

Abstract
Basic fibroblast growth factor (Fgf2) is required for the generation of founder cells within the dorsal pseudostratified ventricular epithelium, which will generate the cerebral cortex, but the ganglionic eminences are not affected. We report here that the Fgf2 null mutant mice show an ∼40% decrease in cortical glutamatergic pyramidal neurons. In contrast, no change in pyramidal or granule cell number is detected in the hippocampus of Fgf2 −/− mice. In addition, the soma of the pyramidal cells in the frontal and parietal cortices are smaller in Fgf2 knock-out mice. The decrease in the number and size of glutamatergic neuronal population affects all cortical layers but is restricted to the frontal and parietal cortices without any change in the occipital cortex, indicating that Fgf2 is necessary to regulate cell number and size in the anterior cerebral cortex. In contrast to pyramidal neurons, cortical GABA interneurons are unaffected by the lack of Fgf2. The resulting imbalance between the excitatory and inhibitory neurotransmission in the cerebral cortex is reflected by an increased duration of sleep when the animals receive a GABA receptor agonist. Thus, Fgf2 signaling may contribute to the regional specification of the cerebral cortex and may play a role in increasing the size of anterior cortical regions during vertebrate evolution.
Keywords: fibroblast growth factor, knock-out, glutamate, GABA, pyramidal neurons, GABA interneurons, null mutation, neurogenesis, mouse, pseudostratified ventricular epithelium, neuronal progenitor, growth, cerebral cortex, regional specification
Basic fibroblast growth factor (Fgf2) is one of the most potent mitogenic factors in the CNS (Gensburger et al., 1987; Kilpatrick and Bartlett, 1993; Vaccarino et al., 1995; Vicario-Abejon et al., 1995). Neural stem cells from a variety of CNS regions may require Fgf2 or a close homolog for their proliferation and differentiation (Ray et al., 1993; Qian et al., 1997;Palmer et al., 1999; Tropepe et al., 2001). Furthermore, the addition of Fgf2 in vitro increases neuronal survival (Grothe et al., 1989; Knusel et al., 1990; Gensburger et al., 1992; Otto and Unsicker, 1993).
Although Fgf2 acts on the proliferation and survival of a wide variety of cellular systems in vitro, initial phenotypic analyses of mice homozygous for a null Fgf2 allele (Fgf2knock-out mice or Fgf2 −/−) revealed apparently normal organogenesis, with the exception of the cerebral cortex (Dono et al., 1998; Ortega et al., 1998; Vaccarino et al., 1999). The cerebral cortical abnormalities consist of a 45% decrease in the total number of cells, including both neurons and astroglia, at maturity (Vaccarino et al., 1999). This defect may be attributable to a lack of cell specification, because the lack of Fgf2 did not affect cell survival, among either neuroepithelial cells during neurogenesis or postmitotic cortical neurons (Raballo et al., 2000). Furthermore, Fgf2 plays a critical role before the onset of cortical neurogenesis, as demonstrated by a 60% reduction of proliferating cells within the dorsal pseudostratified ventricular epithelium (PVE) of Fgf2knock-out mice at embryonic day 10.5 (E10.5) (Raballo et al., 2000). Because Fgf2 −/− mice lacked 45% of cortical neurons at birth, the progenitor loss is compensated only in part during neurogenesis. Interestingly, the development of the basal telencephalon is unaffected by the lack of Fgf2, and the number of neurons within the basal ganglia at birth is unchanged in Fgf2 knock-out mice (Raballo et al., 2000). Thus, it appears that Fgf2 is necessary for establishing the appropriate number of founder cells within the dorsal PVE, which will then give rise to cells of the cerebral cortex. In contrast, the lack of Fgf2 can be compensated, or is less critical, for the development of the basal ganglia.
The decreased progenitor cell pool indicates that Fgf2 is required for the development of a subset of cortical progenitors within the dorsal PVE, which in turn may differentiate into cortical projection neurons. The dorsal PVE is thought to contribute mainly to glutamatergic pyramidal cells that migrate radially from the dorsal neuroepithelium to the cortical plate (Tan et al., 1998). In contrast, a large percentage of cortical interneurons migrate tangentially to the developing cerebral cortex from the developing basal ganglia (de Carlos et al., 1996; Anderson et al., 1997a,b; Lavdas et al., 1999). Because the dorsal PVE was depleted of progenitors but the ganglionic eminences were not affected in Fgf2 −/− mice, we predicted that these mice should lack a population of pyramidal neurons, but their cortical GABA interneurons should have remained unchanged. In the present study, we estimated the number, size, and layout of phenotypically identified cortical neurons in Fgf2 null mutant mice. Fgf2 was found to be necessary for the regulation of both the number and size of pyramidal neurons, but only in the anterior regions of the cerebral cortex. The results suggest that Fgf2 is essential for the generation of this regional subset of pyramidal cells. Because Fgf2 was not required for the generation of GABAergic interneurons, there is an imbalance between excitatory and inhibitory neuron number in the cerebral cortex of these mutant mice. This imbalance is physiologically significant, because Fgf2 knock-out mice are more sensitive to the effects of GABAergic drugs. We propose that Fgf2 regulates cortical patterning by promoting the generation of regionally specific subsets of pyramidal neurons and enhancing their trophism within the cerebral cortex.
MATERIALS AND METHODS
Animals. Fgf2 knock-out and wild-type mice from two different genetic backgrounds were used in this study. The first is the Fgf2 null allele created into a 129Sv:black Swiss genetic background (Zhou et al., 1998). The second strain ofFgf2 knock-out mice was created in a 129Sv:C57BL /6 background (Ortega et al., 1998). Animals used for the experiments were mainly derived from heterozygous crosses. Each experiment compared −/− versus +/+ littermates from a minimum of two separate litters. Animals were genotyped by PCR (Zhou et al., 1998), after digestion of tail DNA according to routine protocols. Extracts were precipitated with isopropanol, and pellets were washed with 75% ethanol. Purified DNA was resuspended in 200 μl of water and used for PCR at dilutions ranging from 1:10 to 1:100.
Immunohistochemistry. Wild-type and knock-out mice were anesthetized using ketamine–xylazine (100 and 10 mg/kg, respectively) and perfused transcardially with 0.1 m PBS, followed by chilled fixative solution containing either 4% paraformaldehyde (in 0.1 m PBS, pH 7.4) or a mixture of 4% paraformaldehyde and 1.0% glutaraldehyde (in 0.1m PBS, pH 7.4). Fixative containing 4% paraformaldehyde was used for all of the labeling procedures, with the exception of glutamate immunolabeling, in which we used a mixed aldehyde fixative. Brains were removed and post-fixed for 2–4 hr in the respective fixatives. Brains were then briefly washed in PBS and cryoprotected in graded series of sucrose. The brains were then placed in OCT mounting compound (Miles, Elkhart, IN) and frozen on dry ice. Cryostat sections (50 μm) of brains were cut coronally in a series of 10 in which every 10th section was collected per each series. Before immunolabeling, sections from both knock-outs and wild-type mice were coded so as to mask the identity of their genotypes and were processed simultaneously for immunolabeling so that the same experimental conditions were applied for both the genotypes.
For each antibody, one series of sections was stained free floating. Sections were blocked in 10% serum and incubated overnight at 4°C in primary antibodies diluted in 10% normal serum as follows: glutamate (1:6000; mouse monoclonal from DiaSorin, Stillwater, MN); calbindin (1:1000; rabbit antibody from Chemicon, Temecula, CA); parvalbumin (1:1000;mouse monoclonal from Sigma, St. Louis, MO); GABA (1:2000; guinea pig; polyclonal from Eugene Tech International Inc., Ridgefield Park, NJ); SMI-32 (1:4000; mouse monoclonal from Sternberger Monoclonals, Lutherville, MD); neuronal-specific nuclear protein (NeuN) (1:500; mouse monoclonal from Chemicon, Temecula, CA); and latexin (undiluted culture supernatant; mouse monoclonal antibody; gift from Dr. Arimatsu, Mitsubishi Kasei Institute of Life Sciences, Tokyo, Japan). On the following day, sections were washed three times for 20 min each in PBS, treated with suitable biotinylated secondary IgG, and processed using the avidin–biotin–peroxidase complex (Vectastain ABC elite kit; Vector Laboratories, Burlingame, CA) as described previously (Raballo et al., 2000). The reaction was visualized using a solution containing 0.0125% diaminobenzidine (DAB) and 0.0005% hydrogen peroxide. Few sections at a time were developed in DAB, and identical developing times were maintained for all of the samples. Sections were mounted on gelatin-subbed slides, dehydrated, and coverslipped. Adjacent series of sections from each animal were stained with cresyl violet for volumetric stereological assessment and the definition of cortical layers. Images were taken using a Zeiss (Oberkochen, Germany) Axiocam video camera on a Zeiss 2M Axioskope.
