

Abstract
Neurotrophins have profound effects on synaptic function and structure. They can be derived from presynaptic, as well as postsynaptic, sites. To date, it has not been possible to measure the release of neurotrophins from axon terminals in intact tissue. We implemented a novel, extremely sensitive assay for the release and transfer of anterogradely transported neurotrophin-3 (NT-3) from a presynaptic to a postsynaptic location that uses synaptosomal fractionation after introduction of radiolabeled NT-3 into the retinotectal projection of chick embryos. Release of the anterogradely transported NT-3 in intact tissue was assessed by measuring the amount remaining in synaptosomal preparations after treatment of whole tecta with pharmacological agents. Use of this assay reveals that release of NT-3 from axon terminals is increased by depolarization, calcium influx via N-type calcium channels, and cAMP analogs, and release is most profoundly increased by excitation with kainic acid or mobilization of calcium from intracellular stores. NT-3 release depends on extracellular sodium, CaM kinase II activity, and requires intact microtubules and microfilaments. Dantrolene inhibits the high potassium-induced release of NT-3, indicating that release of calcium from intracellular stores is required. Tetanus toxin also inhibits NT-3 release, suggesting that intact synaptobrevin or synaptobrevin-like molecules are required for exocytosis. Ultrastructural autoradiography and immunolabel indicate that NT-3 is packaged in presumptive large dense-core vesicles. These data show that release of NT-3 from axon terminals depends on multiple regulatory proteins and ions, including the mobilization of local calcium. The data provide insight in the mechanisms of anterograde neurotrophins as synaptic modulators.
Keywords: neurotrophic factors, secretion, presynaptic terminals, calcium, axonal transport, neurotrophin, synapse, synaptic transmission
Neurotrophins are important regulators of neuronal differentiation and synaptic plasticity (Lohof et al., 1993; Snider, 1994; Kang and Schuman, 1995; Thoenen, 1995;McAllister et al., 1999; Poo, 2001). Traditionally, neurotrophins were believed to be derived by retrograde axonal transport from target cells, but recent studies revealed that neurotrophins are also transported anterogradely along axons (von Bartheld et al., 1996; Zhou and Rush, 1996; Altar et al., 1997; Conner et al., 1997; Heymach and Barres, 1997; Smith et al., 1997; Yan et al., 1997; Fawcett et al., 2000; von Bartheld and Butowt, 2000) and may act as extremely potent excitatory neurotransmitters (Kafitz et al., 1999). Anterograde signals provide trophic support from afferents (Linden, 1994; von Bartheld et al., 1996; Altar et al., 1997), may mediate fast, local effects at synaptic sites (Lohof et al., 1993; Kang and Schuman, 1995; Thoenen, 1995; Kafitz et al., 1999; Poo, 2001), and regulate dendritic growth, neuronal cytoarchitecture, and phenotypes (Altar et al., 1997;McAllister et al., 1999; Fawcett et al., 2000).
Neurotrophin 3 (NT-3) enhances synaptic transmission in neuromuscular junctions (Lohof et al., 1993) and the brain (Kim et al., 1994), alters short-term and long-term plasticity (Kang and Schuman, 1995; Kokaia et al., 1998), and modifies the morphology of cerebellar and cortical neurons (Lindholm et al., 1993; McAllister et al., 1999). In the retinotectal system, endogenous and exogenous NT-3 are moved by anterograde axonal transport (von Bartheld and Butowt, 2000), and exogenous NT-3 is released from retinotectal terminals and taken up by tectal dendrites, promoting the survival of postsynaptic neurons (von Bartheld et al., 1996). Release mechanisms are crucial for an understanding of the action of neurotrophins (Lo, 1995; Thoenen, 1995;Fawcett et al., 1997; Aloyz et al., 1999; Berninger and Poo, 1999;Schinder and Poo, 2000), but mechanisms of the release of anterogradely transported neurotrophins are completely unknown.
Neuropeptides are released by a variety of mechanisms, presumably by packaging into different types of vesicles that are released by different mechanisms (Bartfai et al., 1988; Hille et al., 1999; Kasai, 1999). Therefore, release mechanisms of neurotrophins cannot be predicted on the basis of what is known for other neuropeptides. The role of anterogradely transported neurotrophins has been particularly difficult to assess because of the absence of an assay that localizes the trophic factors within synapses and is sufficiently sensitive to measure the excessively small amounts released from axon terminals.
We developed a technique that allows us to measure the release and transynaptic transfer of picomolar amounts of anterogradely transported NT-3. A novel combination of autoradiographic, biochemical, and experimental pharmacological techniques in an in vivo model system revealed which molecules and events regulate the release of anterogradely transported NT-3 from axon terminals. This technology will define the molecular mechanisms regulating neurotrophin release and help to better understand the role of neurotrophins in neurotransmission and synaptic plasticity.
Parts of this work have been published previously in abstract form (Wang et al., 1999).
MATERIALS AND METHODS
Sources of materials
4-Aminopyridine (4-AP), cadmium chloride, colchicine, cytochalasin D, cytochrome c, kainic acid, KN-93, strophanthidin, tetrodotoxin (TTX), and thapsigargin were from Sigma (St. Louis, MO). 8-Br-cAMP, dantrolene, and forskolin were from ICN Biomedicals (Cleveland, OH). BAPTA, BAPTA AM, ω-conotoxin-GVIA, KN-92, tetanus toxin, and tetanus toxin C fragment were from Calbiochem (La Jolla, CA). Nerve growth factor (NGF) was from Mark Bothwell (Seattle, WA) or Alomone Labs (Jerusalem, Israel). Brain-derived neurotrophic factor (BDNF) and NT-3 were from Regeneron (Tarrytown, NY). NT-3 antibody was from Robert Rush (Adelaide, Australia). Calcium electrodes and calibration buffers were from World Precision Instruments (Sarasota, FL). Fertilized chicken eggs were from California Golden Eggs (Sacramento, CA). A total of 3800 chicken eggs were used in this study; all embryos were staged (Hamburger and Hamilton, 1951). Protocols were approved by the local animal care committee, and surgical procedures were done after anesthesia with sodium pentobarbital (Nembutal; 5 mg/embryo).
Injection of 125I-labeled proteins, trafficking, and release
Neurotrophins, cytochrome c, and tetanus toxin were radio-iodinated with lactoperoxidase (von Bartheld, 2001), and substance P (SP) was iodinated with the Bolton-Hunter reagent (NEN Life Sciences, Boston, MA). Incorporation for NT-3 was ∼90–95%, and specific activity was 90–150 cpm/pg. Specific activities for NGF were 75–189 cpm/pg, for cytochrome c were 36–72 cpm/pg, for tetanus toxin were 40–61 cpm/pg, and for substance P were 18 cpm/pg. Fertilized chicken eggs were incubated at 37°C. Embryos were windowed on embryonic day 15 (E15) to E16, injected with 40–80 ng of125I-labeled NT-3 in the eye in ovo on E16–E17 (von Bartheld et al., 1996), and were perfused intracardially 20 hr later (at E17–E18) with ice-cold nondepolarizing buffer [low K+ Krebs' buffer (in mm): 134 NaCl, 5 KCl, 1 MgSO4 · 7 H2O, 1.25 KH2PO4, 2.0 CaCl2 · 2 H2O, 16 NaHCO3, and 10 glucose, pH 7.5 (after gassing with 95% O2, 5%CO2)] to remove radioactivity that had leaked into systemic circulation. The perfusion was also used to deliver pharmacological agents directly to the brain and to improve penetration. The tectum was removed and placed in cold low K+ buffer, and its radioactivity was counted in a gamma counter. Only tecta containing >6000 cpm (n = 694) were used for subsequent release experiments, because such tecta typically generated >200 cpm in synaptosomes and we found that ∼200 cpm were necessary to reliably quantify synaptosomal peaks. Each dissected tectum was incubated in 3 ml of oxygenated low K+ buffer (6 mm K+) or oxygenated high K+ buffer (56 mm K+) (in mm: 84 NaCl, 55 KCl, 1 MgSO4 · 7 H2O, 1.25 KH2PO4, 2 CaCl2 · 2 H2O, 16 NaHCO3, and 10 glucose) at room temperature with or without experimental drugs for 60 min, unless indicated otherwise. Maximal transport capacity in the retinotectal projection was tested by injection of 2–400 ng (∼0.5–30 × 106 cpm in the eye at the time the animal was killed) and gamma-counting of tecta after 20 hr. The amount of injected 125I-labeled NT-3 in the eye and the amount of anterogradely transported NT-3 in the tectum was quantified; in addition, the amount of the radioactivity in the synaptosomal peaks was taken as a measure for the amount and percentage of 125I-labeled NT-3 within the presynaptic terminals [excluding the amount in the general membrane fraction containing the bulk of tectal radioactivity: axons and dendrites (von Bartheld et al., 1996)].
Synaptosome preparation, data collection, and analysis
Synaptosomes were prepared to measure how much125I-labeled NT-3 remained in presynaptic terminals. The protocol of Viglietti et al. (1977) was used with slight modifications. The incubation buffer was replaced with 5 ml of 0.32m sucrose and 5 mm Tris, pH 7.4 (buffer B); tecta were homogenized by trituration using a 1 ml syringe with a 20 G 1 1/2 gauge needle. The suspension was centrifuged at 4°C for 10 min at 1500 × g to remove the crude nuclear fraction (P1). The pellet was washed two times with 1 ml buffer B, and supernatants were removed and centrifuged at 17,000 × g for 20 min to pellet the membrane fraction (P2). Pellet 1 and supernatant (S2) were counted in a gamma counter. P2 was resuspended in 10 ml of 0.32m sucrose and loaded on a two-step discontinuous ficoll–sucrose gradient in Quick-Seal tubes containing 15 ml of 13% (w/v) ficoll in 0.32 m sucrose and overlaid with 15 ml of 7.5% (w/v) ficoll in 0.32 m sucrose. The samples were centrifuged at 53,000 × g for 90 min at 4°C. The ultracentrifuge tubes were fractionated (50 × 0.8 ml fractions), and each fraction was counted for 10 min in a gamma counter. The radioactivity within the synaptosomal peak (defined by visual inspection of the printed plots) was calculated as the ratio (divided by the total radioactivity in the ultracentrifuge tube). The average ratio of the synaptosomal peak in low K+ buffer was defined as 100% unless indicated otherwise. The ratio of the synaptosomal peak/tube in the experimental condition was divided by the average ratio of the synaptosomal peak/tube in the low K+buffer to calculate the percentage of NT-3 released from retinotectal terminals. Statistical significance was determined by unpaired Student's t test or, for multiple comparisons, by one-way ANOVA with Prism 2.01 software.
Proteins extracted from synaptosomal fractions derived from125I-NT-3-loaded tecta were examined by SDS-PAGE (15% gels). To examine whether release of NT-3 from synaptosomal fractions (obtained from125I-NT-3-loaded tecta) can be measured, synaptosomes were centrifuged and resuspended in either high or low K+ buffer, warmed to 37°C, gassed for 60 min, and centrifuged again in either a microcentrifuge or the ultracentrifuge. The pellets and supernatants were gamma counted to determine release of 125I-NT-3 into the buffer.
Pharmacological manipulations of release
A total of 694 successful release experiments were performed to test molecules potentially involved in release of NT-3. Control experiments (low K+ or high K+) were included in each experimental series of up to eight centrifugation tubes for calibration of NT-3 release (“quality control”). For manipulation of release in vitro, the dissected tecta were incubated with 3 ml of high K+ buffer containing one of the following agents: 4-aminopyridine (1 mm), 8-Br-cAMP (50 μm), cadmium chloride (100 μm), colchicine (175 μm), cytochalasin D (0.2 μm), dantrolene (50 μm), forskolin (10 μm), KN-93 (10 μm), KN-92 (10 μm), nifedipine (10 μm), ω-conotoxin-GVIA (0.1 μm), strophanthidin (500 μm), tetrodotoxin (3 μm), or thapsigargin (5 μm). When drugs required dissolving in DMSO or ethanol, control tecta were tested with identical vehicle solution.
