

Abstract
Cerebrovascular pathology is common in Alzheimer's disease (AD) and is considered to contribute to cerebral malfunction. However, distinct antiangiogenic proteins that accumulate in AD brains have not yet been identified. Endostatin is a 20 kDa C-terminal fragment of collagen XVIII that, when added exogenously, inhibits endothelial proliferation and migration in vitro and angiogenesis and tumor growth in vivo by inducing apoptosis in endothelial cells.
We produced a monoclonal antibody directed against endostatin and observed significantly more (p < 0.0001) immunoreactive cortical neurons in AD brains compared with age-matched neuropathologically unaltered controls. High numbers of extracellular and frequently perivascular endostatin deposits were detected in the cerebral hemispheres. Double-labeling experiments revealed colocalization of endostatin in amyloid-β(1–40)(Aβ(1–40)), tau protein, and periodic acid–Schiff stain-positive plaques that were surrounded by focal gliosis. Western blotting revealed more 20 kDa endostatin in an AD patient compared with a control. In unstimulated SKNSH supernatants, endostatin was detected that increased predominantly after hypoxia in supernatants and cellular lysates. Aβ(1–40) (80 μg/ml) supplementation to SKNSH neurons for 24 hr completely abolished the release of endostatin.
These data show that endostatin is released by neurons to accumulate in amyloid plaques in Alzheimer's disease. Induction by hypoxia and complete abrogation of endostatin release after Aβ(1–40)challenge reveals intricate interactions between the two proteins and opens new avenues for the development of novel treatment strategies of AD patients.
Keywords: endostatin, amyloid, Alzheimer's disease, deposits, neurons
Alzheimer's disease (AD) is a leading cause of disability and decreased quality of life of the elderly (Kawas and Brookmeyer, 2001). Predominant pathophysiological hallmarks are the formation of plaques and blood vessels with amyloid angiopathy. Both contain aggregates of α-, β-, γ-, and δ-secretase-mediated 40- to 42-residue amyloid precursor protein fragments (Glenner and Wong, 1984; Masters et al., 1985; Beyreuther et al., 1991; Bayer et al., 2001). In addition, neurofibrillary tangles are found in neurons and consist predominantly of abnormally phosphorylated tau protein and ubiquitin (Goedert et al., 1991; Lowe et al., 1993). As a consequence, low amyloid-β42(Aβ42) and high tau protein levels in the cerebrospinal fluid are characteristic of AD (Motter et al., 1995;Galasko et al., 1998; Kanai et al., 1998). Although many factors have been identified to influence the course of AD, close interactions between the individual pathophysiological processes and even proteins are thought to orchestrate the disease (Takashima et al., 1993).
Recently, impaired cerebral blood flow has been identified as both cause and consequence of AD. Several studies have shown chronic cerebral hypoperfusion in AD patients (De Jong et al., 1997; De la Torre, 1997), and it was suggested that taken together with endothelial and smooth muscle cell abnormalities, hypoperfusion is a key factor in the development of AD. At least one-third of AD cases may exhibit significant cerebrovascular pathology, which is summarized as small vessel disease (Vinters et al., 1996; Moody et al., 1997). In addition, macro- and microinfarctions, hemorrhages, lacunes, and ischemic white matter changes are also present in AD (Mandybur 1986). Accordingly, AD patients have a higher prevalence of vascular diseases and vascular abnormalities (Brown et al., 2000).
Although Aβ itself is considered to be predominantly neurotoxic (Hardy, 1994; Hardy et al., 1998), upregulation of cytokines and activation of astrocytes and microglial cells contribute to further neuronal degeneration in the vicinity of Aβ deposits (Griffin et al., 1995, 1998; Frautschy et al., 1998) by mediating inflammation (Mattiace et al., 1990; McGeer and McGeer, 1999). Inflammation in turn results in increased formation of reactive oxygen species (ROS), a major mediator of impaired cerebral blood flow. However, in vitroexperiments revealed that Aβ deposits themselves cause cerebrovascular dysfunction in the rat brain by induction of ROS (Price et al., 1997; Mattson et al., 1999) and lead to impairment in cerebral blood flow and neuronal dysfunction (Iadecola et al., 1999). β-amyloid interacts with endothelial cells on blood vessels to produce an excess of superoxide radicals, with attendant alterations in endothelial structure and function (Thomas et al., 1996). As a consequence, cerebral amyloid angiopathy, microvascular degeneration affecting the cerebral endothelium and smooth muscle cells, basal lamina alterations, hyalinosis, and fibrosis are frequent alterations observed in postmortem AD brains. However, accumulation of distinct antiangiogenic proteins in brains of AD patients has not yet been described.
Endostatin is a 20 kDa C-terminal fragment of collagen XVIII, and extracellular administration inhibits endothelial proliferation and migration in vitro and angiogenesis and tumor growthin vivo by inducing apoptosis in endothelial cells (O'Reilly et al., 1997; Dhanabal et al., 1999; Yamaguchi et al., 1999). Endostatin is nontoxic and does not induce acquired drug resistance and therefore has become a potent new therapy strategy in solid neoplasias (Boehm et al., 1997). Biochemical studies revealed that the ability of endostatin to inhibit neoangiogenesis at least in part is mediated by Zn2+ binding and elastase processing (Boehm et al., 1998; Ding et al., 1998; Wen et al., 1999), and widespread endostatin expression was found in elastic fibers in vessel walls (Miosge et al., 1999). Interestingly, endostatin reduces intimal neovascularization and plaque growth in apolipoprotein E (ApoE)–deficient mice (Moulton et al., 1999). These findings are of particular interest, because ApoE is a risk factor not only for atherosclerosis but also for AD (Roses, 1995). In the latter study, however, only descending aortas, but not the brain, were analyzed.
