

Abstract
Generalized epilepsy with febrile seizures plus type 1 is an inherited human epileptic syndrome, associated with a cysteine-to-tryptophan (C121W) mutation in the extracellular immunoglobin domain of the auxiliary β1 subunit of the voltage-gated sodium channel. The mutation disrupts β1 function, but how this leads to epilepsy is not understood. In this study, we make several observations that may be relevant for understanding why this β1 mutation results in seizures. First, using electrophysiological recordings from mammalian cell lines, coexpressing sodium channel α subunits and either wild-type β1 or C121Wβ1, we show that loss of β1 functional modulation, caused by the C121W mutation, leads to increased sodium channel availability at hyperpolarized membrane potentials and reduced sodium channel rundown during high-frequency channel activity, compared with channels coexpressed with wild-type β1. In contrast, neither wild-type β1 nor C121Wβ1 significantly affected sodium current time course or the voltage dependence of channel activation. We also show, using a Drosophila S2 cell adhesion assay, that the C121W mutation disrupts β1–β1 homophilic cell adhesion, suggesting that the mutation may alter the ability of β1 to mediate protein–protein interactions critical for sodium channel localization. Finally, we demonstrate that neither functional modulation nor cell adhesion mediated by wild-type β1 is occluded by coexpression of C121Wβ1, arguing against the idea that the mutant β1 acts as a dominant-negative subunit. Together, these data suggest that C121Wβ1 causes subtle effects on channel function and subcellular distribution that bias neurons toward hyperexcitabity and epileptogenesis.
Keywords: voltage-gated sodium channel, β1 subunit, epilepsy, channelopathy, patch clamp, cell adhesion, Drosophila S2 cells
Idiopathic epilepsies are widely believed to involve polymorphisms in multiple susceptibility genes (Steinlein, 2001). Genetic studies of rare monogenic, inherited epilepsies have identified candidate susceptibility genes and suggest how changes in the function of their protein products cause seizures (Gardiner, 2000; Steinlein and Noebels, 2000; Lerche et al., 2001;Meisler et al., 2001). For example, mutations in genes encoding voltage-gated sodium channels are associated with generalized epilepsy with febrile seizures plus (GEFS+) (Wallace et al., 1998, 2001; Escayg et al., 2000, 2001; Abou-Khalil et al., 2001; Sugawara et al., 2001), an autosomal dominant epileptic syndrome characterized by febrile seizures, which may persist beyond 6 years of age, as well as afebrile generalized seizures in some affected individuals (Scheffer and Berkovic, 1997). How mutations in sodium channel genes alter channel function to cause epilepsy is an area of considerable interest and importance.
Brain sodium channels consist of a central, pore-forming α subunit of 260 kDa and auxiliary subunits of ∼30–40 kDa, designated β1, β2, and β3 (Catterall, 2000; Isom, 2001). β subunits comprise an N-terminal extracellular segment, containing a single Ig domain, a transmembrane segment, and a short C-terminal intracellular segment. β subunits are not required for sodium channel function; however, they modulate the expression levels and functional properties of the α subunit (Isom et al., 1992, 1995a,b) and through their Ig domains may mediate interactions between sodium channels and other proteins (Srinivasan et al., 1998; Xiao et al., 1999; Malhotra et al., 2000).
Somewhat surprisingly, the first identified GEFS+ mutation (GEFS plus 1) was in the β1 gene SCN1B, resulting in substitution of tryptophan for a critical cysteine residue (mutant C121W) in the Ig domain of the β1 subunit (Wallace et al., 1998). The effect of this mutation on sodium channel function was assessed usingXenopus oocytes. Cloned α subunits, expressed in oocytes, form sodium channels that inactivate abnormally slowly (Krafte et al., 1990), whereas coexpression of β1 speeds channel inactivation greater than fivefold (Isom et al., 1992). In contrast, the C121W mutation results in loss of this functional modulation (Wallace et al., 1998). The prevalent hypothesis for how loss of β1 function causes epilepsy is based on the assumption that sodium channels in mammalian neurons behave like cloned sodium channels expressed in frog oocytes. However, several lines of evidence argue against this idea. First, the expression of α subunits in various cultured mammalian cell lines results in the expression of fast sodium channels, even in the absence of heterologously expressed or endogenous β1 subunits (Ukomadu et al., 1992; West et al., 1992; Meadows et al., 2002). Second, coexpression of β1 in mammalian cells does not speed the rate of inactivation of expressed sodium channels (Isom et al., 1995b). Thus,Xenopus oocytes may not be well suited for understanding either the effects of β1 or the pathophysiological consequences of C121Wβ1. These considerations prompted us to reassess the C121W mutation using electrophysiological and biochemical approaches. In this study, we report several novel findings that may be relevant for understanding how C121Wβ1 causes epilepsy.
MATERIALS AND METHODS
Mutation of β1 subunit cDNA. The C121W mutation (to avoid confusion, we have chosen to use the C121W nomenclature, although the original numbering designates this residue as C102) (Isom et al., 1992) was introduced into cDNAs for rat β1 (in vector pCR2.1) and human β1 (in vector pCIH1) using standard PCR mutagenesis (Barek, 1993) with either Pwo (Roche Diagnostics, Laval, Quebec) or AmpliTaq (Perkin-Elmer, Boston, MA) DNA polymerase. The mutant PCR products were subcloned into the full-length rat or human β1 constructs and sequenced to confirm the presence of the mutation and rule out the introduction of spurious mutations during PCR amplification.
Electrophysiological recording in Xenopusoocytes. For oocyte recordings, RNA was transcribed from cDNAs encoding rat Nav1.2a, wild-type β1, and C121Wβ1 (all in vector pSP64T) using the Message Machine RNA synthesis kit (Ambion, Austin, TX). RNA concentrations were estimated from the intensities of bands on RNA gels, relative to the intensities of RNA bands of known concentration. Nav1.2a and wild-type or mutant β1 RNA were mixed at various molar ratios and microinjected into Xenopus oocytes isolated from femaleXenopus frogs (Boreal, St. Catherine, Ontario), as described previously (Li et al., 1999). Sodium currents were examined 2–5 d after injection by two-electrode voltage clamp. The details of voltage-clamp recording of sodium currents in Xenopusoocytes have been described previously (Li et al., 1999).
Electrophysiological recording in mammalian cells. Voltage-activated sodium currents were recorded in CNahIII-12 cells (Chen et al., 2000), a Chinese hamster ovary (CHO)-derived line stably expressing the human Nav1.3 (hNav1.3) sodium channel, and in SNaIIA cells (Isom et al., 1995b), a Chinese hamster lung (CHL)-derived cell line stably expressing the rat Nav1.2a sodium channel, using the whole-cell configuration of the patch-clamp technique (Hamill et al., 1981). The extracellular bath solution contained (in mm): 130 NaCl, 4 KCl, 1.5 CaCl2, 1 MgCl2, 5 glucose, and 10 HEPES, pH 7.4 (with NaOH). The intracellular pipette solution used for CHO cells contained (in mm): 10 NaCl, 10 CsCl, 105 Cs-Aspartate, 10 EGTA, and 10 HEPES, pH 7.4 (with CsOH). The intracellular solution for CHL cells contained (in mm): 10 NaCl, 105 CSF, 10 CsCl, 10 EGTA, and 10 HEPES, pH 7.4 (with CsOH). Capacitive and leak currents were subtracted using TTX subtraction or the P/4 procedure (Bezanilla and Armstrong, 1977). Other details of voltage-clamp recordings were as described previously (Meadows et al., 2002).
The voltage dependence of channel activation was determined from the peak currents recorded during 90-msec-long test pulses to potentials ranging from −50 to +65 mV in 5 mV increments. Conductance (g) was calculated from peak current amplitude (I) according to g =I/(V − Vrev), where V is the test potential andVrev is the measured reversal potential. The voltage dependence of inactivation was assessed by applying 100 msec prepulses to potentials ranging from −100 to −5 mV in 5 mV increments, followed by a test pulse to 0 mV. Normalized voltage conductance and inactivation curves were fit with the Boltzmann equation: 1/[1 + exp (V −V1/2)/k], whereV1/2 is the membrane potential corresponding to the midpoint of the curve, and k is a slope factor. To determine the time course of recovery from inactivation, sodium channels were inactivated with a 5-msec-long pulse to 0 mV, which was followed by a recovery prepulse of variable duration to −80 mV, and a subsequent test pulse to 0 mV to determine the fraction of recovered channels. Recovery data were fit with a single exponential to determine the time constant for recovery from inactivation. Statistical significance between groups was determined using Student'st test or one-way ANOVA, followed by Tukey post hoc tests. Differences were considered significant whenp < 0.05.
Expression of wild-type β1 and C121Wβ1 in cultured mammalian cells. The effects of human wild-type β1 or C121Wβ1 subunits on human Nav1.3 sodium channels in CNahIII-12 cells were assessed using both transient and stable expression. For transient expression of wild-type or mutant β1, CNahIII-12 cells were transfected following the manufacturer's instructions using 7 or 9.5 μl of Fugene 6 (Roche, Hertforshire, UK) or Polyfect (Qiagen, Valencia, CA), respectively, with 4 μl of β1 subunit DNA. One-half microgram of green fluorescent protein (GFP) DNA (vector pEGFPC1; Clonetech, Palo Alto, CA) was also added in each reaction to serve as a marker for identifying transfected cells. After transfection, cells were cultured overnight and split the following day onto 35 mm dishes for electrophysiological recording. Stable cell lines coexpressing human Nav1.3 and either wild-type β1 or C121Wβ1 were obtained after transfection using standard cell cloning procedures (Freshney, 1983) and coselection with G418 (for Nav1.3) and hygromycin (for β1). We obtained similar results using either transient or stable expression of β1. In the figure legends, we explicitly state which procedure was used to obtain a particular set of data.
For experiments involving inducible expression of wild-type β1 or C121Wβ1 in SNAIIA cells, cDNAs were subcloned into the pIND vector (Invitrogen, Carlsbad, CA). SNaIIA cells, a Chinese hamster lung-derived cell line stably expressing the rat Nav1.2a sodium channel (a gift from W. A. Catterall, University of Washington), were cotransfected with pVgRxR (Invitrogen), which constitutively expresses the heterodimeric ecdysone receptor, and either pIND.β1 or pIND.C121Wβ1, using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. pIND, pVgRxR, and ponasterone hormone were obtained from Invitrogen. Stable lines (SNaIIA-pIND.β1 and SNaIIA-pIND.C121Wβ1) were established in the presence of 400 μg/ml zeocin and hygromycin, respectively. β1 or C121Wβ1 subunit protein expression was induced by the treatment of 80% confluent monolayers of cells with 20 μm ponasterone (or ethanol as a control) for 48 hr in a cell culture incubator set at 37°C and 5% CO2 (the ponasterone-containing medium was replaced after the first 24 hr of incubation). Expression of β1 was then determined by Western blot.
