phospho-tau. Phospho-JNK immunoreactivity (Fig.5B,E) colocalizes with that of deposited amyloid (Fig.5A,D). On top of a background of light JNK neuropil staining and within the mass of deposited amyloid, there were defined profiles detected with the phospho-JNK antibody (Fig. 5E). Phospho-JNK immunoreactivity, detected as early as 4 months, was located around amyloid deposits at all ages, with increased staining corresponding to increased amyloid burden. This degree of induction at ages <4 months may be below the sensitivity of the Western blot method because there was no detectable increase at 3 months (Fig. 1) on the Western blot. When the serial phospho-JNK (Fig.6B,E) and phospho-tau (Fig. 6, pthr231, A, D; pser202,C, F) sections are compared, profiles of the same size and shape colocalize, although they are not completely superimposable because of the small diameter of the neurites (∼3 μm) and the thickness of the sections (10 μm). No detectable phospho-JNK localized to neuronal cell bodies in the brains of the Tg2576/PS1P264L mice, which appear unstained (Fig. 5E) compared with the surrounding neuropil.
Fig. 5.
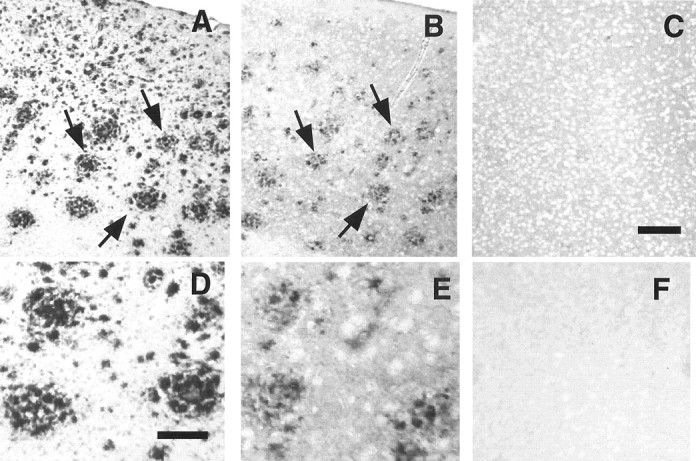
Activated JNK is localized to amyloid deposits. Tg2576/PS1P264L cortex at 12 months, stained with anti- Aβ1–28, recognizing deposited amyloid (A, D) or phosphorylated JNK (B, E). pJNK immunoreactivity is localized to defined profiles within the amyloid deposits.C, Twelve-month-old control without amyloid deposits treated with the phospho-JNK antibody. F, No primary antibody control on Tg2576/PS1P264L cortex, as inB. Arrows indicate amyloid deposits and phospho-JNK staining that colocalize on adjacent sections. Scale bars: (in C) A–C, F, 100 μm; (in D) D, E, 30 μm.
Fig. 6.
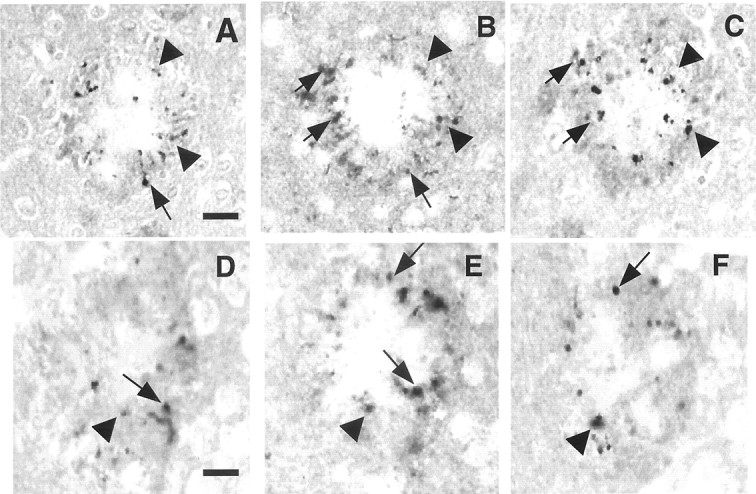
Activated JNK within amyloid deposits is localized to neurites containing phospho-tau. Tg2576/PS1P264L cortex at 12 months, stained with an antibody to phospho-thr231 tau (A, D) phospho-JNK (B, E), or phospho-ser202 tau (C, F). A–C andD–F constitute serial sections through two separate amyloid deposits. Arrows indicate some of the neurites that overlap between two serial sections; arrowheadsindicate neurites that overlap between all three sections. Scale bars: (in A) A–C, 15 μm; (inD) D–F, 12 μm.
Activation of p38 in AD model cortex
p38 activation was examined by immunoblot in the Tg2576/PS1P264L mouse cortex at 7 and 12 months using a phospho-specific, activity-dependent antibody. Again, wild-type cortex was compared at 12 months. p38 phosphorylation (Fig.7A, top) was increased significantly in a progressive manner, whereas total p38 protein was constant with age (Fig. 7A, bottom). The active fractions of phospho-p38/total p38 were increased threefold and eightfold, respectively, at 7 and 12 months in Tg2576/PS1P264L in cortex (Fig.7B). Specific phospho-p38 staining was not detected immunohistochemically in the animal brain with available reagents.