We therefore hypothesized that endostatin might play a role in AD. We produced a monoclonal antibody and analyzed endostatin immunoreactivity in eight previously described AD patients and seven neuropathologically unaltered controls. Double-labeling experiments were used to identify the cellular source of endostatin and reveal associations with know AD pathophysiology. One AD and one control brain sample were analyzed by Western blotting to confirm endostatin expression in vivo. Western blotting of SKNSH neuronal lysates and supernatants was then used to analyze endostatin expression and release after CoCl2 challenge, an in vitro model of hypoxia, after H2O2challenge, to simulate ROS stimulation, and after Aβ(1–40) supplementation.
MATERIALS AND METHODS
Postmortem brain preparations of AD patients. Human brains of eight cases, aged 63–90 years, with varying degrees of Alzheimer-related pathology, were obtained from five different departments of pathology, 12–72 hr postmortem (see Table 1). The brains were fixed in a 4% aqueous solution of formaldehyde. Samples of the anterior entorhinal cortex were dissected coronally and embedded in paraffin. Ten micrometer sections were cut, and Aldehydefuchsin-Darrow red staining was used for topographical orientation. The Gallyas silver technique (Braak and Braak, 1991b) was used to stain neurofibrillary tangles, neuritic plaques, and neuropil threads, and the Campbell-Switzer silver technique was used for the detection of Aβ deposits (Braak and Braak, 1991b; Iqbal et al., 1991). All cases were staged for the distribution of neurofibrillary tangles and neuropil threads (Braak and Braak, 1991a) as well as for the distribution pattern of Aβ deposits (Thal et al., 2000). Cases fulfilling the criteria for the diagnosis of definite AD were used as AD cases; all other cases with AD-related pathology were used as cases having AD-related pathology. All seven age-matched controls were free of any AD-related changes as confirmed by three independent neuropathologists (Mittelbronn et al., 2001).
Table 1.
Endostatin immunoreactive cells in AD brains and controls
| Case | Entry number | Sex | Age | Diagnosis | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | E | N | E | N | E | N | E | N | E | N | E | N | E | N | E | N | E | N | E | |||||
| 1 | OF110/97 | Male | 71 | M. Alzheimer | 46 | 12 | 57 | 15 | 35 | 13 | 53 | 24 | 70 | 31 | 67 | 18 | 36 | 13 | 49 | 13 | 59 | 20 | 52 | 21 |
| 2 | OF116/97 | Male | 78 | M. Alzheimer | 48 | 25 | 44 | 28 | 63 | 30 | 50 | 30 | 68 | 40 | 49 | 23 | 65 | 39 | 44 | 29 | 52 | 25 | 62 | 33 |
| 3 | OF124/97 | Female | 63 | M. Alzheimer | 39 | 23 | 33 | 11 | 41 | 20 | 53 | 24 | 44 | 29 | 37 | 13 | 44 | 16 | 40 | 14 | 36 | 14 | 46 | 21 |
| 4 | OF139/97 | Male | 66 | M. Alzheimer | 96 | 19 | 66 | 19 | 61 | 15 | 74 | 22 | 67 | 32 | 61 | 17 | 88 | 20 | 68 | 21 | 75 | 20 | 65 | 28 |
| 5 | ES230/98 | Female | 67 | M. Alzheimer | 44 | 16 | 61 | 21 | 62 | 21 | 42 | 21 | 49 | 25 | 41 | 14 | 52 | 23 | 56 | 27 | 77 | 34 | 63 | 31 |
| 6 | ES17/01 | Female | 60 | M. Alzheimer | 33 | 16 | 31 | 12 | 32 | 15 | 32 | 18 | 28 | 20 | 29 | 14 | 30 | 15 | 45 | 17 | 31 | 16 | 37 | 13 |
| 7 | ES110/97 | Male | 68 | M. Alzheimer | 52 | 8 | 48 | 16 | 62 | 19 | 87 | 27 | 56 | 17 | 59 | 18 | 69 | 13 | 58 | 24 | 41 | 8 | 44 | 12 |
| 8 | ES19/88 | Male | 38 | Cardiac infarction | 58 | 1 | 56 | 0 | 67 | 2 | 63 | 4 | 56 | 3 | 59 | 2 | 78 | 3 | 53 | 1 | 54 | 2 | 48 | 2 |
| 9 | E171/96 | Female | 37 | Respiratory failure | 51 | 4 | 49 | 2 | 49 | 1 | 27 | 2 | 54 | 4 | 59 | 0 | 49 | 2 | 51 | 3 | 45 | 2 | 52 | 3 |
| 10 | ES143/97 | Female | 76 | Hemorrhagic shock | 41 | 4 | 42 | 5 | 43 | 6 | 45 | 10 | 41 | 9 | 33 | 5 | 45 | 5 | 45 | 12 | 27 | 7 | 42 | 8 |
| 11 | ES97/98 | Female | 92 | Heart failure | 53 | 2 | 65 | 6 | 50 | 6 | 61 | 5 | 68 | 10 | 69 | 10 | 59 | 7 | 63 | 8 | 74 | 5 | 97 | 4 |
| 12 | ES200/97 | Female | 82 | Hemorrhagic shock | 47 | 2 | 38 | 3 | 47 | 2 | 36 | 2 | 29 | 1 | 60 | 0 | 30 | 1 | 38 | 2 | 32 | 1 | 38 | 0 |
| 13 | ES99/98 | Male | 72 | Lung embolism | 45 | 6 | 42 | 4 | 61 | 2 | 38 | 4 | 50 | 4 | 54 | 3 | 45 | 1 | 39 | 3 | 30 | 10 | 39 | 10 |
| 14 | ES201/95 | Female | 56 | Hemorrhagic shock | 29 | 0 | 32 | 0 | 30 | 1 | 29 | 0 | 23 | 1 | 29 | 0 | 30 | 3 | 26 | 0 | 22 | 0 | 24 | 0 |
Immunohistochemical labeling results shown as total numbers of counted cells in a 400× magnification field. Numbers 1–10 indicate the 10 magnification fields counted. N, Total number of counterstained nuclei, E, total number of endostatin-labeled neurons with a counterstained nucleus; M., Morbus.
