

Abstract
In response to injury and inflammation of the CNS, brain cells including microglia and astrocytes secrete tumor necrosis factor-α (TNF). This pro-inflammatory cytokine has been implicated in both neuronal cell death and survival. We now provide evidence that TNF affects the formation of neurites. Neurons cultured on astrocytic glial cells exhibited reduced outgrowth and branching of neurites after addition of recombinant TNF or prestimulation of glial cells to secrete TNF. This effect was absent in neurons of TNF receptor-deficient mice cultured on prestimulated glia of wild-type mice and was reverted by blocking TNF with soluble TNF receptor IgG fusion protein. TNF activated in neurons the small GTPase RhoA. By inactivating Rho with C3 transferase, the inhibitory effect of TNF on neurite outgrowth and branching was abolished. These results suggest that glia-derived TNF, as part of an injury or inflammatory process, can inhibit neurite elongation and branching during development and regeneration.
Keywords: neurite, morphogenesis, Rho, GTPases, TNF, cytokine, TNF receptor, glia
During development and regeneration, vertebrate neurons undergo profound morphological changes that include substantial cytoskeletal restructuring. These changes are induced by contact with other cells and diffusible extracellular signals (Hu and Reichardt, 1999). Contact-dependent modulation involves membrane-bound molecules such as cadherins, integrins, and cell adhesion molecules of the immunoglobulin superfamily. Diffusible signal molecules include neurotransmitters, neurotrophic factors, and guidance molecules such as semaphorins (Chisholm and Tessier-Lavigne, 1999). In particular, neurotrophins have remarkable effects on neurite outgrowth and arborization (Bibel and Barde, 2000). Recently, a direct link between Rho GTPases and neurotrophin signaling was revealed in experiments demonstrating an interaction between RhoA and the p75 neurotrophin receptor (p75NTR). Members of the Rho family of small GTPases, including Rac, Cdc42, and Rho, have been shown to be crucial intracellular regulators of the cytoskeleton in neurons (Luo et al., 1997; Threadgill et al., 1997; Luo, 2000). In transfected cells, p75NTR was demonstrated to constitutively activate RhoA, whereas neurotrophin binding rapidly reduced the activation of the GTPase (Yamashita et al., 1999). The downregulation of RhoA mediated by p75NTRwas proposed to be part of a mechanism accelerating neurotrophin-induced axonal elongation (Yamashita et al., 1999; Tucker et al., 2001). The tumor necrosis factor receptor (TNFR) and the p75NTR receptors are members of the same receptor family. Interestingly, previous experiments have implicated these receptors and their ligand TNF in modulating the cytoskeleton in non-neuronal cells. TNF induced stress fibers and focal adhesion formation mediated by the GTPase Rho in endothelial cells (Wojciak-Stothard et al., 1998), epithelial cells (Koukouritaki et al., 1999), and fibroblasts (Puls et al., 1999).
In the CNS, in contrast to neurotrophins that are mainly produced by neurons, TNF is preferentially produced by activated glial cells, microglia, and astrocytes during brain injury or inflammatory processes (Hopkins and Rothwell, 1995). For instance, TNF protein has been localized in glial cells after experimental autoimmune encephalomyelitis (Renno et al., 1995). TNF protein was also detected in neurons by immunohistochemistry after mechanical (Tchelingerian et al., 1993) or ischemic brain injury (Bruce et al., 1996). Furthermore, TNF gene transcription and protein production have been observed during development in total brain tissue extracts (Munoz-Fernandez and Fresno, 1998; Zhao and Schwartz, 1998). In culture, astrocytes secrete TNF at low level, but cytokine secretion is strongly increased after treatment with inflammatory stimuli such as IFN-γ combined with IL-1β (Chung and Benveniste, 1990). TNF is produced as a propeptide, which integrates into the cell membrane as a stable homotrimer or can be clipped off at the transmembrane domain by the metalloproteinase TNF-α-converting-enzyme [TACE, ADAM-17 (Blobel, 1997)]. The biological activity of TNF is mediated via either the TNFRI (p55 TNF receptor, CD120a) or the TNFRII (p75 TNF receptor, CD120b), which have overlapping signaling capabilities (Rothe et al., 1994; Hsu et al., 1996).
To elucidate the significance of TNF on neuronal morphology, we cocultured hippocampal neurons at low density with glial cells. After treatment with recombinant TNF or stimulation of glial cells to elicit TNF, we observed a marked reduction in length and branching of neurites. Furthermore, we found that RhoA becomes activated in cultures of enriched neurons by TNF and provide evidence that this GTPase is causally involved in the inhibitory effect of recombinant or secreted TNF.
MATERIALS AND METHODS
Mice. C57BL/6 mice were obtained from the animal house facilities of the Max-Planck Institutes of Neurobiology and Biochemistry (Martinsried, Germany). C57BL/6 TNFRI- plus TNFRII-deficient mice were obtained by crossing C57BL/6 TNFRII-deficient mice (back crossed for 10 generations with C57BL/6; The Jackson Laboratory, Bar Harbor, ME) with TNFRI-deficient mice (backcrossed nine generations with C57BL/6; Klaus Pfeffer, Technical University, Munich). Homozygous mice deficient for both TNF receptors (TNFRI plus TNFRII) were confirmed by established PCR protocols (K. Pfeffer, Technical University, Munich, Germany/The Jackson Laboratory, Bar Harbor, ME) and then maintained as homozygotes in our animal house facility.
Murine hippocampal neurons. Primary hippocampal neuronal cell cultures were prepared as described previously (Neumann et al., 1995). Briefly, hippocampi were isolated from whole brains of embryonic day 16 mice, and the meninges were removed. The trimmed tissue was dissociated by trituration through a fire-polished Pasteur pipette. Cells (5 × 103/ml) were plated on astrocyte-enriched glia culture or into dishes that had been pretreated with poly-l-ornithine (0.5 mg/ml; Sigma, St. Louis, MO) in 0.15 m boric acid. Cells were cultured in chemically defined medium containing basal medium Eagle (BME; Invitrogen, Gaithersburg, MD) with B27 supplement [2% (v/v), Invitrogen] and glucose [1% (v/v) 45%; Sigma]. Cells were treated with TNF (murine recombinant TNF, 10 ng/ml; Roche Products, Hertforshire, UK), TNF receptor IgG fusion protein (recombinant human TNF receptor p55IgG1 fusion protein, 20 μg/ml; gift from Dr. Werner Lesslauer, Roche), or human control IgG (20 μg/ml; Dianova, Hamburg) as indicated in Results.