Fig. 7.

Age-dependent activation of p38 in the Tg2576/PS1P264L cortex. A, Thetop demonstrates 100 μg of cortical protein, stained with an antibody selective for activated p38 phosphorylated by an upstream kinase such as MKK3/6. The bottom depicts total p38 protein, which is relatively constant with age. Bdepicts the active fraction of p38 that increases with age and amyloid burden. Six samples per group were each assayed in triplicate on immunoblot. *p ≤ 0.05 indicates significant differences.
ERK is not activated in AD model cortex
Whereas the stress-related pathways JNK and p38 demonstrated increased activity consistent with a role in the response of the brain to deposited amyloid, the trophic arm of the MAPK pathways was not induced with age and amyloid deposition. The two molecular weight forms of phospho-ERK, p42 and p44 (Fig.8A, top), were not elevated when normalized to the corresponding total ERK protein (Fig. 8A, bottom). In fact, the active fraction of the higher molecular weight form, p44 ERK, was significantly decreased at 12 months in Tg2576/PS1P264L cortex attributable to relatively constant levels of phospho-p44 ERK and increased levels of p44 ERK (Fig. 8B). Although there was a subtle increase in p42 activity at 7 months with a decrease at 12 months, these effects were not statistically significant.
Fig. 8.

ERK pathway is not activated in the Tg2576/PS1P264L cortex. A, Thetop demonstrates 100 μg of cortical protein, stained with anti-phospho-ERK. A very small increase in p44-ERK phosphorylation was present in cortical samples from Tg2576/PS1P264Lmice compared with the increase in total ERK from the same blot (A, bottom). The active fraction of p44 is reduced in an age-dependent manner (B). The active fraction of p42 is not changed significantly. Six samples per group were each assayed by immunoblot in triplicate. *p ≤ 0.05 indicates significant differences.WT 12, Wild-type mice at 12 months of age.
Increased tau phosphorylation in AD model cortex
Tau phosphorylation increases with amyloid deposition in a number of AD models (Moechars et al., 1999; Sturchler-Pierrat and Staufenbiel, 2000; Masliah et al., 2001; Tomidokoro et al., 2001). Similar to the AD brain, phospho-tau was localized to amyloid-associated neurites (Fig. 6) in the brain of Tg2576/PS1P264L animals at 12 months. Elevated phospho-tau staining was not detected in neuronal cell bodies compared with wild-type cortex. Increased tau phosphorylation was evident on Western blots with AT8, recognizing phosphorylated serine 202 (Fig. 2A, top). Confirming the phosphorylation at this site, steady-state levels of the tau-1 epitope (dephosphorylated serine 202) were reduced (data not shown). There was also increased phosphorylation at threonine 231 (Fig.9, top), whereas total tau protein levels were relatively constant (Fig. 9, bottom). As expected after increased phosphorylation, the form of total tau comigrating with the phosphorylated forms (Fig. 9, bottom,thinner top band) was increased in the depositing animals. Although the increase in the higher molecular weight species of phospho-tau is apparent at 7 months, there is no corresponding increase in the specific thr231 phospho-tau epitope at this age, suggesting that another site(s) contributes primarily to the shift in molecular weight.
Fig. 9.

Increased phosphorylation of tau occurs with age in Tg2576/PS1P264L cortex. Whereas concentrations of total tau are relatively constant with increasing amyloid burden (bottom blot), phosphorylated tau at thr231 increases in intensity (top blot), as does the higher molecular weight species stained with the antibody to total tau in Tg2576/PS1P264L mice (bottom,arrow). This higher molecular weight tau species comigrates with phosphorylated tau stained with both phospho-tau antibodies used in this study. The phosphorylated tau epitope ser202 increases as well in 12-month-old Tg2576/PS1P264Lcortex, as seen in Figure 2. WT 12, Wild-type mice at 12 months of age.
Progressive neuronal loss is not present in the cortex or CA1
Neuronal cell counts were performed in the cortex (excluding piriform and entorhinal) at 12 months in both wild-type and Tg2576/PS1P264L littermates. There were no differences in numbers of cortical neurons between these groups (WT, 1.35 × 107;n = 5; Tg2576/PS1P264L, 1.45 × 107, n = 3). Neuronal counts were also performed in hippocampal CA1 at 12 months in wild-type, PS1P264L, and Tg2576/PS1P264L littermate brains and at 2 months in wild-type and Tg2576/PS1P264Llittermates. Although there were no significant differences, there was a small reduction of 17% in neuronal number at 12 months in Tg2576/PS1P264L compared with wild-type mice (Fig. 10). A similar magnitude of reduction was present at 2 months when amyloid deposition was virtually undetectable in the Tg2576/PS1P264Lbrains. In addition, animals with the PS1-P264L mutation alone had an equivalent reduction at 12 months of age compared with wild type. Therefore, the small reduction in CA1 neuronal number in this model was independent of age or amyloid deposition and most likely attributable to a developmental reduction driven by either the PS1-P264L mutation or the reduction in PS1 endoproteolytic cleavage products in this model (Flood et al., 2001).