BAPTA. Buffers were prepared as described, except that CaCl2 was replaced with MgCl2 at the same concentration. BAPTA AM (10 μm) or BAPTA (20 or 200 μm) was added to the calcium-free buffers overnight before use. Higher BAPTA concentrations were necessary because tecta were a source of calcium that leaked into the incubation solution. Calcium concentrations in the incubation buffer were measured with and without tecta, and with normal calcium, or calcium-free buffers containing 20 μm BAPTA, 200 μm BAPTA, or 10 μmBAPTA AM. Calcium electrodes and calibration buffers were used as described previously (Baudet et al., 1994).
N-Methyl-d-glucamine. N-Methyl-d-glucamine (NMG) was used to replace extracellular sodium in high K+buffer (in mm: 100 NMG-HCl, 39 KCl, 1 MgSO4, 1.25 KH2PO4, 2 CaCl2, 16 KHCO3, and 10 glucose). Chick embryos were perfused with high K+ buffer containing NMG, dissected optic tecta were incubated in NMG buffer for 60 min, and the tissue was further processed as described. Some NMG-treated tecta were incubated in normal (Na-containing) high K+ buffer to determine whether the NMG effect was reversible.
Tetanus toxin. Tetanus toxin (1–4 μg) was injected into the eyes 1 d before NT-3 injection to target the toxin by anterograde axonal transport to the retinotectal nerve terminals. Whereas 4 μg was lethal, 1–2 μg was tolerated. The tetanus toxin C fragment (responsible for trafficking of the molecule) was radio-iodinated with lactoperoxidase to a specific activity of 40–61 cpm/pg. Free iodide was removed by membrane centrifugation (von Bartheld, 2001). Radiolabeled toxin C fragment (300 ng) was injected into the eye of chick embryos, and the tecta were processed for autoradiography to confirm that the toxin was transported anterogradely by retinal ganglion cell (RGC) axons to the optic tectum.125I-NT-3 was coinjected with (cold) tetanus toxin to test whether the toxin interfered with the normal anterograde transport of NT-3. The tecta, loaded with tetanus toxin and125I-NT-3, were treated with high K+ buffer and processed as described.
Control experiments
To determine whether NT-3 was released constitutively during the incubation in low K+ buffer, the amount of NT-3 in retinotectal terminals was compared between tecta processed immediately after killing, and tecta were incubated for 60 min in oxygenated low K+ buffer. To determine whether NT-3 or other neurotrophins or peptides bound to terminals during the preparation of synaptosomes, the tectal homogenates of normal (not injected) chick embryos were incubated with 0.1–0.3 ng/ml radio-iodinated NT-3, BDNF, NGF, SP, or cytochrome c and subjected to ficoll–sucrose gradient centrifugation, and gamma counts were plotted after fractionation. Some NT-3 preparations were incubated with 500-fold excess cold NT-3 to determine specific, receptor-mediated binding. Total protein content of synaptosomal peaks was measured by UV absorption [Bio-Rad (Hercules, CA) standard protein assay].
Anterograde transport and release of other proteins
To compare the transport and release of NT-3 with that of other proteins, we measured NGF, tetanus toxin C fragment, and cytochromec. These proteins were chosen because, unlike substance P, they can be transported in the retinotectal system after intraocular injection. The amount of radiolabeled protein injected in the eye was measured 20 hr later to assess retention in the eye. The transport efficiency was measured by comparing the amount in the eye at the time the animal was killed with the amount contained in the contralateral tectum. By competing with excess cold protein (20- to 50-fold), it was examined whether anterograde transport was receptor mediated. Accumulation of the transported protein in retinotectal terminals was assessed by measuring the radioactivity in the synaptosomal peak and comparing it with the total radioactivity in the contralateral tectum.
In vivo release of NT-3
Kainic acid (8 μl, 5 mm) was injected into the eye (estimated concentration in the eye, 0.625 mm) of125I-NT-3 treated chick embryos 45 min before dissection of the tecta. To determine whether the effect of the kainic acid could be blocked by TTX, 5 μl of TTX (100 μm) or PBS (as a control for the TTX application) was injected into the eyes, followed by 8 μl of kainic acid (5 mm) 15 min later. The embryos were anesthetized 45 min later and perfused intracardially, and the tecta were processed as described.
Release of substance P from tectal terminals
Radio-iodinated NT-3 was injected in the eye 20 hr before homogenization of the tecta to identify the fractions containing the synaptosomal peak. Tecta were treated with either low or high K+ buffer as described. Fractions were counted in the gamma counter for 1 min. HCl was added to 3 ml fractions containing synaptosomes, and samples were stored immediately at −80°C. Samples of 500 μl were analyzed by radioimmunoassay for the content of SP (Chen et al., 1996).
Vesicle purification
Tecta from E17 chick embryos loaded with125I-labeled NT-3 were dissected and homogenized at 4°C in 15 ml of buffered sucrose (4 mmHEPES and 320 mm sucrose, adjusted to pH 7.3 with NaOH). Vesicles were purified according to Huttner et al. (1983) and Han and Fischbach (1999). After centrifugations, resuspended vesicles (6.7 ml) were loaded on sucrose step gradients in 40 ml Quick-Seal centrifuge tubes, loaded with 6.75 ml each of 50, 200, 400, 550, and 800 mm sucrose (from bottom to top), and centrifuged at 24,500 rpm (42.1 rotor; 65,000 × g) for 5 hr. Fractions were collected, counted, and plotted as described for synaptosome fractionations.
Electron microscopic autoradiography and immunocytochemistry
Localization and quantification of anterogradely transported125I-NT-3 in tectal tissue was performed by autoradiography as described previously (von Bartheld et al., 1996;von Bartheld, 2001). Five hundred thirty silver grains from two E17 tecta with 125I-NT-3-loaded retinotectal terminals were analyzed. Silver grains overlying presynaptic or postsynaptic compartments were quantified, and the percentages of silver grains were calculated. In addition, 87 silver grains within the stratum opticum were analyzed for association with either endosomes or large dense-cored vesicles (LDCVs) within axons. Synaptosomes from125I-NT-3-loaded tecta were fixed in 2.5% glutaraldehyde and centrifuged at 10,000 × g at 4°C, embedded in Spurr, and sectioned at 80–90 nm. Sections were coated with a monolayer of Ilford L4 emulsion and exposed for 8–10 weeks at 4°C. Developed sections were examined on a Philips CM10 transmission electron microscope. The distribution of silver grains was photographed, and 94 random silver grains were quantified at a magnification of 15,000–20,000×. For immunolabeling, tectal tissue from E19–E20 embryos was fixed in 4% paraformaldehyde and 0.1% glutaraldehyde by intracardial perfusion. Standard pre-embedding immunolabeling techniques were used. Tecta were sectioned with a vibratome at 100–150 μm. Endogenous peroxidase activity was quenched by pretreatment with 3% H2O2. The sections were incubated for 24 hr at 4°C with 20 μg/ml purified polyclonal antibodies to NT-3 (Zhou and Rush, 1993) (courtesy of R. Rush, Flinders University, Adelaide, Australia) and processed with the ABC kit (Vector Laboratories, Burlingame, CA). Sections were embedded in Spurr and thin sectioned at 80–90 nm. In control sections, the primary antibody was omitted.
RESULTS
Anterogradely transported 125I-NT-3 in tectal terminals can be quantified
Injections of 40–80 ng of 125I-NT-3 in the eye resulted in a total of ∼100 pg of125I-NT-3 in the contralateral tectum. The synaptosomal peaks contained 5.94 ± 0.36% (SEM;n = 19) of the total radioactivity measured in the contralateral tectum, equivalent to 15–20% of the radioactivity in the ultracentrifugation tube (Fig.1A). Much of the radioactivity was found in the general membrane fraction, containing125I-NT-3 in retinal axons en route to their terminals in the tectum (von Bartheld et al., 1996). There was no radioactive peak in the synaptosomes from the ipsilateral (control) tecta, indicating that accumulation of125I-NT-3 in tectal synaptosomes was not attributable to diffusion or uptake after leakage into systemic circulation, and 125I-NT-3 arrived in the tectum intact. The localization of anterogradely transported125I-NT-3 in synaptosomes (after fractionation) was confirmed by quantitative autoradiography at the ultrastructural level. Synaptosomes were identified in expected quality and quantity (≈60–70% purity) (Viglietti et al., 1977), and >60% of silver grains were located over clearly identified synaptosomes (Fig. 1B,C). The identity of synaptosomal fractions was verified by increased levels of synaptic vesicle protein (SV2) in Western blots (data not shown). The association of radiolabeled NT-3 in synaptosomal peaks with retinotectal terminals (rather than other tectal terminals) can be concluded because of the fact that the highest level of autoradiographic labeling in the tectum in situ occurs in the layer known to contain the retinotectal terminals (von Bartheld et al., 1996). To determine whether 125I-NT-3 in multivesicular bodies (MVBs) of postsynaptic dendrites may contribute to the radioactivity in the synaptosomal peak, silver grains over MVBs were quantified. Only ∼2% of silver grains labeled MVBs (Fig. 1C), ruling out significant contamination by these and other postsynaptic elements. These data confirm that synaptosomal fractionation measures the content of anterogradely transported125I-NT-3 in retinotectal terminals.
Fig. 1.
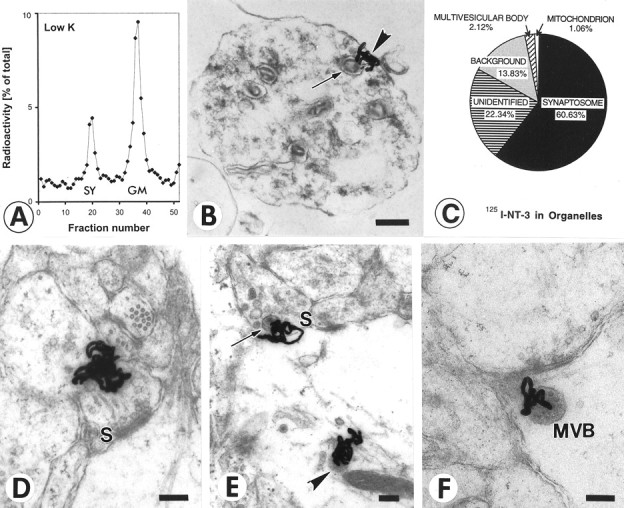
Quantification of radiolabeled NT-3 in retinotectal terminals after anterograde axonal transport.A, Quantification of 125I-NT-3 in the synaptosomal peak after centrifugation on ficoll-sucrose gradients and fractionation. The synaptosomal peak (SY) and the general membrane fraction (GM) are indicated. B, Synaptosome shows a silver grain (arrowhead) next to a large dense core vesicle (arrow). C, Quantification of silver grains over synaptosomes shows majority of grains over synaptosomes. Silver grains (n = 94) were randomly analyzed. D, Synapse before release of NT-3. Cluster of silver grains over a presynaptic terminal in layer f of the stratum griseum et fibrosum superficiale (SGFSf) of the optic tectum after anterograde transport. Note the synaptic profile (S). E, Synapse in the process of releasing NT-3. Silver grains over a presynaptic profile (S) containing a large dense core vesicle (arrow) and over a postsynaptic organelle reminiscent of a multivesicular body (arrowhead). F, Synapse after release of NT-3. Silver grain on the postsynaptic side of the synapse (S) overlying a multivesicular body (MVB). Scale bars, 200 nm.