Murine astrocyte-enriched glial cells. Hippocampi of embryonic day 16 C57BL/6 mice were isolated and dissociated into single-cell suspensions as described for neuronal hippocampal preparations. Cells were plated in 50 ml tissue culture flasks that had been pretreated with poly-l-lysine (5 μg/ml; Sigma). Cells were cultured in serum containing medium with MEM-d-valine (Invitrogen), 10% heat-inactivated FCS (Pan System, Wuerzburg, Germany), and 1%l-glutamine. Microglial cells were removed by several rounds of shaking on a rotary shaker. Astrocyte-enriched glial cells were cultured for 10–20 d and then plated in BME with B27 supplement [2% (v/v), Invitrogen] and glucose [1% (v/v), 45%; Sigma] at a density of 2 × 104/ml before experimentation. In total, 94% (±3% SD) of cells were astrocytes as determined by immunolabeling with rabbit antibodies directed against GFAP (10 μg/ml; Dako, Glostrup, Denmark). Murine recombinant IFN-γ (100 U/ml; HyCult Biotechnology, Amsterdam, The Netherlands) and murine recombinant Il-1β (10 ng/ml; Biosourceand Roche Molecular Biochemicals) were added to the cells for 24 hr as indicated in Results.
RT-PCR for TNFRI, TNFRII, and TNF. Single-cell RT-PCR was performed as described previously (Neumann et al., 1995). Cytoplasm was sampled from individual hippocampal neurons with a micropipette after whole-cell recording configuration of the patch-clamp technique. Samples of the extracellular fluid and pipette solution served as negative controls. The content of the micropipette was transferred into a test tube and directly used for reverse transcription. Reverse transcription of RNA was performed with dithiothreitol (10 mm; Invitrogen), ribonuclease inhibitor (20 U;Promega, Madison, WI), hexamer random primer (1 μl; Roche Molecular Biochemicals), the four deoxyribonucleotide triphosphates (0.5 mm; Amersham Biosciences), and Moloney murine leukemia virus transcriptase (100 U; Invitrogen) for 1 hr at 42°C. As a control, cytoplasmic mRNA of myelin oligodendrocyte glycoprotein peptide-specific CD4-positive T lymphoblasts was collected through the micropipette and reverse transcribed as described above. Cytoplasmic RNA of astrocytes was collected with RNAzol B (Wak-Chemie, Bad Soden, Germany) and reverse transcribed as described above. Oligonucleotide sequences were selected with the program Primer3 (Whitehead Institute, MIT, Cambridge, MA). Forward and reverse primer pairs were chosen from two different exons and were, respectively, 5′-CCCGGTGGAGGC- CCGAAG-3′ and 5′-GCTGGGGAGGGGGCTGGA-3′ for TNFRI; 5′-AGTGCGGCCCTGGCTTCGG-3′ and 5′-GTTGGGGACTCGGGC- GCAC-3′ for TNFRII; 5′-GGGGTGATCGGTCCCCAAAGG-3′ and 5′-CGGGGCAGCCTTGTCCCTTG-3′ for TNF; and 5′-TCCGCCCC- TTCTGCCGATG-3′ and 5′-CACGGAAGGCCATGCCAGTGA-3′ for glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
PCR reaction was performed in a final volume of 50 μl containing 1 μl of the transcribed cDNA probe, the four deoxyribonucleotide triphosphates (0.2 mm each; Amersham Biosciences), 2.5 U Ampli Taq polymerase (Perkin-Elmer, Roche Molecular Systems), and 1× PCR buffer (Perkin-Elmer, Roche Molecular Systems) covered with two drops of mineral oil (Sigma). PCR amplification was performed on a programmed thermocycler (MultiCycler, MJ Research Inc.). After denaturing of the cDNA at 95°C for 3 min, denatured primers (100 pmol each) were added at 80°C. PCR was performed for 50 cycles (93°C for 1 min; 60°C for 1 min; 72°C for 1 min) and followed by one final interval at 72°C for 5 min. Ten microliters of the PCR reaction were loaded in parallel with the molecular weight marker (ΦX 174, HaeIII digested; Amersham Biosciences) on a 1.7% Agarose gel containing ethidium bromide. The enzyme GAPDH served as a control for the quality and quantity of cDNA. Individual T lymphoblasts analyzed as a positive control for the single-cell RT-PCR approach showed gene transcripts of both TNF receptors. PCR products derived from individual neurons were purified from ethidium bromide-stained Agarose gels using a gel extraction method (Quiagen, Hilden, Germany) and directly sequenced (MediGenomix, Martinsried, Germany).
Immunofluorescence labeling of neurons for TNFRI and TNFRII. Hippocampal neurons cultured without glial cells for 1 week were washed with PBS, fixed in 4% paraformaldehyde, incubated with rat monoclonal antibodies directed against TNFRI (2 μg/ml; HM-104; Biermann, Bad Nauheim, Germany) and TNFRII (2 μg/ml; HM-102; Biermann) followed by secondary fluorochrome Cy3-conjugated goat antibody directed against rat IgG (10 μg/ml; Dianova). Subsequently, after extensive washing, cells were incubated with the neuron-specific mouse monoclonal antibody specific for β-tubulin III (1 μg/ml; Sigma) and secondary dichlorotriazinyl aminofluorescein-conjugated goat antibody directed against mouse IgG (10 μg/ml; Dianova). Optical sections along the z-axis were scanned with a confocal laser-scanning microscope (Leica) equipped with a 63× oil objective. Baseline labeling was determined with rat control antibodies and secondary fluorochrome Cy3-conjugated goat antibodies directed against rat IgG.
Precipitation and Western blot analysis of GTP–RhoA. Primary hippocampal neurons cultured without glial cells were lysed in 50 mm Tris, pH 7.2, 1% Triton X-100, 150 mm NaCl, 10 mm MgCl2, with leupeptin (10 μg/ml), aprotinin (10 μg/ml), and PMSF (1 mm). Sepharose beads conjugated with a GST-tagged Rho binding domain of Rhotekin were produced as described (Ren et al., 1999). Cell lysates were clarified by centrifugation at 13,000 × g at 4°C for 5 min, and the supernatants were incubated with ∼20 μg of Rhotekin conjugated to beads at 4°C for 45 min (Ren et al., 1999). The beads were washed four times with washing buffer [50 mm Tris, pH 7.2, 1% Triton X-100, 150 mm NaCl, 10 mm MgCl2, leupeptin (10 μg/ml), aprotinin (10 μg/ml), and PMSF (1 mm)]. Bound Rho proteins were detected by gel electrophoresis followed by Western blotting using a monoclonal antibody directed against and specifically recognizing RhoA (2bc4, 0.1 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA) and secondary peroxidase-conjugated antibodies directed against mouse IgG and the ECL detection system (Amersham Biosciences).