Fig. 10.

Neuronal cell bodies were not lost in the hippocampal CA1 subfield at 12 months in Tg2576/PS1P264L. Cell counts were performed in littermate brains after cresyl violet staining of Tg2576/PS1P264L, PS1P264L, or wild-type frozen sections. Animals were either 2 or 12 months of age. A 17% reduction in CA1 neuronal number present at 12 months in Tg2576/PS1P264L CA1 was also present at 12 months in PS1P264L mice and at 2 months in Tg2576/PS1P264L mice. These data indicate that any reduction is not attributable to age/amyloid burden but may be developmental and attributable to the PS1P264Lmutation. Five subjects in each group, except for the PS1P264L group with three subjects.
Synaptophysin is reduced with age and amyloid deposition
Synaptophysin was measured in the four littermate-controlled genotypes at 12 months as an indicator of synaptic loss or dysfunction. Synaptophysin levels were significantly reduced by 50% in the Tg2576/PS1P264L cortex at 12 months compared with wild type, whereas the PS1P264L and Tg2576 groups demonstrated no significant reduction at this age (Fig, 11A, top blot, B). The actin blot demonstrates comparable protein loads per lane (Fig. 11A, bottom blot). When the same measurements were performed in Tg2576/PS1P264L animals at increasing ages and levels of amyloid deposition, there is a progressive loss of synaptophysin (Fig. 11C). Although there is a nonsignificant reduction of 22% at 3 months, cortical synaptophysin is significantly reduced at both 7 and 12 months to 60 and 40% of wild-type values, respectively.
Fig. 11.
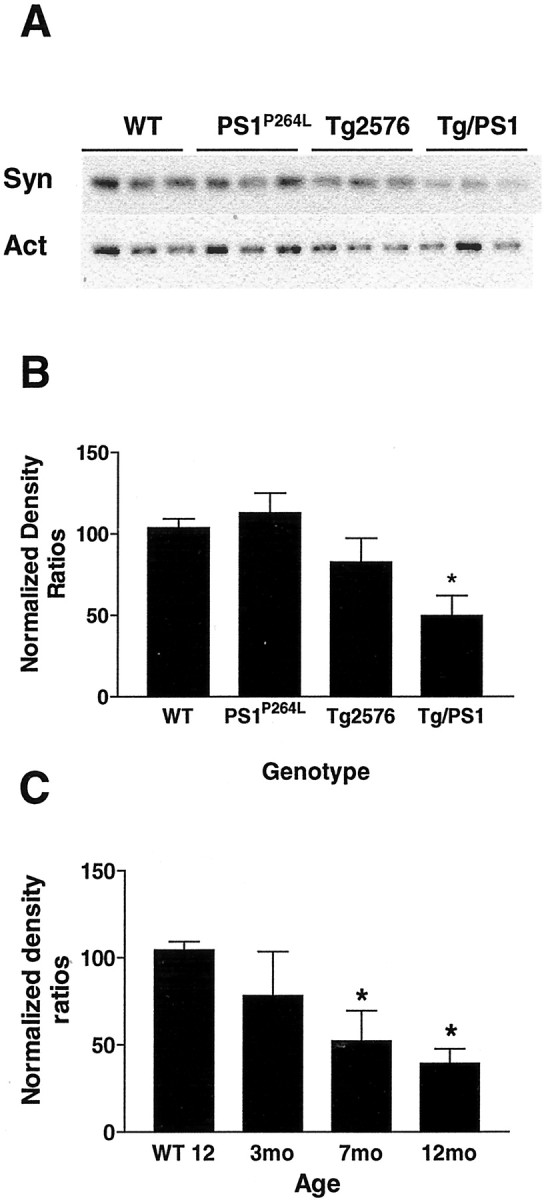
Cortical synaptophysin is significantly reduced in aged Tg2576/PS1P264L mice, and the reduction is genotype dependent. A, The topdemonstrates 3 μg of cortical protein per sample stained with an antibody recognizing synaptophysin (Syn). At 12 months, a reduction was seen only in the Tg2576/PS1P264Lcortex compared with wild type. The bottom demonstrates approximately equivalent actin (Act) protein per well on the same blot, as a control. Graphs depict the density ratios of synaptophysin/actin. B, A 50% reduction is found in the Tg2576/PS1P264L cortex compared with the wild-type controls at 12 months. Three subjects per group with seven immunoblot assay replicates. C, Progressive reductions in the synaptophysin/actin ratio exist at 7 and 12 months in the Tg2576/PS1P264L mice compared with wild-type controls. Six samples per group with duplicate assays. *p ≤ 0.05 indicates significant differences.WT 12, Wild-type mice at 12 months of age.