Fluorescence labeling. For SMI-32 fluorescence labeling, an anti-mouse IgG conjugated to Alexa-488 (1:500; Molecular Probes, Eugene, OR) secondary antibody was used. Nuclei were counter stained with TO-PRO3 iodide (1:2000; Molecular Probes). For SMI-32 and GABA double immunolabeling in postnatal day 7 (P7) brains, after treating the sections with primary antibodies overnight at 4°C, sections were rinsed in PBS and reacted with biotinylated anti-guinea pig IgG for 1 hr at room temperature (Vector Laboratories). Sections were then treated simultaneously with anti-mouse IgG conjugated to Alexa-488 and Texas Red avidin DCS (1:200; Vector Laboratories) for 1 hr. Images were captured using a Zeiss Axiovert 100M confocal microscope.
Stereological analyses. Volume and total cell numbers were assessed using stereological techniques on coded brains for which the experimental condition was not known to the investigator. The details of the optical dissector method we used were described by Vaccarino et al. (1999). Volume and cell number of hippocampus and cerebral cortex were analyzed in a series of one every 6–10 sections spanning the whole region. Cell counts were done at 100× magnification in three-dimensional areas (3 × 10−6mm3), and an average of 10–45 areas depending on the region were counted per brain.
Bin counts. For the determination of cell density in various layers of the cerebral cortex, a series of one every 10 sections immunostained with glutamate or SMI-32 were counted within a 100-μm-wide bin extending from the pial surface to the white matter border. This bin was further subdivided into 50-μm-thick sectors. Cell density was scored within each sector from the pial surface to the white matter. The correspondence between the various sectors and cortical layers was assessed using adjacent sections stained with cresyl violet. Counts were made blindly to the genotype of the animals. We chose landmarks to distinguish different regions of cortex as follows: for frontal cortex, cortical areas above the caudate nucleus and anterior commissure; for parietal cortex, the area just above the hippocampal fimbria; and for occipital cortex, the region above the hippocampal granule cell layer. Sections from different animals were matched by comparing these landmarks in cresyl violet-stained sections.
Analyses of cell size. Cells were randomly selected within the frontal, parietal, or occipital regions in sections immunostained with SMI-32. Landmarks were used to distinguish various regions of the cortex as explained above. A total of 50–60 randomly selected neurons from all cortical layers were entered for this analysis per animal. The soma perimeter and area were determined using a Macintosh-based analysis program based on NIH Image. Cells were viewed on a ZeissAxioplan microscope equipped with a Sony (Tokyo, Japan) DXC 9000 video camera and a New Vista Plus frame grabber board. Cell perimeters were traced using a WACOM drawing tablet and high-resolution mouse. Cell areas were calculated with the program automatically. Only the cells that displayed a minimum of three neurites were included in this analysis to avoid assaying cells that were cut tangentially to the cell membrane.
Sodium pentobarbital-induced righting reflex loss. The duration of the sodium pentobarbital (PTB)-induced loss of the righting reflex was measured after intraperitoneal injections of PTB-Na (50 mg/kg body weight) in Fgf2 −/− and wild-type littermates (wild type, n = 9; Fgf2 −/−,n = 13) as described previously (Matsumoto et al., 1996). For each animal, we measured the time lapse between the administration of the drug and the onset of righting reflex loss and the period of time between the loss of the righting reflex and its return. The latter time period was the duration of the righting reflex loss (belly-up time).
RESULTS
The number of hippocampal neurons is unchanged in Fgf2 knock-out mice
We demonstrated previously that, in Fgf2 null mutant mice, the number of progenitor cells in the dorsal PVE is significantly decreased (Raballo et al., 2000). The hippocampus arises from the dorsomedial portion of the PVE, and it could potentially be affected by these changes. Furthermore, it has been shown that, when injected peripherally, Fgf2 increases thymidine incorporation in the hippocampus at P1 (Tao et al., 1996). To see whether the loss of Fgf2 affects hippocampal development, we estimated the volume and neuron number for the pyramidal and granule cell layers of the hippocampus in NeuN-stained tissue sections from adult knock-out and wild-type mice (n = 4). These analyses revealed that the total number of granule neurons is 3.86 × 105 in wild-type mice and 4.08 × 105 inFgf2 knock-out mice. These values are not significantly different (p > 0.5). Similarly, the number of hippocampal pyramidal neurons was 4.8 × 105 and 5.5 × 105 for wild-type and knock-out mice, respectively (p > 0.1). Neither the neuronal soma size nor the volume of the hippocampal granule and pyramidal layers showed significant differences between wild-type and mutant mice (data not shown). These data suggest that, under normal conditions, Fgf2 may not be necessary for the development or maintenance of hippocampal neurons.
The number of cortical pyramidal neurons is decreased in Fgf2 null mutants, whereas that of interneurons is unchanged
We showed previously that the total number of NeuN-stained cortical neurons is decreased by 25% in adult Fgf2knock-out mice (Vaccarino et al., 1999). To determine whether this decrease is restricted to any particular neuronal subclass, we used immunocytochemical markers for excitatory and inhibitory neurons. Antibodies to calbindin, parvalbumin, and GABA were used to identify cortical interneurons, whereas SMI-32, latexin, and glutamate antibodies were used to identify pyramidal neurons (Conti et al., 1987;Campbell et al., 1991; Hof and Morrison, 1995; Arimatasu et al., 1999a,b). In the dorsolateral prefrontal and parietal cortices, the density of glutamate-immunoreactive cells was markedly lower and cell somata of pyramidal neurons were visibly smaller in Fgf2 null mutant mice compared with littermate controls (Figs. 1, 2). In contrast, no changes in glutamate cell density were found in occipital cortex (data not shown).
Fig. 1.
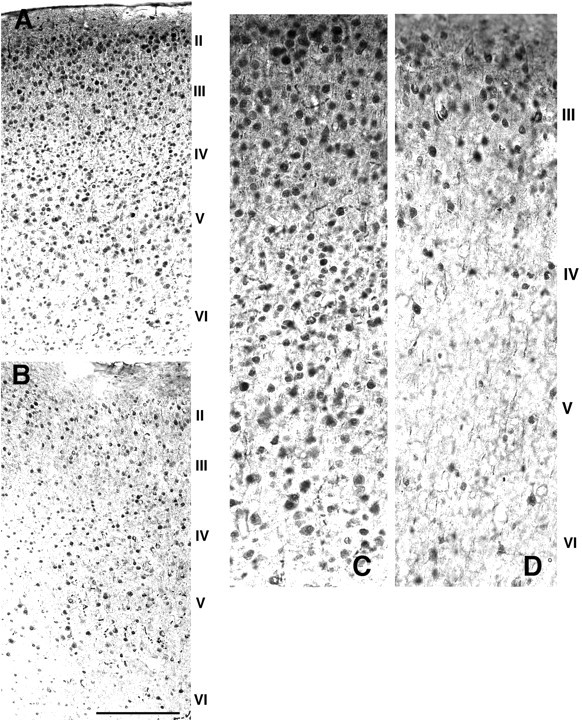
Glutamate immunostaining in dorsolateral prefrontal cortex. Coronal sections from the dorsolateral prefrontal cortex of an adult wild-type (A, C) andFgf2 knock-out (B, D) mice immunostained for glutamate. Roman numerals indicate cortical layers. Whereas the different layers of the cortex are clearly distinguishable in the wild-type mouse (A), the laminar organization looks disrupted in the knock-out animal (B) attributable to many missing cells in all layers. In addition, cell somata are smaller in knock-out compared with the wild-type mice (compare C, D). Scale bar: A, B, 200; C,D, 400 μm.
Fig. 2.
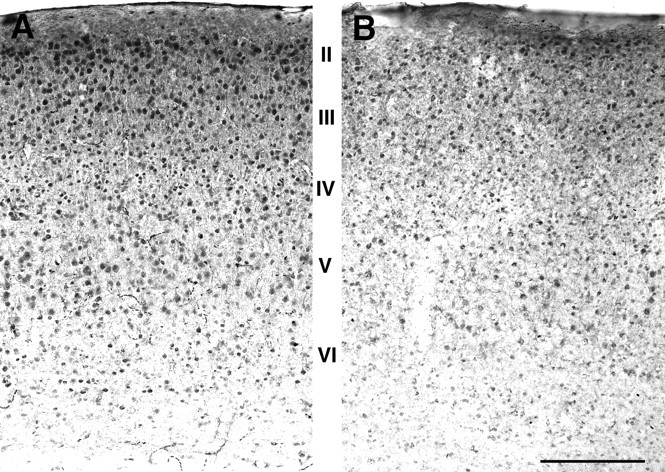
Glutamate immunostaining in parietal cortex. Coronal sections from the parietal cortex of an adult wild-type (A) and Fgf2 knock-out mouse (B) immunostained for glutamate. Roman numerals indicate cortical layers. A clear decrease in the glutamate-stained somata was evident in the knock-outs compared with wild-type mice. Scale bar, 400 μm.