Neurite morphometry of β-tubulin III-immunolabeled neurons. Primary hippocampal neurons were cultured at a low density on glial cells to allow morphological analysis of single neurons. The cells were fixed in 4% paraformaldehyde and incubated with primary mouse monoclonal antibody directed against β-tubulin III (1 μg/ml, Sigma) and secondary fluorochrome Cy3-conjugated goat antibody directed against mouse IgG (10 μg/ml; Dianova). Images of β-tubulin III-labeled neurons were captured with a 20× or 40× objective with a laser scanning microscope (Leica). Neurites of scanned cells were traced using Optimas software. Data of the morphometry of individual neurons were collected and scored for total neurite length per cell, axonal length per cell, number of primary processes per cell, and branch points per cell. Neuronal survival was analyzed by determination of the density of β-tubulin III-positive cells that survived per each experimental group. Primary processes were defined as neurites originating at the neuronal somata. Axons were defined as the longest primary process per cell. Experiments were repeated at least three times, and data are presented as mean ± SEM of independent experiments.
Trituration of neurons together with C3 transferase. The C3 exoenzyme from Clostridium botulinum (C3 transferase) was produced in bacteria as glutathione S-transferase (GST) fusion protein and purified as described (Nobes and Hall, 1995). Trituration together with C3 transferase was performed as described previously (Borasio et al., 1989; Jin and Strittmatter, 1997). Briefly, primary hippocampal neurons were suspended in 25 mm Tris-HCl, pH 7.5, 150 mmNaCl, 5 mm MgCl2, with C3 transferase at 0.1 mg/ml directly after isolation from the brain tissue, and passed 50 times through a 200 μl pipette tip. After trituration, neurons were plated on the glial cells as described above.
Flow cytometry for membrane-bound TNF. The astrocyte-enriched glial cells were collected from the culture by short trypsinization (trypsin-EDTA solution; Invitrogen). Cells were washed with PBS, and unspecific binding sites were blocked with 2% bovine serum albumin (Sigma). Then, cells were incubated with polyclonal rabbit anti-TNF antibody (1:100, Genzyme) followed by dichlorotriazinyl aminofluorescein-conjugated goat anti-rabbit antibody (10 μg/ml, Dianova). Polyclonal rabbit IgG (Dianova) followed by dichlorotriazinyl aminofluorescein-conjugated secondary antibody was used as a control. Cells were incubated with 0.5 μg/ml propidium iodide to distinguish living from dead cells and analyzed with a flow cytometer (FACScan, Becton Dickinson).
Measurement of released TNF. To measure TNF secreted by astrocyte-enriched glia, cells were kept in serum-free medium for 6 hr. The supernatant was collected and assayed via ELISA according to the instructions of the manufacturer (OptEIA mouse TNF mono/mono, PharMingen).
RESULTS
TNF receptor expression in primary hippocampal neurons
Primary neurons were cultured from hippocampal tissue derived from embryonic mice. TNF receptor gene transcripts were analyzed in primary neuronal cultures by single-cell RT-PCR of individual neurons. Neurons were first unequivocally identified by whole-cell patch-clamp electrophysiology, and then the cell cytoplasm was collected with the micropipette. Gene transcripts for TNFRI as well as TNFRII were detected in the majority of single neurons (Fig.1) and confirmed by sequencing. Specifically, 21 of 25 analyzed neurons were positive for TNFRI and 19 of 25 were positive for TNFRII, whereas in 18 of 25 neurons both TNF receptor types were detected.
Fig. 1.

TNF receptor gene transcription in hippocampal neurons. Single-cell RT-PCR of cultured primary hippocampal neurons. Gene transcripts for TNFRI and TNFRII were detected in the majority of neurons. Co-amplification of gene transcripts for GAPDH served as a control. M, Molecular weight marker ΦX174/HaeIII; C, negative PCR control;N1–N9, samples of single neurons.
Protein expression of TNF receptors in primary hippocampal neurons was analyzed by immunohistochemistry. Neurons were immunolabeled with rat monoclonal antibodies directed against mouse TNFRI or TNFRII and subsequently double labeled with antibodies directed against the neuronal cytoskeleton protein β-tubulin III. Background labeling was determined with control rat immunoglobulin. Intracellular granular staining for TNFRI and TNFRII was obtained in the neuronal somata and processes of neurons (Fig. 2). In total, 81.8% (±7.9% SEM) of hippocampal neurons identified by β-tubulin III immunohistochemistry showed TNFRI labeling, and 77.6% (±5.7% SEM) showed TNFRII labeling. Immunolabeling for TNFRI as well as TNFRII gave only background staining in hippocampal neurons derived from TNFRI- plus TNFRII-deficient mice.
Fig. 2.

Immunodetection of TNFRI and TNFRII molecules in primary hippocampal neurons. Neurons were immunolabeled with rat monoclonal antibodies directed against TNFRI or TNFRII and rat control antibodies. Neurons were subsequently double labeled with antibody directed against β-tubulin III. Scale bar, 10 μm.
Reduced neurite outgrowth and branching of primary hippocampal neurons in response to TNF
Low-density cultures of primary hippocampal neurons were performed by culturing neurons together with glial cells. This low-density culture system allows detailed morphometric analysis of individual neurons. Astrocyte-enriched glial cells were obtained from primary hippocampal brain cells after removal of microglia. Hippocampal neurons derived from E16 embryonic C57BL/6 or TNF receptor-deficient C57BL/6 mice were cultured on these astrocyte-enriched glial cells in chemically defined medium at low density, resulting in individual neurons without neuron–neuron contacts. Within a few hours, the neurons showed outgrowth of neurites up to 100 μm on the glial cell layer.
To analyze the effects of TNF on neurons, recombinant mouse TNF was added a few hours after plating to the neurons on the glial cell layer. After 16 hr, cells were fixed and immunolabeled with β-tubulin III. Neurites were traced and analyzed with an image analysis system. Neurite morphology of TNF-treated cell cultures was completely different from untreated cultures (Fig.3), whereas neuronal survival was not altered by the TNF treatment (Table 1). Neurons treated with TNF showed reduced branching of their processes, and most neurites were almost equal in length without clear distinction of an axon-like process. The total neurite length per neuron and the axonal length, defined as the length of the longest neurite per neuron, were determined. Treatment with TNF (10 ng/ml, 16 hr) decreased the total neurite length per cell from 453 μm (±18.9 μm SEM) to 230 μm (±27.6 μm SEM) as well as the axonal length from 148 μm (±24 μm SEM) to 70 μm (±15.6 μm SEM) (Fig.4). No change in the number of primary processes originating from the neuronal somata was detectable after treatment with TNF. However, there was a reduction in the number of branch points per cell in response to TNF. Untreated neurons showed on average 5.5 (±0.46 SEM) branch points per cell, whereas neurons treated with TNF exhibited 1.8 (±0.31 SEM) branch points per cell (Fig. 4). TNF affected neurite length and morphology via neuronal TNF receptors. Hippocampal neurons derived from TNFRI- plus TNFRII-deficient mice were cultured on glial cells derived from C57BL/6 wild-type mice. In this case the neurons did not show alterations in total neurite length, axonal length, and number of branch points after treatment with TNF (Fig. 4). Furthermore, we cultured hippocampal neurons derived from C57BL/6 wild-type mice on glial cells derived from TNFRI- plus TNFRII-deficient mice. TNF again reduced neurite outgrowth and branching of neurons (Fig. 4), indicating that the effect of TNF on neurite morphology was directly mediated via the TNF receptors of the neurons.