DISCUSSION
In this report, activation of both JNK and p38 is demonstrated in the cortex of the Tg2576/PS1P264L mouse model of AD. A third MAP kinase family member, ERK, was not activated.
JNK activation was age dependent and was associated with both amyloid deposition and tau phosphorylation. Increased phospho-JNK immunoreactivity was detected both on Western blot and in tissue sections, despite decreased levels of total JNK. Thus, the JNK activation was likely to be mediated by activation of an upstream kinase. This was confirmed by an increase in the active fraction of MKK4 in the Tg2576/PS1P264L cortex. MKK4 and MKK7 are both JNK-activating MAPK kinases, which can be activated by a number of MAPKK kinases, such as mixed-lineage kinase, apoptosis signal-regulating kinase-1, MAPK kinase kinase-1, and transforming growth factor-β-activated kinase-1 (Kyriakis and Avruch, 2001). MKK4 activity was increased eightfold and sixfold at 7 and 12 months, respectively. Because the phospho-MKK4 immunostaining did not colocalize with phospho-JNK in the neurites (data not shown) and demonstrated greater activation at 7 months compared with 12 months, unlike that of JNK, the direct role of MKK4 in the neuritic JNK activation is unclear. The localization of phospho-MKK4 in AD brain has not been reported.
The species of JNK activated in this model was not determined because activation-specific reagents do not distinguish between JNK isoforms. Twelve isoforms result from the alternative splicing of three JNK genes, JNK1–JNK3 (Kyriakis and Avruch, 2001). Two molecular weight forms, p54 and p46, were detected with the JNK antibodies used here. Both forms were activated in the Tg2576/PS1P264L cortex at 12 months, although p54 exhibited 5.8-fold activation compared with twofold for p46. JNK activation was not attributable to either the PS1-P264L mutation or the overexpression of SwAPP695 alone, because cortical samples at 12 months from the individual genotypes did not demonstrate significant activation on the blot compared with wild-type littermates.
JNK pathway activation in AD brain was demonstrated previously, in which levels of both phospho-JNK (Shoji et al., 2000; Zhu et al., 2001a) and c-Jun (Anderson et al., 1994, 1996) were measured. In AD, JNK activation was localized to both neuronal cell bodies and neurites around amyloid deposits. In the Tg2576/PS1P264L brain, JNK activation was also localized to reactive neurites containing phospho-tau, consistent with a role for amyloid in inducing this activation. The neuritic activation of JNK colocalized with increased tau phosphorylation at thr231 and ser202 in serial sections. In a cell-free system, JNK can phosphorylate tau at these sites that are hyperphosphorylated in AD tau (Reynolds et al., 2000). In the brains of mice exposed to starvation-induced stress, increased phospho-tau (phospho-ser202) also correlated with JNK activation (Planel et al., 2001). Chemical agents that disrupt microtubule structure can also activate JNK (Wang et al., 1998), suggesting, perhaps, that tau phosphorylation could lead to JNK activation. Interestingly, stress-activated protein kinase pathway activation colocalizes with tau pathology in a number of neurodegenerative diseases (Atzori et al., 2001).
Consistent with JNK activation in vivo in both AD and animal model brains, treatment of primary rat cortical neurons with Aβ25–35 or treatment of neuronal-like PC12 cells with Aβ1–42 activated the JNK pathway (Bozyzcko-Coyne et al., 2001; Morishima et al., 2001; Troy et al., 2001). In addition, MKK4 activation was demonstrated both in cell culture after treatment with toxic Aβ species (Bozyzcko-Coyne et al., 2001) and here in the Tg2576/PS1P264Lcortex with increased amyloid burden. Compared with the AD brain and the cell culture systems, however, there is no loss of neurons or nuclear activation of either JNK or c-Jun in the Tg2576/PS1P264L cortex. This lack of c-Jun activation is consistent with the absence of neuronal loss.
c-Jun induction need not follow JNK activation. In a recent paper examining the distribution of active JNK in cerebellar granule neurons (CbG) (Coffey et al., 2000), most of the activity was not involved in stress-activated gene transcription. The cytoplasmic JNK was instead developmentally upregulated during neurite outgrowth in parallel with increased MKK4/7 activity in the absence of c-Jun activation; similar biochemical changes are reported here. Also, JNK inhibition led to an increase in neurite outgrowth in the CbG. In another report using dorsal root ganglia, JNK inhibition also promoted the extension of neurites (Borasio et al., 1998), although JNK inhibition in NGF-induced PC12 cells was associated with a delay in neurite outgrowth (Maroney et al., 1999). Long-lasting JNK activation has also been reported after axotomy in the CNS (Herdegen et al., 1998) or peripheral nervous system (PNS) (Kenney and Kocsis, 1998). JNK activation persisted for days after axotomy in both studies and was downregulated after reestablishment of target contact in the PNS. In both studies, despite persistently increased induction of both JNK and c-Jun, there was no neuronal death. JNK activation may function to modulate regenerative neurite outgrowth after axotomy, during development, and in the neuritic response to amyloid deposition.
