

Abstract
Traumatic axonal injury (TAI), a consequence of traumatic brain injury (TBI), results from progressive pathologic processes initiated at the time of injury. Studies attempting to characterize the pathology associated with TAI have not succeeded in following damaged and/or disconnected axonal segments back to their individual neuronal somata to determine their fate. To address this issue, 71 adult male Sprague Dawley rats were subjected to moderate central fluid percussion injury and killed between 30 min and 7 d after injury. Antibodies to the C terminus of β-amyloid precursor protein (APP) identified TAI in continuity with individual neuronal somata in the mediodorsal neocortex, the hilus of the dentate gyrus, and the dorsolateral thalamus. These somata were followed with immunocytochemical markers of neuronal injury targeting phosphorylated 200 kDa neurofilaments (RMO-24), altered protein translation (phosphorylated eukaryotic translation initiation factor 2α), and cell death [terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)], with parallel electron microscopic (EM) assessment. Despite the finding of TAI within 20–50 μm of the soma, no evidence of cell death, long associated with proximal axotomy, was seen via TUNEL or routine light microscopy/electron microscopy. Rather, there was rapid onset (<6 hr after injury) subcellular change associated with impaired protein synthesis identified by EM, immunocytochemical, and Western blot analyses. When followed 7 d after injury, these abnormalities did not reveal dramatic progression. Rather, some somata showed evidence of potential reorganization and repair. This study demonstrates a novel somatic response to TAI in the perisomatic domain and also provides insight into the multifaceted pathology associated with TBI.
Keywords: traumatic brain injury, traumatic axonal injury, axotomy, axon reaction, rat, protein translation, TUNEL
Traumatic axonal injury (TAI), as well as its clinical manifestation, diffuse axonal injury (DAI), is a common sequela of traumatic brain injury (TBI) (Cordobes et al., 1986;Adams et al., 1989) and is characterized by axonal swelling and disconnection (Maxwell et al., 1997). Once thought to occur immediately as a result of tearing evoked by TBI, it is now known that tearing rarely occurs (Maxwell et al., 1997). Rather, TAI typically involves a more progressive response involving a transient, traumatically induced disruption of the axonal membrane, allowing for unregulated calcium entry (Maxwell et al., 1995, 1999; Pettus et al., 1994; Pettus and Povlishock, 1996; Wolf et al., 2001). This calcium influx initiates calpain activation (Buki et al., 1999; Shields et al., 2000) and mitochondrial injury/swelling (Okonkwo and Povlishock, 1999) with cytochrome c release and caspase activation (Buki et al., 2000), leading to further axonal injury and detachment over time.
Insight into the pathology of TAI has not been accompanied, however, by parallel information on the fate of the related neuronal somata. It is unknown whether the cell bodies of origin of traumatically injured axons eventually die, atrophy, or reorganize. This uncertainty stems from the fact that the study of TAI has focused on long tract axons (Buki et al., 2000), remote from their somata. These tracts assessed within various white matter domains offer the advantage of relative homogeneity and high axonal density. Identification of TAI within or near the gray matter, close to neuronal somata, is limited by the complex cytoarchitecture.
Although no studies have identified neuronal somata directly linked to TAI, some have attempted to characterize this somatic response to injury. Maxwell and colleagues (1994), using optic nerve stretch to generate TAI near the chiasm, demonstrated onset of chromatolysis 72 hr after injury in select retinal ganglion cells, with potentially related cellular loss at 7–14 d. Other studies of TAI have reported increased somatic accumulation of β-amyloid precursor protein (APP) (Gentleman et al., 1993; Bramlett et al., 1997; Van den Heuvel et al., 1998). In these studies, however, affected neuronal somata could not be linked to TAI. Thus, it was uncertain whether these responses were the result of TAI or a generalized response to trauma.
In this communication, we characterize, for the first time, the neuronal somatic response to TAI. Using an antibody to the C terminus of APP (Stone et al., 2000), we show TAI in continuity with adjacent somata. Via the utilization of immunocytochemical markers of somatic injury, including antibodies to phosphorylated neurofilament subunits, antibodies to phosphorylated translation factors, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) methodologies, and electron microscopic (EM) analyses, we followed these somata over 7 d after injury. These studies yielded the unanticipated finding that TAI, even in immediate proximity to the neuronal soma, did not result in death during the period assessed, inconsistent with previous studies of primary axotomy produced by transection, crush, or stretching (Barron, 1983). These findings illustrate the differences between TAI and primary axotomy and also demonstrate the complexity of TBI pathobiology.
MATERIALS AND METHODS
Surgical preparation and injury induction. The procedures by which rats were subjected to central fluid percussion injury (FPI) were consistent with those described previously (Sullivan et al., 1976; Dixon et al., 1987). Briefly, 71 male Sprague Dawley rats weighing 315–413 gm were surgically prepared for the induction of central FPI. Each animal was anesthetized in a bell jar with 4% isoflurane in 70% N2O and 30% O2. After induction, each animal's head was shaved and placed in a stereotactic frame (David Kopf Instruments, Tujunga, CA) fitted with a nose cone to maintain anesthesia with 1–2% isoflurane in 70% N2O and 30% O2. A thermostatically controlled heating pad (Harvard Apparatus, Holliston, MA) was then placed under the animal and set to monitor the rectal temperature and, via feedback control, maintain the body temperature at 37°C during the surgery. A midline sagittal incision was made to expose the skull from bregma to lambda. The skull was cleaned and dried, and two 1 mm holes were drilled in the right frontal and occipital bones 1 mm rostral and caudal to bregma and lambda, respectively, for the subsequent insertion of fixation screws. A 4.8 mm circular craniotomy was then made along the sagittal suture midway between bregma and lambda, taking care not to damage the underlying dura. A Leur-Loc syringe hub was then cut away from a 20 gauge needle and affixed to the craniotomy site using cyanoacrylate. After confirming the integrity of the seal between the hub and the skull, two fixation screws (round machine screws; 3/16 inch long) were inserted into the 1 mm holes. Dental acrylic was then applied over the screws and around the hub to provide stability during the induction of injury. After the dental acrylic hardened, the skin was closed over the hub with sutures, topical Bacitracin ointment was applied, and the animal was removed from anesthesia and monitored in a warmed cage until fully recovered. The animal was then returned to the central housing facility and allowed to recover for 24 hr before injury. Before the induction of injury, each animal was again anesthetized for 4 min in a bell jar with 4% isoflurane in 70% N2O and 30% O2. After removal from the jar, an incision was quickly made to expose the craniotomy site and the male end of a spacing tube was inserted into the hub. The hub-spacer assembly was filled with normal saline and the female end of the spacer was inserted on to the male end of the fluid percussion apparatus ensuring that no air bubbles were introduced into the system. The animals were then injured at a magnitude of 2.00 ± 0.05 atmospheres, corresponding to a brain injury of moderate severity (Dixon et al., 1987). The pressure pulse measured by the transducer was displayed on a storage oscilloscope (Tektronix 5111, Beaverton, OR), and the peak pressure was recorded. Injury preparation and induction were completed before recovery from anesthesia. After injury, the animals were monitored for recovery of spontaneous respiration and, if necessary, ventilated to ensure adequate post-injury oxygenation until spontaneous respiration was regained. The hub, dental acrylic, and screws were removed en bloc, and the incision was closed quickly with sutures before recovery from unconsciousness. The duration of transient unconsciousness was determined for all animals by measuring the time it took each to recover the following reflexes: toe pinch, tail pinch, corneal blink, pinnal, and righting. After recovery of the righting reflex, animals were placed in a holding cage with a heating pad to ensure the maintenance of normothermia and monitored during recovery. For animals receiving a sham injury, all of the above steps were followed minus the release of the pendulum to induce the injury. Animals with a short post-injury survival time (≤2 hr) were removed from the holding cage for perfusion, and animals with longer survival times (≥4 hr) were returned to the animal housing facility after recovery until the designated time of perfusion. Central FPI caused transient unconsciousness in all injury groups when compared with sham-injured animals (p < 0.001; df between groups = 9; df within groups = 62; data not shown), as determined by a one-way ANOVA of righting reflex times with Tukey post hoc analysis. No significant difference in transient unconsciousness was noted among injury groups (p > 0.05; data not shown), indicating that all groups received injuries of comparable severity.