Fig. 3.

Computer-assisted tracing of β-tubulin III-labeled neurites. Primary hippocampal neurons were cultured for 16 hr on astrocyte-enriched glial cells. Neurons were immunolabeled with antibodies against β-tubulin III, and neurites of captured images were traced with image analysis software. Treatment with 10 ng/ml TNF reduced neurite outgrowth and branching of neurites. Scale bar, 10 μm.
Table 1.
Neuronal survival in relation to untreated cultures
| Untreated cultures | TNF-treated cultures | |
|---|---|---|
| Wild type | 1 ± 0 | 0.84 ± 0.15 |
| TNFR −/− neurons | 1 ± 0 | 1.07 ± 0.17 |
| TNFR −/− glia | 1 ± 0 | 1.11 ± 0.14 |
Fig. 4.

Morphometric analyses of primary hippocampal neurons cultured for 16 hr on astrocyte-enriched glia. Neurons and glial cells were derived from E16 C57BL/6 mice (Wild-type) or C57BL/6 TNFRI- plus TNFRII-deficient mice (Neurons TNFR −/−, Glia TNFR −/−). The cultures were either untreated (gray columns) or treated for 16 hr with 10 ng/ml TNF (black columns). TNF reduced the total neurite length, axonal length, and number of branch points of neurons derived from C57BL/6 mice, but not TNFR −/− mice. The reduction in neurite outgrowth and branching was independent of the TNF receptor expression of the glia. Data are presented as mean ± SEM of three independent experiments.
TNF production by cytokine-stimulated glial cells
In contrast to normal glial cells, reactive glial cells, in particular activated microglia and astrocytes of the injured brain, have been shown to produce TNF. In culture, gene transcription of TNF in astrocytes can be induced by combined treatment with the pro-inflammatory cytokines IL-1β and IFN-γ. Therefore, we pretreated our astrocyte-enriched glial cells with IFN-γ (100 U/ml) and ILl-1β (10 ng/ml) for 24 hr. After pretreatment, gene transcripts for TNF were detected via RT-PCR (Fig.5). In contrast, no TNF gene transcripts were amplified from untreated glial cell culture. Treatment with IL-1β or IFN-γ alone resulted in low but detectable gene transcription of TNF.
Fig. 5.
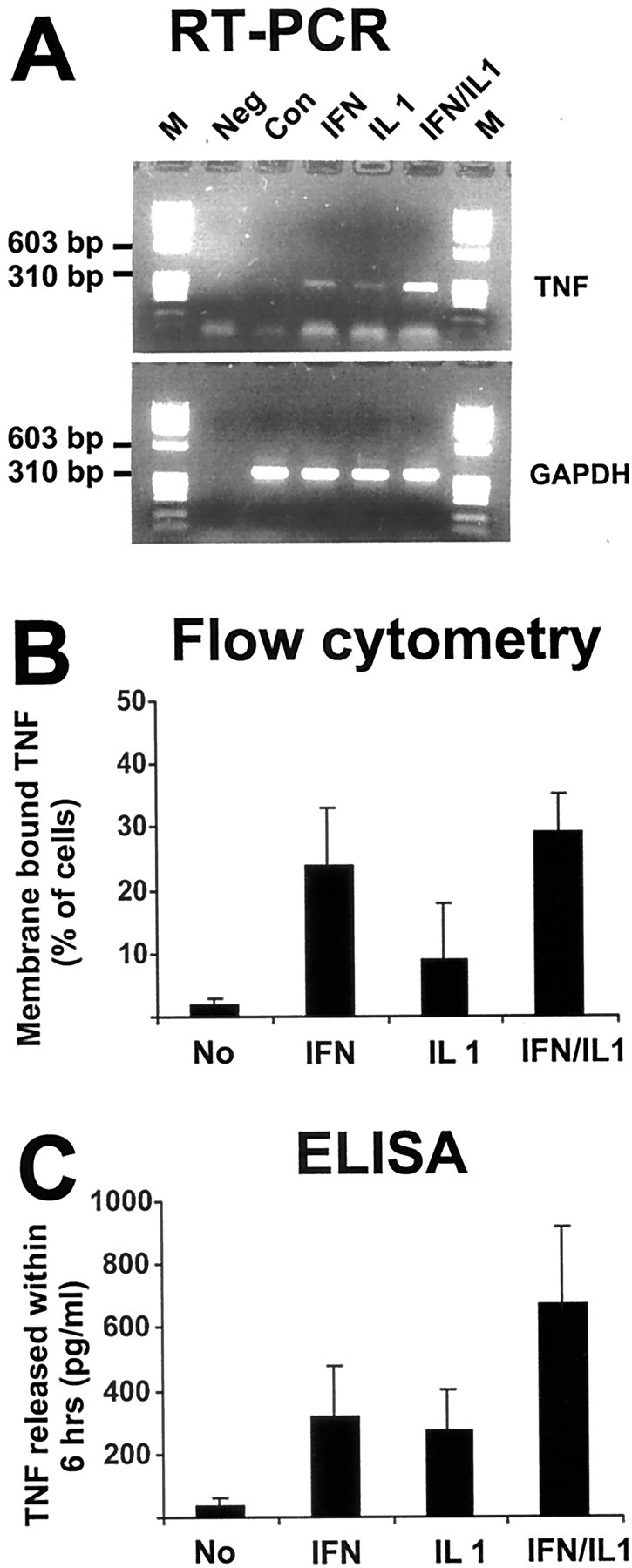
Detection of TNF gene transcripts, membrane-bound TNF, and TNF released in response to cytokine stimulation of glia. Astrocyte-enriched glial cells were untreated (Con) or treated with 100 U/ml IFN-γ (IFN), 10 ng/ml IL-1β (IL1), or IFN-γ plus IL-1β (IFN/IL1) for 24 hr. A, Analysis of TNF gene transcripts by RT-PCR. Glia showed low amounts of TNF gene transcripts after treatment with IFN-γ or IL-1β alone, whereas high amounts of TNF gene transcripts were detected after combined treatment with IFN-γ and ILl-1β. Coamplification of gene transcripts for GAPDH served as a control. M, Molecular weight marker ΦX174/HaeIII; Neg, negative PCR control. B, Membrane-bound TNF was detected on astrocyte-enriched glial cells with fluorescence-labeled rabbit antibody directed against TNF and flow cytometry. Data are presented as mean ± SEM of three independent experiments. C, Soluble TNF released within 6 hr was detected in the supernatant of cytokine-stimulated glia. Data are presented as mean ± SEM of three independent experiments.