Cloning and production of recombinant mouse endostatin and control peptides. The cDNA encoding the C-terminal endostatin fragment of mouse collagen XVIII was amplified by PCR from liver RNA and subcloned into a pET expression vector (Angewandte Gentechnologie Systeme, Heidelberg, Germany). Recombinant endostatin was produced in BL21(DE3) Escherichia coli cells (Angewandte Gentechnologie Systeme) by induction with isopropylthio-β-d-galactoside for 4 hr. Recombinant endostatin was purified by nickel-chelate chromatography (Strik et al., 2001). Protein concentration was determined by the Bradford assay with bovine serum albumin (BSA) as a standard (Bio-Rad, Munich, Germany). Recombinant control peptides were produced as described previously (Schluesener et al., 1997).
Production of monoclonal antibody. For generation of monoclonal antibodies, BALB/c mice were immunized with 50 μg of recombinant endostatin, and hybridoma cell lines were established by standard procedures. Cell culture supernatants were screened by ELISA, Western blotting, and immunohistology, and positive clones were subcloned. The antibody was then affinity purified (BMA Biomedicals, Augst, Switzerland), again tested for specificity, adapted to a concentration of 1 μg/μl, and used in all consecutive experiments.
Immunohistochemistry and evaluation. Rehydrated sections were boiled (in a 600 W microwave oven) four times for 5 min in citrate buffer (2.1 gm sodium citrate per liter, pH 6). Endogenous peroxidase was inhibited with 1% H2O2in methanol for 15 min. Sections were incubated with 10% unspecific porcine serum (Biochrom, Berlin, Germany) to block nonspecific binding of immunoglobulins. The primary mouse anti-endostatin antibody was added at a concentration of 10 μg/ml and applied overnight at 4°C. Antibody binding was detected by biotinylated rabbit anti-mouse IgG F(Ab)2antibody fragment (1:400 for 30 min; Dako, Hamburg, Germany), followed by incubation with a peroxidase-conjugated streptavidin-biotin complex (Dako). The enzyme was visualized with diaminobenzidine as a substrate (Fluka, Neu-Ulm, Germany). Sections were counterstained with Mayer's Hemalaun. Controls included absence of the primary antibody, irrelevant monoclonal antibodies, and blocking experiments.
In double-labeling experiments, slices were pretreated as described above, and then the differentiating antibodies directed against neurofilament (Dako), GFAP (Chemicon, Temecula, CA), and cluster differentiation (CD)68 (Dako) were applied at a dilution of 10 μg/ml in 10% TBS/BSA to identify the cellular origin of endostatin. To provide information for the localization of extracellular endostatin deposits, antibodies directed against Aβ(1–40)(Sigma, Deisenhofen, Germany) and tau protein (Dako) were applied. Visualization was achieved by biotinylated rabbit anti-mouse IgG in TBS/BSA and alkaline phosphatase-conjugated ABC both diluted 1:400 in TBS/BSA. Consecutively, we developed with Fast Blue BB (Boehringer Mannheim, Mannheim, Germany) salt chromagen substrate solution, yielding a blue reaction product, and then slices were irradiated once more in a microwave (Lan et al., 1996). Complete inhibition of alkaline phosphatase function was achieved as described previously (Deininger and Meyermann, 1998). Alternatively, a periodic acid–Schiff (PAS) stain was performed, and then endostatin was immunolabeled as described above.
The number of endostatin-labeled cells was counted in 10 magnification (400×) fields per patient and displayed as percentage of all counterstained nuclei. Means were calculated and compared with the controls using Mann–Whitney U test.
Cell culture and stimulation. Human SKNSH neuroblastoma cells were obtained from the American Type Culture Collection (Manassas, VA). Cells were raised in RPMI 1640 medium with Glutamax II (Invitrogen, Paisley, UK) containing 10% fetal calf serum (FCS; Invitrogen) and 1.2% penicillin/streptomycin (Fluka) at 37°C and 5% CO2. At near confluency, cells were harvested by trypsinization and processed for further analyses.
In stimulation experiments, cells were washed twice with PBS and resubstituted with serum-free media. Then, cells were incubated with Aβ(1–40) (Bachem) for 24 hr at a concentration of 80 μg/ml (Busciglio et al., 1992). Before the experiment, Aβ(1–40) was dissolved in distilled water at a concentration of 6 mg/ml. Then, the solution was diluted to a concentration of 1 mg/ml in PBS without Ca2+, pH 7.4, and incubated at 37°C for 3 d. Hypoxia was mimicked using 2 μm cobalt chloride for 24 hr (Chandel et al., 1998), and reactive oxygen challenge was performed by 20 mmH2O2 incubation for 15 min (Mates and Sanchez-Jimenez, 2000). Then, cells were incubated for 24 hr in serum-free medium and analyzed as described below.
Flow cytometry. Cells were trypsinized, washed twice, and stained with mouse anti-endostatin monoclonal antibody at a concentration of 10 μg/ml in PBS/BSA for 1 hr at 4°C. Visualization was achieved by adding FITC-conjugated rabbit-anti mouse IgG (Serotech, Oxford, UK) for 30 min. Cells were analyzed using a FACScan Cytometer (Becton Dickinson, Überlingen, Germany) and CellQuest software. Controls included preparations lacking the primary antibody or irrelevant primary antibodies.
Protein preparation and Western blotting. Cells were lysed in a buffer containing 125 mm Tris base, 20% glycerol, 2% SDS, 1% bromophenol blue, 2% 2-mercaptoethanol, and protease inhibitors. Supernatants were precipitated by acetone and resuspended in running buffer. All samples were sonicated and boiled. Approximately 30 μg of total protein was loaded per lane, electrophoresed on a 12% SDS-polyacrylamide gel, and transferred to polyvinylidene difluoride membranes (Bio-Rad) by semidry blotting. Membranes were blocked with FCS, and the primary monoclonal antibody directed against endostatin was visualized using HRP-conjugated avidin-biotin complex and ECL visualization. Controls included blocking experiments, lacking primary antibody or irrelevant antibodies.