p38 is part of a proinflammatory signal transduction pathway activated in response to a number of cytokines (Foltz et al., 1997). p38 activation increased with age and amyloid burden in the Tg2576/PS1P264L cortex. Like the JNK pathway, activated p38 was detected using antibodies to phospho-specific epitopes that result from upstream activation by MAPK kinases, in this case, MKK3/6. The increase in phospho-p38 did not result from an increase in p38 protein, which remained stable with age. By 12 months, p38 was activated by eightfold in the Tg2576/PS1P264L cortex compared with wild type. The localization of phospho-p38 was not determined here; however, activation of both p38 and MKK6 has been localized in the AD brain to neuronal cell bodies and abnormal neurites surrounding amyloid deposits (Hensley et al., 1999; Zhu et al., 2000, 2001b). Like JNK, p38 can phosphorylate tau in a cell-free system (Reynolds et al., 2000) and was coimmunoprecipitated with PHF-1 tau (Zhu et al., 2000) from AD brain. In addition, cell culture experiments demonstrated p38 activation after Aβ treatment of primary rat microglia (McDonald et al., 1998; Pyo et al., 1998). JNK was not activated in this microglial model (McDonald et al., 1998).
There was no significant ERK activation in the Tg2576/PS1P264L cortex. In fact, there was a significant, age-dependent reduction in the active fraction of p44 to 30% of wild-type levels by 12 months. p42 was not significantly affected; however, there was a slight increase in the active fraction at 7 months and a decrease at 12 months. This is similar to the findings of Dineley et al. (2001) in the CA1 of Tg2576 hippocampus. They reported a subtle yet significant increase in phospho-ERK2 (p42) at 13 months in CA1 accompanied by a comparable increase in total ERK2. By 20 months, levels of both total and phospho-ERK were significantly reduced. Tg2576 animals at 20 months have a cortical amyloid burden comparable with the Tg2576/PS1P264L at 12 months (24%) (Flood et al., 2001). These results together suggest that chronic exposure to high levels of amyloid downregulate ERK signaling in AD animal models. This could lead to memory deficits because ERK is required for some aspects of long-term potentiation (English and Sweatt, 1997; Impey et al., 1998). ERK was activated in the AD brain (Perry et al., 1999; Ferrer et al., 2001) and transiently in the dentate gyrus of Tg2576 (Dineley et al., 2001). Whether this activation is present before end-stage AD is unknown.
Although neurons were not lost as a result of increased amyloid burden in our model, synaptophysin levels were reduced in an age- and genotype-dependent manner. The loss of synapses in other mouse models of Aβ deposition has been demonstrated previously (Hsia et al., 1999;Wong et al., 1999; Mucke et al., 2000), and the loss of neurons was reported in two models (Calhoun et al., 1998; Hsia et al., 1999). In two studies (Hsia et al., 1999; Mucke et al., 2000), the reported reductions in synapse number likely resulted from increased soluble Aβ rather than insoluble amyloid because the reductions were present earlier than the deposits and there was little progressive loss with increasing plaque burden. Increased soluble Aβ may also play a role in reducing the synaptophysin/actin ratio in the Tg2576/PS1P264L cortex; however, the loss did progress with age to 50–60% of wild-type levels with increasing plaque burden. The role of JNK or p38 in this loss is unknown; however, the progressive loss in synaptophysin is associated with the concomitant increase in JNK and p38 activity. The behavioral and electrophysiological consequences of this reduction in synaptophysin are presently unknown in this model.
The intersection of MAP kinase pathways with APP and tau biology has been suggested by the reports already mentioned and by a number of additional reports: (1) presenilin activity inhibits JNK (Kim et al., 2001); (2) JNK phosphorylates APP (Standen et al., 2001); (3) JNK binding-inhibitory protein binds APP (Matsuda et al., 2001); and (4) Dishevelled increases APP secretion via JNK (Mudher et al., 2001). Regarding the role of JNK and p38 in AD pathology, one possibility is that a stressor(s) activates JNK/p38, leading to changes in APP metabolism, tau phosphorylation, synaptic/neuritic dystrophy, or glial activation. Alternatively, Aβ, phospho-tau, or cytokines may be the stressors that activate JNK/p38. Positive feedback loops may be present in the AD brain whereby the initial stressor is amplified via MAP kinase pathway activation.
In addition to reproducing amyloid deposits and tau hyperphosphorylation in common with AD brain, the Tg2576/PS1P264L mouse also incorporates brain activation of the JNK and p38 pathways. This reinforces the likely importance of stress-activated protein kinase pathways in AD because pathway activation has now been demonstrated after Aβ toxicity in vitro, in an AD animal model, and in the AD brain. The Tg2576/PS1P264L mouse model of AD will allow for the testing of JNK and p38 pathway inhibitors to determine their role in AD pathology.