S2 cell aggregation assay. For expression in S2 cells, C121Wβ1 was cloned into the Drosophila expression vector pRmHa3 (a gift from M. Hortsch, University of Michigan).Drosophila S2 cells (American Type Culture Collection, Manassas, VA) were transfected with pRmHa3.β1C121W using Lipofectin (Invitrogen). The cells were cotransfected with pPC4 to confer α-amanitin resistance as a selectable marker as described previously (Malhotra et al., 2000). A stable line expressing wild-type β1 subunits has been established previously (Malhotra et al., 2000). Individual cell clones were induced overnight in the presence of 0.7 mm CuSO4 with mechanical shaking, as described previously (Malhotra et al., 2000), and analyzed by Western blot for wild-type β1 or C121Wβ1 protein expression, and by phase-contrast microscopy for cell aggregation. For immunocytochemical determination of wild-type β1 or C121Wβ1 distribution, S2 cells were fixed with 2% paraformaldehyde and permeabilized with 0.5% Triton X-100. A polyclonal antiserum to an extracellular domain of β1 (KRRSETTAETFTEWTFR) (anti-β1EX) was used as the primary antibody followed by incubation with fluorescein isothiocyanate-conjugated anti-rabbit antibody. Slides were then viewed with a Bio-Rad Medical Research Council 600 confocal scanning laser microscope in the Microscopy and Image Analysis Laboratory at the University of Michigan.
Western blot analysis of mammalian and Drosophilacells. For Western blot analysis, cells were solubilized in 5% SDS and boiled in SDS-PAGE sample buffer containing 5% β-mercaptoethanol. Rat brain membranes, prepared as described previously (Isom et al., 1995b), were also solubilized and used as positive controls for sodium channel expression. Samples were separated by 10% acrylamide SDS-PAGE and transferred to nitrocellulose. Western blots were probed as described previously (Malhotra et al., 2000) with anti-β1EX at 1:500 dilution and then with horseradish peroxidase-conjugated goat anti-rabbit antibody (1:100,000). Immunoreactive bands were visualized with Westdura chemiluminescent substrate (Pierce, Rockford, IL).
Coimmunoprecipitation of sodium channel α subunits with β1 or C121Wβ1. Association of induced wild-type β1 or C121Wβ1 with α subunits in SNaIIA cells (after treatment with ponasterone or ethanol) or stably introduced wild-type or mutant β1 subunits in CNahIII-12 cells was assessed by coimmunoprecipitation. The details of cell membrane preparation, immunoprecipitation, and Western blotting have been described previously (Malhotra et al., 2000; Meadows et al., 2001). Briefly, membranes were solubilized with Triton X-100, immunoprecipitated with antibodies specific for the sodium channel Nav1.3 or Nav1.2a (Alomone Labs, Jerusalem, Israel) subunits or with nonimmune serum, electrophoresed in a 10% SDS-polyacrlyamide gel, and electrophoretically transferred to nitrocellulose. The blot was incubated in β1EXantibody (1:500) and then in horseradish peroxidase-conjugated goat anti-rabbit antibody (1:100,000). The blot was enhanced for visualization with Westdura chemiluminescent substrate (Pierce) and developed using ECL Hyperfilm.
Surface biotinylation of sodium channels. SNaIIApIND.β1 or SNaIIApIND.C121Wβ1 cells were induced with ponasterone or ethanol as described above. Cells on tissue culture plates were washed briefly with wash buffer (PBS containing 1 mmMgCl2 and 0.1 mmCaCl2) and incubated in 15 mg/ml sulfo-N-hydroxysuccinimide-biotin (sulfo-NHS-biotin; Pierce) in PBS for 30 min at 4°C. The cells were quenched twice for 10 min with 10 ml of quenching buffer (wash buffer plus 25 mm lysine monohydrochloride) and washed again briefly in wash buffer. The cells were then scraped into 15 ml conical tubes and divided into two equal aliquots, and each tube was centrifuged at 4°C at 2500 rpm for 10 min. In one tube, the wash buffer was aspirated, and the cell pellet was transferred to a microfuge tube containing 300 μl of dilution buffer plus 50 mm glycine. The cells were centrifuged again at 5000 rpm for 5 min at 4°C, and the supernatant was transferred to a fresh microfuge tube containing 5 μl (15 μg) of anti-Nav1.2a antibody. The tube was rotated for 4 hr at 4°C. Prewashed Protein A Sepharose beads (50 μl) were added, and the sample was rotated at 4°C overnight. On the following day, the cells were pelleted, the supernatants were removed, and the beads were washed three times with wash solution containing 0.1% Triton X-100 followed by one wash with solution that did not contain Triton X-100. The supernatants were aspirated, and the beads were boiled in 300 μl of 0.5% SDS for 5 min to release the immunoprecipitated proteins. The samples were recentrifuged for 1 min, and 50 μl of streptavidin beads was added to the supernatants to purify the biotinylated fraction of the proteins immunoprecipitated by anti-sodium channel antibody. The samples were then rotated at 4°C for 2–3 hr and centrifuged at 1000 rpm for 1 min. The supernatant was removed and washed three times with 500 μl of PBS. SDS-PAGE sample buffer was then added, and the samples were boiled for 5 min.
The contents of the second tube were immunoprecipitated with anti-Nav1.2a antibody as described above, but not treated with streptavidin agarose, to compare the total number of sodium channels in the cell with the number of cell-surface channels labeled with biotin (tube 1). Both samples were loaded onto a 5% SDS-polyacrylamide gel and transferred to nitrocellulose. The blot was probed with anti-Nav1.2a antibody (1:200), followed by horseradish peroxidase-conjugated goat anti-rabbit antibody (1:100,000), and visualized with Westdura chemiluminescent substrate.
RESULTS
C121W disrupts functional modulation of sodium channels by β1 in oocytes
The first step in our analysis was to confirm the effects described previously of β1 and C121Wβ1 subunits on cloned sodium channels expressed in Xenopus oocytes (Isom et al., 1992;Wallace et al., 1998). Figure 1 shows the effects of rat wild-type β1 or C121Wβ1 subunits on the time course of whole-cell sodium currents in oocytes expressing the rat Nav1.2a subtype of the sodium channel α subunit. Consistent with previous findings (Isom et al., 1992), coinjection of RNA encoding wild-type β1 and Nav1.2a, at equimolar concentrations, resulted in whole-cell sodium currents that inactivated approximately five times faster than sodium currents in oocytes injected with Nav1.2a RNA alone (Fig. 1A,left-hand traces). In contrast, injection of a 10-fold-higher concentration of C121Wβ1 RNA did not cause detectable modulation of current time course (Fig. 1A,right-hand traces), as described previously (Wallace et al., 1998). Interestingly, however, the C121W mutation did not completely abolish β1 function. Indeed, the mutant β1 subunit fully modulated the sodium current time course but only with injection of ∼100-fold more RNA than was necessary for functional modulation with β1 (Fig.1A,B). These data indicate that the mutation does not destroy the determinants required for modulation of sodium channels expressed in oocytes but instead lowers the efficacy of β1-mediated functional modulation. The data presented below suggest that this lower efficacy was caused by reduced affinity of the mutant β1 subunit for the α subunit.
Fig. 1.

The C121W mutation reduces the efficacy of β1-mediated modulation of brain sodium channels expressed inXenopus oocytes. A, Typical whole-cell sodium currents in oocytes expressing the rat Nav1.2a subtype of the sodium channel α subunit, either alone or with different concentrations of wild-type β1 (left-hand traces) or C121Wβ1 (right-hand traces). The values given for each trace correspond to moles of β1 RNA per moles of α RNA injected into each oocyte. The currents were evoked by depolarization to 0 mV, from a holding voltage of −90 mV. The traces were normalized with respect to the peak currents to enable comparison of inactivation time course. B, The proportion of fast decay, plotted as a function of moles of wild-type β1 (○,n = 6–8) or C121Wβ1 (●, n= 6–8) per mole of α. The proportion of fast decay for each experiment was determined by fitting inactivation of whole-cell currents elicited at 0 mV with the sum of two exponentials and then assessing the fraction of inactivation described by the faster of the two time constants. The fast and slow time constants were fairly constant over a range of β1 concentrations (τfast, ∼1 msec; τslow, ∼5–10 msec), whereas the proportion of the fast and slow decay varied as a function of β1 concentration. In this and subsequent figures, the data points correspond to means ± SEM. Data for α alone (▴, n = 7) are shown for comparison.
C121Wβ1 does not affect the time course of sodium currents in mammalian cells
The data from oocytes suggest that the C121W mutation results in a dramatic reduction in β1 function, but how does this cause epilepsy? As discussed above, Xenopus oocytes may distort sodium channel function and thus may not be the best cell system for addressing this question. In contrast, cultured mammalian cells provide a background that, compared with frog oocytes, is closer to mammalian neurons and in heterologous expression studies may more accurately reconstitute neuronal sodium channel behavior. For these reasons, we examined the effects of expression of β1 and C121Wβ1 subunits on rat Nav1.2a and human Nav1.3 sodium channels stably expressed in cultured mammalian cells. The Nav1.2a channel is a major brain isoform, whereas Nav1.3 is expressed at high levels during brain development (Beckh et al., 1989) and thus may be especially relevant for understanding childhood febrile seizures.