RESULTS
In AD brains, we observed prominent immunoreactivity of endostatin in all cortical layers (Fig.1A). Interestingly, most endostatin+ cells showed morphological characteristics of neurons (Fig. 1A,bottom inset). Double-labeling experiments with an antibody directed against neurofilament confirmed that most endostatin+ cells were neurons (top inset). Furthermore, extracellular endostatin deposits were detected (Fig. 1B, inset), occasionally in the immediate vicinity of blood vessels (Fig. 1B). Antibody labeling specificity was confirmed by blocking experiments and Western blotting experiments using the recombinant peptide. False-positive immunolabeling of lipofuscin was excluded by Fast Blue BB salt development in combination with the alkaline phosphatase AB complex and labeling experiments lacking the primary antibody. To determine the origin and pathological topology of endostatin immunoreactivity, double-labeling experiments with antibodies directed against GFAP and CD68 were performed to identify reactive astrocytes and macrophages/microglial cells, respectively. Although there were no endostatin+ astrocytes, extracellular endostatin deposits were detected in areas of focal gliosis (Fig.1C). To provide information for the localization of extracellular endostatin deposits, additional double-labeling experiments showed frequent colocalization of endostatin with Aβ(1–40) (Fig. 1C,inset), tau protein (Fig. 1D), and PAS-positive plaques (Fig. 1D, top inset). Even when colocalization of tau protein+and endostatin+ deposits was detected, tau protein+ neurons with neurofibrillary tangles did not show endostatin immunoreactivity (Fig. 1C,bottom inset). Singular microglial cells were occasionally double labeled.
Fig. 1.
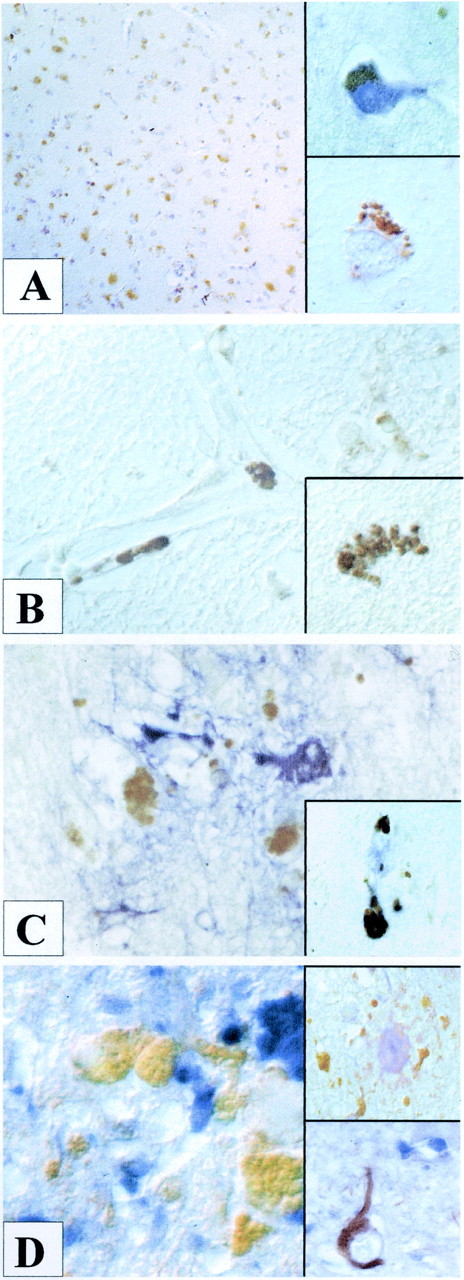
Prominent endostatin deposition (brown) was observed in neurons of all cortical layers in AD brains (A). Counterstain was hemalaun. The bottom inset shows intraneuronal deposition of endostatin (brown). A double-labeling experiment with neurofilament (blue) confirmed the neuronal origin of endostatin+ cells (brown). Paracellular endostatin deposition (brown) was detected in the immediate vicinity of blood vessels in an Aβ(1–40)/endostatin double-labeled section (B). The inset shows a paracellular intraparenchymal endostatin deposit (brown). Extracellular endostatin deposits (brown) were detected in areas of focal glioses (blue) (C), colocalized with Aβ(1–40) fragments (blue) (inset) and tau protein (blue) (D). No endostatin immunoreactivity (blue) was observed in tau+ neurons (brown) with neurofibrillary tangles (bottom inset). Endostatin+ deposits (brown), however, colocalized in PAS-positive plaques (top inset).
In controls, only few endostatin-expressing cells were observed. Accordingly, statistical analysis revealed a significantly (p < 0.0001) higher number of endostatin+ cells in AD brains (mean ± SEM, 40.47 ± 3.83%) compared with controls (mean ± SEM, 7.60 ± 2.19%), as confirmed by Student's ttest (Table 1). In one case, however, we observed more endostatin-immunoreactive cells than in the other controls. Interestingly, this patient had died of hemorrhagic shock. Nevertheless, no extracellular deposits of endostatin were detected in any of the analyzed control brains.
To provide more evidence for expression, release, and function of endostatin in AD, we analyzed SKNSH neurons by flow cytometry and Western blotting. Using flow cytometry, we detected an increase in the number of endostatin-labeled cells after both H2O2 and CoCl2 challenge (Fig.2A). In unstimulated SKNSH cellular lysates, no bands were visible (Fig.2A). After CoCl2 challenge, however, we detected a 20 kDa band and, interestingly, an ∼10 kDa endostatin-immunoreactive band. Surprisingly, we observed a prominent 20 kDa band and again the 10 kDa band in unstimulated SKNSH supernatants that increased after CoCl2stimulation (Fig. 2A). We then analyzed one brain preparation of an AD patient and one of a neuropathologically unaltered control patient (Fig. 2B). Here, a more accentuated ∼20 kDa band was observed in the AD patient, suggesting more endostatin than in the control tissue. To analyze the relationship of endostatin expression and release and amyloid load, we supplemented SKNSH neurons with 80 μg/ml Aβ(1–40) for 24 hr and again analyzed cellular lysates and supernatants separately (Fig. 2C). In both unstimulated and Aβ(1–40)- stimulated cellular lysates, we observed a weak 10 kDa band. Surprisingly, release of the 20 kDa band to the SKNSH supernatants was completely abolished by Aβ(1–40).