Unbiased stereological analyses of the entire cerebral cortex confirmed that there was an overall 38% decrease in number of excitatory neurons, identified by glutamate immunostaining, in Fgf2knock-out mice compared with wild-type mice (p< 0.05) (Table 1). In contrast, the number of interneurons immunostained by calbindin, parvalbumin, or GABA did not differ between the Fgf2 −/− and wild-type animals (Table 1). Interestingly, the sum of calbindin- and parvalbumin-positive cells approached that of GABA-positive cells, confirming that GABA immunostaining encompasses all interneurons, whereas calcium-binding proteins identify specific interneuron subsets. The number of GABA-positive cells was approximately one-third the number of glutamate cells in wild-type mice, which is the normal cortical GABA/glutamate ratio. In contrast, the number of GABA interneurons was one-half the number of glutamate cells in theFgf2 knock-outs, reflecting a striking imbalance in these neurotransmitters (Table 1).
Table 1.
Fgf2 −/− mutant mice show a significant decrease in the pyramidal neurons of the cortex, whereas the interneurons are unchanged
| Cell number (millions ± SEM) | ||||
|---|---|---|---|---|
| Glutamate | GABA | Calbindin | Parvalbumin | |
| Wild-type | 6.478 ± 0.624 | 2.473 ± 0.755 | 1.69 ± 0.306 | 1.035 ± 0.108 |
| Fgf2−/− | 4.017 ± 0.399 | 2.533 ± 0.251 | 1.765 ± 0.165 | 0.995 ± 0.044 |
| % Wild type | 62% | 102% | 104% | 96% |
Fgf2 null mutant mice (Fgf2 −/−) show a significant decrease in number of glutamate-positive pyramidal neurons in the cerebral cortex, whereas the number of GABA-, calbindin-, and parvalbumin-containing interneurons remain unchanged. Cell numbers in the neocortex were estimated by stereologial analysis using the optical dissector method. No statistically significant differences in cortical volume were observed between genotypes. ANOVA showed that the decrease in glutamate-containing cells was statistically significant (genotype main effect, F = 9.95; p < 0.001).n = 6 mice per genotype: three on C57BL/6 and three on 129Sv:black Swiss background. The glutamate/GABA ratio is approximately 3:1 in wild-type mice and 2:1 in Fgf2 knock-outs.
To verify whether the decrease in the number of glutamate-immunostained cells is attributable to a decrease in glutamate content or whether it reflects a lack of pyramidal cells, we used additional markers to identify pyramidal neurons in the cerebral cortex. The SMI-32 monoclonal IgG reacts with a nonphosphorylated epitope in neurofilament H present in a subset of cortical pyramidal cells (Campbell et al., 1991; Hof and Morrison, 1995). Sections from adult wild-type andFgf2 −/− littermates stained with the SMI-32 antibody were analyzed to assess pyramidal cell density within layers of the parietal cortex. The data indicated a significant decrease (p < 0.05) in SMI-32-positive cells inFgf2 knock-outs compared with controls, both in infragranular and supragranular layers of the parietal cortex (Fig.3). Neuronal density in the supragranular layers was decreased by 60% in the mutant mice compared with controls. A similar decrease in SMI-32-positive cell density (45%) was found in the infragranular layers. In contrast, adjacent sections stained with calbindin did not reveal any change in the density of these interneurons in Fgf2 knock-out mice (Fig. 3). SMI-32 fluorescence immunostaining revealed a decrease in the neuropil staining of the frontal and parietal cortices of Fgf2 −/− mutant mice compared with their wild-type littermates (Fig.4). In wild-type mice, the primary dendrites could be seen as continuous long stained processes extending from the cell body. In addition, there was dense punctate staining in the neuropil, which reflected the cross section of pyramidal cell dendrites. In contrast, knock-out animals showed an apparent reduction of staining in the primary dendritic processes and much less punctate staining in the neuropil (Fig. 4).
Fig. 3.

The disruption of the Fgf2 gene decreases the density of cortical pyramidal cells but not that of calbindin-positive interneurons. Density of pyramidal and nonpyramidal cells in layers I–IV (supragranular; SG) and layers V–VI (infragranular; IG) of the parietal cortex in adult wild-type mice (wt) and Fgf2 knock-outs (Fgf2 −/−). Pyramidal cells are identified by SMI-32 and nonpyramidal cells by calbindin immunostaining.n = 3–6 animals per group. Fgf2 null mutant mice show a significant decrease in the SMI-32-stained pyramidal cells in both the infragranular and supragranular layers of the cortex (p < 0.05; Student's ttest), whereas the nonpyramidal cell densities are not affected.
Fig. 4.
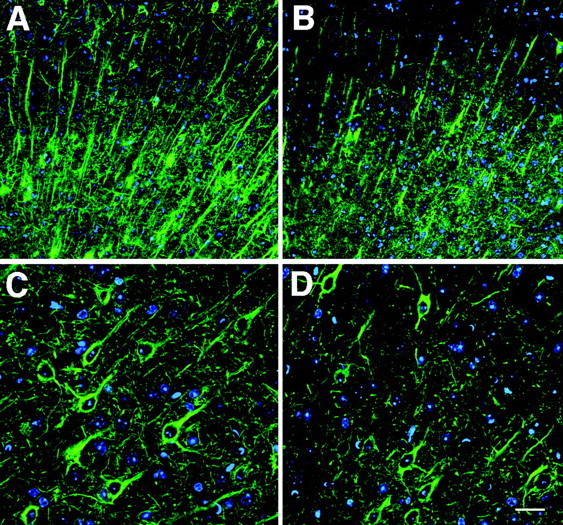
Pyramidal cell dendritic staining is decreased inFgf2 −/− mutant mice. Confocal images of SMI-32 fluorescence-stained sections from the medial prefrontal cortex region of wild-type (A, C) and Fgf2knock-out (B, D) mice show a clear decrease in the proximal dendritic and neuropil staining of the pyramidal cells in the knock-out mice (B). Note that, in addition to fewer cells, the pyramidal cell somata are smaller in the knock-out mice. Scale bar: A, B, 40 μm; C, D, 20 μm.
To rule out the possibility that the decrease in SMI-32-positive neurons in Fgf2 knock-out mice was attributable to any phosphorylation-mediated masking of the epitope recognized by SMI-32 antibody, we used an antibody against latexin. This carboxypeptidase inhibitor is contained within pyramidal cells restricted primarily to layer VI and, to a lesser extent, layer V within the temporoparietal cortex (Arimatasu et al., 1999a). We observed a clear decrease in the number of latexin-immunoreactive neurons in the temporoparietal cortex of the Fgf2 −/− mice (Fig.5). In addition, we again noticed a significant decrease in the staining of the neuropil. The punctate staining in the neuropil of wild-type mice was so dense that the proximal dendritic staining was masked (Fig. 5A). In contrast, the neuropil staining in Fgf2 knock-out mice was less dense and revealed the dendritic architecture (Fig. 5B). In conclusion, the examination of three different markers for pyramidal cells and three additional markers for interneurons suggest that pyramidal cell number was markedly decreased in mice lacking Fgf2, whereas no alterations were noticed in the number of nonpyramidal cells.
Fig. 5.
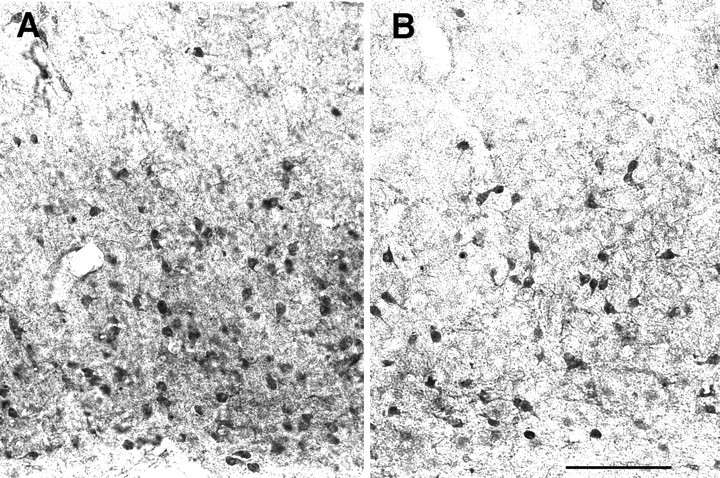
Pyramidal neurons in parietal areas are decreased in number in Fgf2 knock-out mice. Immunostaining for the carboxypeptidase inhibitor latexin, which is contained within pyramidal cells of layers V and VI of the temporoparietal cortex. Latexin-immunoreactive cell bodies and neuropil are markedly decreased in the parietal cortex of Fgf2 knock-out mice. Scale bar, 200 μm.