CNahIII-12 cells are a CHO-derived cell line, which stably expresses the human Nav1.3 α subunit (Chen et al., 2000). The parent CHO cell line expresses extremely low levels of endogenous sodium current (<65 pA) and does not express detectable sodium channel β subunits as assessed by reverse transcription-PCR analysis (Meadows et al., 2002). Therefore, this cell line is well suited for studying sodium channel function and modulation by β1 subunits. Coimmunoprecipitation data demonstrated that C121Wβ1, like wild-type β1, was expressed in transfected CNahIII-12 cells and associated with the hNav1.3 α subunit (Fig.2). Thus, the mutation did not prevent α-β1 dimerization in CNahIII-12 cells. We examined how coexpression of wild-type or mutant β1 with hNav1.3 affected hNav1.3 sodium channel function using whole-cell voltage-clamp recordings. Figure 3,A and B, shows mean normalized sodium currents elicited by depolarization to 0 mV in CNahIII-12 cells expressing hNav1.3 alone, hNav1.3 plus β1, or hNav1.3 plus C121Wβ1. These traces illustrate several important differences between sodium channels heterologously expressed in mammalian cells and sodium channels expressed in oocytes. First, current inactivation was fast in CNahIII-12 cells, even in the absence of β1 subunits (Fig. 3A), with a small persistent current remaining at the end of a 90-msec-long depolarization (Fig.3B). Second, neither β1 nor C121Wβ1 significantly altered the inactivation time course or the level of persistent current. The inactivation time course was best fit by the sum of two exponentials, reflecting prominent fast and smaller slow components of inactivation. Both the values of the fast and slow inactivation time constants (Fig. 3C) and their relative contribution to total current decay (Fig. 3D) were unaffected by wild-type or mutant β1 subunits over a broad range of test potentials. The addition of the auxiliary β2 subunit did not affect the current time course or other channel properties, when expressed with hNav1.3 alone, with wild-type β1, or with C121Wβ1 (Meadows et al., 2002) (data not shown). In contrast to the oocyte results, these data argue against the idea that loss of β1-mediated functional modulation causes seizures by slowing sodium current inactivation.
Fig. 2.

Wild-type β1 and C121Wβ1 associate with human Nav1.3 α subunits in CNahIII-12 cells. Coimmunoprecipitation experiments in two different CNahIII-12-derived cell lines, one stably coexpressing the human Nav1.3 α subunit and the human β1 subunit (right-hand blot) and the other stably coexpressing Nav1.3 and C121Wβ1 (left-hand blot). In each experiment, sodium channels were immunoprecipitated from solubilized membranes using an anti-Nav1.3 antibody and then probed using an anti-β1 polyclonal antiserum.
Fig. 3.

Neither wild-type β1 nor C121Wβ1 affect sodium current time course in CNahIII-12 cells. A, Mean time course of currents evoked at 0 mV in cells stably expressing hNav1.3 alone (solid line,n = 5), hNav1.3 plus β1 (dashed line, n = 6), or hNav1.3 plus C121Wβ1 (dotted line,n = 7). For each cell, current elicited by a 90 msec pulse to 0 mV was normalized, and then the normalized traces for each cell type were averaged together. Vertical linesindicate SEM determined at 0.2 msec intervals. B, Averaged traces over the entire 90-msec-long pulse duration, rescaled to show the persistent currents. In this case, the error bars are not shown. C, Current decay for each cell was fit according toAfastexp−t/τfast +Aslowexp−t/τslow +c, in which τfast and τsloware fast and slow time constants andAfast andAslow are scaling factors, respectively. The graph shows fast (filled symbols) and slow (open symbols) time constants for hNav1.3 alone (squares), hNav1.3 plus β1 (diamonds), and hNav1.3 plus C121Wβ1 (triangles), determined over a range of test potentials. D, The proportion of current decay corresponding to the slow time constant.Symbols are the same as in C. For all experiments in this figure, we used TTX subtraction to eliminate capacitive and leak currents.
C121Wβ1 increases sodium channel availability
What other changes in channel function might be responsible for causing the GEFS+ phenotype? One possibility is that channels coexpressed with C121Wβ1 subunits open or inactivate over a different voltage range than channels coexpressed with wild-type β1 subunits. For example, if channels associated with C121Wβ1 activate at more negative voltages, this would increase cell excitability by lowering the action potential threshold. We investigated whether β1 and C121Wβ1 had differing effects on sodium channel activation in CNahIII-12 cells by applying test pulses to a range of test potentials and converting the resulting current–voltage relationships to activation curves (see Materials and Methods). For CNahIII-12 cells expressing Nav1.3 alone, the midpoint of the activation curve was approximately −12 mV (Fig.4A). Neither β1 nor C121Wβ1 significantly altered the voltage dependence of channel activation (Fig. 4A). These data suggest that C121Wβ1 does not cause seizures by altering the voltage range over which sodium channels open.
Fig. 4.
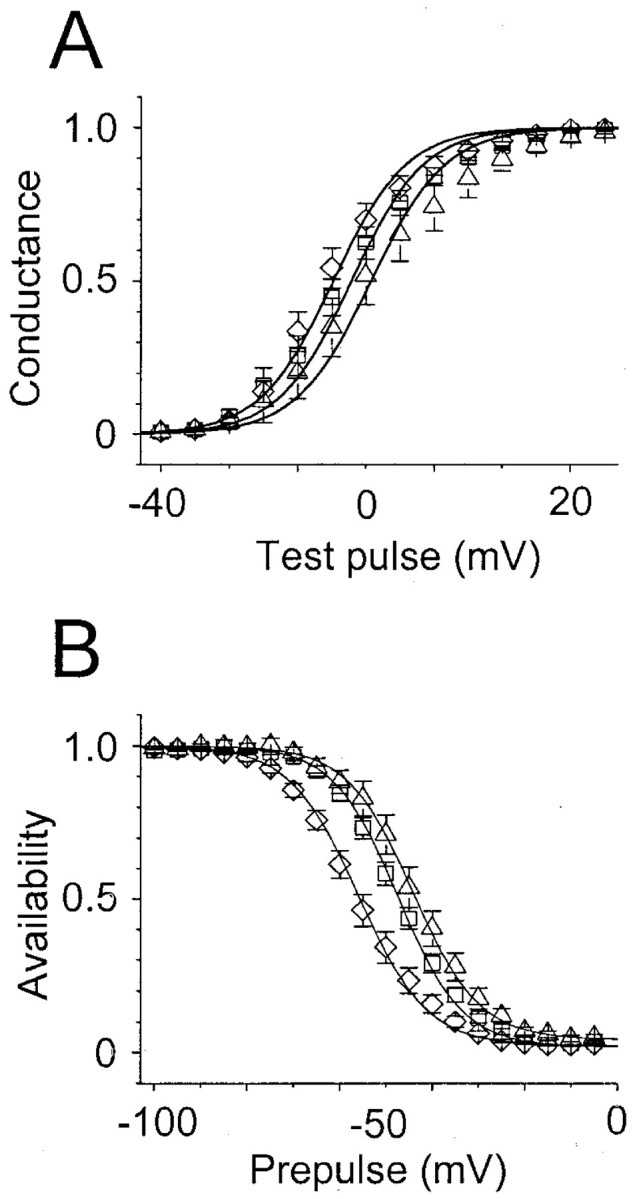
The voltage dependence of sodium channel availability is more positive in cells expressing C121Wβ1 than in cells expressing wild-type β1. A, Activation curves for CNahIII-12 cells, expressing human Nav1.3 alone (■,n = 19) or transiently coexpressing wild-type β1 (⋄, n = 13) or C121Wβ1 (▵,n = 10). Current–voltage relationships were converted to activation curves as described in Materials and Methods. The smooth lines are according to the Boltzmann equation (see Materials and Methods), using the following mean values forV1/2 and k determined from fits of individual experiments: hNav1.3:V1/2 = −12.1 ± 1.6,k = −5.3 ± 0.3; hNav1.3β1: −14.7 ± 1.6, −5.3 ± 0.5; hNav1.3C121Wβ1: −9.2 ± 3, −5.5 ± 0.6. B, Availability curves from the same cells as in A. The data were generated as described in Materials and Methods and fit with the Boltzmann equation as in A, using the following mean values for V1/2 and k: hNav1.3: V1/2 = −47.5 ± 1.2, k = 7 ± 0.2; hNav1.3β1: −55.9 ± 1.7, 7.4 ± 0.4; hNav1.3C121Wβ1: −44.1 ± 2, 7.1 ± 0.4. β1 shifted inactivation significantly negative compared with Nav1.3 alone (p < 0.001) or Nav1.3 with C121Wβ1 (p < 0.001).
Neuronal excitability can also be influenced by the fraction of sodium channels that are available to open at subthreshold membrane potentials. We assessed the voltage dependence of channel availability by applying 100-msec-long conditioning pulses to various potentials, followed by test pulses to 0 mV. For CHO cells expressing hNav1.3 alone, the midpoint of the sodium channel availability curve was approximately −47 mV (Fig.4B). Coexpression of β1 shifted the availability curve ∼10 mV negative (Fig. 4B). In contrast, C121Wβ1 did not cause this negative shift in channel availability (Fig. 4B). The more positive availability curve for sodium channels coexpressed with C121Wβ1 compared with channels coexpressed with wild-type β1 could increase cell excitability by increasing the fraction of sodium channels available to open at subthreshold voltages, resulting in increased sodium current amplitude.
C121W reduces frequency-dependent rundown of sodium channels
Whole-cell sodium currents run down during high-frequency channel activity, reflecting incomplete channel repriming between episodes of channel activation. This cumulative rundown may act as a break on cell excitability during high-frequency firing (Colbert et al., 1997; Jung et al., 1997) and may be important for suppressing pathophysiological hyperexcitability. To examine whether the epileptogenic properties of C121Wβ1 could be caused at least in part by effects on frequency-dependent sodium channel rundown, we examined whole-cell sodium currents over the course of rapid pulse trains. In CNahIII-12 cells expressing β1, whole-cell sodium currents declined by ∼60% by the end of a 100 pulse train of 5-msec-long test pulses to +10 mV applied at a frequency of 80 Hz (Fig.5A). In contrast, in CNahIII-12 cells expressing hNav1.3 alone or hNav1.3 plus C121Wβ1, the currents declined by only ∼20–30% (Fig. 5A). These differences in rundown developed almost entirely from the first to the second pulse in the train, suggesting that they were caused by differences in recovery of channels from fast inactivation between depolarizing test pulses. To test this hypothesis, we examined the recovery time course of channels inactivated by 5-msec-long conditioning pulses to 0 mV. Recovery time constants were ∼4, 5, and 11 msec in cells expressing hNav1.3 alone, hNav1.3 plus C121Wβ1, and hNav1.3 plus wild-type β1, respectively (Fig.5B). These recovery rates predict declines in current of ∼10, 20, and 50% from the first to second pulse at 80 Hz (Fig.5B, dashed line) for cells expressing hNav1.3 alone, hNav1.3 plus C121Wβ1, or hNav1.3 plus wild-type β1, respectively, which are values that match very closely to the observed frequency-dependent rundown for these different cell types. In summary, these data suggest that loss of β1 function caused by the C121W mutation may make neurons more excitable in part by accelerating recovery from fast inactivation and thus reducing sodium current rundown during high-frequency channel activity.