Fig. 2.
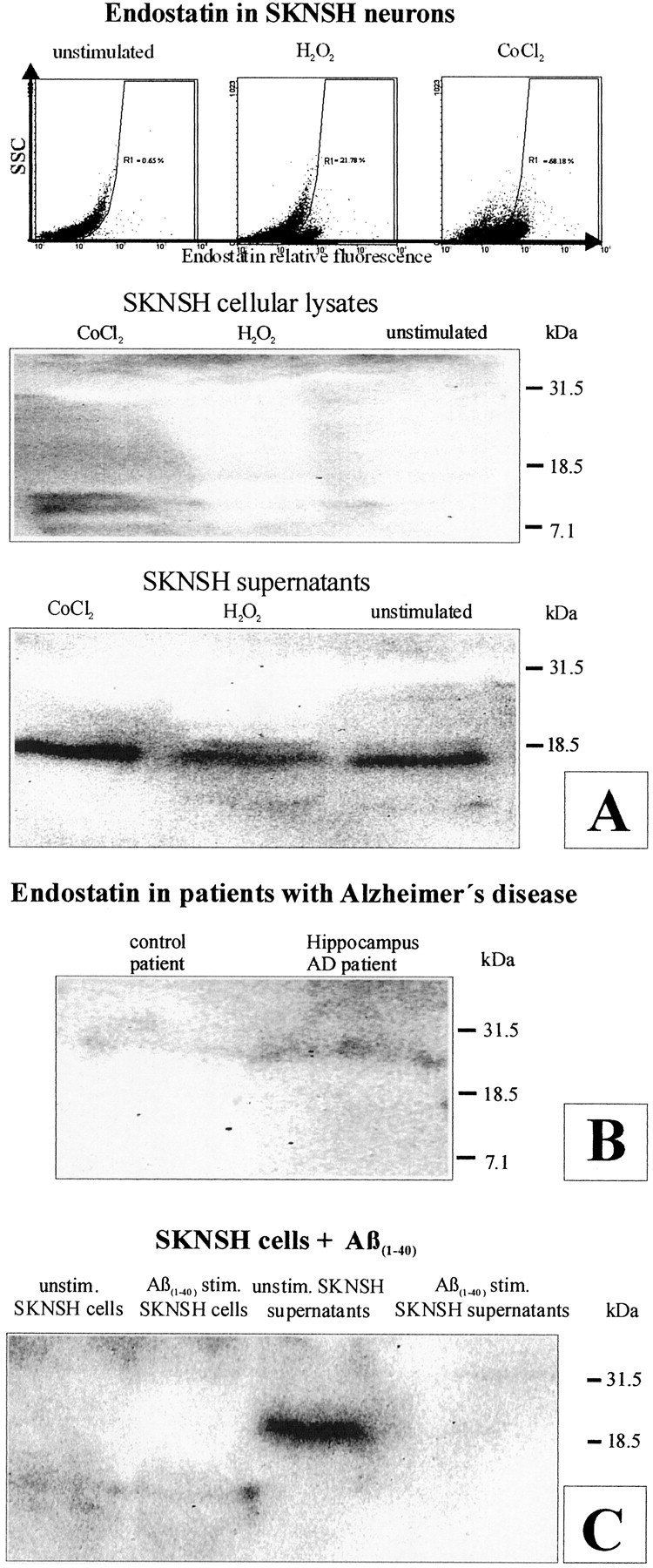
Flow cytometry (top) shows an increase in the number of endostatin-labeled cells after H2O2 and CoCl2 stimulation (A). Western blotting of cellular lysates (middle) and supernatants (bottom) reveals a more accentuated 10 kDa endostatin-immunoreactive band in CoCl2-stimulated than in unstimulated SKNSH cellular lysates and a more accentuated 20 kDa band in their supernatants (A). Western blotting demonstrates a more accentuated 20 kDa endostatin-immunoreactive band in an AD patient compared with a control (B). Surprisingly, Aβ(1–40) (80 μg/ml) supplementation to SKNSH neurons for 24 hr completely abolished the release of the 20 kDa band (C).
DISCUSSION
We observed significantly more (p < 0.0001) immunoreactive cortical neurons in AD brains compared with age-matched neuropathologically unaltered controls. High numbers of extracellular and frequently perivascular endostatin deposits were detected in the cerebral hemispheres. Double-labeling experiments revealed colocalization of endostatin in Aβ(1–40), tau protein, and PAS-positive plaques that were surrounded by focal gliosis. Western blotting revealed more 20 kDa endostatin in an AD patient compared with a control. In unstimulated SKNSH supernatants, endostatin was detected that increased predominantly after hypoxia in supernatants and cellular lysates. Aβ(1–40) (80 μg/ml) supplementation to SKNSH neurons for 24 hr completely abolished the release of endostatin.
The observation that endostatin accumulates in perivascular and cortical plaques of AD patients is the first description of an antiangiogenic protein that deposits in AD brains. However, a wide range of proteins were found to be localized in these structures, which were thought previously to contain only Aβ fragments. These include the growth-associated protein-43 (Masliah et al., 1991), protein kinase C (Masliah et al., 1990), epidermal growth factor receptor, and an abundant amount of others. Interestingly, zinc deposits have also been identified histochemically (Suh et al., 2000). This is of note, because zinc binding of endostatin is essential for its antiangiogenic activity (Boehm et al., 1998). More significant data on the involvement of endostatin in AD have been published previously. The intention of that study was to show that endostatin reduces intimal neovascularization and plaque growth in apolipoprotein E (ApoE)–deficient mice (Moulton et al., 1999). Here, ApoE-deficient mice were chosen as a model for plaque formation in the peripheral vasculature. These findings are of particular interest, because ApoE is a risk factor not only for atherosclerosis but also for AD (Roses, 1995). In humans, the ε4 and ε2 alleles of ApoE have been linked to the severity of cerebral amyloid angiopathy (CAA) (Hyman et al., 1996). Although CAA occurs independently, particularly in the elderly, it is present to some degree in patients with AD. Accordingly, the ApoEε4 allele was identified as a risk factor for AD (Schmechel et al., 1993). In families with increased risk for late-onset AD, the presence of ApoEε4increased the relative risk and dramatically decreased the age of onset (Corder et al., 1993), whereas the ApoEε2genotype had a protective effect (Corder et al., 1994). The observation that endostatin is capable of further reducing neovascularization in ApoE-deficient mice and additional data that showed that endostatin reduces blood vessel formation during wound healing (Bloch et al., 2000) underscore the specific and widespread nature of endostatin-mediated inhibition of blood vessel growth in the process of neovascularization. Our observation that endostatin accumulates in AD brains therefore suggests a novel mechanism of endothelial disruption yet unrelated to classical AD pathology that constitutes a novel and independent contributor to the course of the disease.