Tissue preparation for single-label immunocytochemistry, double-label immunofluorescence, and TUNEL analysis. After injury, animals were allowed to recover for times varying from 30 min to 7 d (sham, n = 7; 30 min, n = 5; 2 hr,n = 3; 4 hr, n = 5; 6 hr,n = 5; 12 hr, n = 3; 24 hr,n = 15; 48 hr, n = 8; 72 hr,n = 8; 7 d, n = 6). At the predetermined times, the rats were intraperitoneally injected with an overdose of sodium pentobarbital and then transcardially perfused with 4% paraformaldehyde and 0.1% glutaraldehyde in Millonig's buffer for immunohistochemistry. After perfusion, the brains were removed and immersed in the perfusion fixative for 1 hr. Each brain was coronally blocked at the optic chiasm and the midbrain to include the parietal and temporal cortices, hippocampus, and thalamus. The blocked brains were then post-fixed in the perfusion fixative for 24 hr. For tissue sectioning, the brain blocks were flat mounted with cyanoacrylate and embedded in agar. Blocks were then coronally sectioned in 0.1m phosphate buffer with a Leica VT1000S (Leica Microsystems, Bannockburn, IL) at a thickness of 40 μm. Sections were serially collected in alternating wells, such that each well consisted of adjacent sections that, after immunocytochemical processing, could be compared to permit spatiotemporal characterization of the chosen markers. The tissue was stored in Millonig's buffer in 12-well culture plates (Falcon, Newark, DE).
Immunocytochemistry. To identify and characterize the response of neuronal somata injured secondary to TAI, established makers of axonal and neuronal injury were used. Parallel light microscopic (LM) and EM immunocytochemical studies using antibodies to APP, a well characterized marker of impaired axonal transport denoting sites of TAI (Stone et al., 2000), were performed to identify injured axonal segments and their somata. Because previous studies using experimental primary axotomy have demonstrated abnormal accumulation of phosphorylated neurofilaments within axotomized neurons (Rosenfeld et al., 1987; Koliatsos et al., 1989; Martin et al., 1999), this possibility was also explored via the use of an antibody targeting a specific phosphorylated epitope on the tail domain of the 200 kDa neurofilament (RMO-24). Additionally, given that other studies of primary axotomy have revealed alteration of the rough endoplasmic reticulum (RER) (Lieberman, 1971; Barron, 1983; Kreutzberg, 1995), which is associated with protein synthesis (Siegel et al., 1999), we used an LM approach that targets a factor involved in the regulation of protein translation. Specifically, we used antibodies targeting a specific serine-51 phosphorylated form of the α-subunit of eukaryotic translation initiation factor 2 [eIF2α(P); eIF2α, unphosphorylated], which has previously been shown to inhibit protein synthesis (Singh et al., 1994) after postischemic reperfusion injury (DeGracia et al., 1997). The antibodies to eIF2α(P) were used in single- and double-label immunocytochemical approaches and in Western blots to identify corresponding changes in neuronal somata with TAI. Furthermore, to address the potential for the above-described change to evolve to nuclear fragmentation and cell death after TAI, the TUNEL method was also used.
Bright-field LM and EM single-labeling protocol. Antibodies targeting the C terminus of the β-APP (Zymed, San Francisco, CA) were used because recent studies from our laboratory have found them to be sensitive and specific markers (Stone et al., 2000) for identifying TAI within gray matter regions. Endogenous peroxidase activity was blocked with 0.5% H2O2 in PBS for 30 min. Sections were then processed using the temperature-controlled microwave antigen retrieval approach described previously (Stone et al., 1999). After microwave antigen retrieval, sections were preincubated for 45 min in 10% normal goat serum (NGS) with 0.2% Triton X-100 in PBS. The tissue was then incubated overnight in a 1:2500 dilution of the APP C terminus primary antibody (rabbit; Zymed)in 1% NGS in PBS. Sections were then incubated for 1 hr in biotinylated goat anti-rabbit secondary antibody (IgG) diluted 1:200 in 1% NGS in PBS (Vector, Burlingame, CA) and then for 1 hr in a 1:200 dilution of an avidin–horseradish peroxidase complex (ABC Standard Elite Kit, Vector). The reaction product was visualized with 0.05% diaminobenzidine, 0.01% hydrogen peroxide, and 0.3% imidazole in 0.1 m phosphate buffer for 10–20 min. Sections were mounted on gelatin-coated slides, dehydrated, and coverslipped. To localize phosphorylated neurofilaments and ascertain whether abnormal somatic accumulation of phosphorylated neurofilaments occurred subsequent to TAI, the above APP protocol was used with the following modifications: 0.1 m Tris, pH 7.4, was used in place of PBS, and 0.1 m Tris, pH 7.6, was used in place of 0.1 m phosphate buffer, given the phosphospecific nature of the antibody. The primary mouse RMO-24 antibody (Zymed) was used at a dilution of 1:3000 in 1% normal horse serum (NHS) in 0.1 m Tris, pH 7.4. The secondary, biotinylated rat-adsorbed horse anti-mouse antibody was used at a dilution of 1:300 in 1% NHS in 0.1 m Tris, pH 7.4. To identify the generalized suppression of protein translation mediated via an injury-induced phosphorylation of eIF2α, the above RMO-24 procedure was used with several changes. The primary, rabbit anti-eIF2α(P) (Biosource, Camarillo, CA) antibody was used at a dilution of 1:7500 in 1% NGS in 0.1 m Tris, pH 7.4, and the secondary, biotinylated goat anti-rabbit antibody was used at 1:400 in 1% NGS in 0.1 m Tris, pH 7.4.
Electron microscopy. Selected APP-labeled neocortical sections from animals killed at 6, 12, 24, 48, 72 hr, and 7 d after injury were subjected to further processing for EM analysis to ascertain the effect of delayed axonal injury on subcellular organization and to survey for any signs indicative of cellular fate. The tissue was osmicated, dehydrated, and flat embedded between plastic slides in medcast resin (Ted Pella, Redding, CA). The embedded slides were then scanned to identify neurons in clear continuity with adjacent, traumatically injured, APP-immunopositive axons. Once identified, these sites were removed, mounted on plastic studs, and thick sectioned to the depth of the immunoreactive sites of interest. Serial 70 nm sections were cut and picked up onto Formvar-coated slotted grids. The grids were then stained in 5% uranyl acetate in 50% methanol for 2 min and in 0.5% lead citrate for 1 min. Ultrastructural analysis was performed using a JEOL 1200 electron microscope.
TUNEL. After perfusion, brains from selected sham injured and injured animals surviving 6, 24, 48, 72 hr, and 7 d were paraffin embedded and sectioned to a thickness of 10–12 μm. To identify nuclear fragmentation within degenerating cells, TUNEL staining was used (ApopTag Peroxidase In Situ Apoptosis Detection Kit; Intergen, Gaithersburg, MD). Sections were deparaffinized, rinsed in PBS for 5 min, and pretreated with 0.5% Triton X-100 (Sigma, St. Louis, MO) for 5 min. The manufacturer's protocol was then followed to apply both the digoxigenin-dUTP label and subsequent anti-digoxigenin peroxidase antibody. Briefly, sections were treated with 3.0% H2O2 for 5 min to quench endogenous peroxidase activity and washed two times for 5 min in PBS. The proprietary equilibration buffer was applied to the samples for 10 min, after which terminal deoxynucleotidyl transferase (TdT) was applied for 60 min and the tissue was incubated in a humidified chamber at 37°C for 1 hr. The slides were then immersed in the proprietary stop/wash buffer for 10 min, and the sections were then incubated at room temperature in a humidified chamber for 45 min in the anti-digoxigenin peroxidase conjugate. After four 2 min rinses in PBS, the reaction product was developed with 0.05% diaminobenzidine (Sigma) and 0.01% H2O2 in 0.1m phosphate buffer for 10 min. After rinsing three times for 1 min in double-distilled H2O, slides were counterstained with hematoxylin and dehydrated, cleared, and coverslipped for routine LM analysis. Thymic sections from naïve animals (unoperated controls) were used as positive controls, and omission of the TdT enzyme and/or anti-digoxigenin antibody was used to generate negative controls.
Double-label immunofluorescence microscopy. To better define the spatiotemporal linkage between neuronal perisomatic axotomy and impairment in protein translation, double-label immunofluorescence was performed using antibodies to APP and eIF2α(P). Because both of these primary antibodies were generated in rabbit hosts, direct fluorescent labeling via tyramide signal amplification (TSA) (New England Nuclear, Boston, MA) was used with the eIF2α(P) antibody. Control experiments were performed to ensure that the concentration of primary eIF2α(P) antibody used was sufficient to generate a signal using direct green tyramide amplification while also ensuring that the concentration was low enough to prevent any detection when cross-reacting secondary antibodies to APP were applied (Teramoto et al., 1998; Wang et al., 1999).