Fig. 5.
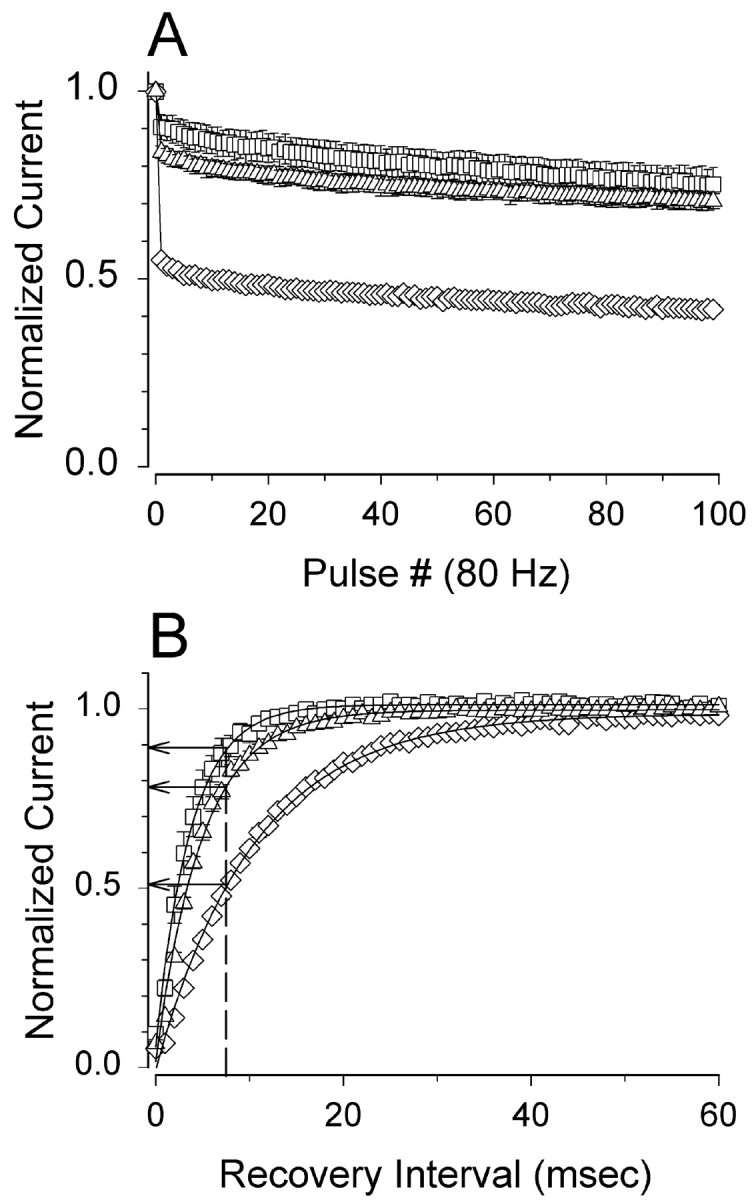
Sodium currents in CNahIII-12 cells expressing C121Wβ1 show reduced frequency-dependent rundown and faster recovery from inactivation, compared with currents in cells expressing wild-type β1. A, Mean amplitudes of currents elicited by 80 Hz pulse trains in CNahIII-12 cells expressing hNav1.3 alone (■, n = 5) and for cells stably coexpressing wild-type β1 (⋄, n = 5) or C121W β1 (▵,n = 6). The pulse trains consisted of 100 pulses, each 5 msec long, to +10 mV, from a holding voltage of −80 mV. Current amplitudes in each experiment were normalized with respect to the current evoked by the first pulse. B, Mean time course of recovery from inactivation for the same cells as inA. Recovery time course was assessed as described in Materials and Methods. The smooth lines are means of exponential fits of the data, with time constants of 3.7 ± 0.5, 4.9 ± 0.5, and 10.5 ± 0.3 msec, for hNav1.3 alone, hNav1.3 plus C121Wβ1, and hNav 1.3 plus β1, respectively. The vertical dashed line shows the extent of recovery after 7.5 msec, the duration between pulses in the 80 Hz trains. Both rundown and recovery time course were significantly different in cells coexpressing β1 than in cells expressing hNav1.3 alone or with C121Wβ1 (p < 0.0001).
C121Wβ1 does not act as a dominant-negative subunit for modulation of channel function
Loss-of-function mutations are frequently associated with recessive phenotypes, yet GEFS plus 1 shows an autosomal dominant inheritance pattern (Wallace et al., 1998). One way in which a loss-of-function mutation can show dominant inheritance is if the mutated protein acts as a dominant-negative, suppressing the activity of functional protein subunits. For example, C121Wβ1 could act as a dominant-negative subunit by binding to the sodium channel α subunit and occluding association of the functional wild-type β1. We tested this hypothesis by coexpressing both wild-type β1 and C121Wβ1 in the same CNahIII-12 cells and then examining the functional properties of the expressed sodium channels using whole-cell recording. If C121Wβ1 acted as a dominant negative, then we would expect expression of the mutant β1 subunit to at least partially occlude functional modulation by the wild-type β1 subunit; however, this was not the case. Indeed, both the negative shift in inactivation and the increase in frequency-dependent rundown caused by wild-type β1 were unaffected by coexpression of mutant β1 (Fig. 6). Thus, although both the wild-type and mutant β1 subunits associate with α (Fig. 2), wild-type β1 apparently binds to α with much higher affinity and thus displaces β1C121W in competition experiments. These data argue against a dominant-negative effect of β1C121W on current modulation.
Fig. 6.

C121Wβ1 does not act as a dominant-negative subunit. A, Mean V1/2 values of availability curves for CNahIII-12 cells expressing Nav1.3 alone (V1/2 = −47.5 ± 1.2; k = 7 ± 0.2;n = 18), for lines stably coexpressing β1 (−60.7 ± 0.9; 7 ± 0.3; n = 6), or for C121Wβ1 (−47.3 ± 1.5; 7.1 ± 0.2; n = 8), for the stable β1 line transiently coexpressing C121W β1 (−62.1 ± 1.8; 6.9 ± 0.6; n = 6), and for the stable C121Wβ1 line transiently coexpressing β1 (−60.4 ± 2; 7.1 ± 0.6; n = 8). β1 caused significant negative shifts in V1/2(p < 0.00001), even when coexpressed with C121Wβ1. B, Frequency-dependent rundown for CNahIII-12 cells expressing hNav1.3 alone (■) or hNav1.3 plus β1 (⋄; same data as in Fig. 5) and for the stable β1 line transiently coexpressing C121Wβ1 (○, n = 4).
C121W causes loss of β1 functional modulation of rat Nav1.2a sodium channels
Brain neurons express at least five different α subtypes (Goldin et al., 2000). Can the differences in functional modulation of Nav1.3 channels by wild-type β1 and C121Wβ1 in mammalian cells be generalized to other channel subtypes? To begin to address this question, we examined rat brain Nav1.2a sodium channels stably expressed in SNaIIA cells, a Chinese hamster lung-derived cell line (Isom et al., 1995b). To examine the effects of wild-type and mutant β1 subunits on rat Nav1.2a channels, we made SNaIIA-derived cell lines stably coexpressing either rat β1 or C121Wβ1 under the control of an ecdysone-inducible promoter. Figure7A shows Western blots demonstrating that β1 or C121Wβ1 subunit protein expression was induced in SNaIIA-pIND.β1 and SNaIIA-pIND.β1C121W cells, respectively, after 48 hr of treatment with 20 μm ponasterone (+), whereas there was no β1 subunit expression after treatment with vehicle alone (−). Thirty micromolar ponasterone or longer treatment times did not result in additional increases in the degree of β1 subunit expression (data not shown). Coimmunoprecipitatation experiments (Fig. 7B) showed that both wild-type β1 and C121Wβ1 subunits associated efficiently with rat Nav1.2a α subunits in SNaIIA cells. These results are consistent with coimmunoprecipitation data in Figure2, providing additional evidence that the C121W mutation does not prevent α-β1 dimerization in mammalian cells.
Fig. 7.
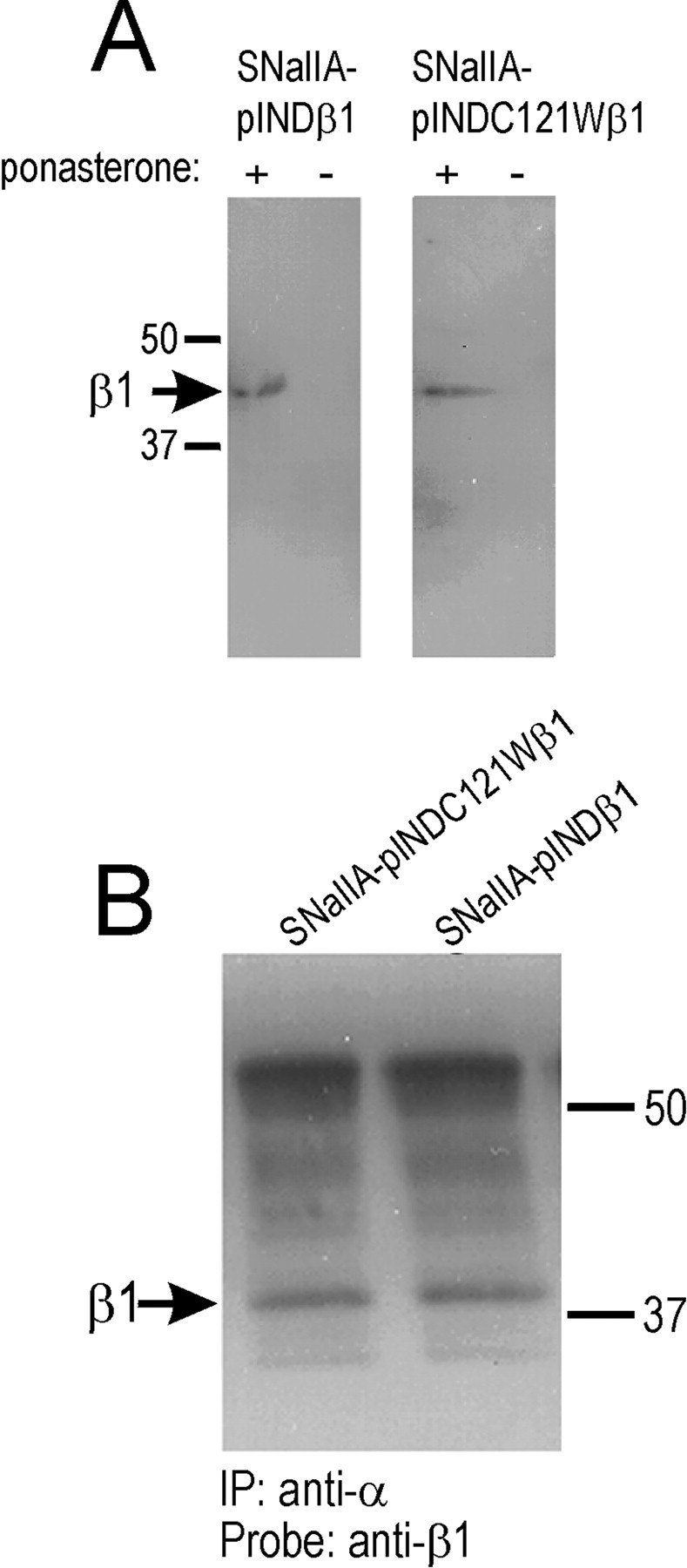
Ecdysone-inducible expression of β1 or C121Wβ1 subunits and association of β1 or C121Wβ1 with rat Nav1.2a α subunits. A, SNaIIA-pIND.β1 or SNaIIA-pIND.C121Wβ1 cells were treated with vehicle (0 ponasterone) or hormone (20 μm ponasterone) for 48 hr in culture, solubilized in 5% SDS, and boiled in SDS-PAGE sample buffer containing 5% β-mercaptoethanol. Samples were separated by 10% acrylamide SDS-PAGE and transferred to nitrocellulose. Western blots were probed with anti-β1EX antibody (1:500 dilution) and then with horseradish peroxidase-conjugated goat anti-rabbit antibody (1:100,000). Immunoreactive bands were visualized with Westdura chemiluminescent substrate. Arrow indicates position of β1 immunoreactive band. B, Equal aliquots of SNaIIA-pIND.β1 or SNaIIA-pIND.C121Wβ1 cells were treated with 20 μm ponasterone for 48 hr in culture, and then equal aliquots of cells were immunoprecipitated with anti-Nav1.2a antibody as described in Materials and Methods. The samples were then separated by SDS-PAGE, transferred to nitrocellulose, and probed with anti-β1EX antibody (1:500), followed by horseradish peroxidase-conjugated goat anti-rabbit antibody (1:100,000). The blot was detected with Westdura chemiluminescent substrate and exposed to ECL Hyperfilm. Arrow indicates migration of immunoreactive β1 subunits.