Fgf2 is necessary for the specification of glutamate cell number in anterior cortical regions
To assess whether pyramidal cell number was uniformly affected throughout cerebral cortical regions and layers in Fgf2−/− mice, the density of glutamate-immunoreactive cells was ascertained in three cortical areas: dorsolateral prefrontal, parietal, and occipital. Cell density accurately reflects changes in cortical cell number in these animals, because no significant differences in cortical volumes were present between wild-type and mutants. This analysis included a total of six mice per genotype (wild-type orFgf2 −/− littermates): three on a 129Sv:black Swiss genetic background and three on a 129Sv:C57BL /6 background. Cell counts were performed in bins of cortical tissue encompassing the whole thickness of the cerebral cortex. The overall glutamate-immunostained cell density (cells per cubic millimeter) was 230,452 ± 4694 for wild type and 145,598 ± 10,565 for Fgf2 knock-outs. Glutamate neuron density (cells per cubic millimeter) for wild-type mice in each area, averaged across all cortical layers, was 229,474 ± 4650 in prefrontal cortex, 247,751 ± 14,054 in parietal, and 214,129 ± 5744 in occipital. In Fgf2knock-outs, the average glutamate neuron density (cells per cubic millimeter) was 119,172 ± 18,429 in prefrontal cortex, 151,605 ± 19,264 in parietal cortex, and 166,017 ± 14,054 in occipital cortex. ANOVA showed that glutamate neuron density was significantly different between wild type and knock-out across the entire cerebral cortex (genotype main effect, F = 42.18; p = 0.0001). However, the lack of Fgf2 affected anterior cortical regions (frontoparietal areas) significantly more than the posterior (occipital) cortical areas (interaction between genotype × cortical areas, F = 5.6;p < 0.05). A Bonferroni post hoc test confirmed that wild-type and knock-out animals differed in glutamate cell density in frontoparietal areas (p < 0.00001) but not in occipital areas (p > 0.05). In conclusion, in wild-type mouse, frontal and parietal cortices have higher glutamate cell densities than occipital cortex. In contrast, inFgf2 knock-out animals, glutamate-immunoreactive cell densities found in frontal and parietal cortex were less than those found in the occipital cortex, suggesting that region-specific differences in pyramidal cell number were reversed.
To better assess the laminar organization of the cerebral cortex inFgf2 knock-out mice, we examined the density of pyramidal cells along different cortical layers. Figure6 shows glutamate cell density averages for each cortical layer for frontal, parietal, and occipital regions. In frontal and parietal regions of Fgf2 knock-out animals, glutamate-immunoreactive cells were substantially decreased in all cortical layers except layer 1. In contrast, no significant differences were found in occipital regions (Fig. 6). ANOVA showed again a strong effect of genotype (p < 0.0001) and a significant genotype × region interaction (p < 0.05) when frontoparietal regions were compared with occipital. There was no significant genotype × layer effect.
Fig. 6.
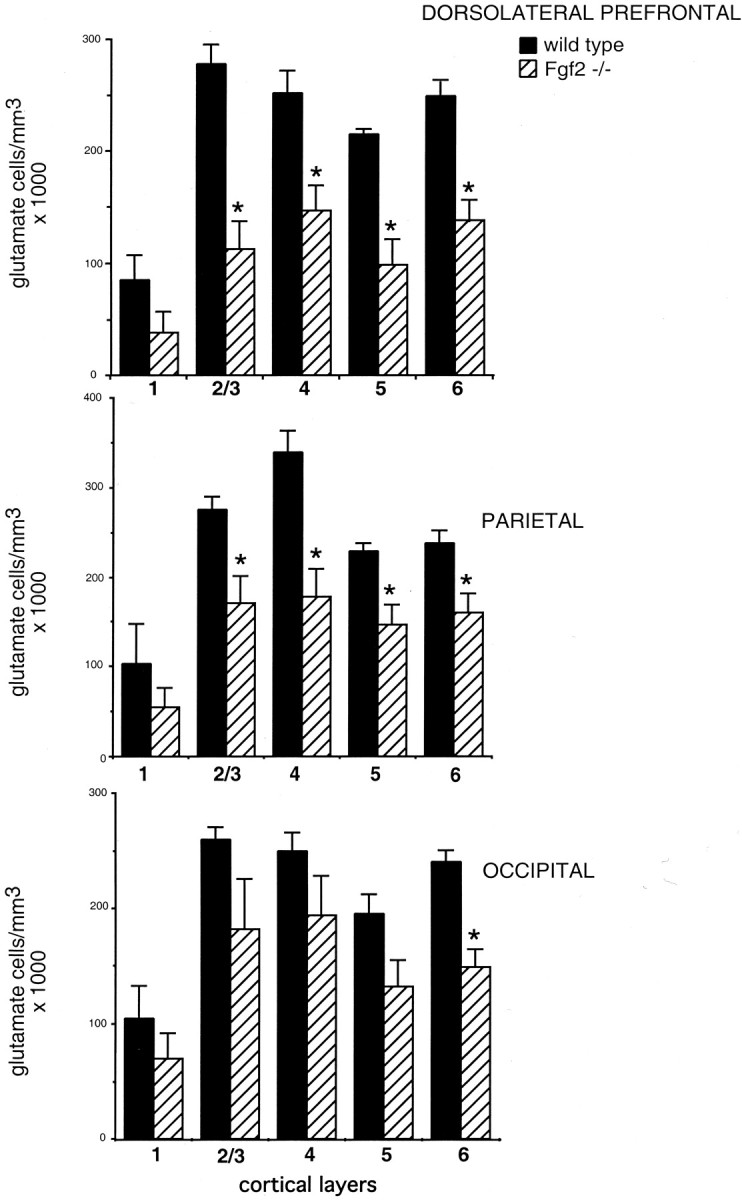
Fgf2 null mutants lack pyramidal cells in all cortical layers. Glutamate-immunoreactive cell densities were assessed in 100 μm bins of cortical tissues within the prefrontal, parietal, and occipital cortices; three nonadjacent sections and two adjacent bins per section were counted per animal. Fgf2knock-out mice had significantly less pyramidal cells in layers 2 through 6 in prefrontal and parietal but no differences in cell density for any of the layers in occipital cortices, with the exception of layer 6. n = 6 mice per genotype: three on C57BL/6 and three on 129Sv:black Swiss background. *p < 0.05 comparing wild-type and knock-out mice; Student's t test.
Despite the decreases in cell density in all layers, peak cell densities within each layer were relatively well preserved in mutant animals, ruling out a primary defect in the layer-specific aggregation of pyramidal cells. Furthermore, the location of layer II/III as assessed by calbindin immunoreactivity (Fig. 3) and of layer V/VI as assessed by latexin (Fig. 5) were not altered. Together, these data suggest that cortical cell migration and aggregation into specific layers is not affected by the lack of Fgf2. The data confirm thatFgf2 knock-out mice have a profound decrease in the number of glutamate-positive neurons in all layers of the cerebral cortex. This loss in pyramidal cells is restricted to anterior cortical regions, suggesting that Fgf2 signaling contributes to the anteroposterior specification of the cerebral cortex.
Fgf2 is critical for the size of the pyramidal cell soma
In addition to the decrease in the number of pyramidal cells, cell size was visibly smaller in the knock-out animals compared with wild-type mice in glutamate-immunostained sections. The decrease in the apparent volume of the cell somata was particularly evident in areas in which pyramidal cells are bigger in size, such as the medial prefrontal cortex (Fig. 7), but was also noticeable in dorsolateral prefrontal (Fig. 1) and parietal (Fig. 2) cortices. In contrast, no difference in the size of glutamatergic neurons was evident in the occipital cortex (data not shown). To understand whether this effect on size was attributable to a decrease in glutamate content versus an actual smaller cellular volume, we examined sections immunostained with SMI-32, which recognizes a structural component of the cell. Quantitative assessments of the area of SMI-32-immunostained cell profiles revealed complex patterns that differed between cortical regions (Fig. 8). In the frontal cortex of wild-type mice, there was a unimodal distribution of cell sizes centered on values in the range of 151–200 μm2; ∼65% of the cells were larger than 151 μm2. In the Fgf2 mutants, this distribution shifted to the left, with only 38% of cells in the same range (Fig. 8). In wild-type parietal cortex, there was a bimodal distribution of cell areas centered around two peaks: 101–150 μm2 (34% of the cells) and 201–250 μm2 (28% of the cells). In contrast, a unimodal distribution with areas centered on the 101–150 μm2 range (45% of cells) replaced these two populations in the Fgf2 −/− parietal cortex, and the proportion of cells over 201–250 μm2decreased to <11% compared with 36% in the wild-type mice. In the wild-type occipital cortex, a unimodal distribution of pyramidal cells was present, with the greatest number of cells centered on values of 101–150 μm2 area. There was no change in the Fgf2 knock-outs. In sum, the largest pyramidal cells in both frontal and parietal cortices represented by the populations with soma areas above 200 μm2 decreased in the Fgf2 knock-out animals (Fig. 8). As a result, the diversity in pyramidal cell size present in the wild-type cerebral cortex was replaced in the knock-outs by a uniform pattern throughout the cortex (Fig. 8), which was remarkably similar to that normally present in posterior cortical regions. Thus, Fgf2 is necessary for upregulating pyramidal cell size in prefrontal and parietal regions of the cerebral cortex. In its absence, all cortical regions show a “default” pattern similar to what is normally present in the occipital cortex.