Footnotes
We thank Drs. Donna Bozyczko-Coyne and Craig Dionne for helpful comments on this manuscript, Renee Simmons and Edwin McCabe for excellent care of the animals, and James Knabb for excellent technical assistance.
Correspondence should be addressed to Dr. Mary Savage, Cephalon Inc., 145 Brandywine Parkway, West Chester, PA 19380. E-mail:msavage@cephalon.com.
REFERENCES
- 1.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson AJ, Cummings BJ, Cotman CW. Increased immunoreactivity for Jun- and Fos-related proteins in Alzheimer's disease. Exp Neurol. 1994;125:286–295. doi: 10.1006/exnr.1994.1031. [DOI] [PubMed] [Google Scholar]
- 3.Anderson AJ, Su JH, Cotman CW. DNA damage and apoptosis in Alzheimer's disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J Neurosci. 1996;16:1710–1719. doi: 10.1523/JNEUROSCI.16-05-01710.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atzori C, Ghetti B, Piva R, Srinivasan AN, Zolo P, Delisle MB, Mirra SS, Migheli A. Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J Neuropath Exp Neurol. 2001;60:1190–1197. doi: 10.1093/jnen/60.12.1190. [DOI] [PubMed] [Google Scholar]
- 5.Borasio GD, Horstmann S, Anneser JMH, Neff NT, Glicksman MA. CEP-1347/KT7515 a JNK pathway inhibitor, supports the in vitro survival of chick embryonic neurons. NeuroReport. 1998;9:1435–1439. doi: 10.1097/00001756-199805110-00034. [DOI] [PubMed] [Google Scholar]
- 6.Bozyzcko-Coyne D, O'Kane TM, Wu ZL, Dobrzanski P, Murthy S, Vaught JL, Scott RW. CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Aβ-induced cortical neuron apoptosis. J Neurochem. 2001;77:849–863. doi: 10.1046/j.1471-4159.2001.00294.x. [DOI] [PubMed] [Google Scholar]
- 7.Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Neuron loss in APP transgenic mice. Nature. 1998;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- 8.Campion D, Flaman JM, Brice A, Hannequin D, Dubois B, Margin C, Moreau V, Charbonnier F, Didierjean O, Tardieu S, Penet C, Puel M, Pawquier F, Le Doze F, Bellis G, Calenda A, Heilig R, Martinez M, Mallet J, Bellis M. Mutations of the presenilin I gene in families with early-onset Alzheimer's disease. Hum Mol Genet. 1995;4:2373–2377. doi: 10.1093/hmg/4.12.2373. [DOI] [PubMed] [Google Scholar]
- 9.Chang L, Karin M. Mammalian MAP kinase signaling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 10.Coffey ET, Hongisto V, Dickens M, Davis RJ, Courtney MJ. Dual roles for c-Jun N-terminal kinase in developmental and stress responses in cerebellar granule neurons. J Neurosci. 2000;20:7602–7613. doi: 10.1523/JNEUROSCI.20-20-07602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;19965:417–421. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- 12.Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. β-Amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long-term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 14.Ferrer I, Blanco R, Carmona M, Ribera R, Goutan E, Puig B, Rey MJ, Cardozo A, Vinals F, Ribalta T. Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurons and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer's disease, Pick's disease, progressive supranuclear palsy, and corticobasal degeneration. Brain Pathol. 2001;11:144–158. doi: 10.1111/j.1750-3639.2001.tb00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flood DG, Finn JP, Walton KM, Dionne CA, Contreras PC, Miller MS, Bhat RV. Immunolocalization of the mitogen-activated protein kinases p42MAPK and JNK1, and their regulatory kinases MEK1 and MEK4, in adult rat central nervous system. J Comp Neurol. 1998;398:373–392. doi: 10.1002/(sici)1096-9861(19980831)398:3<373::aid-cne6>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 16.Flood DG, Reaume AG, Dorfman KS, Lin YG, Lang DM, Trusko SP, Savage MJ, Annaert WG, De Strooper B, Siman R, Scott RW. FAD mutant PS-1 gene-targeted mice: increased Aβ42 and Aβ deposition without APP overexpression. Neurobiol Aging. 2001;23:335–348. doi: 10.1016/s0197-4580(01)00330-x. [DOI] [PubMed] [Google Scholar]
- 17.Foltz IN, Lee JC, Young PR, Schrader JW. Hemopoietic growth factors with the exception of interleukin-4 activate the p38 mitogen-activated protein kinase pathway. J Biol Chem. 1997;272:3296–3301. doi: 10.1074/jbc.272.6.3296. [DOI] [PubMed] [Google Scholar]
- 18.Goedert M, Hasegawa M, Jakes R, Lawler S, Cuenda A, Cohen P. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997;409:57–62. doi: 10.1016/s0014-5793(97)00483-3. [DOI] [PubMed] [Google Scholar]