The effects of wild-type β1 and C121Wβ1 on the properties of rat Nav1.2a channels in SNaIIA cells are summarized in Figure 8. Induction of wild-type β1 in SNaIIA-pIND.β1 cells did not significantly alter sodium current time course (Fig. 8A) or the voltage dependence of activation (Fig. 8B, open symbols) but shifted the voltage dependence of inactivation to more negative potentials (Fig. 8B, filled symbols) and increased frequency-dependent rundown (Fig. 8C), compared with uninduced SNAIIA cells. In contrast, induction of C121Wβ1 did not have this effect. These data are qualitatively similar to data obtained using CNahIII-12 cells and thus are consistent with the hypothesis that loss of β1-mediated functional modulation in mammalian cells by the C121W mutation can be generalized to different brain α subtypes.
Fig. 8.
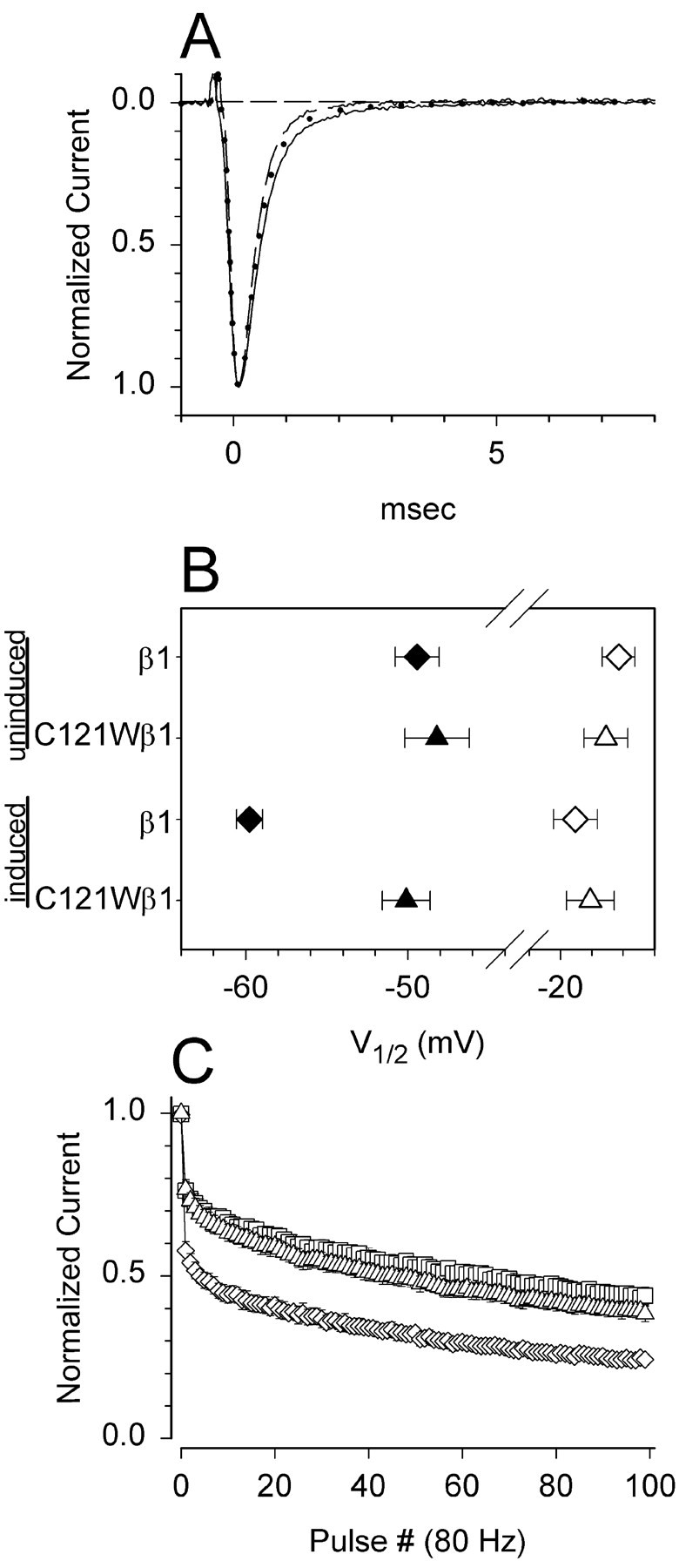
The C121W mutation causes loss of β1-mediated functional modulation of rat Nav1.2a sodium channels expressed in SNaIIA cells. A, Mean current time courses at 0 mV for uninduced SNaIIA-pIND.β1 and SNaIIA-pIND.C121Wβ1 cells, which express rat Nav1.2a alone (solid line,n = 8), and for induced cells expressing Nav1.2a plus β1 (dashed line,n = 6) or Nav1.2a plus C121Wβ1 (dotted line, n = 4). Neither wild-type nor mutant β1 significantly altered current time course.B, Mean V1/2 values for activation (open symbols) and availability (solid symbols) for uninduced SNaIIA cells (SNaIIA-pIND.β1: activation: V1/2 = −16.3 ± 1.1 mV, k = −6.1 ± 0.2; availability: −49.4 ± 1.4, 5.4 ± 0.3, n = 4; SNaIIA-pIND.C121Wβ1: −17.1 ± 1.4, −6.3 ± 0.4, −48.2 ± 2, 5.7 ± 0.4; n = 4) and for induced cells coexpressing β1 (−19 ± 1.4, −6.3 ± 0.3, −59.8 ± 0.8, 5.3 ± 0.2, n = 6) or C121Wβ1 (−18.1 ± 1.5, −6 ± 0.2, −50.1 ± 1.5, 5.6 ± 0.5, n = 4). Induction of wild-type β1 shifted the midpoint of availability significantly negative compared with cells expressing Nav1.2a alone or Nav1.2a with C121Wβ1 (p < 0.001). C, Mean frequency-dependent rundown in uninduced SNaIIA cells (■, n = 8) and in induced cells coexpressing β1 (⋄, n = 6) or C121Wβ1 (▵,n = 4). Wild-type β1 significantly increased frequency-dependent rundown compared with uninduced cells or cells coexpressing C121Wβ1 (p < 0.01).
C121Wβ1 and wild-type β1 promote surface expression of sodium channels
We have shown previously that coexpression of β1 in SNaIIA cells resulted in a twofold to fourfold increase in the level of plasma membrane binding sites for the sodium channel-specific ligand3H-saxitoxin (Isom et al., 1995b; Meadows et al., 2001). In the present study, we used a different biochemical approach, surface biotinylation followed by two rounds of immunoprecipitation, to compare the ability of wild-type β1 or C121Wβ1 to promote the cell surface expression of sodium channels in our ecdysone-inducible cell lines. Figure9A demonstrates that the number of biotin-labeled Nav1.2a sodium channels on the cell surface is extremely low in parental SNaIIA cells and is unaffected by ponasterone or ethanol treatment. In contrast, the level of total sodium channels (intracellular plus extracellular) in SNaIIA cells is abundant. Presumably, in the absence of β subunits, most of these channels never reach the cell surface. In SNaIIA-pIND.β1 and SNaIIA-pIND.C121Wβ1 cells, which express inducible β1 or C121Wβ1 subunits, respectively, vehicle treatment did not change the levels of cell surface sodium channels (Fig. 9B); however, treatment with 20 μm ponasterone, which maximally induces β subunit expression (Fig. 7), promoted the translocation of sodium channels to the cell surface (Fig. 9C). Interestingly, wild-type β1 and C121Wβ1 subunits were equally effective in this assay. Thus, consistent with previous findings (Tammaro et al., 2002), the C121W mutation does not prevent β1 subunit-mediated translocation of α subunits to the plasma membrane. These data provide additional biochemical evidence for interaction between α and C121Wβ1 subunits.
Fig. 9.