To confirm TNF protein synthesis and secretion by astrocyte-enriched glial cells, we analyzed membrane-bound TNF by flow cytometry. Untreated and cytokine-stimulated glial cells were incubated with antibodies directed against TNF. Membrane-bound TNF was detected in 29% (±5.4% SEM) of glial cells pretreated for 24 hr with IFN-γ plus IL-1β (Fig. 5). Membrane-bound TNF was already seen after treatment with IFN-γ alone (24 ± 9%).
The amount of TNF secreted within 6 hr by glial cells was determined by ELISA. Treatment with IFN-γ plus IL-1β stimulated glial cells to release TNF up to 1000 pg/ml within 6 hr (Fig. 5). Thus, the combined treatment with IFN-γ and IL-1β resulted in significant gene transcription, membrane localization, and secretion of TNF.
Inhibition of outgrowth and branching of neurites by TNF from reactive glia
Primary hippocampal neurons derived from E16 embryonic C57BL/6 or TNF receptor-deficient C57BL/6 mice were cultured on astrocyte-enriched glial cells derived from C57BL/6 wild-type mice now pretreated with IFN-γ and IL-1β to produce sufficient amounts of TNF. After 16 hr, these neurons showed decreased total neurite length, decreased axonal length, and decreased number of branch points (Fig.6) compared with neurons cultured on unstimulated glial cells (Fig. 4). To analyze whether this reduced outgrowth and branching of neurons on stimulated glia were indeed induced by TNF, we neutralized the endogenously secreted TNF in the culture.
Fig. 6.

Morphometric analyses of primary hippocampal neurons cultured for 16 hr on reactive glia that has been pretreated for 24 hr with IFN-γ and IL-1β to induce TNF production. Neurons were derived from E16 C57BL/6 mice (Neurons wild-type) or C57BL/6 TNFRI- plus TNFRII-deficient mice (Neurons TNFR −/−). The cultures were treated for 16 hr with control IgG (gray columns) or with TNF receptor IgG fusion protein (TNFR-IgG, black columns). Blockade of TNF with TNFR-IgG antagonized the reduced total neurite length, axonal length, and number of branch points of neurons derived from wild-type, but not TNFR −/− mice. Data are presented as mean ± SEM of three independent experiments.
The effects of both membrane-bound as well as soluble TNF can be blocked by a TNF receptor IgG fusion protein. Therefore, we treated the cultures with TNF receptor IgG fusion protein and control IgG. Reduced total neurite length, axonal length, and number of branch points were completely antagonized by blockade of TNF activity with TNF receptor IgG fusion protein (Fig. 6). Neurons of cultures treated with TNF receptor IgG fusion protein for 16 hr showed total neurite outgrowth per cell of 502 μm (±18.9 μm SEM) and axonal length of 180 μm (±28.7 μm SEM), whereas neurons of cultures treated with control IgG showed total neurite length per cell of 176 μm (±9.8 μm SEM) and axonal length of 55.6 μm (±4.7 μm SEM) (Fig. 6). No change in neuronal survival was detected after treatment with the TNF receptor IgG fusion protein (Table 2). Hippocampal neurons derived from TNFRI- plus TNFRII-deficient mice did not show substantial reduction in total neurite length, axonal length, and number of branch points after culture on TNF-producing astrocytes (Fig.6).
Table 2.
Neuronal survival in relation to control IgG-treated cultures with wild-type neurons
| Glial cells not pretreated | Glial cells pretreated with IFN-γ and IL-1β | |||
|---|---|---|---|---|
| Control IgG | TNFR– IgG | Control IgG | TNFR– IgG | |
| Wild-type neurons | 1 ± 0 | 1 ± 0.15 | 0.94 ± 0.09 | 0.83 ± 0.11 |
| TNFR −/− neurons | 0.79 ± 0.12 | 0.95 ± 0.07 | 0.99 ± 0.16 | 0.84 ± 0.05 |
In the next step we added the TNF receptor IgG fusion protein to hippocampal neurons cultured on unstimulated glial cells not secreting detectable amounts of TNF (Fig. 7). No change in neurite outgrowth and branching was detected after treatment of unstimulated cultures with the TNF receptor IgG fusion protein, indicating that unstimulated glia and the low-density neurons do not secrete sufficient amounts of TNF to modulate neurite outgrowth and branching.
Fig. 7.

Morphometric analyses of primary hippocampal neurons cultured for 16 hr on unstimulated glia. Neurons were derived from E16 C57BL/6 mice (Neurons wild-type) or C57BL/6 TNFRI- plus TNFRII-deficient mice (Neurons TNFR −/−). The cultures were treated for 16 hr with control IgG (gray columns) or with TNF receptor IgG fusion protein (TNFR-IgG, black columns). Blockade of TNF with TNFR-IgG did not change the total neurite length, axonal length, and number of branch points of neurons cultured on unstimulated glial cells. Data are presented as mean ± SEM of three independent experiments.
RhoA activation in hippocampal neurons after TNF treatment
Hippocampal neurons were cultured on poly-l-ornithine-coated dishes without glial cells and were treated with recombinant murine TNF (10 ng/ml). The cells were lysed at different time points after treatment with TNF. The active form of RhoA was precipitated with Sepharose beads conjugated with a GST-tagged binding domain of Rhotekin that specifically binds to GTP–RhoA. The amount of precipitated active RhoA was determined by Western blotting. RhoA was activated by treatment with TNF within 1 hr. The activity of the GTPase gradually increased after treatment with TNF up to 16 hr (Fig. 8).
Fig. 8.

RhoA activation in primary hippocampal neurons after TNF treatment. Neurons were treated with 10 ng/ml TNF for different time periods (15 min, and 1, 4, and 16 hr), and GTP–RhoA was detected by Western blotting after precipitation with Rhotekin. TNF activated RhoA within 1 hr, showing increasing levels up to 16 hr. Activation of RhoA with 10% serum (10 min) served as a positive control.
Reduced neurite outgrowth and branching in response to TNF is mediated by Rho
Next, we analyzed whether the GTPase Rho was involved in the effect of TNF on neurite length and branching of hippocampal neurons cultured on glia. Rho can be inhibited efficiently by trituration of the cells in the presence of C3 transferase, a procedure allowing cell membrane passage of C3 transferase under special buffer conditions (Borasio et al., 1989; Jin and Strittmatter, 1997). The C3 transferase from C. botulinum specifically ADP ribosylates Rho and thus inactivates the protein. Inhibition of neuronal Rho activity by trituration of neurons together with C3 transferase inhibited the effect of TNF on total neurite length, axonal length, and number of branch points (Fig. 9). Specifically, TNF-treated neurons triturated together with the C3 transferase showed total neurite length of 426 μm (±75.2 μm SEM) and axonal length of 172 μm (±47.6 μm SEM) compared with TNF-treated neurons triturated with buffer alone showing total neurite length of 175 μm (±6.1 μm SEM) and axonal length of 59 μm (±12.3 μm SEM) (Fig. 9).