Western blotting revealed lighter and endostatin-immunoreactive bands, most frequently of ∼10 kDa weight. Previously, alternatively spliced endostatin variants have been described in a wide range of human tissues, including liver, heart, kidney, placenta, prostate, ovaries, skeletal muscle, small intestine, and others (John et al., 1999). The endostatin-immunoreactive bands observed here had a molecular weight of 26, 18.5, 30, and 34 kDa using an antibody directed toward an N-terminal endostatin epitope. Interestingly, the authors reported not antiproliferative but significant antimigratory effects of the described endostatins on vessel wall cells, thus underscoring production of cooperating processing intermediates from a common precursor molecule. In this light, expression of a 10 kDa endostatin-immunoreactive band in SKNSH cellular lysates and supernatants may point to additional functions of endostatin variants during brain disease in general and AD in particular.
The precursor of Aβ(1–40), the β-amyloid precursor protein, is an integral membrane glycoprotein (Kang et al., 1987; Lammich et al., 1999). Alternative splicing and processing lead to the production of several isoforms that are expressed in a cell type-specific manner, including in neurons. Chronic neuronal expression of recombinant retroviral vectors harboring normal and mutant cDNAs for human neuron-specific amyloid-β precursor protein 695 in fetal rat brain transplants induced AD-typical plaques (Bayer et al., 1996). In this context, the observed reduction of endostatin by Aβ(1–40) may indicate a last mechanism to escape the endostatin-mediated vicious circle in AD (Fig.3).
Fig. 3.

Schematic diagram demonstrating the observed phenomena. Endostatin and Aβ(1–40) are released by neurons (solid line) and cooperate in the mediation of hypoperfusion that leads to accentuated hypoxia in yet unaffected neurons (dotted line). This in turn accelerates neuronal endostatin release, consequent hypoperfusion, and continuing hypoxia.
In conclusion, our data show that endostatin is released by neurons to accumulate in amyloid plaques in Alzheimer's disease. Induction by hypoxia and complete abrogation of endostatin release after Aβ(1–40) challenge reveal intricate and novel interactions between the two proteins and open new avenues for the development of novel treatment strategies of AD patients.
M.H.D. and B.A.F. contributed equally to this work.
We thank Kubrom Bekure-Nemariam for expert technical assistance.
Correspondence should be addressed to Martin H. Deininger, Institute of Brain Research, University of Tuebingen Medical School, Calwer Strasse 3, D-72076 Tuebingen, Germany. E-mail:martin.deininger@uni-tuebingen.de.
REFERENCES
- 1.Bayer TA, Fossgreen A, Czech C, Beyreuther K, Wiestler OD. Plaque formation in brain transplants exposed to human beta-amyloid precursor protein 695. Acta Neuropathol Berl. 1996;92:130–137. doi: 10.1007/s004010050500. [DOI] [PubMed] [Google Scholar]
- 2.Bayer TA, Wirths O, Majtenyi K, Hartmann T, Multhaup G, Beyreuther K, Czech C. Key factors in Alzheimer's disease: beta-amyloid precursor protein processing, metabolism and intraneuronal transport. Brain Pathol. 2001;11:1–11. doi: 10.1111/j.1750-3639.2001.tb00376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beyreuther K, Bush AI, Dyrks T, Hilbich C, Konig G, Monning U, Multhaup G, Prior R, Rumble B, Schubert W. Mechanisms of amyloid deposition in Alzheimer's disease. Ann NY Acad Sci. 1991;640:129–139. doi: 10.1111/j.1749-6632.1991.tb00204.x. [DOI] [PubMed] [Google Scholar]
- 4.Bloch W, Huggel K, Sasaki T, Grose R, Bugnon P, Addicks K, Timpl R, Werner S. The angiogenesis inhibitor endostatin impairs blood vessel maturation during wound healing. FASEB J. 2000;14:2373–2376. doi: 10.1096/fj.00-0490fje. [DOI] [PubMed] [Google Scholar]
- 5.Boehm T, Folkman J, Browder T, O'Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404–407. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- 6.Boehm T, O'Reilly MS, Keough K, Shiloach J, Shapiro R, Folkman J. Zinc-binding of endostatin is essential for its antiangiogenic activity. Biochem Biophys Res Commun. 1998;252:190–194. doi: 10.1006/bbrc.1998.9617. [DOI] [PubMed] [Google Scholar]
- 7.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol Berl. 1991a;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991b;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 9.Brown WR, Moody DM, Thore CR, Challa VR. Cerebrovascular pathology in Alzheimer's disease and leukoaraiosis. Ann NY Acad Sci. 2000;4:39–45. doi: 10.1111/j.1749-6632.2000.tb06348.x. [DOI] [PubMed] [Google Scholar]
- 10.Busciglio J, Lorenzo A, Yankner BA. Methodological variables in the assessment of beta amyloid neurotoxicity. Neurobiol Aging. 1992;13:609–612. doi: 10.1016/0197-4580(92)90065-6. [DOI] [PubMed] [Google Scholar]
- 11.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 13.Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, Rimmler JB, Locke PA, Conneally PM, Schmader KE. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- 14.Deininger MH, Meyermann R. Multiple epitope labeling by the exclusive use of alkaline phosphatase conjugates in immunohistochemistry. Histochem Cell Biol. 1998;110:425–430. doi: 10.1007/s004180050303. [DOI] [PubMed] [Google Scholar]