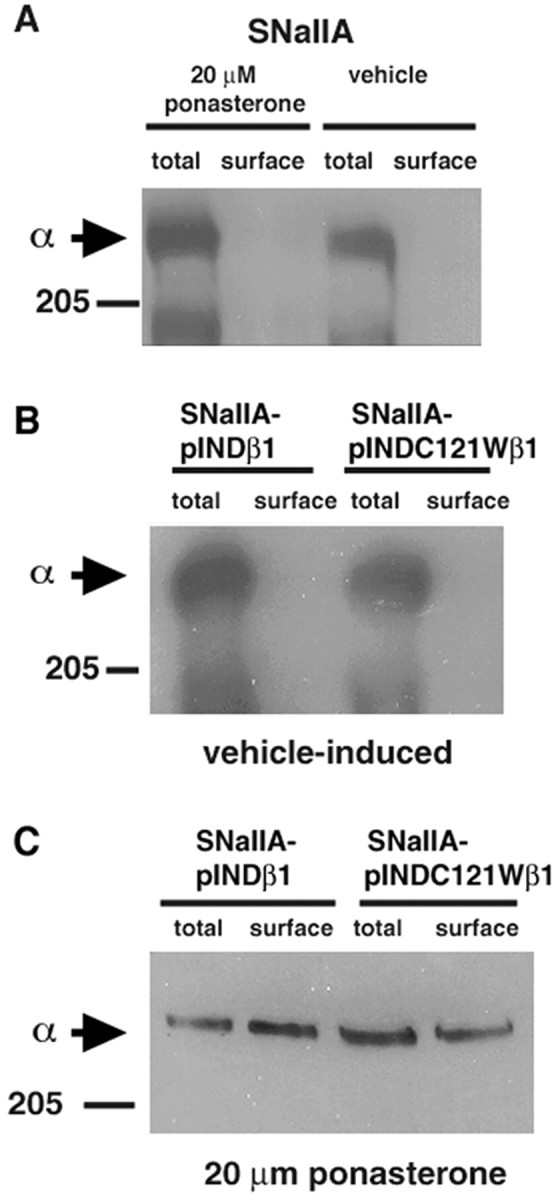
β1 and C121Wβ1 subunits promote translocation of Nav1.2a α subunits to the plasma membrane. SNaIIA, SNaIIA-pIND.β1, or SNaIIA-pIND.C121Wβ1 cells were treated with vehicle or 20 μm ponasterone for 48 hr in culture and treated with sulfo-NHS-biotin as described in Materials and Methods. Each cell sample was immunoprecipitated with anti-SP11-II and divided into two equal aliquots. One aliquot was prepared for SDS-PAGE as described (total). The remaining half was immunoprecipitated with anti-SP11-II antibody, boiled in 5% SDS to release the proteins from the Protein A Sepharose beads, reprecipitated with streptavidin agarose to purify the fraction that was biotinylated, and prepared for SDS-PAGE as described (surface). The samples were then separated by SDS-PAGE, transferred to nitrocellulose, and probed with anti-SP11-II antibody (1:500) followed by horseradish peroxidase-conjugated goat anti-rabbit antibody (1:100,000). The blot was detected with Westdura chemiluminescent substrate and exposed to ECL Hyperfilm. Arrows indicate migration of immunoreactive α subunits. A, An undetectable percentage of Nav1.2a subunits in SNaIIA cells is located at the cell surface. Treatment of cells with ponasterone does not affect cell surface expression of sodium channels. B, Treatment of SNaIIA-pIND.β1 or SNaIIA-pIND.C121Wβ1 cells with vehicle does not result in translocation of Nav1.2a sodium channels to the cell surface. C, Treatment of SNaIIA-pIND.β1 or SNaIIA-pIND.C121Wβ1 cells with 20 μm ponasterone (resulting in β1 or C121β1 subunit expression, as shown in Fig. 7) results in an increase in the percentage of Nav1.2a sodium channels located at the cell surface.
Considering the large increase in sodium channels detected biochemically, we expected to observe a comparably large increase in sodium current amplitude after induction of wild-type β1 or C121Wβ1 subunits. Surprisingly, however, current amplitudes after induction of wild-type or mutant β1 were statistically indistinguishable from currents in uninduced SNaIIA cells (Fig.10A). Similarly, neither wild-type β1 nor C121Wβ1 increased the amplitude of sodium currents in CNahIII-12 cells (Fig. 10B). In oocytes, injection of moderate concentrations of wild-type β1 or C121Wβ1 did not affect current amplitude, whereas injection of high concentrations of C121Wβ1 actually decreased whole-cell currents (Fig.10C). These data are consistent with a previous study in which we found that a large increase in saxitoxin-binding sites in SNaIIA cells constitutively coexpressing β1 did not result in a concomitant increase in whole-cell sodium currents (Meadows et al., 2001). Together, these data suggest that many of the biochemically detected sodium channels brought to the surface by β1 do not open in response to depolarization in whole-cell voltage-clamp experiments. The significance of this observation for neuronal excitability will require further investigation.
Fig. 10.

Neither wild-type β1 nor C121Wβ1 increases sodium current amplitude. A–C, Mean amplitudes of currents elicited by depolarizations to 0 mV from a holding voltage of −90 mV, for SNaIIA cells (A), CNahIII-12 cells (B), and Xenopus oocytes expressing rat Nav1.2a (C).
C121Wβ1 subunits do not induce cellular aggregation
The data presented in the preceding sections suggest that loss of β1 functional modulation caused by the C121W mutation may increase neuronal excitability by subtly altering channel behavior. However, in addition to their effects on the electrophysiological properties of sodium channels, β1 subunits also exhibit cell adhesion properties that may be important for mediating protein–protein interactions involving sodium channels. We investigated the effects of the C121W mutation on β1-mediated cell adhesive interactions by examining the behavior of Drosophila S2 cells expressing wild-type or mutant β1 subunits. S2 cells are a classic model system in which potential cell adhesion molecules (CAMs) have been tested for homophilic and heterophilic interactions (Hortsch and Bieber, 1991;Bieber, 1994). Untransfected S2 cells do not express detectable sodium channel α, β1, or β2 subunits (Malhotra et al., 2000). They show no tendency to adhere to each other or to tissue culture plastic and thus grow as a suspension culture (Bieber, 1994). cDNAs of interest are cloned into the S2 cell expression vector, pRmHa3, under control of an inducible Drosophila metallothionein promoter. S2 cells that have been transfected with CAM cDNAs in pRmHa3 aggregate after induction of protein expression with CuSO4. Using the S2 cell model system, we showed previously that sodium channel β1 subunits cause S2 cells to aggregate and subsequently recruit ankyrin to points of cell–cell contact (Malhotra et al., 2000).
Both wild-type β1 and C121Wβ1 subunits are efficiently expressed in S2 cells as assessed by Western blot analysis (Fig.11A). S2 cells transfected with wild-type β1 subunits formed aggregates after induction with CuSO4 and mechanical shaking (Fig.11B, top right panel). In contrast, C121Wβ1-transfected cells (Fig. 11B, bottom right panel), untransfected cells, or mock-transfected cells (data not shown) treated similarly did not aggregate. Immunocytochemical experiments using an antibody directed to the extracellular domain of β1 (anti-β1EX) (Malhotra et al., 2000) showed that C121Wβ1 subunit protein was expressed on the cell surface of the transfected S2 cells (Fig.11B, bottom left panel). Together, these data suggest that the cysteine-to-typtophan mutation disrupts determinants on the β1 Ig loop that are critical for the adhesive functions of β1. Coexpression of wild-type β1 and C121Wβ1 in S2 cells did not disrupt cell aggregation (Fig.12), indicating that C121Wβ1 did not act as a dominant negative for cell adhesion.
Fig. 11.
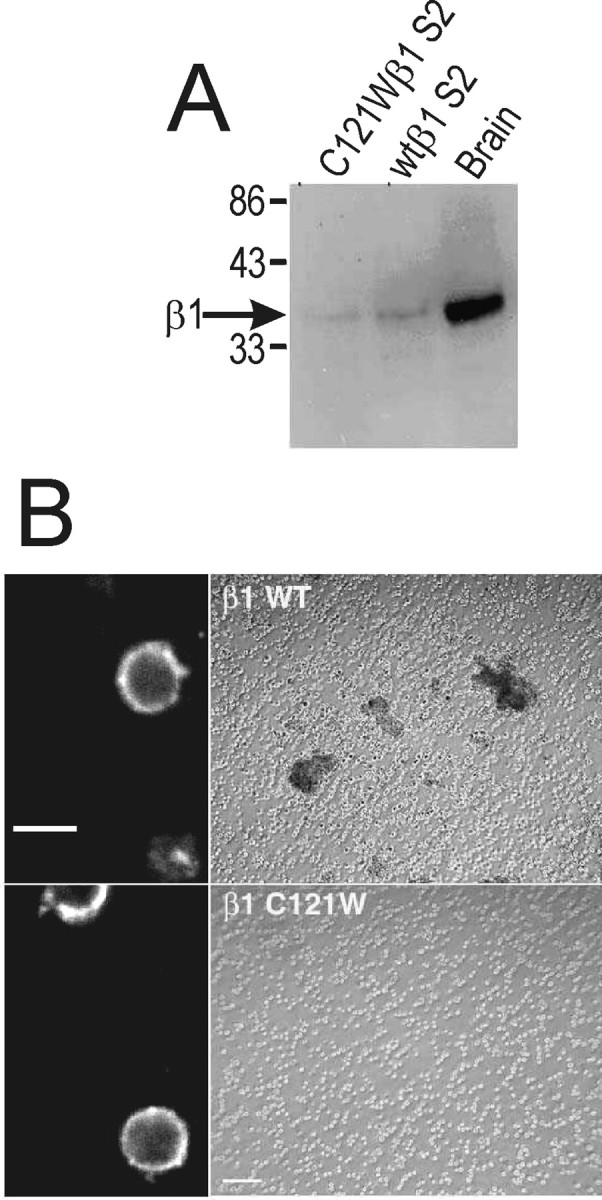
The C121W mutation disrupts β1–β1 homophilic interactions. A, Western blot analysis of β1 subunit expression in transfected S2 cells. Wild-type β1- or C121Wβ1-transfected S2 cells were solubilized in 5% SDS and boiled in SDS-PAGE sample buffer containing 5% β-mercaptoethanol. Samples were separated by 10% acrylamide SDS-PAGE and transferred to nitrocellulose. The Western blot was probed with anti-β1EX antibody (1:500 dilution) and then with horseradish peroxidase-conjugated goat anti-rabbit antibody (1:100,000 dilution). Immunoreactive bands were visualized with Westdura chemiluminescent substrate (Pierce). B, C121Wβ1 subunit expression does not promote S2 cell aggregation. Transfected S2 cells were induced in the presence of 0.7 mmCuSO4. An aliquot of each cell line was removed and stained for β1 or C121Wβ1 expression at the cell surface (left panel). Cells were viewed with a confocal microscope. Scale bar, 10 μm. In the remaining cells, aggregation was induced by rotary shaking. Cells were viewed with a phase-contrast microscope. Scale bar, 100 μm. Aggregation was not observed in cells transfected with C121Wβ1 in any field of view.