125I-NT-3 in presynaptic terminals in situ: quantitative electron microscopy
To verify the amount of 125I-NT-3 in the presynaptic retinotectal terminals in situ, quantitative autoradiography was performed at the ultrastructural level. Typical examples of silver grains over presynaptic and postsynaptic profiles are shown in Figure 1D–F. A total of 530 silver grains from two chick tecta were sampled randomly, and their location was analyzed at a magnification of 24,400×. In many (42.5%) but not all of the labeled terminals, LDCVs were visible in close vicinity (within ≈100 nm) of the silver grains (Fig. 1E), consistent with the notion that anterograde neurotrophins localize in LDCVs (Fawcett et al., 1997; Michael et al., 1997; Holstege et al., 1999). The percentage of silver grains over presynaptic terminals was 5.48% (range, 4.72–6.17%) of the total 530 grains, and 3.15% were over postsynaptic terminals (Fig.1E,F). The percentage for presynaptic localization of 125I-NT-3 (5.48%) is remarkably similar to that obtained by synaptosomal fractionation (5.94%). Although we cannot rule out coincidence, this similarity supports the validity of our assay for the measurement of exogenous NT-3 in presynaptic axon terminals.
125I-NT-3 in synaptosomes is intact by SDS-PAGE
At least some of the NT-3 that arrives in the tectum after anterograde axonal transport from the eye is intact (von Bartheld et al., 1996), but it was not known whether this included the125I-NT-3 within synaptosomes. Protein extracted from synaptosomal fractions was examined by SDS-PAGE. The large majority of 125I-NT-3 recovered from synaptosomes was intact compared with native125I-NT-3 (Fig.2A). When treated with detergents and trypsin, a major fraction of NT-3 in the synaptosomal fraction was degraded, indicating that NT-3 was located within a membrane-confined compartment (data not shown). These results indicate that anterogradely transported NT-3 within synaptosomes remains intact.
Fig. 2.

NT-3 in synaptosomes: K+-induced-release of NT-3 and other proteins from axon terminals and binding of neurotrophins and neuropeptides to synaptosomes. A, SDS gel shows that the large majority of NT-3 recovered from synaptosomes (Synapt.) migrates identically to native NT-3 at ∼14 kDa. The arrow indicates minor degradation products. Three month exposure. B, Plot of anterogradely transported 125I-NT-3 in the contralateral tectum (cpm) as a function of the amount of 125I-NT-3 remaining in the injected eye. C, Plot of the125I-NT-3 in the synaptosomal fraction from the contralateral tectum (filled circles) as a function of the amount of 125I-NT-3 remaining in the injected eye. When the125I-NT-3 in the eye was competed with 20–50-fold excess cold NT-3, only background cpm were found in the synaptosomes (open circles). Note that the amounts of NT-3 in the tectum and in the synaptosomal peak saturate at ∼10 × 106-15 × 106 cpm in the eye.D, Representative examples show decreased size of synaptosomal peaks (SY) from tectum depolarized in high K+ buffer (right panel) compared with low K+ buffer (left panel). E, Quantification of D reveals a 15–20% decrease in NT-3 content in depolarized terminals (HiK, high K+ buffer; ***p ≤ 0.001, ANOVA, error bars indicate SEM). The average of the values for low K+ buffer (LoK, 60 min) was taken as 100%. Comparison of incubation for 60 min versus 0 min in low K+ shows lack of basal (constitutive) release. Numbers of experiments are indicated on bars. F, Release of radiolabeled tetanus toxin C fragment (Tet tox) was not statistically significant in high K+ buffer.G, Radiolabeled cytochrome c(Cyt.C) was not released in high K+ buffer compared with low K+buffer. H, Depolarized tecta showed a 25–50% release of substance P (SP) from tectal terminals, measured by radioimmunoassay in 500 μl samples (1:6 dilution). *p ≤ 0.05, t test, HiK versus LoK, 60 min, error bars indicate SEM. I, When homogenized tecta were incubated with radiolabeled NT-3, NGF, or cytochromec (Cyt C), NT-3 bound to tectal terminals which was competed by excess cold NT-3, whereas NGF and cytochromec showed very little binding. SY, Synaptosomal peak; GM, general membrane fraction.
Direct release from synaptosomes using the conventional assay
We attempted to measure release of anterogradely transported125I-NT-3 directly from synaptosomes by using a conventional static release protocol (Androutsellis-Theotokis et al., 1996), with the modification that125I-NT-3 was loaded into tectal synaptosomes via anterograde transport from the retina in vivo. After injections of 50–100 ng of125I-NT-3 into the eye, ∼3–4 pg (450–600 cpm) of 125I-NT-3 was present in the synaptosomal fraction, but only a fraction of this amount (∼1 pg ≈100–200 cpm) was recovered per tectum after washes, resuspension, and warming to 37°C. The radioactivity in the supernatant after centrifugation ranged from 30 to 60%, and this amount was not increased when synaptosomes were resuspended in high K+ buffer instead of low K+ buffer (data not shown). The radioactivity in the supernatant likely represents125I-NT-3 that leaked out of damaged immature synaptosomes, thus yielding a relatively high and variable background that made it impossible to measure secreted125I-NT-3. The radioactivity levels in this prep were approximately three orders of magnitude lower than what is usually used for synaptosomal release studies (Androutsellis-Theotokis et al., 1996), and it was impossible to increase the amount of anterogradely transported125I-NT-3 per tectum, because its transport into synapses saturated at ∼10 × 106 cpm/eye (Fig.2B,C). We conclude that the measurement of anterogradely transported125I-NT-3 release directly from synaptosomes is impossible with the conventional technique.
Depolarization of tectal terminals induces release of NT-3
Next we tested whether release of NT-3 from terminals in intact tissue can be measured by quantifying the amount remaining within terminals rather than measuring the amount that is released. Tecta whose retinotectal terminals had been loaded with anterogradely transported 125I-NT-3 were incubated in oxygenated buffer containing high K+ (56 mm), whereas control tecta were incubated in low K+ (5 mm), and the amount of125I-NT-3 remaining in terminals was quantified by measuring the amount in the synaptosomal fraction relative to the total amount in the tectum or the total amount in the ficoll–sucrose gradient tube. Depolarization resulted in smaller synaptosomal peaks of 125I-NT-3 (Fig.2D), equivalent to a loss of 15–20% of the radioactivity in the terminals after depolarization (p ≤ 0.001; ANOVA; multiple comparisons). These data were normalized to the total amount of radioactivity in the gradient ultracentrifugation tube. With an input of 40–80 ng of125I-NT-3 in the eye, we found that ∼8000–12,000 cpm was transported to the tectum, and 6% (∼500 cpm) of this amount was reliably measured in the synaptosomal peak (Table1). No basal release of NT-3 was detected during the 60 min incubation step in low K+ buffer (Fig. 2E). To determine whether the release of 125I-NT-3 was specific or merely reflected the loss of 10–15% of presynaptic protein attributable to stimulated release, the total protein content of synaptosomal preparations was compared between depolarized and control tecta. There was no significant difference between the two groups (data not shown), indicating that NT-3 was specifically released. These data show that the novel release assay has significant advantages over the conventional assay in terms of recovery, reliability, and sensitivity, primarily because the synaptosomes are used merely to determine the amount of radiolabeled NT-3. Because the release has occurred in primarily intact tissue (the tectum), the assay does not require the synaptosomes to be functionally intact, which is a major challenge for immature synaptosomes prone to leakage at 37°C.
Table 1.
Behavior of different proteins in the retinotectal system of the chick embryo
| Protein | kDa | Retention in eye 20 hr after injection (%) | Transport efficiency pg/tectum/ng/eye | Receptor-mediated transport? (%) | Synaptic accumulation(% of whole tectum) | Amount in synaptosomal fraction (cpm) |
|---|---|---|---|---|---|---|
| NT-3 | 28 | 30.3 ± 2.4 | 1.34 ± 0.1 | >80 | 5.94 ± 0.36 | 522 ± 48.5 |
| NGF | 26 | 25.1 ± 3.5 | 2.02 ± 0.4 | ≈501-a | 1.15 ± 0.10 | 69.9 ± 1.4 |
| Tet tox | 47 | 36.5 ± 6.9 | 4.04 ± 0.6 | >80 | 1.72 ± 0.16 | 170 ± 35.5 |
| Cyt. C | 12.3 | 47.1 ± 2.1 | 1.78 ± 0.4 | 01-b | 2.06 ± 0.27 | 162 ± 23.2 |
| SP | 1.3 | 1.8 ± 0.30 | 0.52 ± 0.1 | <10 | 0.58 ± 0.11 | 27 ± 2.6 |
Retention in the eye, efficiencies of anterograde axonal transport, and accumulation in retinotectal synapses. Values are ± SEM. Kilodaltons are indicated for monomers (Tet tox, Cyt. C, and SP) and dimers (NT-3 and NGF). Tet tox, Tetanus toxin C fragment; Cyt. C, cytochrome C; SP, substance p.
Data from von Bartheld et al., 1996.
Data from Butowt and von Bartheld, 2001.
Control experiments with other transported proteins: limitations of the new assay
To determine whether the release of125I-NT-3 was specific or whether other proteins were released similarly when measured with the same assay, we compared the transport, synaptic accumulation, and release of NT-3, NGF, tetanus toxin, and cytochrome c. The novel assay requires that the protein is retained in the eye (to be internalized by retinal ganglion cells), that it be transported to the terminals in the tectum with sufficient efficiency, and that it accumulate in the retinotectal terminals. The data showing how these three requirements are met for five different proteins are summarized in Table 1. Cytochrome c showed the highest retention in the eye (47%) after 20 hr, and HSP showed the lowest (2%). The transport efficiencies of NT-3, NGF, and cytochrome c were similar (1–2 pg/ng in the tectum retained in the eye) and was significantly more efficient for tetanus toxin (4 pg/ng in the eye). The greatest differences between the proteins were found with synaptic accumulation: NT-3 accumulated in terminals almost six times more than NGF, whereas tetanus toxin and cytochrome c accumulated only slightly more than NGF (Table 1). These data provide evidence for some remarkable differences in the trafficking of these proteins in the retinotectal system. When we attempted to determine release of NGF in high K+ buffer, it turned out that the counts per minutes amounts in the synaptosomal fractions were too low to be reliably quantified (Table 1). Release of tetanus toxin could be quantified in this system; although there was a trend for lower peaks with depolarization, the difference did not reach statistical significance (Fig. 2F). Release of125I-cytochrome c could also be quantified, and there was no release of this protein with high K+ buffer (Fig. 2G). NGF transport appeared to be more efficient than in a previous study (von Bartheld et al., 1996), probably because here we measured radioactivity exclusively in the tectum, and free iodine and degradation products were not removed by dehydration of the tissue. Together, these data show that 125I-NT-3 is specifically stored in retinotectal terminals, and this capacity makes it possible to quantify release of NT-3, which is released during depolarization, unlike other proteins that can also be transported to retinotectal terminals but do not accumulate there. Requirements and limitations of our novel release assay thus include the following: (1) internalization by projection neurons; (2) anterograde axonal transport; (3) accumulation of the transported protein in presynaptic terminals; and (4) release of the protein from the terminal. The utility of this assay may be useful for any protein and neuronal projection system in which sufficient amounts of protein can be quantified, and the novel assay thus may not be restricted to NT-3 and the retinotectal system of chick embryos.