- 15.De Jong GI, De Vos RA, Steur EN, Luiten PG. Cerebrovascular hypoperfusion: a risk factor for Alzheimer's disease? Animal model and postmortem human studies. Ann NY Acad Sci. 1997;826:56–74. doi: 10.1111/j.1749-6632.1997.tb48461.x. [DOI] [PubMed] [Google Scholar]
- 16.De la Torre JC. Hemodynamic consequences of deformed microvessels in the brain in Alzheimer's disease. Ann NY Acad Sci. 1997;826:75–91. doi: 10.1111/j.1749-6632.1997.tb48462.x. [DOI] [PubMed] [Google Scholar]
- 17.Dhanabal M, Ramchandran R, Waterman MJ, Lu H, Knebelmann B, Segal M, Sukhatme VP. Endostatin induces endothelial cell apoptosis. J Biol Chem. 1999;274:11721–11726. doi: 10.1074/jbc.274.17.11721. [DOI] [PubMed] [Google Scholar]
- 18.Ding YH, Javaherian K, Lo KM, Chopra R, Boehm T, Lanciotti J, Harris BA, Li Y, Shapiro R, Hohenester E, Timpl R, Folkman J, Wiley DC. Zinc-dependent dimers observed in crystals of human endostatin. Proc Natl Acad Sci USA. 1998;95:10443–10448. doi: 10.1073/pnas.95.18.10443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- 20.Galasko D, Chang L, Motter R, Clark CM, Kaye J, Knopman D, Thomas R, Kholodenko D, Schenk D, Lieberburg I, Miller B, Green R, Basherad R, Kertiles L, Boss MA, Seubert P. High cerebrospinal fluid tau and low amyloid β42 levels in the clinical diagnosis of Alzheimer disease and relation to apolipoprotein E genotype. Arch Neurol. 1998;55:937–945. doi: 10.1001/archneur.55.7.937. [DOI] [PubMed] [Google Scholar]
- 21.Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 22.Goedert M, Spillantini MG, Crowther RA. Tau proteins and neurofibrillary degeneration. Brain Pathol. 1991;1:279–286. doi: 10.1111/j.1750-3639.1991.tb00671.x. [DOI] [PubMed] [Google Scholar]
- 23.Griffin WS, Sheng JG, Roberts GW, Mrak RE. Interleukin-1 expression in different plaque types in Alzheimer's disease: significance in plaque evolution. J Neuropathol Exp Neurol. 1995;54:276–281. doi: 10.1097/00005072-199503000-00014. [DOI] [PubMed] [Google Scholar]
- 24.Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer's disease: the potential role of a “cytokine cycle” in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardy J. ApoE, amyloid, and Alzheimer's disease. Science. 1994;263:454–455. doi: 10.1126/science.8290946. [DOI] [PubMed] [Google Scholar]
- 26.Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer's disease and related dementias: amyloid and its relationship to tau. Nat Neurosci. 1998;1:355–358. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- 27.Hyman BT, Gomez-Isla T, Rebeck GW, Briggs M, Chung H, West HL, Greenberg S, Mui S, Nichols S, Wallace R, Growdon JH. Epidemiological, clinical, and neuropathological study of apolipoprotein E genotype in Alzheimer's disease. Ann NY Acad Sci. 1996;802:1–5. doi: 10.1111/j.1749-6632.1996.tb32592.x. [DOI] [PubMed] [Google Scholar]
- 28.Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- 29.Iqbal K, Braak E, Braak H, Zaidi T, Grundke-Iqbal I. A silver impregnation method for labeling both Alzheimer paired helical filaments and their polypeptides separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Neurobiol Aging. 1991;12:357–361. doi: 10.1016/0197-4580(91)90022-c. [DOI] [PubMed] [Google Scholar]
- 30.John H, Preissner KT, Forssmann WG, Standker L. Novel glycosylated forms of human plasma endostatin and circulating endostatin-related fragments of collagen XV. Biochemistry. 1999;38:10217–10224. doi: 10.1021/bi990787+. [DOI] [PubMed] [Google Scholar]
- 31.Kanai M, Matsubara E, Isoe K, Urakami K, Nakashima K, Arai H, Sasaki H, Abe K, Iwatsubo T, Kosaka T, Watanabe M, Tomidokoro Y, Shizuka M, Mizushima K, Nakamura T, Igeta Y, Ikeda Y, Amari M, Kawarabayashi T, Ishiguro K. Longitudinal study of cerebrospinal fluid levels of tau, Aβ1–40, and Aβ1–42(43) in Alzheimer's disease: a study in Japan. Ann Neurol. 1998;44:17–26. doi: 10.1002/ana.410440108. [DOI] [PubMed] [Google Scholar]
- 32.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 33.Kawas CH, Brookmeyer R. Aging and the public health effects of dementia. N Engl J Med. 2001;344:1160–1161. doi: 10.1056/NEJM200104123441509. [DOI] [PubMed] [Google Scholar]
- 34.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lan HY, Mu W, Ng YY, Nikolic-Paterson DJ, Atkins RC. A simple, reliable, and sensitive method for nonradioactive in situ hybridization: use of microwave heating to improve hybridization efficiency and preserve tissue morphology. J Histochem Cytochem. 1996;44:281–287. doi: 10.1177/44.3.8648089. [DOI] [PubMed] [Google Scholar]
- 36.Lowe J, Mayer RJ, Landon M. Ubiquitin in neurodegenerative diseases. Brain Pathol. 1993;3:55–65. doi: 10.1111/j.1750-3639.1993.tb00726.x. [DOI] [PubMed] [Google Scholar]
- 37.Mandybur TI. Cerebral amyloid angiopathy: the vascular pathology and complications. J Neuropathol Exp Neurol. 1986;45:79–90. [PubMed] [Google Scholar]