Fig. 12.

C121Wβ1 does not exert a dominant-negative effect on cell adhesion in Drosophila S2 cells. Stable S2β1 cell lines (Malhotra et al., 2000) were transfected with increasing amounts of C121Wβ1 plasmid, as indicated above, using Fugene reagent. Selection (250 μg/ml hygromycin) was started 48 hr after transfection and continued for 30 d. Transfected or untransfected S2 cells were induced in the presence of 0.7 mm CuSO4, and then aggregation was induced by rotary shaking. Cells were viewed with a phase-contrast microscope. Representative fields of view are presented for 0 (S2β1), 1, and 5 μg of transfected C121Wβ1 plasmid, respectively. Untransfected S2 cells are presented for comparison. Scale bar, 100 μm.
DISCUSSION
Since the discovery of the first channelopathies linked to epilepsy and other neurological diseases, the race to identify new disease-linked mutations in ion channel genes has proceeded at an ever-increasing pace. Although our understanding of how these mutations cause disease has lagged somewhat behind, recent functional studies have given important insights into the relationship among gene mutations, altered channel function, and disease phenotypes. In the case of GEFS+ type 1, the first sodium channelopathy associated with epilepsy, Wallace et al. (1998) showed originally in a large Australian family that the disease was caused by a cysteine-to-tryptophan substitution in the β1 subunit of the voltage-gated sodium channel and further demonstrated that this mutation resulted in attenuation of β1 function. Nevertheless, precisely how this causes epilepsy remains unclear. In this study, we have identified several effects of the C121Wβ1 mutation that could contribute to neuronal pathophysiology. First, using two different mammalian cell lines, one stably expressing the human Nav1.3 sodium channel and the other stably expressing the rat Nav1.2a sodium channel, we show that sodium channels in cells expressing C121Wβ1 have subtly different functional properties than channels in cells expressing wild-type β1. Second, using a Drosophila S2 cell adhesion assay, we show that the C121W mutation disrupts the cell adhesive properties of β1. Finally, we show that C121Wβ1 does not occlude β1-mediated functional modulation or cell adhesion in competition experiments. The potential significance of these findings for epilepsy is discussed below.
C121Wβ1 alters sodium channel function
GEFS+, like other epilepsies, is a paroxysmal disorder, characterized by brief seizures separated by long periods of ostensibly normal behavior (Scheffer and Berkovic, 1997). Considering that sodium channels are the main mediators of intrinsic excitability in brain neurons, one might expect, a priori, that mutations in sodium channel genes linked to epilepsy would cause subtle changes in channel function, because dramatic changes in channel behavior would likely result in more severely deleterious or lethal phenotypes (Kearney et al., 2001). Consistent with this hypothesis, GEFS+ mutations characterized previously in genes encoding sodium channel α subunits cause small alterations in channel activation, inactivation, frequency-dependent rundown, and persistent current (Alekov et al., 2000, 2001; Spampanato et al., 2001; Lossin et al., 2002). The data presented here suggest that loss of β1 function, caused by the C121W mutation, results in comparably subtle alterations in sodium channel behavior compared with channels associated with wild-type β1. These differences include a positive shift in the voltage dependence of channel availability and a reduction in current rundown during high-frequency channel activation. We suggest that these subtle changes in sodium channel function permit normal neuronal behavior under most circumstances but slightly destabilize neurons and neuronal networks, so that a small external perturbation, such as fever, is sufficient to cause transient breakdown of electrophysiological homeostasis, resulting in a seizure. In contrast, and consistent with previous findings using the skeletal muscle sodium channel expressed in human embryonic kidney cells (Tammaro et al., 2002), we did not observe an effect of wild-type β1 or C121Wβ1 on sodium current time course. The difference between β1 and C121Wβ1 in frequency-dependent rundown is especially intriguing, because similar effects have been observed for the GEFS+ type 2 mutations R1648H (Spampanato et al., 2001) and D188V (our unpublished observations) in the Nav1.1 sodium channel α subunit. Thus, changes in the frequency response of sodium channels may be a common mechanism contributing to the GEFS+ phenotype.
C121W disrupts β1-mediated cell adhesion
Sodium channel β1 subunits are multifunctional proteins. In addition to modulating channel function, they also act as cell adhesion molecules, in both the presence and absence of α subunits (Srinivasan et al., 1998; Xiao et al., 1999; Malhotra et al., 2000). We proposed previously that β1-mediated adhesion and subsequent ankyrin recruitment may play roles in sodium channel subcellular localization and thus may be important determinants of cell excitability (Malhotra et al., 2000). The C121W mutation disrupts a conserved disulfide bond that is thought to be critical for interaction between the two β-sheets of the Ig fold (Williams and Barclay, 1987), a prerequisite for homophilic and heterophilic interaction involving Ig domains (Zhang and Filbin, 1994). Mutations that disrupt homologous disulfide bonds in Ig domain-containing proteins L1CAM and myelin Podisrupt homophilic and heterophilic interactions and are associated with human diseases (De Angelis et al., 1999; Fabrizi et al., 1999). Thus, we suggest that disruption of homophilic β1–β1 interactions or heterophilic interaction between β1 and contactin (Kazarinova-Noyes et al., 2001) or neurofascin (Ratcliffe et al., 2001), caused by the C121W mutation, contributes to the GEFS+ phenotype, perhaps through abnormal subcellular channel localization. Elucidating precisely how abnormal β1-mediated adhesion may contribute to epilepsy will require a more complete understanding of the role of normal β1 interactions in sodium channel localization and neuronal electrophysiology.
C121Wβ1 is not a dominant-negative subunit for modulation of channel function
Mutagenesis analysis has identified key residues in the A/A′ face of the β1 Ig loop that are required for its interaction with the sodium channel α subunit (McCormick et al., 1998). The C121W mutation is expected to disrupt the structure of the Ig loop, yet, surprisingly, our data indicate that the mutant β1 can still associate with α and, at least in oocytes, can still modulate channel function, albeit only with expression of high levels of mutant subunit. In CHO cells, on the other hand, functional modulation by the mutant β1 subunit appears to be lost completely, although α and C121Wβ1 associate in coimmunoprecipitation assays. These data suggest that β1 functional modulation may involve different molecular mechanisms in mammalian cells than in oocytes. Because mutant β1 retains the ability to associate with the α subunit, we speculated that it might act as a dominant-negative subunit, binding to α and occluding binding of the functional wild-type β1. However, competition experiments suggest that, compared with wild-type β1, the mutant β1 binds to α with much lower affinity. The relative levels of RNA required to achieve functional modulation in oocytes suggest that β1 and C121Wβ1 differ by roughly two orders of magnitude in their affinity for α. Furthermore, C121Wβ1 does not suppress β1–β1-mediated homophilic interactions in the S2 cell adhesion assay, suggesting that it does not act as a dominant negative for the cell adhesion properties of β1. In summary, these data argue against a dominant-negative effect of C121Wβ1 and suggest that haploin sufficiency of the wild-type β1 may be a more likely explanation for the dominant inheritance of the GEFS+ phenotype.
Conclusion
In summary, our data suggest that loss of β1-mediated functional modulation and cell adhesion, caused by the C121W mutation, results in subtle changes in the function and subcellular distribution of brain sodium channels, which in turn increases neuronal excitability and predisposes individuals with the mutant allele toward seizures. In contrast, our data argue against the prevalent idea that slowing of sodium current inactivation time course, as seen in Xenopusoocytes, is responsible for the disease phenotype. The data also argue against a dominant-negative effect of the C121W mutation. The key unresolved question remains whether these in vitroobservations apply to sodium channels in brain neurons. Future studies with transgenic mice expressing mutant β1 subunits will address this issue.
L.S.M. and J.M. contributed equally to this work.
This work was supported by Canadian Institutes of Health Research Grant MT-13485 (D.S.R.), Grant 001654 from the Michigan Life Sciences Corridor (L.L.I.), and Grant 1R01MH59980 from the National Institutes of Health (L.L.I.). L.S.M. was supported by a fellowship from Le Fonds de la Recherche en Santé du Québec. J.M. was supported by an advanced postdoctoral fellowship from the National Multiple Sclerosis Society. We thank Y. H. Chen, A. J. Powell, and J. J. Clare (GlaxoSmithKline Pharmaceuticals) for supplying us with CNahIII-12 cells and the human β1 clone, W. A. Catterall for SNaIIA cells, and J. Nichols and L. De Leon for technical assistance.
Correspondence should be addressed to either of the following: Dr. Lori L. Isom, Department of Pharmacology, University of Michigan Medical School, 1301E MSRB III, Ann Arbor, MI 48109-0632, E-mail:lisom@umich.edu; or Dr. David S. Ragsdale, Montreal Neurological Institute, 3801 University Street, Montreal, Quebec H3A 2B4, Canada, E-mail: dragsdale@mni.mcgill.ca.