Control experiments with substance P
A fraction of retinotectal projections in chicken use SP as a neurotransmitter (Yamagata and Sanes, 1995), and release of SP from synaptosomes can be induced by treatment with high K+ (Lembeck et al., 1977). Radiolabeled SP was very poorly transported anterogradely by RGCs after injection in the eye (Table 1), presumably attributable to the lack of SP receptors on RGCs that transport endogenous SP, and therefore it was not possible to measure release of (exogenous) SP from retinotectal terminals. To determine whether depolarization of tecta induces release of endogenous SP from tectal terminals, similar to that of anterogradely transported NT-3, synaptosomal fractions were prepared from125I-NT-3-loaded tecta (to identify the synaptosomal peaks), and the amount of SP in the synaptosomal peak after depolarization (or maintenance of tecta in low K+ buffer as controls) was measured by radioimmunoassay (Chen et al., 1996). Approximately 25–50% of SP was released by depolarization (Fig. 2H), which is similar to amounts of other neuropeptides released from synaptosomes (25%) (Verhage et al., 1991b). These data confirm the validity of our release protocol by comparison with a classical neuropeptide.
Control experiments with added neurotrophins or cytochrome c
Release of neuropeptides exclusively from terminals is conventionally examined by measuring the release from synaptosomes (Androutsellis-Theotokis et al., 1996) rather than examining the release in intact tissue and then preparing synaptosomes to measure the amount remaining. To determine whether neurotrophins and other small peptides bind to synaptosomes prepared from E17 chick tecta, homogenized tecta were incubated for 15–20 min with radiolabeled neurotrophins or radiolabeled cytochrome c, and the tissue was processed as described. Significant amounts of BDNF and NT-3 accumulated in the synaposomal peak, but very little NGF or cytochromec accumulated (Fig. 2I). To test whether binding of BDNF and NT-3 to synaptosomes was receptor mediated, radiolabeled BDNF and NT-3 were incubated in the presence of 500-fold excess cold same trophic factor. Much of the BDNF (data not shown) and NT-3 (Fig. 2I) bound to synaptosomes in a receptor-mediated manner. These data indicate that NT-3 receptors [presumably tyrosine kinase receptor C (trkC)] are present on tectal terminals (not necessarily retinotectal terminals). These data also show that release of NT-3 from synaptosomes in a static release system likely would fail or grossly underestimate release, because the released NT-3 would bind to receptors present on synaptosomal membranes, thus confounding the results.
Calcium dependence of NT-3 release
To determine whether the depolarization-induced release of NT-3 required physiological concentrations of extracellular calcium,125I-NT-3-loaded tecta were incubated in calcium-free buffer containing the calcium chelator BAPTA. High concentrations of BAPTA (200 μm) abolished the high K+-induced release of NT-3 (Fig.3A), whereas lower concentrations (20 μm) did not (Table2), indicating that the lower concentration of BAPTA was not sufficient to chelate the calcium that may leak from the dissected tecta into the buffer (volume of 3 ml). To determine whether this was indeed the case, calcium was measured in buffers with and without tecta in four conditions (0 μm, 20 μm, and 200 μm BAPTA and 10 μmBAPTA AM) in calcium-free buffers and in addition in normal calcium buffers as shown in Table 2. Calcium leakage from tecta increased the calcium concentration in calcium-free buffers from ∼3 to ∼6 μm, and this increase was not significantly attenuated by 20 μm BAPTA. BAPTA at 200 μm reliably reduced the calcium concentration in these buffers to <1 μm calcium. BAPTA AM at 10 μm did not reduce the extracellular calcium concentration significantly. We will refer to the 2 mm calcium concentration as “normal calcium,” to the 2–10 μm calcium concentration as “low calcium,” and to buffers with ≤ 1 μmcalcium concentration as “calcium-free” buffers. It should be noted that the effective calcium concentration at the nerve terminals within the tectum likely differs from the calcium concentration that was measured in the incubation buffer.
Fig. 3.

Manipulation of NT-3 release from axon terminals by modulation of calcium, other ions, or second messengers.A, High K+-induced release of NT-3 was abolished by BAPTA (BAP), BAPTA/AM (B/AM), cadmium chloride (CAD), ω-conotoxin GVIA (CON), dantrolene (DAN), but not nifedipine (NIF). Release of NT-3 was elicited by thapsigargin (THA), both in high and low K+, and to a lesser extent by 4-aminopyridine (4AP). B, Release of NT-3 was induced by 8-bromo-cAMP (8BR) or forskolin (FOR). High K+-induced release of NT-3 was reduced by the CaM kinase II inhibitor KN-93 (KN93), but not by the inactive analog KN-92 (KN92). Replacing extracellular sodium with N-methyl glucamine (NMG) significantly reduced release of NT-3 induced by high K+. High K+-induced release of NT-3 was unaffected by tetrodotoxin (TTX) treatment. The Na/K ATPase inhibitor strophanthidin (STR) induced release in low K+ buffer. The number of independent experiments is indicated on bars. Error bars indicate SEM. All experimental data points were calibrated in the same batch by obtaining control data without the pharmacological agent. r., release;n.r., no release. Statistical significance compared with LoK (•) or HiK (*) was determined by ANOVA with p ≤ 0.05 (* or •), p ≤ 0.01 (** or ••), andp ≤ 0.001 (*** or •••).
Table 2.
Calcium concentrations in incubation buffers with (+ tect) and without (− tect) a chick optic tectum as a function of the concentration of the calcium chelator BAPTA and resulting release of NT-3 from retinotectal terminals
| Chelator | No Ca2+added to high K+ buffer | Normal Ca2+ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 μmBAPTA | 20 μm BAPTA | 200 μm BAPTA | 10 μm BAPTA AM | 0 μm BAPTA | ||||||
| Condition | − tect | + tect | − tect | + tect | − tect | + tect | − tect | + tect | − tect | + tect |
| [Ca2+] in μm | 3.32 | 6.91 | 2.63 | 4.29 | 0.55 | 0.77 | 4.18 | 5.64 | 2550 | 2760 |
| SEM | 0.30 | 0.75 | 0.44 | 0.95 | 0.073 | 0.067 | 0.15 | 1.28 | 40 | 125 |
| pvalue | p> 0.21 (vs 0 BAPTA) | p ≤ 0.04 (vs 20 BAPTA) | p > 0.51 (vs 20 BAPTA) | |||||||
| Designation | Low calcium | Low calcium | Calcium-free | Low calcium | Normal calcium | |||||
| NT-3 release (%) | N/A | 16.68 | 1.84 | 3.54 | 16.85 | |||||
| SEM | N/A | 3.53 | 2.02 | 2.86 | 2.26 | |||||
| pvalue | p> 0.05 (vs high K, normal Ca) | p ≤ 0.01 (vs high K, normal Ca) | p ≤ 0.05 (vs high K, normal Ca) | p ≤ 0.001 (vs low K, normal Ca) | ||||||
All data are the mean from three to eight determinations with SEM. p values were calculated by unpaired ttest.
To examine whether functional voltage-gated calcium channels were required for depolarization-induced NT-3 release, tecta were incubated in normal calcium buffer containing 100 μm cadmium chloride, a nonselective blocker of voltage-gated Ca2+ channels. Cadmium significantly reduced the K+-evoked NT-3 release (Fig.3A). To determine whether functional N-type calcium channels were required for NT-3 release, tecta were incubated in buffer containing 0.1 μm ω-conotoxin, an N-type Ca2+ channel blocker. ω-Conotoxin significantly reduced NT-3 release (Fig. 3A), consistent with the localization of N-type calcium channels on presynaptic terminals (Westenbroek et al., 1992). Nifedipine (10 μm), a blocker of L-type Ca2+ channels, showed no significant effect on release (Fig. 3A). These data demonstrate that high K+-induced release of NT-3 depends on extracellular calcium, which, during depolarization, enters the synapse via N-type calcium channels.
Release of neurotrophins from brain slices and cultured neurons depends on mobilization of calcium from intracellular stores (Blöchl and Thoenen, 1995; Thoenen, 1995; Griesbeck et al., 1999). To test whether NT-3 release from axon terminals also required intracellular calcium release, tecta were incubated in low calcium buffer containing the membrane-permeable calcium chelator BAPTA AM at 10 μm(calcium concentrations of 4–6 μm) (Table 2). BAPTA AM effectively chelates calcium only after membrane penetration, cleavage by intracellular esterases, and intracellular accumulation (Tsien, 1981). The amount of extracellular calcium (4–6 μm) remaining in the BAPTA AM-treated solution would have been sufficient to allow for the release of NT-3 (compare with 20 μmBAPTA in Table 2). Addition of the relatively small amount of 10 μm BAPTA AM significantly attenuated the release of NT-3 (Fig. 3A). To verify that mobilization of calcium from intracellular stores affects NT-3 release, three experiments were performed. Dantrolene, an inhibitor of calcium release from caffeine-sensitive intracellular stores (Blöchl and Thoenen, 1995), was used at 50 μm in both low calcium and normal calcium high K+ buffer to determine whether it would prevent the release of NT-3. In both low calcium and in normal calcium buffer (as defined in Table 2), dantrolene completely prevented the release of NT-3 (data shown for low calcium in Fig. 3A). These experiments indicate that high K+-induced NT-3 release requires the release of calcium from intracellular stores (calcium-induced calcium release). The role of intracellular calcium was further tested by incubation in 1 mm 4-aminopyridine (4-AP) in normal calcium buffer, which increases intracellular (cytosolic) calcium and has been suggested to cause the release of trophic factors (Grimaldi et al., 2001). 4-AP caused significant NT-3 release (Fig.3A). Finally, to verify that mobilization of calcium from intracellular stores and subsequent increase in intracellular calcium is sufficient to induce release of NT-3, tecta loaded with125I-NT-3 were incubated in high K+ buffer containing 5 μm thapsigargin. Thapsigargin induced a greater release of NT-3 than depolarization alone (Fig. 3A). When thapsigargin was tested in low K+ buffer, the evoked NT-3 release was of a similar magnitude as seen with thapsigargin in high K+ buffer (Fig.3A). It should be noted that thapsigargin acutely induces a transient calcium spike by mobilizing calcium from intracellular stores. This acute effect is distinct from the chronic effects of thapsigargin because of the depletion of intracellular calcium stores that can be seen in dynamic (superfusion) release assays. Such long-term effects of thapsigargin would not be seen in our release assay, because NT-3 is not replenished as shown by colchicine experiments (see below). Such considerations have to be kept in mind when interpreting data obtained with our novel release assay. To verify that the mobilization of calcium from intracellular stores by thapsigargin is immediate and does not require 60 min of incubation, we tested whether NT-3 was released within <10 min incubation. As expected, the effect of thapsigargin on NT-3 release was virtually identical within 10 min (data not shown) and 60 min. Together, these data indicate that mobilization of calcium from intracellular stores is sufficient to trigger release of NT-3 from terminals.
Role of cAMP and CaM kinase II
cAMP has been implicated in the regulation of release of transmitters and neurotrophins (Edwards et al., 1988; Thoenen, 1995;Goodman et al., 1996). To determine whether cAMP is involved in NT-3 release, we used the cAMP analog 8-Br-cAMP and forskolin. Tecta were incubated in low K+ buffer containing 50 μm 8-Br-cAMP. This drug induced NT-3 release (Fig.3B), consistent with the notion that cAMP is involved in the release mechanism. NT-3 release was also induced by 10 μm forskolin, an adenylate cyclase activator (Fig. 3B). Thus, two distinct ways to elevate cAMP levels showed the same result, namely release of NT-3, further supporting the notion that cAMP indeed is involved in the regulation of NT-3 release. Protein kinases regulate synaptic transmission, with the most direct evidence for Ca2+/calmodulin-dependent protein kinase II (CaM kinase II) (Llinas et al., 1991; Kelly, 1993). To test whether CaM kinase II affects NT-3 release, we used the selective inhibitor, KN-93, at doses which primarily abolish the activity of CaM kinase II, but not of other kinases (10 μm) (Sumi et al., 1991). As a control, we used the inactive analog, KN-92 (10 μm). KN-93 significantly reduced high K+-induced NT-3 release, whereas KN-92 had no effect (Fig. 3B). These data indicate that cAMP and CaM kinase II may act downstream of the depolarization induced by high K+.