Sections were rinsed three times for 10 min in 0.1 m Tris, pH 7.4, after which endogenous peroxidase activity was first blocked by a 30 min incubation in 0.5% H2O2 in Tris, followed by a second incubation in 3% H2O2 in Tris for 10 min. Sections were again rinsed three times for 10 min in Tris, before microwave antigen retrieval as described above. Next, the sections were allowed to cool for 20 min and then rinsed rapidly three times in Tris. They were then incubated for 60 min in 10% NGS in Tris with 0.2% Triton X-100 and rinsed once for 10 min in 1% NGS in Tris. Rabbit anti-eIF2α(P) antibody was diluted 1:10,000 in 1% NGS in Tris and applied for an overnight incubation. The eIF2α(P) primary antibody was then removed, and the sections were rinsed three times for 10 min in 1% NGS in Tris. Sections were then incubated for 60 min in 1:200 biotinylated goat anti-rabbit secondary antibody. After three 10 min rinses in Tris, a 1:200 dilution of avidin–horseradish peroxidase complex (ABC Standard Elite Kit, Vector) was applied for 60 min. For TSA, using the TSA Fluorescein System (NEL701A; NEN, Boston, MA), sections were first rinsed in TSA blocking buffer for 20 min, then rinsed two times in Tris 2 for 10 min. Sections were incubated in a 1:300 dilution of the fluorescein-tyramide reagent in 1:5 amplification diluent and Tris for 10 min. At all times during and after TSA, every effort was made to ensure that the tissue was kept in the dark to prevent photobleaching. The tissue was then rinsed three times for 10 min in Tris and then incubated for 60 min in 10% NGS in Tris. The tissue was then incubated overnight in a 1:200 dilution of the APP antibody in 1% NGS in Tris. After removal of the APP solution, sections were rinsed three times for 10 min in 1% NGS in Tris. The tissue was then incubated for 2 hr in a 1:200 dilution of Alexa 594 goat anti-Rabbit IgG (Molecular Probes, Eugene, OR) in 1% NGS in Tris and then rinsed three times for 10 min in Tris.
To determine whether DNA fragmentation and cell death occurred in those neuronal somata linked to traumatically injured axonal segments, TUNEL staining was combined with fluorescent labeling for APP. Selected sections from sham-injured animals, as well as from animals surviving 24, 48, and 72 hr and 7 d after injury, were labeled first with a fluorescein TUNEL tag and then stained for APP. Using the fluorescent TUNEL label (ApoTag Fluorescein In Situ Apoptosis Detection Kit), the tissue was rinsed two times for 5 min in PBS and then preincubated in 0.5% Triton X-100 in PBS for 10 min. The proprietary equilibration buffer was applied for 10 min, after which the sections were incubated in the TdT enzyme for 1 hr in a 37°C, humidified chamber. After rinsing in the proprietary stop/wash buffer for 10 min, sections were rinsed in PBS four times for 2 min. The fluorescein anti-digoxigenin antibody was then applied to the tissue for 30 min. This and all subsequent steps were performed in the dark to prevent photobleaching of the fluorescent dyes. Sections were then rinsed four times for 5 min in PBS and then prepared for microwave antigen retrieval as described previously. After retrieval, sections were allowed to sit for 10 min and then were quickly rinsed three times in PBS. The tissue was incubated for 60 min in 10% NGS in PBS with 0.5% Triton X-100 and subsequently rinsed three times for 10 min in PBS. The sections were then incubated overnight in a 1:200 dilution of the APP antibody in 1% NGS in PBS. After removal of the APP solution, sections were rinsed three times for 10 min in 1% NGS in PBS. The tissue was then incubated for 2 hr in a 1:200 dilution of Alexa 594 Goat anti-Rabbit IgG (Molecular Probes) in 1% NGS in PBS. Sections were then rinsed three times for 10 min in PBS. After processing, all sections were mounted on glass slides, coverslipped using an antifade reagent (Prolong Antifade Kit; Molecular Probes), and sealed with nail polish.
Digital image acquisition and analysis. All qualitative single- and double-label light microscopic analysis and capture were performed using a Nikon Eclipse 800 (Tokyo, Japan) fitted with a Spot-RT digital camera (Diagnostic Instruments, Sterling Heights, MI). Appropriate excitation/emission filters were used with immunofluorescent specimens. Fluorescent images were then analyzed, and overlays were performed using Image-Pro Plus (Media Cybernetics, Silver Spring, MD). Selected APP-stained sections from rats killed at 30 min (n = 3), 2 hr (n = 3), 4 hr (n = 3), 12 hr (n = 3), 24 hr (n = 3), and 48 hr (n = 3) after injury were also subjected to quantitative LM analysis to characterize injured axons and their connected neuronal somata. Four nonadjacent mid-dorsal hippocampal sections, −3.3 to −4.8 mm posterior to bregma (Paxinos and Watson, 1986), were selected and evaluated with a Nikon Labophot microscope fitted with an ocular micrometer. Neuronal somata demonstrating unequivocal connectivity to adjacent injured axons were studied at 400×. In these, three axonal parameters were measured at varying time points after injury: (1) the length of the APP-containing swelling, (2) the length of the axonal segment connecting the neuronal soma to the swelling, and (3) the total length of the axon, including both the swollen and connecting portions. The measurements were made in the mediodorsal neocortex and thalamus, but not the hilus of the dentate gyrus, because the area of this region is much smaller than the previous two and did not provide a large enough sample size for statistical analysis. Three hundred and twenty-two APP-immunopositive swellings connected to neuronal somata were measured in the neocortex, and 300 were characterized in the thalamus. The data were evaluated for significance via one-way ANOVA with subsequent pairwise analysis using Tukey post hoc comparisons at α = 0.05.
Western blot. The method used in this experiment was a modified version of one described previously (DeGracia et al., 1997). Briefly, 24 hr after central FPI or sham-injury, rats (n = 6; sham = 3, injury = 3) were anesthetized for 4 min in 4% isoflurane in 70% N2O/30% O2. Animals were then quickly decapitated, and the brain was removed and placed on ice for subregion isolation. The brain was then sagittally blocked, and the mediodorsal neocortex, hippocampus, and thalamus from both sides were quickly isolated. Each tissue block was homogenized in 5 vol of ice-cold 20 mm Tris, pH 7.6, 2 mm EGTA, 1 mm EDTA, 200 mm sucrose, 50 mm KCl, 100 mm NaF, 5 mm magnesium acetate, 1 mm DTT, 7 μg/ml pepstatin A, 10 μg/ml leupeptin, and 1 μg/ml aprotinin in a Dounce homogenizer. The homogenates were centrifuged for 15 min at 15,800 × gat 4°C, the supernatants was removed, and after 5 μl samples were collected for protein assay (Bio-Rad, Hercules, CA), each supernatant was aliquotted and frozen at −80°C. Total protein concentration in each supernatant was then determined via the Bradford method using duplicate samples of each supernatant assayed in triplicate. Samples of the specific region of interest from each animal were then prepared for SDS-PAGE (Novex NuPage; Invitrogen, Carlsbad, CA) by mixing 10 μg of protein with appropriate volumes of 4× sample buffer and heating to 70°C for 10 min. Samples were then loaded in separate lanes on a 4–12% Bis-Tris gel and electrophoresed. The gel was transferred to nitrocellulose (0.2 μm pore size; Novex), which was fixed in 25% isopropanol and 12.5% acetic acid for 20 min. Gels were subsequently stained for total protein with Coomassie Blue (Sigma) to ensure complete transfer. After six 5 min rinses in Tris-buffered saline (TBS), blots were incubated for 1 hr in 5% nonfat dry milk. Blots were then incubated for another hour in a 1:1000 dilution of eIF2α(P) antibody in 5% milk in TBS with 1% Tween 20 (TTBS) and rinsed six times for 5 min in TTBS. A 1:10,000 dilution of horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch, West Grove, PA) was then added to the blot for 30 min. The blot was then rinsed again six times for 5 min in TTBS, and immunoreactive bands were identified using an enhanced chemoluminescence (ECL) system (Amersham Biosciences, Piscataway, NJ). Given that traumatic insult can affect multiple cellular systems and thus potentially alter the expression of common marker proteins used as internal controls (Bareyre et al., 2001), confirmation of equal loading was performed using Ponceau S solution (Sigma) to stain for total protein in the transfer membrane after ECL was completed. Any lanes that showed unequal protein staining were excluded from analysis. Densitometry was performed (Image Pro Plus) to quantitate the optical density of each band and to detect any injury-induced increase in eIF2α(P) relative to sham-injured animals. Data were analyzed by a two-tailed independent samples t test for significance.