REFERENCES
- 1.Abou-Khalil B, Ge Q, Desai R, Ryther R, Bazyk A, Baily R, Haines JL, Sutcliff JS, George AL., Jr Partial and generalized epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology. 2001;57:2265–2272. doi: 10.1212/wnl.57.12.2265. [DOI] [PubMed] [Google Scholar]
- 2.Alekov AK, Rahman M, Mitrovic N, Lehmann-Horn F, Lerche H. A sodium channel mutation causing epilepsy in man exhibits subtle defects in fast inactivation and activation in vitro. J Physiol (Lond) 2000;529:533–539. doi: 10.1111/j.1469-7793.2000.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alekov AK, Rahman M, Mitrovic N, Lehmann-Horn F, Lerche H. Enhanced inactivation and acceleration of activation of the sodium channel associated with epilepsy in man. Eur J Physiol. 2001;13:2171–2176. doi: 10.1046/j.0953-816x.2001.01590.x. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barek S. Site-directed mutagenesis by double polymerase chain reaction: megaprimer method. In: White BA, editor. Methods in molecular biology, Vol 15: PCR protocols, current methods and applications. Humana; Totowa, NJ: 1993. pp. 277–286. [DOI] [PubMed] [Google Scholar]
- 6.Beckh S, Noda M, Lübbert H, Numa S. Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. Nature. 1989;8:3611–3616. doi: 10.1002/j.1460-2075.1989.tb08534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bieber AJ. Analysis of cellular adhesion in cultured cells. Methods Cell Biol. 1994;44:683–696. doi: 10.1016/s0091-679x(08)60938-3. [DOI] [PubMed] [Google Scholar]
- 8.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 9.Chen YH, Dale TJ, Romanos MA, Whitaker WR, Xie XM, Clare JJ. Cloning, distribution and functional analysis of the type III sodium channel from human brain. Eur J Neurosci. 2000;12:4281–4289. [PubMed] [Google Scholar]
- 10.Colbert CM, Magee JC, Hoffman DA, Johnston D. Slow recovery from inactivation of Na+ channels underlies the activity-dependent attenuation of dendritic action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 1997;17:6512–6521. doi: 10.1523/JNEUROSCI.17-17-06512.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Angelis E, Macfarlane J, Du J-S, Yeo G, Hicks R, Rathjen FG, Kenwick S, Brummendorf T. Pathological missense mutations of neural cell adhesion molecule L1 affect homophilic and heterophilic binding activities. EMBO J. 1999;18:4744–4753. doi: 10.1093/emboj/18.17.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+ 2. Nat Genet. 2000;24:343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- 13.Escayg A, Heils A, MacDonald BT, Haug K, Sander T, Meisler MH. A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus and prevalence of variants in patients with epilepsy. Am J Hum Genet. 2001;68:866–873. doi: 10.1086/319524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fabrizi GM, Cavallaro T, Morbin M, Simonati A, Taioli F, Rizzuto N. Novel mutation of the P0 extracellular domain causes a Dejerine-Sottas syndrome. J Neurol Neurosurg Psychiatry. 1999;66:386–389. doi: 10.1136/jnnp.66.3.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freshney RI. Culture of animal cells. Alan R. Liss; New York: 1983. [Google Scholar]
- 16.Gardiner RM. Impact of our understanding of the genetic aetiology of epilepsy. J Neurol. 2000;247:327–340. doi: 10.1007/s004150050598. [DOI] [PubMed] [Google Scholar]
- 17.Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 18.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 19.Hortsch M, Bieber AJ. Sticky molecules in not-so-sticky cells. Trends Biol Sci. 1991;16:283–287. doi: 10.1016/0968-0004(91)90116-d. [DOI] [PubMed] [Google Scholar]
- 20.Isom LL. Sodium channel beta subunits: anything but auxiliary. Neuroscientist. 2001;7:42–54. doi: 10.1177/107385840100700108. [DOI] [PubMed] [Google Scholar]
- 21.Isom LL, De Jongh KS, Patton DE, Reber BFX, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- 22.Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BF, Scheuer T, Catterall WA. Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 1995a;83:433–442. doi: 10.1016/0092-8674(95)90121-3. [DOI] [PubMed] [Google Scholar]
- 23.Isom LL, Scheuer T, Brownstein AB, Ragsdale DS, Murphy BJ, Catterall WA. Functional co-expression of the β1 and type IIA α subunits of sodium channels in a mammalian cell line. J Biol Chem. 1995b;270:3306–3312. doi: 10.1074/jbc.270.7.3306. [DOI] [PubMed] [Google Scholar]
- 24.Jung H-Y, Mickus T, Spruston N. Prolonged sodium channel inactivation contributes to dendritic action potential attenuation in hippocampal pyramidal neurons. J Neurosci. 1997;17:6639–6646. doi: 10.1523/JNEUROSCI.17-17-06639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kazarinova-Noyes K, Malhotra JD, McEwen DP, Mattei LN, Berglund EO, Ranscht B, Levinson SR, Schachner M, Shrager P, Isom LL, Xiao ZC. Contactin associates with Na+ channels and increases their functional expression. J Neurosci. 2001;21:7517–7525. doi: 10.1523/JNEUROSCI.21-19-07517.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, Goldin AL, Meisler MH. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience. 2001;102:307–317. doi: 10.1016/s0306-4522(00)00479-6. [DOI] [PubMed] [Google Scholar]
- 27.Krafte DS, Goldin AL, Auld VJ, Dunn RJ, Davidson N, Lester HA. Inactivation of cloned sodium channels expressed in Xenopus oocytes. J Gen Physiol. 1990;96:689–706. doi: 10.1085/jgp.96.4.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lerche H, Jurkat-Rott K, Lehmann-Horn F. Ion channels and epilepsy. Am J Med Genet. 2001;106:146–159. doi: 10.1002/ajmg.1582. [DOI] [PubMed] [Google Scholar]
- 29.Li HL, Galue A, Meadows L, Ragsdale DS. A molecular basis for the different local anesthetic affinities of resting versus open and inactivated states of the sodium channel. Mol Pharmacol. 1999;55:134–141. doi: 10.1124/mol.55.1.134. [DOI] [PubMed] [Google Scholar]
- 30.Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL. Molecular basis of an inherited epilepsy. Neuron. 2002;34:877–884. doi: 10.1016/s0896-6273(02)00714-6. [DOI] [PubMed] [Google Scholar]
- 31.Malhotra JD, Kazen-Gillespie K, Hortsch M, Isom LL. Sodium channel beta subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J Biol Chem. 2000;275:11383–11388. doi: 10.1074/jbc.275.15.11383. [DOI] [PubMed] [Google Scholar]
- 32.McCormick KA, Isom LL, Ragsdale D, Smith D, Scheuer T, Catterall WA. Molecular determinants of Na+ channel function in the extracellular domain of the beta1 subunit. J Biol Chem. 1998;273:3954–3962. doi: 10.1074/jbc.273.7.3954. [DOI] [PubMed] [Google Scholar]
- 33.Meadows L, Malhotra JD, Stetzer A, Isom LL, Ragsdale DS. The intracellular segment of the sodium channel beta 1 subunit is required for its efficient association with the channel alpha subunit. J Neurochem. 2001;76:1871–1878. doi: 10.1046/j.1471-4159.2001.00192.x. [DOI] [PubMed] [Google Scholar]
- 34.Meadows LS, Chen YH, Powell AJ, Clare JJ, Ragsdale DS. Functional modulation of human brain Nav1.3 sodium channels, expressed in mammalian cells, by auxiliary β1, β2 and β3 subunits. Neuroscience. 2002;114:745–753. doi: 10.1016/s0306-4522(02)00242-7. [DOI] [PubMed] [Google Scholar]
- 35.Meisler MH, Kearney J, Ottman R, Escayg A. Identification of epilepsy genes in human and mouse. Annu Rev Genet. 2001;35:567–588. doi: 10.1146/annurev.genet.35.102401.091142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ratcliffe CF, Westenbroek RE, Curtis R, Catterall WA. Sodium channel beta1 and beta3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol. 2001;154:427–434. doi: 10.1083/jcb.200102086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain. 1997;120:479–490. doi: 10.1093/brain/120.3.479. [DOI] [PubMed] [Google Scholar]
- 38.Spampanato J, Escayg A, Meisler MH, Goldin AL. Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2. J Neurosci. 2001;21:7481–7490. doi: 10.1523/JNEUROSCI.21-19-07481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Srinivasan J, Schachner M, Catterall WA. Interaction of voltage-gated sodium channels with the extracellular matrix molecules tenascin-C and tenascin-R. Proc Natl Acad Sci USA. 1998;95:15753–15757. doi: 10.1073/pnas.95.26.15753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steinlein OK. Genes and mutations in idiopathic epilepsy. Am J Med Genet. 2001;106:139–145. doi: 10.1002/ajmg.1571. [DOI] [PubMed] [Google Scholar]
- 41.Steinlein OK, Noebels JL. Ion channels and epilepsy in man and mouse. Curr Opin Genet Dev. 2000;10:286–291. doi: 10.1016/s0959-437x(00)00079-4. [DOI] [PubMed] [Google Scholar]
- 42.Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, Mazaki-Miyazaki E, Nagafuji H, Noda M, Imoto K, Wada K, Mitsudome A, Kaneko S, Montal M, Nagata K, Hirose S, Yamakawa K. A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci USA. 2001;98:6384–6389. doi: 10.1073/pnas.111065098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tammaro P, Conti F, Moran O. Modulation of sodium current in mammalian cells by an epilepsy-correlated beta1-subunit mutation. Biochem Biophys Res Commun. 2002;291:1095–1101. doi: 10.1006/bbrc.2002.6570. [DOI] [PubMed] [Google Scholar]
- 44.Ukomadu C, Zhou J, Sigworth FJ, Agnew WS. μ1 Na+ channels expressed transiently in human embryonic kidney cells: biochemical and biophysical properties. Neuron. 1992;8:663–676. doi: 10.1016/0896-6273(92)90088-u. [DOI] [PubMed] [Google Scholar]
- 45.Wallace RH, Wang DW, Singh R, Scheffer IE, George AL, Jr, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel β1 subunit gene SCN1B. Nat Genet. 1998;19:366–370. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- 46.Wallace RH, Scheffer IE, Barnett S, Richards M, Dibbens L, Desai RR, Lerman-Sagie T, Lev D, Mazarib A, Brand N, Ben-Zeez B, Goikhman I, Singh R, Kremmidiotis G, Gardner A, Sutherland GR, George AL, Jr, Mulley JC, Berkovic SF. Neuronal sodium-channel α1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2001;68:859–865. doi: 10.1086/319516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.West JW, Scheuer T, Maechler L, Catterall WA. Efficient expression of rat brain type IIA Na+ channel alpha subunits in a somatic cell line. Neuron. 1992;8:59–70. doi: 10.1016/0896-6273(92)90108-p. [DOI] [PubMed] [Google Scholar]
- 48.Williams AF, Barclay AN. The immunoglobulin superfamily—domains for cell surface recognition. Annu Rev Immunol. 1987;6:381–405. doi: 10.1146/annurev.iy.06.040188.002121. [DOI] [PubMed] [Google Scholar]
- 49.Xiao ZC, Ragsdale DS, Malhotra JD, Mattei LN, Braun PE, Schachner M, Isom LL. Tenascin-R is a functional modulator of sodium channel beta subunits. J Biol Chem. 1999;274:26511–26517. doi: 10.1074/jbc.274.37.26511. [DOI] [PubMed] [Google Scholar]
- 50.Zhang K, Filbin M. Formation of a disulfide bond in the immunoglobulin domain of the myelin P0 protein is essential for its adhesion. J Neurochem. 1994;63:367–370. doi: 10.1046/j.1471-4159.1994.63010367.x. [DOI] [PubMed] [Google Scholar]