Role of extracellular sodium
The role of extracellular sodium for the release of neurotrophins is controversial. Initial studies on the release of neurotrophins concluded that extracellular sodium is required for regulated release of neurotrophins from dendritic and somal compartments (Blöchl and Thoenen, 1995), but this may have been attributable to an independent blocking effect of the sodium substitute,N-methyl-d-glucamine (NMG) (Hoener, 2000). Release of neuropeptides from terminals (rather than whole cells) does seem to require sodium influx (Stuenkel and Nordmann, 1993; Thirion et al., 1999). To determine whether low extracellular sodium may attenuate NT-3 release from axon terminals,125I-NT-3 loaded tecta were incubated in buffer containing high K+, and NMG to replace > 90% of the extracellular sodium. As seen in Figure3B, replacement of sodium reduced the release of NT-3. When NMG was washed out and the buffer replaced with normal sodium, high K+ buffer induced NT-3 release (data not shown), indicating that the effect of NMG was reversible. To determine whether the opposite treatment, increased influx of sodium rather than decreased influx as with NMG, will have the opposite effect on NT-3 release, we used strophanthidin, a sodium-potassium ATPase inhibitor (Meyer and Cooper, 1981). As expected, incubation of tecta with 500 μm strophanthidin induced release of NT-3 (Fig.3B). Release was not attenuated by incubation with tetrodotoxin (TTX, 3 μm) (Fig. 3B), indicating that TTX-sensitive sodium channels are not required for the release process when induced by high K+. Together, these results may suggest that an unconventional sodium-dependence of neurotrophin release may be a general feature for neuropeptide release from axon terminals (Stuenkel and Nordmann, 1993;Thirion et al., 1999).
Structural proteins involved in NT-3 release
Microtubules and microfilaments have been implicated in the movement of vesicles from the axon shaft to the membrane at nerve terminals for exocytosis (Westrum et al., 1983; Hirokawa et al., 1989;Ashton and Dolly, 1991). Microfilaments are thought to act as a barrier to small synaptic vesicle (SSV) fusion, but they may aid in the translocation of LDCVs (Doussau and Augustine, 2000) and LDCVs are known to concentrate outside the active zone (Verhage et al., 1991a). To determine whether microfilaments are required for NT-3 release, tecta were incubated with 0.2 μm cytochalasin D. This drug significantly reduced NT-3 release (Fig.4A). Microtubules are present in tectal nerve terminals (Bird, 1989). To determine whether intact microtubules are required for evoked NT-3 release,125I-NT-3 loaded tecta were incubated in high K+ buffer containing 175 μm colchicine. Colchicine abolished the release of NT-3 (Fig. 4A), indicating that intact microtubules are necessary for evoked NT-3 release from axon terminals. The colchicine experiments also show that there is no significant replenishment of NT-3 from axons into terminals during the 60 min release period. If the released NT-3 was rapidly replenished by anterograde flow from the axon, one would not expect a net loss of NT-3 in terminals. Because colchicine blocks all anterograde axonal transport of NT-3 (von Bartheld et al., 1996), we can exclude this possibility.
Fig. 4.

Manipulation of NT-3 release from axon terminals with toxins to structural proteins and examination of upstream and downstream events. A, High K+-induced release of NT-3 (HiK) was abolished by cytochalasin D (CYD), colchicine (COL), and pretreatment with tetanus toxin (TET). Tetanus toxin was targeted specifically to retinotectal terminals by injection of 1 μg of tetanus toxin in the eye 24 hr before NT-3 injection. B, Autoradiography shows that radiolabeled tetanus toxin (125I-TET) was transported anterogradely by retinal ganglion cells to the optic tectum. Scale bar, 500 μm. C, Anterograde transport of125I-NT-3 was not reduced by doses of TET (1–2 μg) in the eye that were used to target the toxin to retinotectal terminals.CTL, control. D, Simultaneous treatment withN-methyl-glucamine (NMG) and thapsigargin (THA) did not reduce the amount of NT-3 that was released, indicating that sodium influx may be upstream of THA action. Cotreatment with BAPTA (BAP) in calcium-free buffer did not attenuate NT-3 release. Simultaneous treatment with colchicine (COL) and THA significantly reduced the release of NT-3, indicating that COL was acting downstream of THA. The number of independent experiments is indicated on each bar. Error bars indicate SEM. Statistical significance compared with LoK (•), HiK (*) THA only (§, D) was determined by ANOVA with p≤ 0.05 (*, §), p ≤ 0.01 (**), andp ≤ 0.001 (*** or •••).
Role of synaptobrevin/synaptobrevin analogs in NT-3 release
Synaptobrevin and synaptobrevin analogs are required for release of neurotrophins from cell bodies and/or dendrites (Blöchl, 1998). Synaptobrevin can be cleaved by tetanus toxin (Schiavo et al., 2000). To determine whether tetanus toxin treatment affects NT-3 release from terminals, tecta were pretreated by intraocular injection of the whole tetanus toxin (1–2 μg) which is subsequently transported anterogradely to the retinotectal terminals (Manning et al., 1990). To verify this mode of drug delivery, 300 ng of radiolabeled tetanus toxin C-fragment was injected into the eye of chick embryos. Within 24–48 hr, 1 ng of the toxin accumulated in the retinotectal axons and terminals (Fig. 4B). Subsequent anterograde transport of125I-NT-3 to the tectum was not affected by intraocular injection of 1–2 μg tetanus toxin (Fig.4C). Tetanus toxin in the retinotectal terminals (estimated 5 ng/tectum) reduced the release of NT-3 from tectal terminals (Fig.4A). This suggests that intact synaptobrevin (or synaptobrevin analogs) are required for the release of NT-3 from axon terminals, and that at least some of the release is accomplished by synaptobrevin-mediated vesicle trafficking and/or exocytosis.
Requirements for the effect of thapsigargin on NT-3 release
Are extracellular sodium, calcium, or intact microtubules required for the effect of thapsigargin? When tecta were cotreated with NMG and thapsigargin, there was no significant reduction in the amount of NT-3 that was released (Fig. 4D), indicating that the effect of thapsigargin did not require extracellular sodium. Thapsigargin did not require the influx of extracellular calcium for triggering release, because incubation in calcium-free solution containing 200 μm BAPTA did not reduce the magnitude of release (Fig. 4D). When colchicine treatment was combined with thapsigargin, there was a significant reduction in the amount of NT-3 that was released (Fig.4D), indicating that colchicine may act downstream of the mobilization of calcium from internal stores. These data indicate that mobilization of calcium from intracellular stores is sufficient to trigger NT-3 release, but requires intact microtubules, presumably for vesicle translocation (Doussau and Augustine, 2000).
Kainic acid induces NT-3 release in vivo
Kainic acid and tetrodotoxin increase and decrease, respectively, the electrical activity of RGCs (Catsicas and Clarke, 1987; Karlsson and Hallböök, 1998). To determine whether stimulation of RGCs in vivo induces release of NT-3 similar to the depolarization of retinotectal terminals in vitro, 1 μg kainic acid or PBS vehicle control was injected in the eyes of chick embryos in ovo, 19 hr after intraocular injection of125I-NT-3. The final concentration of kainic acid in the eye (≈ 80 μl volume) was estimated to be 62 μm. Animals were killed 45 min after the intraocular injection of kainic acid, and their tecta were processed immediately for synaptosomal fractionation. Kainic acid induced the release of ∼30% of the exogenous NT-3 present in the terminals (Fig.5). To determine whether this effect of kainic acid could be blocked by treatment with tetrodotoxin (TTX), the eyes of embryos were pretreated with TTX (0.16 μg injected, final concentration estimated at 6.25 μm), 20 min before the injection of kainic acid. TTX treatment abolished the effect of kainic acid (Fig. 5B). These data confirm in an in vivo experiment the results from our exposure to drugs by bath application of whole tecta.
Fig. 5.

Kainic acid induced release of NT-3 from axon terminals in vivo. Injection of kainic acid (KA) in the eye increased the amount of NT-3 that was released from retinotectal terminals compared with PBS-treated controls (+PBS). The KA-induced release of NT-3 was prevented by injection of tetrodotoxin (TTX) in the eye 15 min before KA injection when compared with PBS injections as a control for TTX. The number of independent experiments is indicated on each bar. Error bars indicate SEM. Statistical significance compared with +PBS(••) or the KA+PBS condition (§§) was determined by ANOVA withp ≤ 0.01 (••, §§).
Is NT-3 packaged in large dense core vesicles?
Our ultrastructural autoradiography localized125I-NT-3 in the vicinity of large dense core vesicles (LDCVs) (Fig. 1E), but the resolution of the 125I-autoradiography was not sufficient for unambiguous identification of the types of vesicles which contain NT-3 in presynaptic terminals. Therefore, vesicles of different sizes were purified by differential sedimentation in sucrose gradients (Huttner et al., 1983; Han and Fischbach, 1999). Vesicle purification showed that only the anterogradely transported125I-NT-3 accumulated in fractions known to contain LDCVs (Han and Fischbach, 1999), but not the125I-NT-3 that was incubated with synaptosomes (Fig. 6A). After incubation, 125I-NT-3 was marginally increased in fractions containing SSVs. Because the LDCVs are heterogeneous in size and the fractions containing LDCVs also contain endosomes, these data do not prove that the anterogradely transported NT-3 is exclusively associated with LDCVs, but our data are consistent with this notion. When the superficial layer of the optic tectum of E17 chick embryos was immunolabeled for endogenous NT-3 and examined at the electron microscopic level, immunolabel was detected in LDCVs (Fig.6B). We estimate that ∼25% of terminals in retinorecipient layers of the tectum contained LDCVs, and ∼5–6% of such terminals contained LDCVs with NT-3-like immunoreactivity. No labeled LDCVs were detected when the primary antibody was omitted (data not shown). If 125I-NT-3 was present in LDCVs within retinal axons, one would expect to see silver grains over LDCVs in the stratum opticum (SO) within the tectum. Indeed, we found silver grains over and adjacent to dense-cored vesicles within axons of the SO (Fig. 6C). Because of the relatively small size of the LDCVs and the fact that many silver grains may obscure them, we did not attempt to quantify the correlation of silver grains and LDCVs. Taken together, these results further support the idea that anterogradely transported neurotrophins are packaged in LDCVs.
Fig. 6.