Fig. 9.

Morphometric analyses of primary hippocampal neurons cultured for 16 hr on astrocytes. Neurons were derived from E16 C57BL/6 mice and either triturated together with C3 transferase (Neurons C3 treated) or with buffer alone (Neurons buffer). The cultures were untreated (gray columns) or treated for 16 hr with 10 ng/ml TNF (black columns). TNF-mediated reduction in total neurite length, axonal length, and number of branch points was antagonized in C3-treated but not control buffer-treated neurons. Data are presented as mean ± SEM of three independent experiments.
Subsequently, the effects of C3 transferase were analyzed on neurons cultured on astrocyte-enriched glial cells and pretreated with IFN-γ and IL-1β (Fig. 10, Table3). Again, inhibition of neuronal Rho activity by trituration of neurons together with C3 transferase prevented the effect of TNF-producing astrocytes on neurite length and morphology (Fig.10).
Fig. 10.

Morphometric analyses of primary hippocampal neurons cultured for 16 hr on glia that has been pretreated for 24 hr with IFN-γ and IL-1β to induce TNF production. Neurons were derived from E16 C57BL/6 mice and either triturated together with C3 transferase (Neurons C3 treated) or with buffer alone (Neurons buffer). The cultures were treated for 16 hr with control IgG (gray columns) or with TNF receptor IgG fusion protein (TNFR-IgG, black columns). Trituration of neurons together with C3 transferase antagonized the reduced total neurite length, axonal length, and number of branch points. Data are presented as mean ± SEM of three independent experiments.
Table 3.
Neuronal survival in relation to untreated cultures with buffer-treated neurons
| Glial cells not pretreated | Glial cells pretreated with IFN-γ and IL-1β | |||
|---|---|---|---|---|
| Untreated | TNF treated | Control IgG | TNFR–IgG | |
| Buffer-treated neurons | 1 ± 0 | 1.2 ± 0.13 | 1.07 ± 0.28 | 1.04 ± 0.05 |
| C3-treated neurons | 1.1 ± 0.1 | 1.03 ± 0.22 | 0.94 ± 0.09 | 1.08 ± 0.06 |
DISCUSSION
This study demonstrates that TNF profoundly modulates neurite branching and extension in hippocampal neurons in vitro via a mechanism that requires the small GTPase Rho. Recently, it has become clear that the modulation of the activity of Rho GTPases is an important link between signal transduction by membrane proteins and the actin cytoskeleton (Hall, 1998). Activated Rho family GTPases stimulate specific kinases (p65PAK or ROCK) to phosphorylate one of the two isoforms of LIM kinase (LIMk1, LIMk2; Sumi et al., 1999), and the actin polymerization is then regulated through phosphorylation of cofilin and actin depolymerizing factor by the LIM kinases (Arber et al., 1998). In neurons, Rho GTPases act as important check points in the regulation of neuronal morphogenesis, axonal growth and guidance, dendrite elaboration and synapse formation (Luo et al., 1997; Luo, 2000). For instance, inactivation of Rho with C3 transferase stimulates neurite growth of PC12 cells (Lehmann et al., 1999), and Toxin B blocks axonal outgrowth of primary hippocampal neurons (Bradke and Dotti, 1999). These in vitro findings are of relevance in an in vivo context, because crushed optic nerves treated with C3 transferase send numerous axons through the lesion site and even elongate in the distal white matter (Lehmann et al., 1999).
Recently, RhoA has been identified in neurons as a signaling node linking the neurotrophin receptor p75NTR, a member of the TNF receptor superfamily, to the cytoskeleton (Yamashita et al., 1999). The experiments established that p75NTR acts as a constitutive RhoA activator, whereas ligand binding causes rapid loss of RhoA activity (Yamashita et al., 1999). We now show that other members of the TNF receptor superfamily, the classical TNF receptors, stimulate RhoA in primary hippocampal neurons. Interestingly, the direction and kinetics of the effect are opposite between NGF and TNF. Transfection experiments revealed that unligated p75NTRconstitutively activated RhoA, whereas NGF binding to the receptor inactivated RhoA activity within 3 min (Yamashita et al., 1999). This effect was stable for several hours of sustained NGF stimulation. In contrast, TNF leads to a delayed activation of RhoA reaching higher levels of RhoA–GTP within 4–16 hr. No further increase was observed after longer treatment periods with TNF (our unpublished observation). The p75NTR seems to act by directly binding the GTPase RhoA (Yamashita et al., 1999), which might explain the rapid effect of NGF on RhoA activity. In contrast, RhoA activation in neurons by TNF might be indirect, as in a fibroblast cell line, where TNF activated the GTPases in a highly ordered manner: first Rac, followed by Cdc42, and finally Rho (Puls et al., 1999).
TNF reduced the neurite outgrowth and branching of the hippocampal neurons. This effect was mediated by Rho and abolished after blocking Rho with C3 transferase. Furthermore, two lines of evidence suggest that TNF acts directly on neuronal TNF receptors to modulate neurite morphology and not indirectly via glial cells. First, neurons derived from TNFRI- plus TNFRII-deficient mice and cultured on normal astrocytes showed almost no change in neurite morphogenesis. Second, the inhibitory effect of TNF on neurite outgrowth and branching was still detected in cultures of hippocampal neurons on glia derived from TNFRI- plus TNFRII-deficient mice.
Glia stimulated to secrete TNF reduced neurite outgrowth and branching of cocultured neurons. Although blockade of TNF activity with the receptor fusion protein increased outgrowth and branching of neurons cultured on stimulated glial cells, we did not observe a significant change in neurite morphogenesis by this treatment in nonstimulated glial cells. Thus, nonreactive gial cells and neurons do not produce sufficient amounts of TNF to inhibit neurite outgrowth and branching of neurons. However, we cannot exclude the possibility that our neuronal density was too low to detect such possible effects of neurons.
Gene transcripts and protein expression of both TNF receptors (TNFRI and TNFRII) were detected in cultured hippocampal neurons by single-cell RT-PCR and immunohistochemistry, respectively. In situ, neurons of the hippocampus of adult rats showed no or very low gene transcription of TNFRII under basal conditions, whereas TNFRI was induced and TNFRII upregulated by treatment with inflammatory stimuli or after ischemia (Botchkina et al., 1997; Nadeau and Rivest, 1999). Furthermore, gene transcription and protein expression of TNF and TACE (ADAM-17) have been observed in the CNS during embryonic and different postnatal developmental stages (Munoz-Fernandez and Fresno, 1998; Dziegielewska et al., 2000; Karkkainen et al., 2000). In particular, TNF was localized by immunohistochemistry in cells with neuronal morphology during early embryonic development of the neocortex (Dziegielewska et al., 2000). However, it has not been shown that TNF detected in the CNS during development is biologically active and whether the level is comparable to inflammatory processes or injury of the CNS.