RESULTS
General findings
All sections processed for LM immunocytochemical analyses revealed a pattern of macroscopic and microscopic change consistent with that routinely described with moderate central FPI (Dixon et al., 1987). Typically, the brain sites assessed showed no evidence of contusional change or cavitation. The brain parenchyma was devoid of hemorrhage, with the exception of isolated petechial hemorrhages in the corpus callosum. Also, limited subarachnoid bleeding was found over the dorsal convexity incident to the site of injury. In multiple brain loci including the mediodorsal neocortex, the dentate hilus, and dorsolateral thalamus, axonal swellings consistent with TAI were found approximating the neuronal somata, and in many cases these injured axonal segments were found in continuity with their adjacent cell bodies of origin (Fig. 1). Although detailed counts of neurons linked to TAI were not performed, these cells were consistently identified in all injured animals in the above-mentioned regions, indicating a generalized response to injury rather than an isolated occurrence.
Fig. 1.

Neuronal somata linked to TAI can be observed in multiple brain loci after central FPI. Representative photomicrographs from the mediodorsal neocortex (A, 6 hr after injury), hilar region of the dentate gyrus (B, 4 hr after injury), and dorsolateral thalamus (C, 12 hr after injury) illustrate neuronal somata in each region connected to adjacent, APP-immunoreactive segments sustaining TAI. Somata in continuity with injured axonal processes are indicated (arrowheads). Scale bars, 50 μm.
Qualitative and quantitative LM findings for APP
After APP immunocytochemistry, sections from sham-injured animals exhibited limited background APP staining within the gray matter of the neocortex, hippocampus, and thalamus during LM examination. Within these regions, isolated neuronal somata immunoreactive for APP were observed, particularly within laminae II–III of the neocortex. However, no APP-positive axons were discerned adjacent to these immunoreactive somata, nor was staining observed in the white matter tracts of either the corpus callosum or the internal capsule of sham-injured animals.
Thirty minutes after moderate central FPI, small APP-immunoreactive axonal swellings were observed in lamina V of the neocortex, the hilus of the dentate gyrus, and the dorsolateral thalamus. Although many of these spheroidal swellings were isolated, some were observed to be in direct continuity with their cell body of origin (Fig.2A). With light microscopy, these neuronal somata displayed axons exhibiting a proximodistal increase in APP immunoreactivity, ultimately culminating in intensely immunoreactive, lobulated axonal swellings. In the neocortex, the length of these connected axonal swellings at this time was 5.1 ± 1.1 μm (mean ± SEM), whereas in the thalamus the average swelling length was 4.6 ± 0.5 μm (Table1). The mean length of the unswollen portion of the axon connecting the neuronal somata to the axonal swellings and the total axonal length (sum of the swollen and unswollen lengths of the axon) were similar in these two regions, measuring 23.4 ± 5.4 and 28.5 ± 5.4 μm, respectively, in the neocortex and 18.2 ± 4.4 and 23.2 ± 3.7 μm, respectively, in the thalamus. At this time point, some of the swellings remained connected to their downstream segments, whereas others were disconnected. Neuronal somata with attendant TAI exhibited no LM signs of pathologic change or any increase in APP relative to surrounding, nonaxotomized neurons at this time.
Fig. 2.

Axonal swellings connected to adjacent neuronal somata evolve over time. Neocortical neurons with adjacent TAI in animals surviving 30 min (A), 4 hr (B), 12 hr (C), and 48 hr (D) after injury. Representative progressive changes in the length of the unswollen portion of the axon (indicated in A by white line a), length of swollen portion of the axon (indicated in A by black line b), and total axonal length (sum of a andb in A) are illustrated. Scale bars, 20 μm.
Table 1.
Quantitation of injured axonal features in neurons with TAI
| Time after injury (hr) | Neocortex | Thalamus | ||||
|---|---|---|---|---|---|---|
| Swollen axon length (μm) | Unswollen axon length (μm) | Total axonal length (μm) | Swollen axon length (μm) | Unswollen axon length (μm) | Total axonal length (μm) | |
| 0.5 | 5.1 ± 1.1 | 23.4 ± 5.4 | 28.5 ± 5.4 | 4.6 ± 0.5 | 18.2 ± 4.4 | 23.2 ± 3.7 |
| 2 | 7.2 ± 1.4 | 23.6 ± 5.0 | 30.3 ± 5.4 | 7.9 ± 0.9 | 21.3 ± 4.6 | 29.9 ± 4.7 |
| 4 | 8.7 ± 1.7 | 20.1 ± 5.1 | 28.8 ± 5.8 | 9.5 ± 1.4 | 17.1 ± 5.1 | 26.2 ± 3.9 |
| 12 | 15.6 ± 4.51-b | 20.2 ± 4.1 | 34.7 ± 6.2 | 15.7 ± 4.61-a | 23.3 ± 7.9 | 38.0 ± 10.81-a |
| 24 | 14.7 ± 4.51-b | 19.8 ± 6.3 | 34.5 ± 8.1 | NA | NA | NA |
| 48 | 8.7 ± 2.5 | 42.6 ± 8.11-d | 51.5 ± 8.51-c | NA | NA | NA |
NA, Unable to measure at this time point.
Significantly different from 0.5 hr after injury.
Significantly different from 0.5 and 2 hr after injury.
Significantly different from 0.5, 2, and 4 hr after injury.
Significantly different from all other time points after injury.
At 2 hr after injury, few traumatically injured axons were continuous with their downstream segments. With increasing survival (4–6 hr after injury), all visualized reactive axonal swellings were disconnected, with no continuity seen between the APP-positive swellings and distal axonal segments. At this time, multiple immunoreactive neuronal somata with axonal swellings were evident in the neocortex, dentate hilus, and thalamus (Fig. 2B). Intense APP immunoreactivity was observed within the cytoplasm of many neuronal somata with associated swellings, although other APP-immunoreactive neuronal somata were also seen, despite the fact that they shared no relationship to adjacent axotomized profiles. These axotomized neuronal somata continued to exhibit normal morphology at the LM level, with no obvious cellular shrinkage, swelling, or nuclear alteration.
At 12 hr after injury, many neuronal somata in continuity with damaged/swollen APP-positive axons remained visible within the neocortex, hilus of the dentate gyrus, and thalamus. Consistent with the described progression of secondary TAI in pontomedullary white matter (Stone et al., 2001), the axonal swellings now took on an elongate form, with the once spheroidal, ball-like swellings now appearing as truncated, club-like profiles (Fig. 2C). In the neocortex, swelling length was 15.6 ± 4.5 μm at this time, which was significantly greater than the swelling length noted at both 30 min and 2 hr after injury (p < 0.05). Likewise, in the thalamus, swelling length was 15.7 ± 4.6 μm, significantly increased in comparison to 30 min after injury (p < 0.05) (Table 1). In the thalamus, total axonal length measured 38.0 ± 10.8 μm, a significant increase in comparison to the 30 min post-injury time point (p < 0.05). In addition to this finding, an apparent increase in the number of APP-immunoreactive axonal fibers was also noted in the thalamus. However, most of these immunoreactive fibers were not observed in continuity with adjacent neuronal cell bodies. Within all analyzed fields, neuronal morphology appeared unchanged at the LM level, wherein axotomized neurons exhibited no alteration in comparison to adjacent neurons without associated axonal injury. As in the previous time points, axotomized neuronal somata continued to demonstrate increased APP immunoreactivity.
By 24 hr after injury, the distribution and morphological features of the axotomized neuronal somata did not appear to differ from earlier time points (data not shown). A further increase in disconnected APP-immunoreactive profiles was noted in the thalamus at this time. The intensity of the immunoreactivity in this region interfered with clear and reliable identification of neurons with adjacent TAI, excluding this and subsequent time points from further quantitative analysis. At 48 hr after injury, however, a reduction in the length of the axonal swellings in direct continuity with their sustaining neuronal somata to 8.7 ± 2.5 μm was noted in the neocortex (Fig.2D). The average length of the unswollen portion of the axon at this time in the neocortex was significantly elevated over all previous time points after injury, increasing to 42.6 ± 8.1 μm (p < 0.05). The total axonal length was also significantly greater than earlier time points, measuring 51.5 ± 8.5 μm (p < 0.05).
Few isolated APP-positive axons were noted by 72 hr after injury, and fewer were observed in continuity with their neuronal somata in the neocortex and hilar region of the dentate gyrus (data not shown). Similarly, in the thalamus, neuronal somata with axonal injury were infrequently noted, although numerous isolated APP-immunoreactive segments and swellings persisted in this locus. Again, at this time point, the morphology of scattered axotomized neurons appeared normal at the LM level, with no overt cell shrinkage or nuclear condensation.