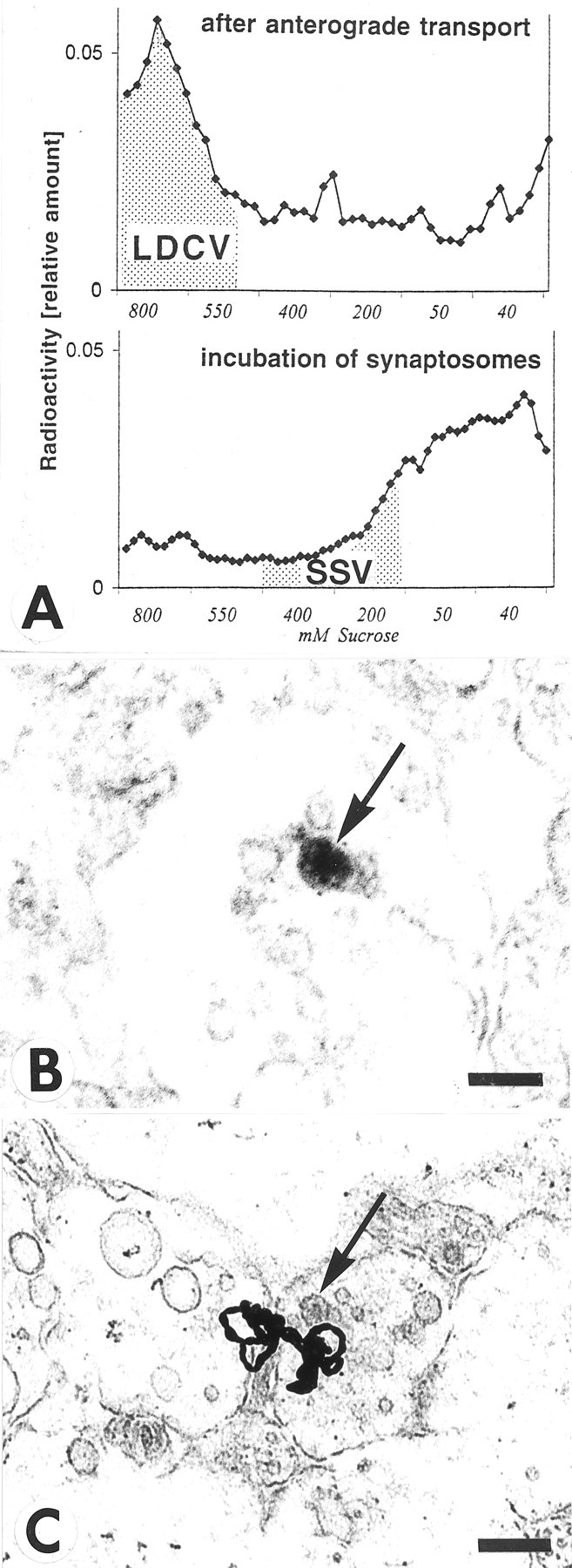
Association of NT-3 with large dense core vesicles (LDCV) rather than small synaptic vesicles (SSV) in retinotectal terminals. A, Vesicle purification of tecta containing anterogradely transported125I-NT-3 shows higher levels of radioactivity in sedimentation fractions known to contain LDCVs in chick embryo brains (Han and Fischbach, 1999), but not in fractions known to contain SSVs (top panel). When homogenized tecta were incubated with125I-NT-3 (bottom panel), radioactivity was seen in sedimentation fractions known to accumulate SSVs (Han and Fischbach, 1999). B, Section through a retinotectal terminal from a 17-d-old chick embryo immunolabeled with an antibody to NT-3, processed with horseradish-peroxidase, and visualized with diaminobenzidine. Note reaction product in a large dense core vesicle (arrow). Scale bar, 100 nm. C, Section through the stratum opticum (SO) of the optic tectum after injection of125I-NT-3 in the eye. Note a cluster of silver grains in the vicinity of a large dense core vesicle (arrow) in sectioned retinotectal fibers. Scale bar, 500 nm.
DISCUSSION
Our study provides first direct evidence for mechanisms of release of anterogradely transported neurotrophins from axon terminals. We found that NT-3 is released by depolarization, that this release depends on intracellular and extracellular calcium, and can be evoked by mobilization of calcium from intracellular stores. Release is further regulated by cAMP and CaM kinase II. We show that release occurs by exocytosis of presumptive large dense-core vesicles (LDCVs). Anterogradely transported and presynaptically located neurotrophins are released by changes in local, intracellular calcium concentrations.
Technical considerations
Previous studies used immunological techniques to measure the amounts of neurotrophins released from entire cells (cell bodies, dendrites and axons) into medium or perfusate (Blöchl and Thoenen, 1995; Goodman et al., 1996), but this technology was not sufficiently sensitive to measure the excessively small amounts (molecules per synapse) exclusively released from axon terminals. Therefore, it is important to consider whether we accurately measured the amount of 125I-NT-3 in retinotectal terminals by synaptosomal fractionation and gamma counting. When synaptosomal measurements were compared with quantitative analysis of the in situ distribution of silver grains after autoradiography at the ultrastructural level, the morphological and the biochemical/pharmacological data showed a remarkable agreement for the estimates of exogenous NT-3 in axon terminals (5.48% vs. 5.94%).
Another important issue is whether anterogradely transported exogenous NT-3 is released by the same mechanisms as anterogradely transportedendogenous NT-3. Do endogenous and exogenous neurotrophins pass through the same organelles? RGCs in chicks express NT-3, and endogenous NT-3 is anterogradely transported to the terminals in the optic tectum (von Bartheld and Butowt, 2000). Proteins destined for anterograde transport have to be processed in the Golgi system (Hammerschlag et al., 1982). Exogenous NT-3 (but very little BDNF) accumulates in the Golgi system of RGCs (Butowt and von Bartheld, 2001;von Bartheld et al., 2001) and transcytoses RGCs in a receptor-mediated manner, possibly by sequential binding to trkC and p75 receptors (Butowt and von Bartheld, 2001). Therefore, it is likely that exogenous NT-3 joins the pathway normally taken by endogenous NT-3. Alternatively, there may be parallel pathways of release, one for newly synthesized NT-3, and one for transcytosed NT-3 (von Bartheld et al., 2001).
Our release assay has significant advantages (sensitivity and synapse specificity) as well as limitations compared with conventional release assays. It requires sufficient internalization, anterograde axonal transport, and accumulation of the protein in presynaptic terminals, and it is not a dynamic assay with replenishment of the source. Nevertheless, its utility is not necessarily restricted to the retinotectal system or to neurotrophins.
Molecular determinants of NT-3 release
Our results discern 3 major steps that lead to the release of NT-3 from axon terminals (Fig. 7). These conclusions are based on data obtained with a variety of pharmacological agents at concentrations commonly used for tissue slices. The first step involves depolarization of the axon terminus with influx of calcium through N-type calcium channels. The second step may be induced by calcium influx and/or mobilization of calcium from intracellular stores. Increased calcium concentrations in the presynaptic compartment (Verhage et al., 1991a), as well as kinase activation (cyclic AMP-dependent kinases and CaM kinase II), may provide signals for vesicle translocation via microtubules to the active zone at the synaptic cleft. The vesicle then fuses with the membrane, presumably mediated by synaptobrevin, because release is tetanus-toxin-sensitive. The neurotrophin is released and may diffuse in the synaptic cleft to bind to presynaptic and/or postsynaptic neurotrophin receptors (Levine et al., 1995; Wu et al., 1996; Kafitz et al., 1999; von Bartheld et al., 2001). The role of sodium influx for neurotrophin release from whole cells is controversial (Blöchl and Thoenen, 1995; Hoener, 2000). We find that NT-3 release from terminals requires sodium influx, apparently via TTX-insensitive sodium channels, consistent with reports of TTX-insensitive, sodium-evoked release of neuropeptides from axon terminals (Stuenkel and Nordmann, 1993) and sodium influx into docked vesicles (Thirion et al., 1999). Sodium influx may also interact with mobilization of calcium from internal stores (Berridge, 1998).
Fig. 7.

Diagram summarizes the proposed mechanisms of release of anterogradely transported NT-3 from axon terminals. Depolarization by high potassium (K+) opens N-type calcium channels. Calcium influx induces calcium release from internal stores. Increased intraterminal calcium concentrations are sufficient to induce release of NT-3 containing vesicles. Release can also be induced by cAMP and requires CaM kinase II activity. Second messengers and kinases may act directly on vesicle release or indirectly via calcium mobilization from internal stores (Hille et al., 1999). Translocation of the NT-3-containing vesicle requires intact microtubules and microfilaments, and fusion of the vesicle depends on synaptobrevin or synaptobrevin-like molecules. Once released, NT-3 diffuses across the synaptic cleft, binds to trkC receptors, is internalized, and accumulates postsynaptically in multivesicular bodies (MVB).
Release from different compartments is regulated by different mechanisms
Potassium-stimulated release of neuropeptides is typically mediated by activation of L-type calcium channels (Trimble et al., 1991; Meir et al., 1999), but release from terminals is often mediated via N-type channels, consistent with their localization in terminals (Westenbroek et al., 1992; Simmons et al., 1995). Our study is the first to examine mechanisms of the release of anterogradely transported neurotrophins. Previous studies on the release of neurotrophins could not discriminate between different compartments (dendrites, soma, and axons) (Blöchl and Thoenen, 1995; Goodman et al., 1996;Blöchl, 1998; Griesbeck et al., 1999), or the exogenous neurotrophin was loaded to unidentified compartments within synaptosomes (Androutsellis-Theotokis et al., 1996). The release from dendritic/somal compartments showed a constitutive and regulated release, with the constitutive release primarily restricted to the soma and proximal dendrites (Blöchl and Thoenen, 1995, 1996; Goodman et al., 1996). In contrast, anterogradely transported NT-3 from retinotectal terminals was not released constitutively, or such release was below the detection limit of our assay. Release of neurotrophins from cell bodies/dendrites was induced by mobilization of calcium from intracellular stores, and appeared to require extracellular calcium only when examined in a static release system, but not in a perfusion release system (Griesbeck et al., 1999). Release of NT-3 from axon terminals was regulated by intracellular calcium, but was also triggered by influx of extracellular calcium (present study) (Fig. 3), similar to the release of anterogradely transported IGF-I (Nieto-Bona et al., 1993). It is not clear whether targeting and release differs between neurotrophins and whether overexpression may change release mechanisms attributable to rate-limiting sorting steps (Mowla et al., 1999; Farhadi et al., 2000), or whether all neurotrophins are processed and released similarly (Blöchl and Thoenen, 1995; Goodman et al., 1996; Heymach et al., 1996; Canossa et al., 1997; Griesbeck et al., 1999).
Localization of anterogradely transported neurotrophins in presynaptic organelles
Because of the low levels of expression and small number of molecules which are anterogradely transported, it has been difficult to determine in which types of vesicles (if any) the anterogradely transported neurotrophins are packaged. BDNF has been localized to LDCVs in terminals of the spinal cord (Michael et al., 1997; Holstege et al., 1999) and to LDCV fractions of the hippocampus after vesicle purification (Fawcett et al., 1997). Anterogradely transported NT-3 did not accumulate within small synaptic vesicles in retinotectal terminals, rather, our data indicate that NT-3 was associated with LDCVs (Figs. 1E, 6A–C). Thus, increasing evidence indicates that anterogradely transported neurotrophins are contained within LDCVs and are released by conventional exocytosis.
Amount of released exogenous NT-3 is a few molecules/synapse
The amount of exogenous NT-3 that was transferred from a presynaptic to a postsynaptic site in retinotectal terminals during depolarization was calculated to be ∼1–1.2 pg. The number of RGCs in the chick embryo at E16–17 is 2–3 million (Rager, 1980), the approximate average number of axon branches of individual RGCs in the tectum is 20 at this age (Thanos and Bonhoeffer, 1987), and thus the number of RGC terminals in the tectum, and the number of released NT-3 molecules can be estimated. On average, one RGC terminal released ∼0.5 molecules 125I-NT-3, assuming that all RGCs transported NT-3 anterogradely, and that NT-3 was distributed among all RGC terminals. This appears to be a small amount, but one molecule of NT-3 in the synaptic cleft (10×20×200 nm3 = 40,000 nm3) is equivalent to a concentration of 40 μm. This concentration is ∼4 orders of magnitude higher than what is thought to be sufficient for saturation of neurotrophin receptors. Thus, a small number of molecules/synaptic cleft may be sufficient for activation of postsynaptic trk receptors.