- 38.Masliah E, Cole G, Shimohama S, Hansen L, DeTeresa R, Terry RD, Saitoh T. Differential involvement of protein kinase C isozymes in Alzheimer's disease. J Neurosci. 1990;10:2113–2124. doi: 10.1523/JNEUROSCI.10-07-02113.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Masliah E, Mallory M, Hansen L, Alford M, Albright T, DeTeresa R, Terry R, Baudier J, Saitoh T. Patterns of aberrant sprouting in Alzheimer's disease. Neuron. 1991;6:729–739. doi: 10.1016/0896-6273(91)90170-5. [DOI] [PubMed] [Google Scholar]
- 40.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mates JM, Sanchez-Jimenez FM. Role of reactive oxygen species in apoptosis: implications for cancer therapy. Int J Biochem Cell Biol. 2000;32:157–170. doi: 10.1016/s1357-2725(99)00088-6. [DOI] [PubMed] [Google Scholar]
- 42.Mattiace LA, Davies P, Dickson DW. Detection of HLA-DR on microglia in the human brain is a function of both clinical and technical factors. Am J Pathol. 1990;136:1101–1114. [PMC free article] [PubMed] [Google Scholar]
- 43.Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S. Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer's and Parkinson's diseases. Ann NY Acad Sci. 1999;893:154–175. doi: 10.1111/j.1749-6632.1999.tb07824.x. [DOI] [PubMed] [Google Scholar]
- 44.McGeer PL, McGeer EG. Inflammation of the brain in Alzheimer's disease: implications for therapy. J Leukoc Biol. 1999;65:409–415. doi: 10.1002/jlb.65.4.409. [DOI] [PubMed] [Google Scholar]
- 45.Miosge N, Sasaki T, Timpl R. Angiogenesis inhibitor endostatin is a distinct component of elastic fibers in vessel walls. FASEB J. 1999;13:1743–1750. doi: 10.1096/fasebj.13.13.1743. [DOI] [PubMed] [Google Scholar]
- 46.Mittelbronn M, Dietz K, Schluesener HJ, Meyermann R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol Berl. 2001;101:249–255. doi: 10.1007/s004010000284. [DOI] [PubMed] [Google Scholar]
- 47.Moody DM, Brown WR, Challa VR, Ghazi-Birry HS, Reboussin DM. Cerebral microvascular alterations in aging, leukoaraiosis, and Alzheimer's disease. Ann NY Acad Sci. 1997;826:103–116. doi: 10.1111/j.1749-6632.1997.tb48464.x. [DOI] [PubMed] [Google Scholar]
- 48.Motter R, Vigo-Pelfrey C, Kholodenko D, Barbour R, Johnson-Wood K, Galasko D, Chang L, Miller B, Clark C, Green R. Reduction of β-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer's disease. Ann Neurol. 1995;38:643–648. doi: 10.1002/ana.410380413. [DOI] [PubMed] [Google Scholar]
- 49.Moulton KS, Heller E, Konerding MA, Flynn E, Palinski W, Folkman J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation. 1999;99:1726–1732. doi: 10.1161/01.cir.99.13.1726. [DOI] [PubMed] [Google Scholar]
- 50.O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 51.Price JM, Sutton ET, Hellermann A, Thomas T. beta-Amyloid induces cerebrovascular endothelial dysfunction in the rat brain. Neurol Res. 1997;19:534–538. doi: 10.1080/01616412.1997.11740853. [DOI] [PubMed] [Google Scholar]
- 52.Roses AD. On the metabolism of apolipoprotein E and the Alzheimer diseases. Exp Neurol. 1995;132:149–156. doi: 10.1016/0014-4886(95)90019-5. [DOI] [PubMed] [Google Scholar]
- 53.Schluesener HJ, Seid K, Zhao Y, Meyermann R. Localization of endothelial-monocyte-activating polypeptide II (EMAP II), a novel proinflammatory cytokine, to lesions of experimental autoimmune encephalomyelitis, neuritis and uveitis: expression by monocytes and activated microglial cells. Glia. 1997;20:365–372. doi: 10.1002/(sici)1098-1136(199708)20:4<365::aid-glia8>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 54.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strik HM, Schluesener HJ, Seid K, Meyermann R, Deininger MH. Localization of endostatin in rat and human gliomas. Cancer. 2001;91:1013–1019. [PubMed] [Google Scholar]
- 56.Suh SW, Jensen KB, Jensen MS, Silva DS, Kesslak PJ, Danscher G, Frederickson CJ. Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer's diseased brains. Brain Res. 2000;852:274–278. doi: 10.1016/s0006-8993(99)02096-x. [DOI] [PubMed] [Google Scholar]
- 57.Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K. Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc Natl Acad Sci USA. 1993;90:7789–7793. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thal DR, Rueb U, Schultz C, Sassin I, Ghebremedhin E, Del Tredici K, Braak E, Braak H. Sequence of Aβ-protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol. 2000;59:733–748. doi: 10.1093/jnen/59.8.733. [DOI] [PubMed] [Google Scholar]
- 59.Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. beta-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996;380:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- 60.Vinters HV, Wang ZZ, Secor DL. Brain parenchymal and microvascular amyloid in Alzheimer's disease. Brain Pathol. 1996;6:179–195. doi: 10.1111/j.1750-3639.1996.tb00799.x. [DOI] [PubMed] [Google Scholar]
- 61.Wen W, Moses MA, Wiederschain D, Arbiser JL, Folkman J. The generation of endostatin is mediated by elastase. Cancer Res. 1999;59:6052–6056. [PubMed] [Google Scholar]
- 62.Yamaguchi N, Anand-Apte B, Lee M, Sasaki T, Fukai N, Shapiro R, Que I, Lowik C, Timpl R, Olsen BR. Endostatin inhibits VEGF-induced endothelial cell migration and tumor growth independently of zinc binding. EMBO J. 1999;18:4414–4423. doi: 10.1093/emboj/18.16.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]