After 7 d, immunoreactive neuronal somata in continuity with adjacent reactive axonal segments could not be identified in the neocortex, hilus of the dentate gyrus, or thalamus because of the inability to identify APP immunopositive axonal swellings at this time point. In the neocortex, scattered APP-positive neuronal somata persisted; however, unlike the isolated APP-positive somata identified within neocortical laminae II–III of sham-injured animals, these neurons were found predominantly within lamina V, consistent with that anatomical locus previously recognized to contain neuronal somata linked to APP-immunoreactive swollen axons. Detailed examination of these cells did reveal somatic morphologic change. Some neurons displayed eccentrically displaced nuclei, consistent with a chromatolytic response (data not shown). The isolated APP-immunoreactive segments and swellings noted in the thalamus at previous time points were markedly reduced at this time.
Qualitative ultrastructural analysis
Although the current communication evaluated EM change in neocortical, hippocampal, and thalamic regions and found comparable abnormalities in all, the following description will be restricted to the neocortex for the purposes of clarity and focus. Examination of sections taken from sham-injured animals revealed neocortical neuronal somata with centrally located nuclei, well organized arrays of RER, compact mitochondria, and multiple synaptic contacts (data not shown). These morphologic features are consistent with the features previously described in the neocortex of uninjured animals (Peters et al., 1991).
Within 6 hr of the traumatic injury, EM analyses confirmed the presence of perisomatic axonal injury in addition to the onset of organelle change within the related soma (data not shown). Sites of TAI revealed axonal disconnection, with the axon in continuity with the soma revealing swelling and the local accumulation of APP in vesicular and tubular profiles. These swollen axonal profiles were invested by a thinned myelin sheath, with some showing no myelin investment, most likely caused by myelin slippage. The proximal axonal segment in continuity with the soma was swollen with APP-containing vesicles and tubules consistent with an impairment of axoplasmic transport. The related soma did not manifest overt structural change, revealing an intact cytoskeleton as well as normal-appearing mitochondria and Golgi. Although the RER and its associated ribosomes and polysomes typically revealed normal detail, discrete foci within the soma demonstrated disaggregation of the cytoplasmic polysomes, with dispersal and degranulation of the RER. The perisomatic cell membrane showed limited evidence of glial swelling in addition to the presence of electron-dense, degenerating nerve terminals. In the adjacent neuropil, electron-dense nerve terminals were seen in addition to other electron-dense, axonal debris, all of which were consistent with the onset of anterograde/Wallerian degeneration. Despite the presence of these reactive changes, however, other neurons in the same field showed no evidence of related axonal injury or any other form of reactive change.
By 24 hr after injury (Fig. 3), the above-described changes revealed continued progression, with the injured axon showing swelling and APP accumulation. Typically, the axonal swellings were disconnected and were devoid of myelin investment caused by slippage. In contrast to the 6 hr time frame, the related neuronal soma revealed dramatic alteration of the RER and related polysomes. Now there was widespread dispersal and degranulation of the RER, with disaggregation of the polysomes. Only isolated profiles of RER could be seen. The Golgi was also dispersed, although the related mitochondria appeared intact. Similarly, the cytoskeleton appeared unaltered, with no evidence of neurofilament accumulation. Consistent with the LM finding of intense cytoplasmic APP immunoreactivity, APP electron-dense reaction product was found confined to tubular and vesicular profiles scattered throughout the cytoplasm (Fig.3C). Swollen glial processes as well as electron-dense, degenerating axon terminals now encompassed the perisomatic cell membrane as well as the proximal dendrites. Similar terminal debris, glial swelling, and other forms of anterograde and retrograde axonal change were noted in the surrounding neuropil. Collectively, these findings were consistent with a retrograde response to the observed neocortical axotomy, together with an anterograde response to axotomy of thalamocortical afferents, as evidenced by the finding of TAI within the thalamus coupled with the presence of electron-dense, degenerating boutons in contact with neocortical neurons and their processes. The observed anterograde abnormalities also occurred in relation to other neuronal somata, demonstrating no evidence of cellular/subcellular alteration. Additionally, other scattered neuronal somata, not associated with axotomy, showed evidence of necrotic change reflected in dramatic neuronal mitochondrial damage, increased somatic electron density, and nuclear condensation (Fig. 3D). In relation to these necrotic neurons, scattered neutrophils and macrophages could be recognized.
Fig. 3.
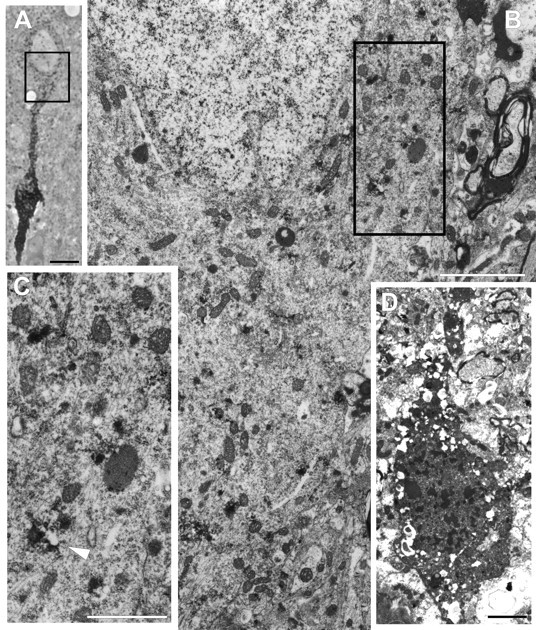
Reactive somatic change occurs in neurons with TAI at 24 hr after injury. A, This 1 μm osmicated semi-thick section taken from tissue reacted with antibodies to APP illustrates a neocortical neuronal soma in continuity with an adjacent disconnected axonal segment. This section was taken from an animal surviving for 24 hr after injury. Scale bar, 50 μm. B, This electron photomicrograph reveals the area outlined inA, demonstrating portions of the neuronal soma and axon hillock. Note the dispersion and loss of RER with the retention of normal mitochondrial and cytoskeletal detail. Scale bar, 2 μm.C, With higher magnification of the area outlined inB, such RER loss is better appreciated. Note the persistence of vesicles with the electron-dense reaction product for APP (arrowhead). Scale bar, 1 μm. D, Also note that at 24 hr after injury, isolated necrotic neurons showing no relation to axonal injury were found within the same sections containing neurons with TAI. Scale bar, 2 μm.
By 48–72 hr after injury (Fig. 4), those somata continuous with injured axons showed persistent ribosomal dispersal and disaggregation and loss of the RER. These somata were reminiscent of those described at 24 hr, with no evidence of further subcellular change suggestive of either necrosis or apoptosis. As before, the neuronal cytoskeleton was unaltered, with no evidence of neurofilamentous hyperplasia. APP immunoreactivity remained confined to tubular and vesicular profiles scattered throughout the cytoplasm. The only distinction at these time points was the fact that the neuropil manifested more dramatic anterograde/Wallerian degeneration reflected in numerous electron-dense axon terminals and degenerating myelinated axonal profiles. Again, scattered necrotic neurons, not associated with axotomy, were also identified, as were scattered neutrophils and macrophages.
Fig. 4.
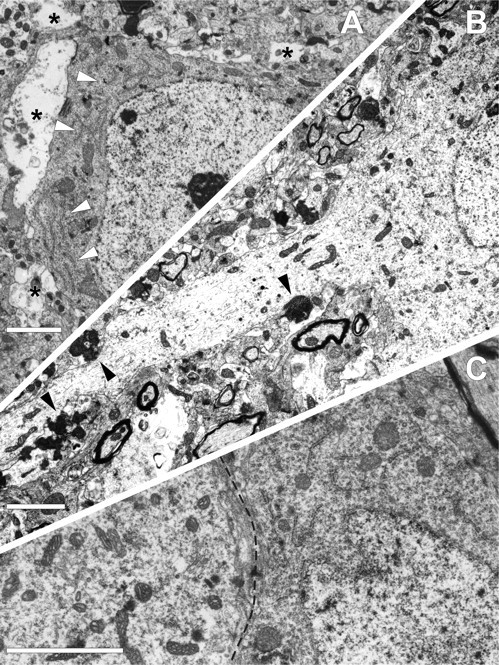
TAI results in both anterograde synaptic loss and retrograde reactive somatic change. At 48 hr after injury, TAI is seen to result in anterograde and retrograde deafferentation without subcellular alteration. A, Neocortical neuron from lamina V of the mediodorsal region reveals no evidence of TAI and displays well organized arrays of RER in the cytoplasm (arrowheads). However, in this otherwise unaltered soma, anterograde synaptic loss, most likely caused by TAI of thalamocortical afferents, results in perisomatic ensheathment by glial processes (asterisks). B illustrates at 72 hr after injury, in the same anatomical locus, a soma definitively linked to TAI. Note that this neuron and proximal dendritic shaft appear electron lucent because of RER dispersion and degranulation. Also note that this same axotomized soma reveals electron-dense axon terminals (arrowheads) in relation to the soma and its dendritic shaft as the likely consequence of afferent thalamocortical fiber damage. C, This micrograph taken from the same neuron shown in B reveals striking differences between the soma linked to TAI and another adjacent uninjured neuron. Note that the injured soma to the left of the dotted line reveals RER loss, whereas the uninjured soma to theright displays intact RER. Scale bars, 2 μm.