Implications of release mechanisms for an understanding of synaptic plasticity
Neurotrophins also stimulate the release of neurotrophins (Canossa et al., 1997; Krüttgen et al., 1998). In the retinotectal system, BDNF is expressed and upregulated by increased neural activity (Karlsson and Hallböök, 1998). BDNF is internalized by RGC axons and retrogradely transported from the tectum to the retina (Herzog and von Bartheld, 1998). Besides a direct effect of NT-3 on axon branching (Inoue and Sanes, 1997) and synapse formation (Martinez et al., 1998), release of NT-3 from retinotectal presynaptic terminals may also trigger subsequent BDNF release from postsynaptic tectal neurons, and thereby stabilize functional synapses (Snider and Lichtman, 1996; Canossa et al., 1997; Boulanger and Poo, 1999; Wang et al., 2000; Poo, 2001; von Bartheld et al., 2001). Recent studies have shifted focus on the source of neurotrophins in the synaptic cleft to include a presynaptic release and a postsynaptic transfer of neurotrophins (Kafitz et al., 1999; Poo, 2001; Lever et al., 2001). The technology developed in the present report will help to elucidate the mechanisms of release of neurotrophins from axon terminals and help to define their roles as extremely potent excitatory neurotransmitters as well as neuromodulators in synaptic plasticity.
Footnotes
This work was supported by National Institutes of Health Grants TW 05700 (R.B.), NS 34159 (M.R.V.), HD 29177, and NS 35931 (C.S.v.B.). We thank Regeneron Inc. (Tarrytown, NY) for neurotrophins and Jared Baeten and Anish Sudra for technical help. NT-3 antibodies were generous gifts from Robert Rush and SV2 antibodies from Steve Carlsson. Bomie Han kindly shared unpublished data and protocols. We thank Don Bers, Mark Bothwell, Jim Kenyon, and John Sutko for helpful comments and discussions.
Correspondence should be addressed to Christopher von Bartheld, Department of Physiology and Cell Biology, Mailstop 352, University of Nevada School of Medicine, Reno, NV 89557. E-mail:chrisvb@physio.unr.edu.
REFERENCES
- 1.Aloyz R, Fawcett JP, Kaplan DR, Murphy RA, Miller FD. Activity-dependent activation of TrkB neurotrophin receptors in the adult CNS. Learn Mem. 1999;6:216–231. [PMC free article] [PubMed] [Google Scholar]
- 2.Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- 3.Androutsellis-Theotokis A, McCormack WJ, Bradford HF, Stern GM, Pliego-Rivero FB. The depolarisation-induced release of [125I]BDNF from brain tissue. Brain Res. 1996;743:40–48. doi: 10.1016/s0006-8993(96)00981-x. [DOI] [PubMed] [Google Scholar]
- 4.Ashton AC, Dolly JO. Microtubule-dissociating drugs and A23187 reveal differences in the inhibition of synaptosomal transmitter release by botulinum neurotoxins types A and B. J Neurochem. 1991;56:827–835. doi: 10.1111/j.1471-4159.1991.tb01998.x. [DOI] [PubMed] [Google Scholar]
- 5.Bartfai T, Iverfeldt K, Fisone G, Serfözö P. Regulation of the release of coexisting neurotransmitters. Annu Rev Pharmacol Toxicol. 1988;28:285–310. doi: 10.1146/annurev.pa.28.040188.001441. [DOI] [PubMed] [Google Scholar]
- 6.Baudet S, Hove-Madsen L, Bers DM. How to make and use calcium-specific mini- and microelectrodes. Methods Cell Biol. 1994;40:93–113. [PubMed] [Google Scholar]
- 7.Berninger B, Poo MM. Exciting neurotrophins. Nature. 1999;401:862–863. doi: 10.1038/44727. [DOI] [PubMed] [Google Scholar]
- 8.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 9.Bird MM. Microtubules and their relationships with other cytoskeletal components at cholinergic tectal synapses in culture. J Anat. 1989;166:1–6. [PMC free article] [PubMed] [Google Scholar]
- 10.Blöchl A. SNAP-25 and syntaxin, but not synaptobrevin 2, cooperate in the regulated release of nerve growth factor. NeuroReport. 1998;9:1701–1705. doi: 10.1097/00001756-199806010-00006. [DOI] [PubMed] [Google Scholar]
- 11.Blöchl A, Thoenen H. Characterization of nerve growth factor (NGF) release from hippocampal neurons: evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur J Neurosci. 1995;7:1220–1228. doi: 10.1111/j.1460-9568.1995.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 12.Blöchl A, Thoenen H. Localization of cellular storage compartments and sites of consitutive and activity-dependent release of nerve growth factor (NGF) in primary cultures of hippocampal neurons. Mol Cell Neurosci. 1996;7:173–190. doi: 10.1006/mcne.1996.0014. [DOI] [PubMed] [Google Scholar]
- 13.Boulanger L, Poo MM. Presynaptic depolarization facilitates neurotrophin-induced synaptic potentiation. Nat Neurosci. 1999;2:346–351. doi: 10.1038/7258. [DOI] [PubMed] [Google Scholar]
- 14.Butowt R, von Bartheld CS. Sorting of internalized neurotrophins into an endocytic transcytosis pathway via the Golgi system: ultrastructural analysis in retinal ganglion cells. J Neurosci. 2001;21:8915–8930. doi: 10.1523/JNEUROSCI.21-22-08915.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canossa M, Griesbeck O, Berninger B, Campana G, Kolbeck R, Thoenen H. Neurotrophin release by neurotrophins: implications for activity-dependent neuronal plasticity. Proc Natl Acad Sci USA. 1997;94:13279–13286. doi: 10.1073/pnas.94.24.13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Catsicas S, Clarke PGH. Spatiotemporal gradients of kainate-sensitivity in the developing chicken retina. J Comp Neurol. 1987;262:512–522. doi: 10.1002/cne.902620405. [DOI] [PubMed] [Google Scholar]
- 17.Chen JJ, Barber LA, Dymshitz J, Vasko MR. Peptidase inhibitors improve recovery of substance P and calcitonin gene-related peptide release from rat spinal cord slices. Peptides. 1996;17:31–37. doi: 10.1016/0196-9781(95)02091-8. [DOI] [PubMed] [Google Scholar]
- 18.Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat brain: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doussau F, Augustine GJ. The actin cytoskeleton and neurotransmitter release: an overview. Biochimie. 2000;82:353–363. doi: 10.1016/s0300-9084(00)00217-0. [DOI] [PubMed] [Google Scholar]
- 20.Edwards RH, Selby MJ, Mobley WC, Weinrich SL, Hruby DE, Rutter WJ. Processing and secretion of nerve growth factor: expression in mammalian cells with a vaccinia virus vector. Mol Cell Biol. 1988;8:2456–2464. doi: 10.1128/mcb.8.6.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farhadi HF, Mowla SJ, Petrecca K, Morris SJ, Seidah NG, Murphy RA. Neurotrophin-3 sorts to the constitutive secretory pathway of hippocampal neurons, is diverted to the regulated secretory pathway by coexpression with brain-derived neurotrophic factor. J Neurosci. 2000;20:4059–4068. doi: 10.1523/JNEUROSCI.20-11-04059.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fawcett JP, Aloyz R, McLean JH, Pareek S, Miller FD, McPherson PS, Murphy RA. Detection of brain-derived neurotrophic factor in a vesicular fraction of brain synaptosomes. J Biol Chem. 1997;272:8837–8840. doi: 10.1074/jbc.272.14.8837. [DOI] [PubMed] [Google Scholar]
- 23.Fawcett JP, Alonso-Vanegas MA, Morris SJ, Miller FD, Sadikot AF, Murphy RA. Evidence that brain-derived neurotrophic factor from presynaptic nerve terminals regulates the phenotype of calbindin-containing neurons in the lateral septum. J Neurosci. 2000;20:274–282. doi: 10.1523/JNEUROSCI.20-01-00274.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- 25.Griesbeck O, Canossa M, Campana G, Gartner A, Hoener MC, Nawa H, Kolbeck R, Thoenen H. Are there differences between the secretion characteristics of NGF and BDNF? Implications for the modulatory role of neurotrophins in activity-dependent neuronal plasticity. Microsc Res Tech. 1999;45:262–275. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<262::AID-JEMT10>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 26.Grimaldi M, Atzori M, Ray P, Alkon DL. Mobilization of calcium from intracellular stores, potentiation of neurotransmitter-induced calcium transients, and capacitative calcium entry by 4-aminopyridine. J Neurosci. 2001;21:3135–3143. doi: 10.1523/JNEUROSCI.21-09-03135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamburger V, Hamilton H. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- 28.Hammerschlag R, Stone GC, Bolen FA, Lindsey JD, Ellisman MH. Evidence that all newly synthesized proteins destined for axonal transport pass through the Golgi apparatus. J Cell Biol. 1982;93:568–575. doi: 10.1083/jcb.93.3.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han B, Fischbach GD. Processing of ARIA and release from isolated nerve terminals. Philos Trans R Soc Lond B Biol Sci. 1999;354:411–416. doi: 10.1098/rstb.1999.0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herzog KH, von Bartheld CS. Contributions of the optic tectum and the retina as sources of brain-derived neurotrophic factor for retinal ganglion cells in the chick embryo. J Neurosci. 1998;18:2891–2906. doi: 10.1523/JNEUROSCI.18-08-02891.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heymach JV, Jr, Barres BA. Neurotrophins moving forward. Nature. 1997;389:789–791. doi: 10.1038/39743. [DOI] [PubMed] [Google Scholar]
- 32.Heymach JV, Jr, Kruttgen A, Suter U, Shooter EM. The regulated secretion and vectorial targeting of neurotrophins in neuroendocrine and epithelial cells. J Biol Chem. 1996;271:25430–25437. doi: 10.1074/jbc.271.41.25430. [DOI] [PubMed] [Google Scholar]
- 33.Hille B, Billiard J, Babcock DF, Nguyen T, Koh DS. Stimulation of exocytosis without a calcium signal. J Physiol (Lond) 1999;520:23–31. doi: 10.1111/j.1469-7793.1999.00023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirokawa N, Sobue K, Kanda K, Harada A, Yorifuji H. The cytoskeletal architecture of the presynaptic terminal and molecular structure of synapsin 1. J Cell Biol. 1989;108:111–126. doi: 10.1083/jcb.108.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoener MC. Role played by sodium in activity-dependent secretion of neurotrophins—revisited. Eur J Neurosci. 2000;12:3096–3106. doi: 10.1046/j.1460-9568.2000.00190.x. [DOI] [PubMed] [Google Scholar]
- 36.Holstege JC, Rooijen-Boot AV, Jongen JLM, Haasdijk E, Neuteboom RF, Vecht CJ. Localization of BDNF and GDNF protein in rat spinal cord using light and electron microscopy immunocytochemistry. Soc Neurosci Abstr. 1999;25:1272. [Google Scholar]
- 37.Huttner WB, Schiebler W, Greengard P, De Camilli P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoue A, Sanes JR. Lamina-specific connectivity in the brain: regulation by N-cadherin, neurotrophins, and glycoconjugates. Science. 1997;276:1428–1431. doi: 10.1126/science.276.5317.1428. [DOI] [PubMed] [Google Scholar]
- 39.Kafitz KW, Rose CR, Thoenen H, Konnerth A. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature. 1999;401:918–921. doi: 10.1038/44847. [DOI] [PubMed] [Google Scholar]
- 40.Kang HJ, Schuman EM. Long-lasting enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 41.Karlsson M, Hallböök F. Kainic acid, tetrodotoxin and light modulate expression of brain-derived neurotrophic factor in developing avian retinal ganglion cells and their tectal target. Neuroscience. 1998;83:137–150. doi: 10.1016/s0306-4522(97)00340-0. [DOI] [PubMed] [Google Scholar]