Fig. 7.
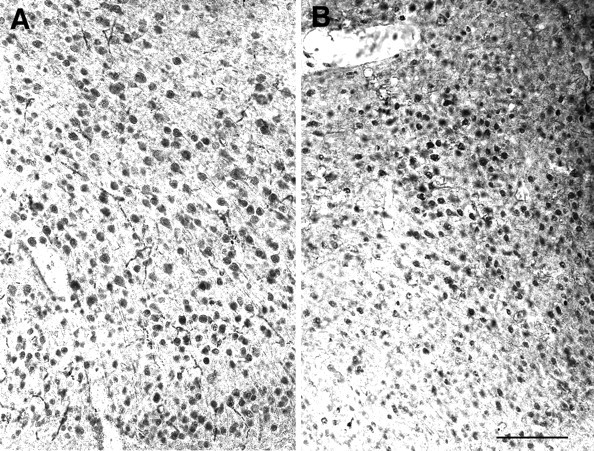
The lack of Fgf2 decreases the size of pyramidal cell somata. Coronal sections from the medial prefrontal cortex of the wild-type (A) and Fgf2 null mutant (B) mice stained for glutamate immunoreactivity. Note a clear decrease in the size of neuronal somata in the knock-out mice (B) compared with that of wild-type mice (A). Scale bar, 200 μm.
Fig. 8.

Decrease in soma size in mice with the Fgf2 null mutation is restricted to the frontoparietal regions of the cortex. Area of neuronal somata in the frontal, parietal, and occipital cortices. In the frontal and parietal cortices of Fgf2knock-out mice, percentages of total cells shift toward the left (decreasing area values), indicating an absence of large-sized pyramidal cells in these areas in the Fgf2 knock-out animals. There is a lack of such differences between the wild-type (wt) and Fgf2 knock-outs in the occipital cortex. Note that there are different cell populations with respect to size in the wild-type cerebral cortex, whereas there is a single population in the mutants.
We then investigated the developmental underpinnings of this phenotype. No differences in somal size were observed among progenitors of the PVE between Fgf2 knock-out mice and wild types (data not shown). To find out whether this difference in soma size is evident during early postnatal development or appears later as the animal attains maturity, we looked at the SMI-32 staining in the P7 brains, the first time period when SMI-32-immunoreactive pyramidal cells are readily identifiable in the cerebral cortex (Fig.9). When we compared series of sections from wild-type and Fgf2 −/− mice at P7, we found no difference in the soma size between the genotypes, whereas differences were easily detected in adult mice stained with the same antibody (compare Figs. 4, 9). These data suggest that, whereas in wild-type mice soma size is gradually upregulated over the course of subsequent postnatal development, knock-out mice may be unable to do so. However, we readily noticed a decrease in the pyramidal cell number in the knock-out mice compared with the wild-type mice at P7 (Fig. 9). This is in agreement with previous observations that, in the Fgf2 mutants, there is a decrease in progenitor cells that give rise to cortical pyramidal neurons early in development (Raballo et al., 2000).
Fig. 9.
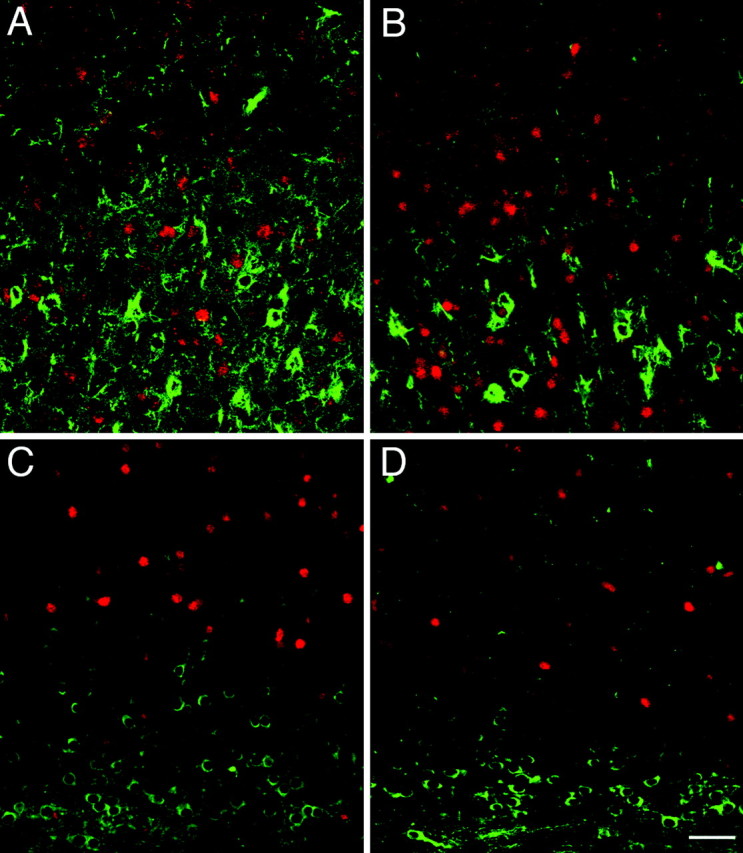
Difference in soma size is not evident during early postnatal development. Confocal images showing double immunolabeling with SMI-32 (green) and GABA (red) from the dorsolateral prefrontal cortex (A, B) and parietal cortex (C, D) of P7 wild-type (A,C) and knock-out (B, D) mice. Although we have not noticed any change in the soma size of the pyramidal neurons (stained in green with SMI-32) between the genotypes at this stage, note that there are fewer SMI-stained neurons and processes in the knock-out mice (B,D). In the parietal cortex, although we can see pyramidal cells arranged in columns with their extended processes in the wild-type mice (C), such cells and processes are not noticeable in the knock-out mice (D). Note that the GABA interneurons (stained in red) are not changed between the genotypes. Scale bar, 50 μm.
Physiological consequences of the imbalance in the excitatory/inhibitory cortical neuron ratio in Fgf2 null mutants
Because of the decreased proportion of glutamatergic versus GABAergic neurons in the cerebral cortex of the Fgf2 null mutants, Fgf2 knock-out mice are likely to have an imbalance between excitatory and inhibitory neurotransmission in a large portion of the cerebral cortex. To find out whether these anatomical abnormalities have a functional counterpart, we stimulated inhibitory synapses using PTB, an agonist at the GABA receptor-gated chloride channel (Olsen and Leeb-Lundberg, 1981; Olsen, 1988). We hypothesized that, if there is a relative excess of inhibitory synapses on the soma of the remaining pyramidal cells, stimulating these synapses should reveal functional abnormalities consistent with an abnormal decrease in cortical excitability. To evaluate this, we measured the duration of the righting reflex loss in response to the administration of PTB inFgf2 knock-out mice and wild-type littermates. Mice were examined at two different ages: 3- and 6-month-old mice. Although the time lapse between the administration of the drug and the induction of sleep remained unchanged among the two groups of animals, we observed that the time lapse between the loss and the subsequent return of the righting reflex (sleeping time) was increased in the mutants. In 3- to 4-month-old male mice, sleeping time was 159 ± 9.4 min in wild-type mice and 218 ± 7 min in Fgf2 knock-outs; in 4–5 months old female mice, sleeping time was 88 ± 15 min in wild type and 118 ± 19 in Fgf2 knock-outs. The main effect of genotype on sleeping time was statistically significant (ANOVA;F = 7.95; p < 0.01; n= 13 wild type; n = 9 knock-outs). There were no differences in body weight or rate of induction of anesthesia, suggesting a comparable tissue distribution of the anesthetic. The data support the conclusion that GABAergic agonists are more potent because of the lack of Fgf2.