As with the LM observations, no axonal swellings could be detected at 7 d after injury (Fig. 5), and thus no axonal reactive change could be definitively linked to the APP-containing somata. Despite this limitation, however, many APP-immunopositive somata were seen in close proximity to trailing Wallerian debris. These showed cellular abnormalities consistent with those described in the axotomized somata in the earlier survival periods. Again, these somata displayed RER dispersion, with some neurons now revealing nuclear eccentricity. In these, the somata frequently displayed areas devoid of significant organelle content, with the remaining organelles clustered in one quadrant of the somata. These changes were observed in both large and small neocortical neuronal populations, which, although not specifically addressed in the current communication, demonstrate that these axotomy-mediated changes occurred in neurons of varying size. Despite the presence of these reactive changes, the neuronal somata involved appeared otherwise unaltered and showed no evidence of overt organelle or cytoskeletal change. Additionally, although not assessed via quantitative analyses, their perisomatic plasmalemma revealed more intact synaptic input in addition to the presence of unanticipated synaptic contacts, typically involving somatodendritic synaptic sites. Collectively these findings do not support a death cascade. Rather, they suggest an attempt at reorganization and repair.
Fig. 5.
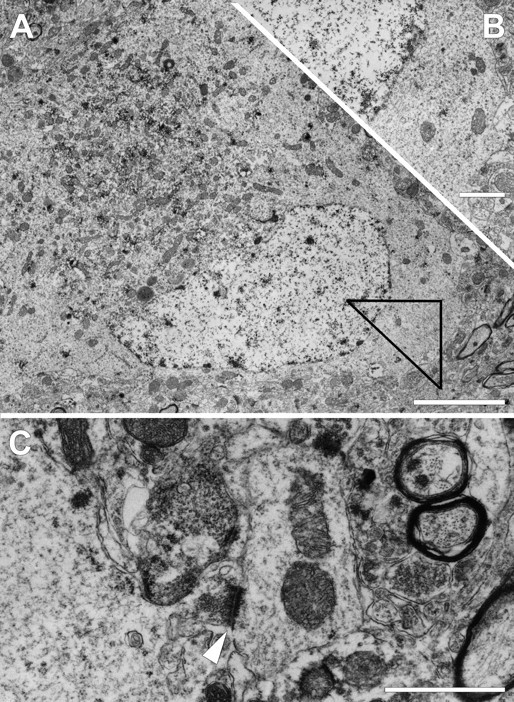
Electron microscopy of chromatolytic neuronal somata observed at 7 d after injury in lamina V of the mediodorsal neocortex. A, Large pyramidal neuron displaying nuclear eccentricity with perinuclear accumulation of organelles with electron-dense staining for APP. Other areas of the somatic cytoplasm are almost entirely devoid of organelles. Scale bar, 4 μm.B, Higher magnification of a serial section of the area outlined in A illustrating the relatively empty, granular cytoplasm lacking rough endoplasmic reticulum. Scale bar, 1 μm. C, A smaller chromatolytic neocortical neuron in the same region can be seen reestablishing connectivity via a somatodendritic synapse (arrowhead).Scale bar, 4 μm.
Neurofilament phosphorylation (data not shown)
In sham-injured animals, LM analysis revealed no RMO-24 staining in the gray matter of the neocortex, hilus of the dentate gyrus, and thalamus. As expected, immunoreactivity was found within the axons of the subcortical white matter and the internal capsule because of the normal presence of phosphorylated neurofilaments within the axons investing these tracts (Brown, 1998). Control sections from brainstems of sham-injured animals also demonstrated intense axonal immunoreactivity within the fiber tracts of the brainstem. In injured animals, comparable to the control condition, no somatic immunoreactivity was seen within 72 hr of injury in neocortical, hilar, and thalamic neurons. However, unlike the controls, RMO-24 immunoreactivity was now found within isolated axons and reactive axonal swellings in the gray matter of the mediodorsal neocortex and thalamus. By 7 d after injury, however, few RMO-24-labeled axonal segments and reactive swellings were observed in both of the above regions. At this time, in contrast to earlier observations, isolated immunoreactive neuronal cell bodies were identified in the mediodorsal neocortex but not the thalamus. These cells were diffusely scattered and revealed no continuity with RMO-24-immunoreactive axonal segments or swellings. No RMO-24-immunoreactive cell bodies were identified in the thalamus 7 d after injury.
Protein translation
Using single-label immunocytochemistry with LM analysis, staining for eIF2α(P) was negligible throughout the brains of sham-injured animals (Fig. 6A). Thirty minutes after central FPI, staining for the marker remained at control levels (data not shown); however, by 4 hr after injury, increased immunoreactivity was seen within scattered neuronal somata of the thalamus and laminae IV–V of the neocortex (Fig.6B). At 24 hr after injury, the staining was more intense in these same regions, with increased numbers of neuronal somata exhibiting intense eIF2α(P) immunoreactivity (Fig.6C). Those cells with increased eIF2α(P) immunoreactivity demonstrated the reaction product throughout the neuronal somata and their dendritic trees, with no staining of axons, a finding not unanticipated because axons lack protein translational machinery (Siegel et al., 1999). eIF2α(P) immunoreactivity was also evident, although attenuated, in the neocortex and thalamus at 48 and 72 hr after injury (data not shown). By 7 d after injury, little to no staining above that seen in sham-injured animals was observed (Fig.6D).
Fig. 6.

Neuronal eIF2α(P) transiently increases after injury. In a sham-injured animal, low basal levels of eIF2α(P) are observed in the mediodorsal neocortex (A). Four hours after injury, scattered neurons intensely staining for eIF2α(P) can be observed in lamina V of the mediodorsal neocortex (B). Many neurons in comparable regions exhibit elevated immunoreactivity for eIF2α(P) 24 hr after injury (C). At 7 d after injury, eIF2α(P) immunoreactivity returns to levels similar to controls (D). Scale bars, 100 μm.
When brain sections from sham-injured animals were double labeled with antibodies targeting both APP and eIF2α(P), little to no signal was seen for eIF2α(P), whereas only background staining was observed for APP (data not shown), consistent with the above-described single-label observations. Sections from animals surviving 24 hr after injury, however, again demonstrated a marked increase in cellular eIF2α(P) in the mediodorsal neocortex and dorsolateral thalamus that colocalized to the cytoplasm of neuronal somata also displaying APP-immunopositive axonal swellings (Fig. 7). All visible axotomized neuronal somata showed increased eIF2α(P). However, in some of the same fields, neuronal somata intensely stained for eIF2α(P) were also seen, despite the fact that they were neither axotomized nor associated with any somatic increase in APP. Forty-eight hours after injury, axotomized neurons revealed dispersed cytoplasmic staining for eIF2α(P), and by 72 hr, few axotomized neurons with increased eIF2α(P) staining were observed.
Fig. 7.

Neurons sustaining TAI demonstrate a somatic increase in eIF2α(P). Double labeling with antibodies to β-APP (red) and eIF2α(P) (green) reveal a striking increase in somatic eIF2α(P) in neurons with axonal injury (two examples are indicated with arrowheads). However, isolated neurons exhibiting no evidence of TAI or somatic increase in APP were also found to demonstrate a somatic increase in eIF2α(P) immunofluorescence (arrow). Scale bar, 50 μm. Inset, Higher magnification of neuronal soma with TAI. Increased immunofluorescence for eIF2α(P) can be seen to colocalize to the cytoplasm of the neuronal cell body. Scale bar, 20 μm.
To provide a semiquantitative assessment of the observed increases in eIF2α(P), Western blotting of specific brain subregions with subsequent densitometry was performed (Fig.8). In tissue taken from the mediodorsal neocortex 24 hr after central FPI, eIF2α(P) was elevated 32% over sham levels, although this change was not significant (p = 0.18). As evidenced by the above immunocytochemistry for eIF2α(P), this result may be attributable to the diffuse nature of the injury within an isolated region of the neocortex. In the hippocampus, the injury-induced increase in eIF2α(P) was greater, rising 125% over sham-injured levels (p < 0.01). No change in eIF2α(P) was observed in the thalamus (data not shown).