- 42.Kasai H. Comparative biology of Ca2+-dependent exocytosis: implications of kinetic diversity for secretory function. Trends Neurosci. 1999;22:88–93. doi: 10.1016/s0166-2236(98)01293-4. [DOI] [PubMed] [Google Scholar]
- 43.Kelly RB. Storage and release of neurotransmitters. Cell [Suppl] 1993;72:43–53. doi: 10.1016/s0092-8674(05)80027-3. [DOI] [PubMed] [Google Scholar]
- 44.Kim HG, Wang T, Olafsson P, Lu B. Neurotrophin 3 potentiates neuronal activity and inhibits gamma-aminobuty ratergic synaptic transmission in cortical neurons. Proc Natl Acad Sci USA. 1994;91:12341–12345. doi: 10.1073/pnas.91.25.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kokaia M, Asztely F, Olofsdotter K, Sindreu CB, Kullmann DM, Lindvall O. Endogenous neurotrophin-3 regulates short-term plasticity at lateral perforant path-granule cell synapses. J Neurosci. 1998;18:8730–8739. doi: 10.1523/JNEUROSCI.18-21-08730.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krüttgen A, Möller JC, Heymach JV, Jr, Shooter EM. Neurotrophins induce release of neurotrophins by the regulated secretory pathway. Proc Natl Acad Sci USA. 1998;95:9614–9619. doi: 10.1073/pnas.95.16.9614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lembeck F, Mayer N, Schindler G. Substance P in rat brain synaptosomes. Naunyn Schmiedebergs Arch Pharmacol. 1977;301:17–22. doi: 10.1007/BF00501259. [DOI] [PubMed] [Google Scholar]
- 48.Lever IJ, Bradbury EJ, Cunningham JR, Adelson DW, Jones MG, McMahon SB, Marvizon JC, Malcangio M. Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J Neurosci. 2001;21:4469–4477. doi: 10.1523/JNEUROSCI.21-12-04469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances neurotransmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linden R. The survival of developing neurons. A review of afferent control. Neuroscience. 1994;58:671–682. doi: 10.1016/0306-4522(94)90447-2. [DOI] [PubMed] [Google Scholar]
- 51.Lindholm D, Castrén E, Tsoulfas P, Kolbeck R, da Penha Berzaghi M, Leingärtner A, Heisenberg C-P, Tesarollo L, Parada LF, Thoenen H. Neurotrophin-3 induced by tri-iodothyronine in cerebellar granule cells promotes Purkinje cell differentiation. J Cell Biol. 1993;122:443–450. doi: 10.1083/jcb.122.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Llinas R, Gruner JA, Sugimori M, McGuinness TL, Greengard P. Regulation by synapsin I and Ca2+-calmodulin-dependent protein kinase II of the transmitter release in squid giant synapse. J Physiol (Lond) 1991;436:257–282. doi: 10.1113/jphysiol.1991.sp018549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lo DC. Neurotrophic factors and synaptic plasticity. Neuron. 1995;15:979–981. doi: 10.1016/0896-6273(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 54.Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 55.Manning KA, Erichsen JT, Evinger C. Retrograde transneuronal transport properties of fragment C of tetanus toxin. Neuroscience. 1990;34:251–263. doi: 10.1016/0306-4522(90)90319-y. [DOI] [PubMed] [Google Scholar]
- 56.Martinez A, Alcantara S, Borrell V, Del Rio JA, Blasi J, Otal R, Campos N, Boronat A, Barbacid M, Silos-Santiago I, Soriano E. TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J Neurosci. 1998;18:7336–7350. doi: 10.1523/JNEUROSCI.18-18-07336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 58.Meir A, Ginsburg S, Butkevich A, Kachalsky SG, Kaiserman I, Ahdut R, Demirgoren S, Rahamimoff R. Ion channels in presynaptic nerve terminals and control of transmitter release. Physiol Rev. 1999;79:1019–1088. doi: 10.1152/physrev.1999.79.3.1019. [DOI] [PubMed] [Google Scholar]
- 59.Meyer EM, Cooper JR. Correlations between Na+-K+ ATPase activity and acetylcholine release in rat cortical synaptosomes. J Neurochem. 1981;36:467–475. doi: 10.1111/j.1471-4159.1981.tb01616.x. [DOI] [PubMed] [Google Scholar]
- 60.Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DL, Yan Q, Priestley JV. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci. 1997;17:8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mowla SJ, Pareek S, Farhadi HF, Petrecca K, Fawcett JP, Seidah NG, Morris SJ, Sossin WS, Murphy RA. Differential sorting of nerve growth factor and brain-derived neurotrophic factor in hippocampal neurons. J Neurosci. 1999;19:2069–2080. doi: 10.1523/JNEUROSCI.19-06-02069.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nieto-Bona MP, Garcia-Segura LM, Torres-Aleman I. Orthograde transport and release of insulin-like growth factor I from the inferior olive to the cerebellum. J Neurosci Res. 1993;36:520–527. doi: 10.1002/jnr.490360504. [DOI] [PubMed] [Google Scholar]
- 63.Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- 64.Rager GH. Development of the retino-tectal projection in the chicken. Adv Anat Embryol Cell Biol. 1980;63:1–92. [PubMed] [Google Scholar]
- 65.Schiavo G, Matteoli M, Montecucco C. Neurotoxins affecting neuroexocytosis. Physiol Rev. 2000;80:717–766. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- 66.Schinder AJ, Poo MM. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23:639–645. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- 67.Simmons ML, Terman GW, Gibbs SM, Chavkin C. L-type calcium channels mediate dynorphin neuropeptide release from dendrites but not axons of hippocampal granule cells. Neuron. 1995;14:1265–1272. doi: 10.1016/0896-6273(95)90273-2. [DOI] [PubMed] [Google Scholar]
- 68.Smith MA, Zang LX, Lyons WE, Mamounas LA. Anterograde transport of endogenous brain-derived neurotrophic factor in hippocampal mossy fibers. NeuroReport. 1997;8:1829–1834. doi: 10.1097/00001756-199705260-00008. [DOI] [PubMed] [Google Scholar]
- 69.Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 70.Snider WD, Lichtman JW. Are neurotrophins synaptotrophins? Mol Cell Neurosci. 1996;7:433–442. doi: 10.1006/mcne.1996.0031. [DOI] [PubMed] [Google Scholar]
- 71.Stuenkel EL, Nordmann JJ. Sodium-evoked, calcium-independent vasopressin release from rat isolated neurohypophysial nerve endings. J Physiol (Lond) 1993;468:357–378. doi: 10.1113/jphysiol.1993.sp019776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun. 1991;181:968–975. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- 73.Thanos S, Bonhoeffer F. Axonal arborization in the developing chick retinotectal system. J Comp Neurol. 1987;261:155–164. doi: 10.1002/cne.902610114. [DOI] [PubMed] [Google Scholar]
- 74.Thirion S, Troadec JD, Pivovarova NB, Pagnotta S, Andrews SB, Leapman RD, Nicaise G. Stimulus-secretion coupling in neurohypophysial nerve endings: a role for intravesicular sodium? Proc Natl Acad Sci USA. 1999;96:3206–3210. doi: 10.1073/pnas.96.6.3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thoenen H. Neurotrophins and synaptic plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 76.Trimble WS, Linial M, Scheller RH. Cellular and molecular biology of the presynaptic nerve terminal. Annu Rev Neurosci. 1991;14:93–122. doi: 10.1146/annurev.ne.14.030191.000521. [DOI] [PubMed] [Google Scholar]
- 77.Tsien RY. A non-disruptive technique for loading calcium buffers and indicators into cells. Nature. 1981;290:527–528. doi: 10.1038/290527a0. [DOI] [PubMed] [Google Scholar]
- 78.Verhage M, McMahon HT, Ghijsen WE, Boomsma F, Scholten G, Wiegant VM, Nicholls DG. Differential release of amino acids, neuropeptides, and catecholamines from isolated nerve terminals. Neuron. 1991a;6:517–524. doi: 10.1016/0896-6273(91)90054-4. [DOI] [PubMed] [Google Scholar]
- 79.Verhage M, Ghijsen WE, Nicholls DG, Wiegant VM. Characterization of the release of cholecystokinin-8 from isolated nerve terminals and comparison with exocytosis of classical transmitters. J Neurochem. 1991b;56:1394–1400. doi: 10.1111/j.1471-4159.1991.tb11437.x. [DOI] [PubMed] [Google Scholar]
- 80.Viglietti CP, Panzica GC, Gremo F. Nerve endings isolated from chick embryonic optic tectum. I. Developmental aspects of intact synaptosomes. Experientia. 1977;33:458–460. doi: 10.1007/BF01922210. [DOI] [PubMed] [Google Scholar]
- 81.von Bartheld CS. Tracing with radiolabeled neurotrophins. Methods Mol Biol. 2001;169:195–216. doi: 10.1385/1-59259-060-8:195. [DOI] [PubMed] [Google Scholar]
- 82.von Bartheld CS, Butowt R. Expression of neurotrophin-3 (NT-3) and anterograde axonal transport of endogenous NT-3 by retinal ganglion cells in chick embryos. J Neurosci. 2000;20:736–748. doi: 10.1523/JNEUROSCI.20-02-00736.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.von Bartheld CS, Byers MR, Williams R, Bothwell M. Anterograde transport and axo-dendritic transfer of neurotrophins in the developing visual system. Nature. 1996;379:830–833. doi: 10.1038/379830a0. [DOI] [PubMed] [Google Scholar]
- 84.von Bartheld CS, Wang XX, Butowt R. Anterograde axonal transport, transcytosis, and recycling of neurotrophic factors: the concept of trophic currencies in neural networks. Invited review. Mol Neurobiol. 2001;24:1–28. doi: 10.1385/MN:24:1-3:001. [DOI] [PubMed] [Google Scholar]
- 85.Wang XX, Butowt R, von Bartheld CS. Mechanisms of the release of anterogradely transported NT-3 from presynaptic axon terminals. Soc Neurosci Abstr. 1999;25:1786. doi: 10.1523/JNEUROSCI.22-03-00931.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang XX, Butowt R, von Bartheld CS. Effects of NT-3 on the number, size and vesicle density of retinotectal synapses in chick embryos. Soc Neurosci Abstr. 2000;26:843. [Google Scholar]
- 87.Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- 88.Westrum LE, Gray EG, Burgoyne RD, Barron J. Synaptic development and microtubule organization. Cell Tissue Res. 1983;231:93–102. doi: 10.1007/BF00215777. [DOI] [PubMed] [Google Scholar]
- 89.Wu K, Xu J-L, Suen PC, Levine E, Huang YY, Mount HTJ, Lin SY, Black IB. Functional trkB neurotrophin receptors are intrinsic components of the adult brain postsynaptic density. Mol Brain Res. 1996;43:286–290. doi: 10.1016/s0169-328x(96)00211-2. [DOI] [PubMed] [Google Scholar]
- 90.Yamagata M, Sanes JR. Target-independent diversification and target-specific projection of chemically defined retinal ganglion cell subsets. Development. 1995;121:3763–3776. doi: 10.1242/dev.121.11.3763. [DOI] [PubMed] [Google Scholar]
- 91.Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennett L, Welcher AA. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience. 1997;78:431–448. doi: 10.1016/s0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]
- 92.Zhou X-F, Rush RA. Localization of neurotrophin-3-like immunoreactivity in peripheral tissues of the rat. Brain Res. 1993;621:189–199. doi: 10.1016/0006-8993(93)90106-w. [DOI] [PubMed] [Google Scholar]
- 93.Zhou X-F, Rush RA. Endogenous brain-derived neurotrophic factor is anterogradely transported in primary sensory neurons. Neuroscience. 1996;74:945–951. doi: 10.1016/0306-4522(96)00237-0. [DOI] [PubMed] [Google Scholar]