- 19.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 20.Hensley K, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X, Pye QN, Stewart CA, Geddes J, Markesberry WR, Patel E, Johnson GVW, Bing G. p38 kinase is activated in Alzheimer's disease brain. J Neurochem. 1999;72:2053–2058. doi: 10.1046/j.1471-4159.1999.0722053.x. [DOI] [PubMed] [Google Scholar]
- 21.Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin M. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 24.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, De Graaff E, Wauters E, Van Baren J. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 25.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Crosstalk between ERK and PKA is required for Ca+2 stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 26.Iqbal K, Grundke-Iqbal I. Molecular mechanism of Alzheimer's neurofibrillary degeneration and therapeutic intervention. Ann NY Acad Sci. 1996;777:132–138. doi: 10.1111/j.1749-6632.1996.tb34411.x. [DOI] [PubMed] [Google Scholar]
- 27.Kenney AM, Kocsis JD. Peripheral axotomy induced long-term c-Jun amino-terminal kinase-1 activation and activator protein-1 binding activity by c-Jun and JunD in adult rat dorsal root ganglia in vivo. J Neurosci. 1998;18:1318–1328. doi: 10.1523/JNEUROSCI.18-04-01318.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JW, Chang TS, Lee JE, Huh SH, Yeon SW, Yang WS, Joe CO, Mook-Jung I, Tanzi RE, Kim TW, Choi EJ. Negative regulation of the SAPK/JNK signaling pathway by presenilin 1. J Cell Biol. 2001;153:457–463. doi: 10.1083/jcb.153.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 30.Mandelkow EM, Schweers O, Drewes G, Biernat J, Gustke N, Trinczek B, Mandelkow E. Structure, microtubule interactions, and phosphorylation of tau protein. Ann NY Acad Sci. 1996;777:96–106. doi: 10.1111/j.1749-6632.1996.tb34407.x. [DOI] [PubMed] [Google Scholar]
- 31.Maroney AC, Finn JP, Bozyczko-Coyne D, O'Kane TM, Neff NT, Tolkovsky AM, Park DS, Yan CYI, Tron CM, Green LA. CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J Neurochem. 1999;73:1901–1912. [PubMed] [Google Scholar]
- 32.Masliah E, Sisk A, Mallory M, Games D. Neurofibrillary pathology in transgenic mice overexpressing V171F beta-amyloid precursor protein. J Neuropathol Exp Neurol. 2001;60:357–368. doi: 10.1093/jnen/60.4.357. [DOI] [PubMed] [Google Scholar]
- 33.Matsuda S, Yasukawa T, Homma Y, Ito Y, Niikura T, Hiraki T, Hirai S, Ohno S, Kita Y, Kawasumi M, Kouyama K, Yamamoto T, Kyriakis JM, Nishimoto I. c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain 1 scaffolds Alzheimer's amyloid precursor protein with JNK. J Neurosci. 2001;21:6597–6607. doi: 10.1523/JNEUROSCI.21-17-06597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald DR, Bamberger ME, Combs CK, Landreth GE. β-Amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP1 monocytes. J Neurosci. 1998;18:4451–4460. doi: 10.1523/JNEUROSCI.18-12-04451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- 36.Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME. β-Amyloid induced neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of fas ligand. J Neurosci. 2001;21:7551–7560. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mudher A, Chapman S, Richardson J, Asuni A, Gibb G, Pollard C, Killick R, Iqbal T, Raymond L, Varndell I, Sheppard P, Makoff A, Gower E, Soden PE, Lewis P, Murphy M, Golde TE, Rupniak HT, Anderton BH, Lovestone S. Dishevelled regulates the metabolism of amyloid precursor protein via protein kinase C/mitogen-activated protein kinase and c-Jun terminal kinase. J Neurosci. 2001;21:4987–4995. doi: 10.1523/JNEUROSCI.21-14-04987.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perry G, Roder H, Nunomura A, Takeda A, Friedlich AL, Zhu X, Raina AK, Holbrook N, Siedlak SL, Harris PLR, Smith MA. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer's disease links oxidative stress to abnormal phosphorylation. NeuroReport. 1999;10:2411–2415. doi: 10.1097/00001756-199908020-00035. [DOI] [PubMed] [Google Scholar]
- 40.Planel E, Yasutake K, Fujita SC, Ishiguro K. Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3β and cyclin-dependent kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J Biol Chem. 2001;276:34298–34306. doi: 10.1074/jbc.M102780200. [DOI] [PubMed] [Google Scholar]
- 41.Pyo H, Jou I, Jung S, Hong S, Joe EH. Mitogen-activated protein kinases activated by lipopolysaccharide and beta-amyloid in cultured rat microglia. NeuroReport. 1998;9:871–874. doi: 10.1097/00001756-199803300-00020. [DOI] [PubMed] [Google Scholar]