Fig. 8.

Western blot analysis of neocortical and hippocampal eIF2α(P) levels in animals surviving for 24 hr after central FPI. A and B display eIF2α(P) levels in the mediodorsal neocortex and hippocampus, respectively, of sham-injured (S) and animals surviving for 24 hr after injury (I). Densitometry revels a modest 32% increase in the neocortex (not significant;p = 0.18) and a greater increase of 125% in the hippocampus after injury (*p < 0.01).
TUNEL
Using single-label TUNEL methodologies (data not shown), control tissue samples from the thymus of naïve rats demonstrated robust TUNEL staining of thymocytes, whereas controls generated via omission of the TdT enzyme and/or anti-digoxigenin antibody showed no reactivity. In sections from sham-injured animals, no TUNEL-positive cells were noted. At 24 hr after injury, only isolated neuronal somata were noted in the CA3 subsector of the hippocampus and the granule cell layer of the suprapyramidal and infrapyramidal blades of the dentate gyrus, as well as the dorsolateral thalamus, with no cortical involvement. Some of these immunoreactive cells displayed nuclear and cytoplasmic shrinkage, consistent with the morphologic phenotype associated with apoptosis. Other TUNEL-positive cells, however, exhibited variations in morphology not consistent with apoptosis. Rather than the shrunken, compact appearance commonly associated with apoptosis, these cells displayed no signs of nuclear condensation, with vacuolation of the TUNEL-positive cytoplasm. Those cells demonstrating cytoplasmic TUNEL staining appeared comparable to the type I TUNEL-positive cells described previously by Rink et al. (1995) that exhibit necrotic rather than apoptotic morphology. No immunopositive cells, however, were identified within either the mediodorsal neocortex or the hilar region of the dentate gyrus, the regions in which axotomy was found.
At 48 hr after injury, the hippocampal and thalamic TUNEL staining remained sparse, with only occasional immunoreactivity observed in the hippocampal CA3 region, the suprapyramidal and infrapyramidal blades of the dentate gyrus, and the dorsolateral thalamus, with no staining noted in the neocortex. No change in the magnitude or localization of TUNEL staining was evident 72 hr after injury when compared with 48 hr. At 7 d after injury, the isolated TUNEL-positive cells noted in previous time points were virtually absent. Double labeling with TUNEL and antibodies to APP confirmed the single-label findings in that no evidence was found to support the colocalization of DNA fragmentation within neuronal somata sustaining TAI.
DISCUSSION
The results of this communication reveal a series of axotomy-mediated neuronal somatic responses in multiple brain foci after TBI and, for the first time, link sites of TAI to their somata of origin. Consistent with the pathobiology of DAI, the somatic injury is diffuse, and the related TAI follows a repertoire of qualitative and quantitative change consistent with that described previously in injured long-tract axons of the brainstem (Stone et al., 2000, 2001). The data provided (Table 1) suggests that TAI occurs at comparable distances from the somata of origin regardless of their cortical or thalamic origin. Why the site of injury is relatively constant is unknown; however, the quantitative values provided are consistent with the length of the initial axonal segment, before the start of the myelin sheath (Conradi, 1969; Conradi and Skoglund, 1969). This suggests that the initiating injury occurs at the junction between the nonmyelinated initial axonal segment and the beginning of the first internode, which may constitute a point of biomechanical vulnerability.
Although we initially hypothesized that the pathology occurring in these somata would parallel that described with primary mechanical/surgical axotomy of CNS tracts, our findings yielded unanticipated differences. Consistent with primary axotomy (Lieberman, 1971; Barron, 1983; Kreutzberg, 1995), neuronal somata axotomized subsequent to TBI did show chromatolytic change. Axonal injury within 20–50 μm of the soma was associated with anterograde and retrograde bouton alteration and degeneration, disruption of the RER, Golgi dispersion, and other alterations observed after primary axotomy (Barron, 1983). Although the current repertoire of change did mimic that seen in cortical neurons after surgical axonal transection (Barron and Dentinger, 1979; Dentinger et al., 1979), distinct differences in both the time of onset and pathologic progression were observed. In this communication, ultrastructural alterations were first recognized at 6 hr after injury, with dramatic cellular change seen at 24 hr. In primary CNS axotomy paradigms, comparable neuronal somatic responses evolve over a more prolonged time course (Barron and Dentinger, 1979;Giehl and Tetzlaff, 1996). The reason for this different temporal response is unknown; however, it may be related to the fact that the TAI occurred perisomatically, in contrast to primary axotomy paradigms that used, by necessity, injury to more remote white matter sites (McBride et al., 1989; Merline and Kalil, 1990; Bonatz et al., 2000). The perisomatic localization of TAI may allow for more rapid retrograde signaling to evoke the reactive change observed (Kreutzberg, 1995).
Despite the proximity of the TAI to the neuronal somata and the rapid onset of reactive change, neuronal cell bodies associated with TAI did not show a pathological progression to cell death, contrary to contemporary thought. A comparable lack of cell loss involving corticospinal neurons after primary axotomy was observed when lesioning was confined to spinal (McBride et al., 1989) or medullary levels (Merline and Kalil, 1990). In contrast, more proximal, internal capsular lesions resulted in significant neuronal loss. Specifically, a 31% decrease in corticospinal neuronal numbers was reported at 5 d after lesioning, progressing to 46% by 7 d, with the remaining neurons manifesting severe atrophic change (Giehl and Tetzlaff, 1996). On the basis of this increased neuronal loss with more proximal axonal lesions, a finding confirmed in other CNS fiber systems (Egan et al., 1977; Villegas-Perez et al., 1993), we anticipated that the observed perisomatic axotomy would result in rapid and dramatic cell death. As noted, neuronal death related to TAI, as assessed by routine LM/EM and TUNEL methodologies, was observed in neither lamina V of the mediodorsal neocortex, which contains corticospinal neuronal somata, nor the hilus of the dentate gyrus. Additionally, unlike the responses seen in neuronal somata subjected to primary axotomy (Rosenfeld et al., 1987; Koliatsos et al., 1989; Martin et al., 1999), neurofilamentous hyperplasia and accumulation of phosphorylated heavy neurofilaments were not found, nor was there evidence of mitochondrial swelling, microvacuolation, nuclear alteration, or condensation.
Not only could we find no evidence of cell death in relation to those neurons revealing TAI, but also some of the changes observed therein suggested a potential neuronal attempt at reorganization/repair. The reduction of eIF2α(P) noted at 7 d after injury suggested reestablishment of protein synthesis. This, coupled with the observation of lengthening of the axotomized process at 48 hr and the overall reduction in axonal swelling size at the same time, argues that those neurons sustaining TAI are not dying but rather may be attempting a reparative response. Using a dissimilar optic nerve stretch injury model, Maxwell et al. (1994) alluded to a similar possibility in a subpopulation of injured neurons, whereas another group (Emery et al., 2000) has recently identified TBI-induced expression of markers associated with neuronal regeneration in the same brain regions found herein to sustain perisomatic TAI.
The reasons for these unanticipated responses to axotomy are unknown. Perhaps they may be linked to the somewhat unique pathogenesis of TAI, because the traumatically induced axonal response to injury is dissimilar from that seen with nerve transection or crush. Specifically, with TAI, axons are diffusely injured in a brain environment that shows no other abnormality in terms of blood–brain barrier alteration, blood flow, and other forms of reactive change (Povlishock et al., 1992). The injured axons, however, show focal changes in axolemmal permeability as well as other cytoskeletal changes that lead to axonal swelling and disconnection over a 4–6 hr period (Maxwell et al., 1997). This pathobiology is dissimilar from that involving mechanical transection, wherein massive focal axonal damage causes an immediate disruption of ionic equilibrium (Balentine, 1985), perhaps triggering retrograde degradation of the soma. Conceivably, the more slow and progressive axonal pathobiology associated with TAI allows for reduced ionic disruption, thereby translating into attenuated retrograde somatic responses or a latency period that allows for the initiation of somatic survival signaling, issues that both merit further consideration.