DISCUSSION
This study demonstrates that the lack of Fgf2 produces a loss in number and a marked decrease in soma size of glutamatergic pyramidal neurons in the neocortex. In contrast, GABAergic interneurons throughout the cerebral cortex remain unchanged. The decreased number and growth of pyramidal neurons is not uniform but affects the cerebral cortex in an anteroposterior gradient; that is, anterior regions are profoundly affected and posterior regions are spared. These data place Fgf2 among a restricted group of molecules that influence regional specificity in the developing neocortex.
Glutamate neurons are thought to arise from the dorsal PVE by radial migration into the cortical plate, whereas GABAergic interneurons may reach the cortex by tangential migration from the developing basal ganglia (Luskin et al., 1988; de Carlos et al., 1996;Anderson et al., 1997a; Tan et al., 1998; Lavdas et al., 1999). To what extent the dorsal PVE may contribute to the GABAergic population is unclear. We demonstrated previously a 60% loss of proliferating progenitor cells in the dorsal PVE of Fgf2 knock-out mice and a similar loss of cortical neurons at birth, although we did not detect any abnormalities in the basal ganglia (Raballo et al., 2000). The selective loss of glutamate-containing pyramidal cells in neonatal and adult Fgf2 −/− animals confirms that the dorsal PVE generates glutamate-containing neurons. The sparing of GABAergic, as well as of calbindin- and parvalbumin-containing, neurons in these mutants suggest that most forebrain GABAergic interneurons originate from the basal telencephalon, the development of which is intact inFgf2 null mutants.
The loss in glutamate-containing neurons and the sparing of GABAergic interneurons in Fgf2 null mice demonstrates that the development of these two cortical neuron types can be dissociated and that GABAergic and glutamatergic neurons are subjected to the influence of different genetic and environmental factors. Fgf2 is not expressed in the medial ganglionic eminence (Raballo et al., 2000). The medial ganglionic eminence is the main source of interneurons for both the lateral ganglionic eminence and the neocortex (Anderson et al., 2001). We showed previously that progenitor cells for GABAergic neurons, differently than glutamate-progenitor cells, do not require Fgf2 for their proliferation and differentiation in primary culture (Vaccarino et al., 1995). Together, these data imply that there is a decreased responsiveness of basal telencephalic cells to Fgf2 because they may rely on other Fgf ligands or different classes of morphogens.
The concerted action of Fgf2 on both cell hypertrophy and cell number is not unexpected. Many morphogenetic proteins, such as members of thewingless/Wnt and Hedgehog/Shh families, regulate cell growth as well as proliferation to pattern the shape of the embryo. Furthermore, the control of cell growth and cell cycle may be intimately interconnected at the molecular level. Two Fgf-activated intracellular signaling systems, the ribosomal protein S6 kinases pp90rsk and pp70S6K (Tan et al., 1996) and the Ras/mitogen-activated protein kinasespp42,44 (Huang et al., 1995; Mohamadi et al., 1996), are able to regulate both cell growth and the cell cycle. Ras and S6 kinases activate the cdk2/4 and their respective D cyclins, promoting reentry into S phase (Leone et al., 1997; Peeper et al., 1997). Ras and its downstream effector Myc, as well as S6 kinases, also regulate cell hypertrophy by inducing protein synthesis, translation initiation factors, and nucleolar structural proteins (Dang, 1999; Kim et al., 2000; Prober and Edgar, 2000, 2001). Fgf2 has been shown to induce an early response gene involved in ribosomal protein synthesis, and it is required for the hypertrophy of cardiac myocites in response to a pressure load (Nelson et al., 2000).
The molecular mechanisms used by Fgf2 to increase cell number and size may be similar, but these functions may be performed at different stages of development. Although it is likely that Fgf2 regulates cortical cell number by increasing the proliferation of cortical founder cells or stem cells before the onset of neurogenesis (Raballo et al., 2000), the effect on pyramidal soma growth may be performed when neurons are postmitotic. Our observation that the lack of Fgf2 does not affect the soma size of progenitors during embryogenesis or postmitotic neurons at P7 indicates that the role of Fgf2 on soma size may be explicated at later times during the postnatal period. In this context, it is interesting to note that Fgf2 is downregulated in progenitor cells at midneurogenesis and expressed by cortical astrocytes after the first postnatal week of development (Kuzis et al., 1995; Raballo et al., 2000). Western blot analyses of cortical Fgf2 protein levels in rats show undetectable Fgf2 levels before P10 and a progressive increase from P10 to P35 (Ganat et al., 2001). We therefore hypothesize that Fgf2 in cortical astrocytes is necessary for upregulating neuronal size over the postnatal period. Extracellular growth factors are responsible for the growth of animal cells whether they are proliferating or not (Conlon and Raff, 1999). Some of these signaling molecules exert their role both as mitogens and cell growth stimulators (Zettenberg et al., 1984). One of the possible mechanisms by which Fgf2 could regulate the cell size is by activating intracellular signaling pathways that stimulate protein synthesis. Fgf2 is shown to be involved in one of these pathways that operates through phosphotidylinositol 3-kinase, resulting in the regulation of cell morphology (Kay et al., 1998). Alternatively, Fgf2 may affect cortical neurons indirectly, through the synthesis of other trophic factors by astroglial cells.
Intriguingly, Fgf2 is critical for the regulation of number and growth of a restricted set of neurons, pyramidal cells in the frontal and parietal cortex. For example, whereas in wild-type mice there is an increased density of pyramidal neurons in the anterior cortical regions compared with posterior regions, in knock-out mice pyramidal cell densities found in frontal and parietal cortices are less than those in occipital cortex. Although frontoparietal neurons comprise the largest cells in the neocortex, the lack of Fgf2 does not seem to affect the growth of other large-sized neurons, such as hippocampal pyramidal cells or brainstem motor neurons. Thus, despite the wide distribution of Fgf2, this factor appears to be critical only for neurons of the anterior cerebral cortex. The lack of morphologic abnormalities in the hippocampal granule and pyramidal cell layers was surprising in view of the previously hypothesized role of Fgf2 in the development of these regions (Tao et al., 1997). These data also suggest that Fgf2 may not be involved in granule cell neurogenesis in the adult, at least under baseline conditions.
The mechanism responsible for the regionally restricted action of this growth factor is presently unknown. The dorsal PVE is composed of radially oriented progenitors that guide their neuronal progeny to topographically corresponding regions of the developing neocortex (Rakic, 1988; Noctor et al., 2001). Fgf2 and Fgf receptor 1 are both expressed by these radially oriented progenitor cells in an anteroposterior decreasing gradient (Wilke et al., 1997; Vaccarino et al., 1999; Raballo et al., 2000; Ragsdale et al., 2000; Vaccarino et al., 2001), suggesting that Fgf2 is a paracrine signal for these cells. To find out whether Fgf2 regulates progenitor cell division preferentially in anterior regions of the PVE, we examined sections from wild-type and Fgf2 knock-out animals in which the whole population of constitutively proliferating cells was labeled by cumulative bromodeoxyuridine (BrdU) injections for 8 hr (Raballo et al., 2000). In E11.5 sections, we found no evidence thatFgf2 knock-out mice have defects in the density or proportion of BrdU-labeled cells in the anterior portion of the PVE with respect to wild-type mice (data not shown). However, we detected previously in the same sections a 45% decrease in the total number of BrdU-labeled cells in the PVE of Fgf2 knock-out mice (Raballo et al., 2000). This is in line with our previous hypothesis that, in the absence of Fgf2, the total progenitor cell pool is smaller, even if the dynamics of cell division are the same among genotypes (Raballo et al., 2000). For example, progenitors for neurons of anterior regions of the cerebral cortex could undergo a premature exit from the cell cycle in Fgf2 knock-outs; alternatively, there could be a smaller pool of stem cells in the anterior PVE. Unfortunately, there is a lack of cell lineage markers that could allow us to reliably demarcate the anterior cortical progenitor pool from the posterior during neurogenesis. Furthermore, although we found no evidence for abnormal cell migration, it is still possible that fewer postmitotic neurons migrate to anterior cortical regions inFgf2 knock-out mice.
The multiple roles of Fgf2 in the regulation of pyramidal cell number and growth may impede compensatory mechanisms and lead to functional defects in Fgf2 knock-out mice. For example, the atrophy of the soma of these characteristically large cells may lead to a decreased capacity to sustain the metabolic demands of their large dendritic tree and a decreased turnover of synaptic proteins. Our findings on the decreased dendritic and neuropil staining in the anterior cortical regions of the Fgf2 knock-out mice further supports this view. This may further exacerbate the loss in pyramidal cell number and create a stronger defect in excitatory neurotransmission in the anterior cerebral cortex. Functional defects attributable to cortical abnormalities in mice are difficult to demonstrate because the cerebral cortex is not essential for survival or even for simple learning tasks in farm-raised small rodent species (Thompson, 1959). However, we show that the sleeping time after barbiturate challenge was greatly prolonged in these mutant mice. Because barbiturates act through the GABA receptor channel (Olsen and Leeb-Lundberg, 1981; Olsen, 1982), this observation suggests that GABAergic transmission is abnormally enhanced in these mice, leading to decreased arousal. It is possible that the excess sleep time may be attributable to a slower breakdown or excretion of barbiturates, although there is currently no evidence for either liver or kidney abnormalities in Fgf2 knock-out mice.