No gross anatomical CNS abnormalities of TNFRI- and TNFRII-deficient mice under normal conditions were observed (our unpublished observation). However, a recent report suggested that TNF might be involved in hippocampal synaptic plasticity. The induction of long-term potentiation in region CA1 after stimulation of Schaffer collateral axons was impaired in mice deficient for both TNF receptors (Albensi and Mattson, 2000).
TNF and its receptors show polymorphism in humans, and recent reports indicate that this polymorphism might be associated with a noninflammatory brain disorder. In particular, a TNF gene polymorphism [TNF2(A) allele] has been associated with schizophrenia (Wassink et al., 2000), and patients homozygous for a TNFRII polymorphism (allele 1) showed significantly enlarged ventricles and smaller frontal lobes (Boin et al., 2001).
In our experiments, ligation of TNF receptors by TNF stimulated RhoA activity, whereas no effect on survival was observed. In theory, TNF could induce cell death of pyramidal neurons, as shown with cultured cerebellar neurons by antagonizing insulin-like growth factor I (Venters et al., 1999). In another study, blockade of TNF by soluble TNFR-I significantly reduced focal cerebral ischemic injury in hypertensive rats, indicating a destructive potential of TNF in this model (Dawson et al., 1996).
TNF can also be “neuroprotective” in pathological situations, because transgenic mice lacking both TNF receptors have increased tissue lesions in response to ischemia (Bruce et al., 1996). Likewise, pretreatment of cultured hippocampal neurons with TNF reduced death of neurons mediated by ischemia (Cheng et al., 1994).
Our finding that TNF affects neurite elongation suggests that TNF might contribute to inhibit outgrowth of CNS axons during inflammation or lesion of the CNS. This finding is of particular importance, because axons do not regenerate after injury of the adult mammalian CNS. Multiple signals converge to regulate neuronal survival and neurite growth and determine the success or failure of axonal regrowth (Goldberg and Barres, 2000). One barrier to regeneration has been shown to be growth inhibition by myelin components such as Nogo (Chen et al., 2000). The synthesis of inhibitory proteins at the glial scar has been revealed as another impediment to axonal growth (Rudge and Silver, 1990; McKeon et al., 1991). Intriguingly, ablation of reactive astrocytes promoted neurite outgrowth after injury in a transgenic model (Bush et al., 1999). In this context, it is interesting to note that TNF is produced by reactive glia after injury (Hopkins and Rothwell, 1995) and deposited in the extracellular matrix where it binds avidly to substrates such as fibronectin and laminin while maintaining full biological activity (Alon et al., 1994; Hershkoviz et al., 1994).
In conclusion, TNF secreted by reactive glia during injury or inflammation might act as an anti-regenerative factor via its ability to activate Rho proteins in neurons.
Footnotes
Work in the group of H.N. was supported by Deutsche Forschungsgemeinschaft (SFB 391) and the Volkswagen-Stiftung. R.S. was supported by a predoctoral Marie-Curie fellowship from the European Commission. We thank Drs. Werner Lesslauer and Hansruedi Loetscher for the gift of TNF receptor IgG fusion protein, Dr. Klaus Pfeffer for TNFRI deficient mice, Dr. Martin-Alexander Schwartz for the RBD plasmid, and Dr. Christine Bandtlow for the C3 transferase expression construct. We are grateful to Lydia Penner for technical assistance. Furthermore, we thank Dr. Isabelle Medana for helpful discussions.
Correspondence should be addressed to Harald Neumann, Neuroimmunology, European Neuroscience Institute Göttingen, Waldweg 33, 37073 Göttingen, Germany. E-mail: hneuman1@gwdg.de.
REFERENCES
- 1.Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse. 2000;35:151–159. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 2.Alon R, Cahalon L, Hershkoviz R, Elbaz D, Reizis B, Wallach D, Akiyama SK, Yamada KM, Lider O. TNF-alpha binds to the N-terminal domain of fibronectin and augments the b1-integrin mediated adhesion of CD4+ T lymphocytes to the glycoprotein. J Immunol. 1994;152:1304–1313. [PubMed] [Google Scholar]
- 3.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM- kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 4.Bibel M, Barde YA. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000;14:2919–2937. doi: 10.1101/gad.841400. [DOI] [PubMed] [Google Scholar]
- 5.Blobel CP. Metalloproteinase disintegrins: links to cell adhesion and cleavage of TNF-alpha and Notch. Cell. 1997;90:589–592. doi: 10.1016/s0092-8674(00)80519-x. [DOI] [PubMed] [Google Scholar]
- 6.Boin F, Zanardini R, Pioli R, Altamura CA, Maes M, Gennarelli M. Assocation between -G308A tumor necrosis factor alpha gene polymorphism, schizophrenia. Mol Psychiatry. 2001;6:79–82. doi: 10.1038/sj.mp.4000815. [DOI] [PubMed] [Google Scholar]
- 7.Borasio GD, John J, Wittinghofer A, Barde Y-A, Sendtner M, Heumann R. Ras p21 protein promotes survival and fiber outgrowth of cultured embryonic neurons. Neuron. 1989;2:1087–1096. doi: 10.1016/0896-6273(89)90233-x. [DOI] [PubMed] [Google Scholar]
- 8.Botchkina GI, Meistrell ME, Botchkina IL, Tracey KJ. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol Med. 1997;3:765–781. [PMC free article] [PubMed] [Google Scholar]
- 9.Bradke F, Dotti CG. The role of local actin instability in axon formation. Science. 1999;283:1931–1934. doi: 10.1126/science.283.5409.1931. [DOI] [PubMed] [Google Scholar]
- 10.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 11.Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- 12.Chen MS, Huber AB, Van der Haar ME, Frank M, Schnell L, Spillmann AA, Christ F, Schwab ME. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000;403:434–439. doi: 10.1038/35000219. [DOI] [PubMed] [Google Scholar]
- 13.Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 14.Chisholm A, Tessier-Lavigne M. Conservation and divergence of axon guidance mechanisms. Curr Opin Neurobiol. 1999;9:603–615. doi: 10.1016/S0959-4388(99)00021-5. [DOI] [PubMed] [Google Scholar]
- 15.Chung IY, Benveniste EN. Tumor necrosis factor-alpha production by astrocytes: induction by lipopolysaccharide, interferon-gamma and interleukin-1. J Immunol. 1990;144:2999–3007. [PubMed] [Google Scholar]