- 42.Reynolds CH, Betts JC, Blackstock WP, Nebreda AR, Anderton BH. Phosphorylation sites on tau identified by nanoelectrospray mass spectroscopy: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase 3β. J Neurochem. 2000;74:1587–1595. doi: 10.1046/j.1471-4159.2000.0741587.x. [DOI] [PubMed] [Google Scholar]
- 43.Savage MJ, Iqbal M, Loh T, Trusko SP, Scott R, Siman R. Cathepsin G: localization in human cerebral cortex and generation of amyloidogenic fragments from the β-amyloid precursor protein. Neuroscience. 1994;60:607–619. doi: 10.1016/0306-4522(94)90490-1. [DOI] [PubMed] [Google Scholar]
- 44.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lehad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid β protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 45.Shoji M, Iwakami N, Takeuchi S, Waragai M, Suzuki M, Kanazawa I, Lippa CF, Ono S, Okazawa H. JNK activation is associated with intracellular β-amyloid accumulation. Brain Res Mol Brain Res. 2000;85:221–233. doi: 10.1016/s0169-328x(00)00245-x. [DOI] [PubMed] [Google Scholar]
- 46.Siman R, Reaume AG, Savage MJ, Trusko S, Lin YG, Scott RW, Flood DG. Presenilin-1 P264L knock-in mutation: differential effects on abeta production, amyloid deposition, and neuronal vulnerability. J Neurosci. 2000;20:8717–8726. doi: 10.1523/JNEUROSCI.20-23-08717.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Standen CL, Brownlees J, Grierson AJ, Kesavapany S, Lau KF, McLoughlin DM, Miller CC. Phosphorylation of thr(668) in the cytoplasmic domain of the Alzheimer's disease amyloid precursor protein by stress-activated protein kinase 1b (Jun N-terminal kinase-3). J Neurochem. 2001;76:316–320. doi: 10.1046/j.1471-4159.2001.00102.x. [DOI] [PubMed] [Google Scholar]
- 48.Sturchler-Pierrat C, Staufenbiel M. Pathogenic mechanisms of Alzheimer's disease analyzed in the APP23 transgenic mouse model. Ann NY Acad Sci. 2000;920:134–139. doi: 10.1111/j.1749-6632.2000.tb06915.x. [DOI] [PubMed] [Google Scholar]
- 49.Tokuda T, Fukushima T, Ikeda S, Sekijima Y, Shoji S, Yanagisawa N, Tamaoka A. Plasma levels of amyloid beta proteins Abeta1–40 and Abeta1–42(43) are elevated in Down's syndrome. Ann Neurol. 1997;4:271–273. doi: 10.1002/ana.410410220. [DOI] [PubMed] [Google Scholar]
- 50.Tomidokoro Y, Harigaya Y, Matsubara E, Ikeda M, Kawarabayashi T, Shirao T, Ishiguro K, Okamoto K, Younkin SG, Shoji M. Brain Abeta amyloidosis in APPsw mice induces accumulation of presenilin-1 and tau. J Pathol. 2001;194:500–506. doi: 10.1002/path.897. [DOI] [PubMed] [Google Scholar]
- 51.Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77:157–164. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- 52.Wang TH, Wang HS, Ichijo H, Giannakakou P, Foster JS, Fojo T, Wimalasena J. Microtubule-interfering agents activate c-Jun N-terminal kinase/stress-activated protein kinase through both Ras and apoptosis signal-regulating kinase pathways. J Biol Chem. 1998;273:4928–4936. doi: 10.1074/jbc.273.9.4928. [DOI] [PubMed] [Google Scholar]
- 53.Wasco W, Pettingell WP, Jondro PD, Schmidt SD, Gurubhagavatula S, Rodes L, DiBlasi T, Romano DM, Guenette SY, Kovacs DM, Growdon JH, Tanzi RE. Familial Alzheimer's chromosome 14 mutations. Nat Med. 1995;1:848. doi: 10.1038/nm0995-848a. [DOI] [PubMed] [Google Scholar]
- 54.West MJ, Gundersen HJ. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol. 1990;296:1–22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- 55.West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 56.Wong TP, Debeir T, Duff K, Cuello AC. Reorganization of cholinergic terminals in the cerebral cortex and hippocampus in transgenic mice carrying mutated presenilin-1 and amyloid precursor protein transgenes. J Neurosci. 1999;19:2706–2716. doi: 10.1523/JNEUROSCI.19-07-02706.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000;59:880–888. doi: 10.1093/jnen/59.10.880. [DOI] [PubMed] [Google Scholar]
- 58.Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, Smith MA. Activation and redistribution of c-Jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem. 2001a;76:435–441. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- 59.Zhu X, Rottkamp CA, Hartzler A, Sun Z, Takeda A, Boux H, Shimohama S, Perry G, Smith MA. Activation of MKK6, an upstream activator of p38, in Alzheimer's disease. J Neurochem. 2001b;79:311–318. doi: 10.1046/j.1471-4159.2001.00597.x. [DOI] [PubMed] [Google Scholar]