The regulation of sleep depends on the interplay between the cerebral cortex and subcortical stations within the basal ganglia, the diencephalon, and the brainstem, particularly the ventral tegmental area (VTA). Cholinergic, noradrenergic, and histaminergic cortical afferents exert a strong activating effect on the cerebral cortex, promoting arousal and modulating attention (Sherin et al., 1996; Saper, 2000). The central stations of this cortical activating system are the lateral hypothalamus and adjacent histaminergic neurons in the tuberomammillary area, which are reciprocally connected with cholinergic and aminergic brainstem areas and in turn reciprocally connect to the entire cerebral cortical mantle, including the cingulate, motor, and sensory areas (Risold et al., 1997; Saper, 2000). No apparent abnormalities in the hypothalamus and the brainstem reticular formation were noticed in the Fgf2 −/− mutants after Nissl staining. Preliminary observations on tyrosine hydroxylase-stained sections from the mutant and wild-type mice do not show any differences in the VTA and adjacent dopaminergic neurons of the substantia nigra. We conclude that the abnormalities in sleep observed in Fgf2 −/− mice most likely represent a consequence of the decreased glutamate/GABA neuronal ratio in the cerebral cortex. Various cortical regions, particularly prefrontal, project to the hypothalamus, the VTA, and the substantia nigra directly via the medial forebrain bundle, and indirectly via the ventral striatum and pallidum, and can conceivably affect the cortical activating system (Risold et al., 1997).
Because of the importance of Fgf2 in pyramidal cells of anterior cortical areas, we speculate that this factor may play a comparatively larger role in the primate species. Furthermore, phylogenetic pressure on the Fgf2 system may have contributed to the evolution of a key characteristic of the primate and human brain, the expansion of the anterior neocortex.
Footnotes
This work was supported by National Institutes of Health Grant PHS R017709 (to F.M.V). We thank J. Rhee for expert technical assistance, Dr. A. Guidotti for suggestions regarding the pharmacological treatment, and Dr. James F. Leckman for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Flora M. Vaccarino, Child Study Center, Yale University, 230 South Frontage Road, New Haven, CT 06520. E-mail: flora.vaccarino@yale.edu.
REFERENCES
- 1.Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997a;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- 2.Anderson SA, Qiu M, Bulfone A, Eisenstat DD, Meneses J, Pedersen R, Rubenstein JLR. Mutations of the homeobox genes Dlx-1 and Dlx-2 disrupt the striatal subventricular zone and differentiation of late born striatal neurons. Neuron. 1997b;19:1–20. doi: 10.1016/s0896-6273(00)80345-1. [DOI] [PubMed] [Google Scholar]
- 3.Anderson SA, Marin O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128:353–363. doi: 10.1242/dev.128.3.353. [DOI] [PubMed] [Google Scholar]
- 4.Arimatasu Y, Kojima M, Ishida M. Area and lamina-specific organization of a neuronal subpopulation defined by expression of latexin in the rat cerebral cortex. Neuroscience. 1999a;88:93–105. doi: 10.1016/s0306-4522(98)00185-7. [DOI] [PubMed] [Google Scholar]
- 5.Arimatasu Y, Ishida M, Takiguchi-Hayashi K, Uratani Y. Cerebral cortical specification by early potential restriction of progenitor cells and later phenotype control of postmitotic neurons. Development. 1999b;126:629–638. doi: 10.1242/dev.126.4.629. [DOI] [PubMed] [Google Scholar]
- 6.Campbell MJ, Hof PR, Morrison JH. A subpopulation of corticocortical neurons is distinguished by somatodendritic distribution of neurofilament protein. Brain Res. 1991;539:133–136. doi: 10.1016/0006-8993(91)90695-r. [DOI] [PubMed] [Google Scholar]
- 7.Conlon I, Raff M. Size control in animal development. Cell. 1999;96:235–244. doi: 10.1016/s0092-8674(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 8.Conti F, Rustioni A, Petrusz P, Towle AC. Glutamate-positive neurons in the somatic sensory cortex of rats and monkeys. J Neurosci. 1987;7:1887–1901. doi: 10.1523/JNEUROSCI.07-06-01887.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Carlos JA, Lopez-Mascaraque L, Valverde F. Dynamics of cell migration from the lateral ganglionic eminence in the rat. J Neurosci. 1996;16:6146–6156. doi: 10.1523/JNEUROSCI.16-19-06146.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ganat Y, Soni S, Chacon M, Schwartz ML, Vaccarino FM (2001) Chronic hypoxia upregulates Fibroblast Growth Factor (Fgf) ligands in the perinatal brain and induces Fgf-responsive radial glial cells in the subependymal zone. Neuroscience, in press. [DOI] [PubMed]
- 13.Gensburger C, Labourdette G, Sensenbrenner M. Brain basic fibroblast growth factor stimulates the proliferation of rat neuronal precursor cells in vitro. FEBS Lett. 1987;217:1–5. doi: 10.1016/0014-5793(87)81230-9. [DOI] [PubMed] [Google Scholar]
- 14.Gensburger C, Capo M, Deloulme JC, Sensenbrenner M. Influence of basic fibroblast growth factor on the development of cholinoceptive neurons from fetal rat cerebrum in culture. Dev Neurosci. 1992;14:278–281. doi: 10.1159/000111672. [DOI] [PubMed] [Google Scholar]
- 15.Grothe C, Otto D, Unsicker K. Basic fibroblast growth factor promotes in vitro survival and cholinergic development of rat septal neurons: comparison with the effects of nerve growth factor. Neuroscience. 1989;31:649–661. doi: 10.1016/0306-4522(89)90430-2. [DOI] [PubMed] [Google Scholar]
- 16.Hof PR, Morrison JH. Neurofilament protein defines regional patterns of cortical organization in the macaque monkey visual system: a quantitative immunohistochemical analysis. J Comp Neurol. 1995;352:161–186. doi: 10.1002/cne.903520202. [DOI] [PubMed] [Google Scholar]
- 17.Huang J, Mohammadi M, Rodrigues GA, Schlessinger J. Reduced activation of RAF-1 and MAP kinase by a fibroblast growth factor receptor mutant deficient in stimulation of phosphatidylinositol hydrolysis. J Biol Chem. 1995;270:5065–5072. doi: 10.1074/jbc.270.10.5065. [DOI] [PubMed] [Google Scholar]
- 18.Kay EP, Park SY, Ko MK, Lee SC. Fibroblast growth factor 2 uses PLC-γ1 for cell proliferation and PI3-kinase for alteration in cell shape and cell proliferation in corneal endothelial cells. Mol Vis. 1998;4:22–31. [PubMed] [Google Scholar]
- 19.Kilpatrick TJ, Bartlett PF. Cloning and growth of multipotential neural precursors: requirements for proliferation and differentiation. Neuron. 1993;10:255–265. doi: 10.1016/0896-6273(93)90316-j. [DOI] [PubMed] [Google Scholar]
- 20.Kim S, Li Q, Dang CV, Lee LA. Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-Myc in vivo. Proc Natl Acad Sci USA. 2000;97:11198–11202. doi: 10.1073/pnas.200372597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knusel B, Michel PP, Schwaber JS, Hefti F. Selective and nonselective stimulation of central cholinergic and dopaminergic development in vitro by nerve growth factor, basic fibroblast growth factor, epidermal growth factor, insulin and the insulin-like growth factors I and II. J Neurosci. 1990;10:558–570. doi: 10.1523/JNEUROSCI.10-02-00558.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuzis K, Reed S, Cherry NJ, Woodward WR, Eckenstein FP. Developmental time course of acidic and basic fibroblast growth factors expression in distinct cellular populations of the rat central nervous system. J Comp Neurol. 1995;358:142–153. doi: 10.1002/cne.903580109. [DOI] [PubMed] [Google Scholar]
- 23.Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci. 1999;19:7881–7888. doi: 10.1523/JNEUROSCI.19-18-07881.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature. 1997;387:422–426. doi: 10.1038/387422a0. [DOI] [PubMed] [Google Scholar]