- 16.Dawson DA, Martin D, Hallenbeck JM. Inhibition of tumor necrosis factor-alpha reduces focal cerebral ischemic injury in the spontaneously hypertensive rat. Neurosci Lett. 1996;218:41–44. doi: 10.1016/0304-3940(96)13116-5. [DOI] [PubMed] [Google Scholar]
- 17.Dziegielewska KM, Moller JE, Potter AM, Ek J, Lane MA, Saunders NR. Acute-phase cytokines IL-1beta and TNF-alpha in brain development. Cell Tissue Res. 2000;299:335–345. doi: 10.1007/s004419900157. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg JL, Barres BA. The relationship between neuronal survival and regeneration. Annu Rev Neurosci. 2000;23:579–612. doi: 10.1146/annurev.neuro.23.1.579. [DOI] [PubMed] [Google Scholar]
- 19.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 20.Hershkoviz R, Cahalon L, Miron S, Alon R, Sapir T, Akiyama SK, Yamada KM, Lider O. TNF-alpha associated with fibronectin enhances phorbol myristate acetate- or antigen-mediated integrin-dependent adhesion of CD4+ T cells via protein tyrosine phosphorylation. J Immunol. 1994;153:554–565. [PubMed] [Google Scholar]
- 21.Hopkins SJ, Rothwell NJ. Cytokines and the nervous system. I. Expression and recognition. Trends Neurosci. 1995;18:83–88. [PubMed] [Google Scholar]
- 22.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 23.Hu S, Reichardt LF. From membrane to cytoskeleton: enabling a connection. Neuron. 1999;22:419–422. doi: 10.1016/s0896-6273(00)80696-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin Z, Strittmatter SM. Rac1 mediates collapsin-1-induced growth cone collapse. J Neurosci. 1997;17:6256–6263. doi: 10.1523/JNEUROSCI.17-16-06256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karkkainen I, Rybnikova E, Pelto-Huikko M, Huovila AP. Metalloprotease-disintegrin (ADAM) genes are widely, differentially expressed in the adult CNS. Mol Cell Neurosci. 2000;15:547–560. doi: 10.1006/mcne.2000.0848. [DOI] [PubMed] [Google Scholar]
- 26.Koukouritaki SB, Vardaki EA, Papakonstanti EA, Lianos E, Stournaras C, Emmanouel DS. TNF-alpha induces actin cytoskeleton reorganization in glomerular epithelial cells involving tyrosine phosphorylation of paxillin and focal adhesion kinase. Mol Med. 1999;5:382–392. [PMC free article] [PubMed] [Google Scholar]
- 27.Lehmann M, Fournier A, Selles-Navarro I, Dergham P, Sebok A, Leclerc N, Tigyi G, McKerracher L. Inactivation of Rho signaling pathway promotes CNS axon regeneration. J Neurosci. 1999;19:7537–7547. doi: 10.1523/JNEUROSCI.19-17-07537.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- 29.Luo L, Jan LY, Jan YN. Rho family GTP-binding proteins in growth cone signalling. Curr Opin Neurobiol. 1997;7:81–86. doi: 10.1016/s0959-4388(97)80124-9. [DOI] [PubMed] [Google Scholar]
- 30.McKeon RJ, Schreiber RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring after CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. 1991;11:3398–3411. doi: 10.1523/JNEUROSCI.11-11-03398.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Munoz-Fernandez MA, Fresno M. The role of tumour necrosis factor, interleukin 6, interferon-gamma and inducible nitric oxide synthase in the development and pathology of the nervous system. Prog Neurobiol. 1998;56:307–340. doi: 10.1016/s0301-0082(98)00045-8. [DOI] [PubMed] [Google Scholar]
- 32.Nadeau S, Rivest S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience. 1999;93:1449–1464. doi: 10.1016/s0306-4522(99)00225-0. [DOI] [PubMed] [Google Scholar]
- 33.Neumann H, Cavalié A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science. 1995;269:549–552. doi: 10.1126/science.7624779. [DOI] [PubMed] [Google Scholar]
- 34.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 35.Puls A, Eliopoulos AG, Nobes CD, Bridges T, Young LS, Hall A. Activation of the small GTPase Cdc42 by the inflammatory cytokines TNF(alpha) and IL-1, and by the Epstein-Barr virus transforming protein LMP1. J Cell Sci. 1999;112:2983–2992. doi: 10.1242/jcs.112.17.2983. [DOI] [PubMed] [Google Scholar]
- 36.Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Renno T, Krakowski M, Piccirillo C, Lin J-Y, Owens T. TNF-a expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. J Immunol. 1995;154:944–953. [PubMed] [Google Scholar]
- 38.Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- 39.Rudge JS, Silver J. Inhibition of neurite outgrowth on astroglial scars in vitro. J Neurosci Res. 1990;10:3594–3603. doi: 10.1523/JNEUROSCI.10-11-03594.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sumi T, Matsumoto K, Takai Y, Nakamura T. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. J Cell Biol. 1999;147:1519–1532. doi: 10.1083/jcb.147.7.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tchelingerian JL, Quinonero J, Booss J, Jacque C. Localization of TNF alpha and IL-1 alpha immunoreactivities in striatal neurons after surgical injury to the hippocampus. Neuron. 1993;10:213–224. doi: 10.1016/0896-6273(93)90312-f. [DOI] [PubMed] [Google Scholar]
- 42.Threadgill R, Bobb K, Ghosh A. Regulation of dendritic growth and remodeling by Rho, Rac, and Cdc42. Neuron. 1997;19:625–634. doi: 10.1016/s0896-6273(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 43.Tucker KL, Meyer M, Barde YA. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- 44.Venters HD, Tang Q, Liu Q, VanHoy RW, Dantzer R, Kelley KW. A new mechanism of neurodegeneration: a proinflammatory cytokine inhibits receptor signaling by a survival peptide. Proc Natl Acad Sci USA. 1999;96:9879–9884. doi: 10.1073/pnas.96.17.9879. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Wassink TH, Crowe RR, Andreasen NC. Tumor necrosis factor receptor-II: heritability and effect on brain morphology in schizophrenia. Mol Psychiatry. 2000;5:678–682. doi: 10.1038/sj.mp.4000807. [DOI] [PubMed] [Google Scholar]
- 46.Wojciak-Stothard B, Entwistle A, Garg R, Ridley AJ. Regulation of TNF-alpha-induced reorganization of the actin cytoskeleton and cell-cell junctions by Rho, Rac, and Cdc42 in human endothelial cells. J Cell Physiol. 1998;176:150–165. doi: 10.1002/(SICI)1097-4652(199807)176:1<150::AID-JCP17>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 47.Yamashita T, Tucker KL, Barde YA. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron. 1999;24:585–593. doi: 10.1016/s0896-6273(00)81114-9. [DOI] [PubMed] [Google Scholar]
- 48.Zhao B, Schwartz JP. Involvement of cytokines in normal CNS development and neurological diseases: recent progress and perspectives. J Neurosci Res. 1998;52:7–16. doi: 10.1002/(SICI)1097-4547(19980401)52:1<7::AID-JNR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]