- 25.Luskin MB, Pearlman AL, Sanes JR. Cell lineage in the cerebral cortex of the mouse studied in vivo and in vitro with a recombinant virus. Neuron. 1988;1:635–647. doi: 10.1016/0896-6273(88)90163-8. [DOI] [PubMed] [Google Scholar]
- 26.Matsumoto K, Ojima K, Watanabe H. Neurosteroidal modulation of social isolation-induced decrease in pentobarbital sleep in mice. Brain Res. 1996;708:1–6. doi: 10.1016/0006-8993(95)01277-x. [DOI] [PubMed] [Google Scholar]
- 27.Mohamadi M, Dikic I, Sorokin A, Burgess WH, Jaye M, Schlessinger J. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol Cell Biol. 1996;16:977–989. doi: 10.1128/mcb.16.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson SA, Aris JP, Patel BKR, LaRochelle WJ. Multiple growth factor induction of a murine early response gene that complements a lethal defect in yeast ribosome biogenesis. J Biol Chem. 2000;275:13835–13841. doi: 10.1074/jbc.275.18.13835. [DOI] [PubMed] [Google Scholar]
- 29.Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- 30.Olsen RW. Drug interactions at the GABA receptor-ionophore complex. Annu Rev Pharmacol Toxicol. 1982;22:245–277. doi: 10.1146/annurev.pa.22.040182.001333. [DOI] [PubMed] [Google Scholar]
- 31.Olsen RW. Barbiturates. Int Anesthesiol Clin. 1988;26:254–261. doi: 10.1097/00004311-198802640-00004. [DOI] [PubMed] [Google Scholar]
- 32.Olsen RW, Leeb-Lundberg F. Convulsant and anticonvulsant drug binding sites related to GABA-regulated chloride ion channels. Adv Biochem Psychopharmacol. 1981;26:93–102. [PubMed] [Google Scholar]
- 33.Ortega S, Ittmann M, Tsang SH, Ehrich M, Basilico C. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci USA. 1998;95:5672–5677. doi: 10.1073/pnas.95.10.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otto D, Unsicker K. FGF-2 modulates dopamine and dopamine-related striatal transmitter systems in the intact and MPTP-lesioned mouse. Eur J Neurosci. 1993;5:927–932. doi: 10.1111/j.1460-9568.1993.tb00943.x. [DOI] [PubMed] [Google Scholar]
- 35.Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH. Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci. 1999;19:8487–8497. doi: 10.1523/JNEUROSCI.19-19-08487.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peeper DS, Upton TM, Ladha MH, Neuman E, Zalvide J, Barnards R, DeCaprio JA, Ewen ME. Ras signaling linked to the cell-cycle machinery by the retinoblastoma protein. Nature. 1997;386:177–181. doi: 10.1038/386177a0. [DOI] [PubMed] [Google Scholar]
- 37.Prober DA, Edgar BA. Ras1 promotes cellular growth in the Drosophila wing. Cell. 2000;100:435–446. doi: 10.1016/s0092-8674(00)80679-0. [DOI] [PubMed] [Google Scholar]
- 38.Prober DA, Edgar BA. Growth regulation by oncogenes: new insights from model organisms. Current Opin Genet Dev. 2001;11:19–26. doi: 10.1016/s0959-437x(00)00151-9. [DOI] [PubMed] [Google Scholar]
- 39.Qian X, Davis AA, Goderie SK, Temple S. FGF2 concentration regulates the generation of neurons and glia from multipotent cortical stem cells. Neuron. 1997;18:81–93. doi: 10.1016/s0896-6273(01)80048-9. [DOI] [PubMed] [Google Scholar]
- 40.Raballo R, Rhee J, Lyn-Cook R, Leckman JF, Schwartz ML, Vaccarino FM. Basic fibroblast growth factor (Fgf2) is necessary for cell proliferation, neurogenesis in the developing cerebral cortex. J Neurosci. 2000;20:5012–5023. doi: 10.1523/JNEUROSCI.20-13-05012.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ragsdale CW, Assimacopoulos S, Fukuchi-Shimogori T, Grove EA. Early patterning of the cerebral cortex may be shaped by gradients of receptors and binding proteins of the FGF, BMP and WNT signaling pathways. Soc Neurosci Abstr. 2000;26:306. [Google Scholar]
- 42.Rakic P. Specification of cerebral cortical areas. Science. 1988;241:170–176. doi: 10.1126/science.3291116. [DOI] [PubMed] [Google Scholar]
- 43.Ray J, Peterson DA, Schinstine M, Gage FH. Proliferation, differentiation, and long-term culture of primary hippocampal neurons. Proc Natl Acad Sci USA. 1993;90:3602–3606. doi: 10.1073/pnas.90.8.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Risold PY, Thompson RH, Swanson LW. The structural organization of connections between hypothalamus and cerebral cortex. Brain Res Rev. 1997;24:197–254. doi: 10.1016/s0165-0173(97)00007-6. [DOI] [PubMed] [Google Scholar]
- 45.Saper CB. Hypothalamic connections with the cerebral cortex. Prog Brain Res. 2000;126:39–48. doi: 10.1016/S0079-6123(00)26005-6. [DOI] [PubMed] [Google Scholar]
- 46.Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–219. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- 47.Tan SS, Kalloniatis M, Sturm K, Tam PPL, Reese BE, Faulkner-Jones B. Separate progenitors for radial and tangential cell dispersion during development of the cerebral cortex. Neuron. 1998;21:295–304. doi: 10.1016/s0896-6273(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 48.Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 kinase and MAPKAP kinase-2. EMBO J. 1996;15:4629–4642. [PMC free article] [PubMed] [Google Scholar]
- 49.Tao Y, Black IB, DiCicco-Bloom E. In vivo neurogenesis is inhibited by neutralizing antibodies to basic fibroblast growth factor. J Comp Neurol. 1997;33:289–296. [PubMed] [Google Scholar]
- 50.Tao YB, Black IB, DiCicco-Bloom E. Neurogenesis in neonatal rat brain is regulated by peripheral injection of basic fibroblast growth factor (bFGF). J Comp Neurol. 1996;376:653–663. doi: 10.1002/(SICI)1096-9861(19961223)376:4<653::AID-CNE11>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 51.Thompson R. Learning in rats with extensive neocortical damage. Science. 1959;129:1223–1224. doi: 10.1126/science.129.3357.1223. [DOI] [PubMed] [Google Scholar]
- 52.Tropepe V, Hitoshi S, Sirad C, Mak TW, Rossant J, van der Kooy D. Direct neural fate specification from embryonic stem cells: a primitive mammalian neural stem cell stage acquired through a default mechanism. Neuron. 2001;30:65–78. doi: 10.1016/s0896-6273(01)00263-x. [DOI] [PubMed] [Google Scholar]
- 53.Vaccarino FM, Schwartz ML, Hartigan D, Leckman JF. Effect of basic fibroblast growth factor on the genesis of excitatory and inhibitory neurons in primary cultures of cells from the mammalian telencephalon. Cereb Cortex. 1995;1:64–78. doi: 10.1093/cercor/5.1.64. [DOI] [PubMed] [Google Scholar]
- 54.Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, Doetschman T, Coffin JD, Wyland JJ, Hung Y-TE. Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci. 1999;2:246–253. doi: 10.1038/6350. [DOI] [PubMed] [Google Scholar]
- 55.Vaccarino FM, Ganat Y, Zhang Y, Zheng W. Stem cells in neurodevelopment and plasticity. Neuropsychopharmacology. 2001;25:805–815. doi: 10.1016/S0893-133X(01)00349-9. [DOI] [PubMed] [Google Scholar]
- 56.Vicario-Abejon C, Johe KK, Hazel TG, Collazo D, McKay RDG. Functions of basic fibroblast growth factor and neurotrophins in the differentiation of hippocampal neurons. Neuron. 1995;15:105–114. doi: 10.1016/0896-6273(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 57.Wilke TA, Gubbels S, Schwartz J, Richman JM. Expression of Fibroblast growth factor receptors (FGFR1, FGFR2, FGFR3) in the developing head and face. Dev Dyn. 1997;210:41–52. doi: 10.1002/(SICI)1097-0177(199709)210:1<41::AID-AJA5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 58.Zettenberg A, Engstrom W, Dafgard E. The relative effects of the different types of growth factors on DNA replication, mitosis, and cellular enlargement. Cytometry. 1984;5:368–375. doi: 10.1002/cyto.990050413. [DOI] [PubMed] [Google Scholar]
- 59.Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, Yin M, Coffin JD, Kong L, Kranias EG, Luo W, Boivin GP, Duffy JJ, Pawlowski SA, Doetschman T. Fibroblast growth factor 2 controls vascular tone. Nat Med. 1998;4:201–207. doi: 10.1038/nm0298-201. [DOI] [PMC free article] [PubMed] [Google Scholar]