To date, those evaluating neuronal injury after TBI have focused on models of contusion involving focal destructive mechanical loading (Lighthall, 1988; McIntosh et al., 1989) and diffuse injury that generates scattered neuronal somatic damage and death (Gennarelli et al., 1981; Meaney et al., 1993; Foda and Marmarou, 1994). Although pathological features are shared between these models, there are distinct differences. It is assumed that contusion-related hemorrhage, coupled with subsequent ischemia, causes localized necrotic and dispersed apoptotic cell death (Fox et al., 1998; Newcomb et al., 1999). Such cell death is assumed to be a major player in the pathobiology and subsequent morbidity inherent to this injury. In contrast, diffuse injury elicits scattered apoptotic or necrotic neuronal death, which has been linked to TBI-induced neuroexcitation (Zipfel et al., 2000). The current work does not invalidate previous studies focusing on cell death; however, it does demonstrate that the neuronal somatic response is more complex than previously assumed and cautions against the impression that a rapid necrotic or apoptotic death is the only sequelae of TBI. Although we did observe scattered necrotic and sparse apoptotic neuronal death in the current study, we could find no evidence linking either to TAI. The semiserial EM analyses used in this work appear to preclude the possibility that even more proximal axotomy occurring within the hillock triggers ultra-rapid neuronal death via apoptosis or necrosis. Conversely, there is no biological rationale as to why distant, undetected TAI would evoke a more rapid and dramatic somatic reaction than the axonal injury and disconnection described herein. McIntosh and Faden and colleagues (Rink et al., 1995; Yakovlev et al., 1997; Conti et al., 1998; Fox et al., 1998) have reported consistent apoptotic neuronal death after TBI; however, unlike the lateral fluid percussion model they used, the central FPI used in this study did not generate contusional change (Dixon et al., 1987). Thus, widespread apoptotic neuronal death may be reserved for more severe forms of injury.
In the current communication, the increase in the phosphorylated form of eIF2α and the RER disruption observed by immunocytochemical and EM studies provides parallel evidence of translational inhibition in neurons sustaining TAI. Although a change in eIF2α(P) in neurons sustaining primary axotomy has not been described previously, numerous investigators have described RER dispersion/loss (Lieberman, 1971;Barron, 1983; Kreutzberg, 1995). Although neurons with TAI consistently demonstrated increases in eIF2α(P), other neurons without visible evidence of axonal injury were sporadically identified in the same fields. Perhaps these cells were axotomized at sites remote from the somata and thus responded in a manner similar to those neurons with perisomatic TAI. Alternatively, these cells may not have sustained TAI but rather incurred injury via an alternate mechanism sufficient to initiate phosphorylation of eIF2α.
The question for future investigations focusing on the neuronal response to TAI must center on the long-term fate of these injured neurons. As noted, the loss of APP immunoreactive axons at 7 d after injury and the inherent difficulties in following Wallerian debris back to the soma of origin at the LM and EM levels suggest that further progress must await the development of new markers for the recognition of TAI and related somata over a prolonged posttraumatic course.
In sum, we believe that the results of the current communication are provocative and force a reevaluation of contemporary thought on the sequelae of TAI. On the basis of the end points used in the current communication, somata associated with TAI do not die rapidly, as would be predicted from the literature, and thus may provide targets for potential therapeutic intervention. Although future quantitative studies assessing total neuronal loss in differing brain regions remains to be performed, the current work does suggest caution on the part of those who use lesioning or crushing injuries to model TAI-associated pathobiology.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grants NS20193 and T32NS007288. We thank Susan Walker, Lynn Davis, Tom Coburn, Raiford Black, and Lesley Harris for their technical assistance.
Correspondence should be addressed to Dr. John T. Povlishock, Professor and Chair, Department of Anatomy, Medical College of Virginia Campus of Virginia Commonwealth University, P.O. Box 980709, Richmond, VA 23298. E-mail: jpovlish@hsc.vcu.edu.
REFERENCES
- 1.Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 2.Balentine JD. Hypotheses in spinal cord trauma research. In: Becker DP, Povlishock JT, editors. Central nervous system trauma status report. Byrd; Richmond, VA: 1985. pp. 455–461. [Google Scholar]
- 3.Bareyre FM, Raghupathi R, Saatman KE, McIntosh TK. DNase I disinhibition is predominantly associated with actin hyperpolymerization after traumatic brain injury. J Neurochem. 2001;77:173–181. doi: 10.1046/j.1471-4159.2001.t01-1-00215.x. [DOI] [PubMed] [Google Scholar]
- 4.Barron KD. Comparative observations on the cytologic reactions of central and peripheral nerve cells to axotomy. In: Kao CC, Bunge RP, Reier PJ, editors. Spinal cord reconstruction. Raven; New York: 1983. pp. 7–40. [Google Scholar]
- 5.Barron KD, Dentinger MP. Cytologic observations on axotomized feline Betz cells. 1. Qualitative electron microscopic findings. J Neuropathol Exp Neurol. 1979;38:128–151. doi: 10.1097/00005072-197903000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Bonatz H, Rohrig S, Mestres P, Meyer M, Giehl KM. An axotomy model for the induction of death of rat and mouse corticospinal neurons in vivo. J Neurosci Methods. 2000;100:105–115. doi: 10.1016/s0165-0270(00)00238-7. [DOI] [PubMed] [Google Scholar]
- 7.Bramlett HM, Kraydieh S, Green EJ, Dietrich WD. Temporal and regional patterns of axonal damage following traumatic brain injury: a beta-amyloid precursor protein immunocytochemical study in rats. J Neuropathol Exp Neurol. 1997;56:1132–1141. doi: 10.1097/00005072-199710000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Brown A. Contiguous phosphorylated and non-phosphorylated domains along axonal neurofilaments. J Cell Sci. 1998;111:455–467. doi: 10.1242/jcs.111.4.455. [DOI] [PubMed] [Google Scholar]
- 9.Buki A, Siman R, Trojanowski JQ, Povlishock JT. The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol. 1999;58:365–375. doi: 10.1097/00005072-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 10.Buki A, Okonkwo DO, Wang KK, Povlishock JT. Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci. 2000;20:2825–2834. doi: 10.1523/JNEUROSCI.20-08-02825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conradi S. Observations on the ultrastructure of the axon hillock and initial axon segment of lumbosacral motoneurons in the cat. Acta Physiol Scand Suppl. 1969;332:65–84. [PubMed] [Google Scholar]
- 12.Conradi S, Skoglund S. Observations on the ultrastructure of the initial motor axon segment and dorsal root boutons on the motoneurons in the lumbosacral spinal cord of the cat during postnatal development. Acta Physiol Scand Suppl. 1969;333:53–76. [PubMed] [Google Scholar]
- 13.Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cordobes F, Lobato RD, Rivas JJ, Cabrera A, Sarabia M, Castro S, Cisneros C, Torres ID, Lamas E. Post-traumatic diffuse axonal brain injury. Analysis of 78 patients studied with computed tomography. Acta Neurochir (Wien) 1986;81:27–35. doi: 10.1007/BF01456261. [DOI] [PubMed] [Google Scholar]
- 15.DeGracia DJ, Sullivan JM, Neumar RW, Alousi SS, Hikade KR, Pittman JE, White BC, Rafols JA, Krause GS. Effect of brain ischemia and reperfusion on the localization of phosphorylated eukaryotic initiation factor 2 alpha. J Cereb Blood Flow Metab. 1997;17:1291–1302. doi: 10.1097/00004647-199712000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Dentinger MP, Barron KD, Kohberger RC, McLean B. Cytologic observations on axotomized feline Betz cells. II. Quantitative ultrastructural findings. J Neuropathol Exp Neurol. 1979;38:551–564. doi: 10.1097/00005072-197909000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- 18.Egan DA, Flumerfelt BA, Gwyn DG. Axon reaction in the red nucleus of the rat. Perikaryal volume changes and the time course of chromatolysis following cervical and thoracic lesions. Acta Neuropathol (Berl) 1977;37:13–19. doi: 10.1007/BF00684534. [DOI] [PubMed] [Google Scholar]
- 19.Emery DL, Raghupathi R, Saatman KE, Fischer I, Grady MS, McIntosh TK. Bilateral growth-related protein expression suggests a transient increase in regenerative potential following brain trauma. J Comp Neurol. 2000;424:521–531. [PubMed] [Google Scholar]
- 20.Foda MA, Marmarou A. A new model of diffuse brain injury in rats. Part II: Morphological characterization. J Neurosurg. 1994;80:301–313. doi: 10.3171/jns.1994.80.2.0301. [DOI] [PubMed] [Google Scholar]
- 21.Fox GB, Fan L, LeVasseur RA, Faden AI. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma. 1998;15:599–614. doi: 10.1089/neu.1998.15.599. [DOI] [PubMed] [Google Scholar]
- 22.Gennarelli TA, Adams JH, Graham DI. Acceleration induced head injury in the monkey. I. The model, its mechanical and physiological correlates. Acta Neuropathol Suppl (Berl) 1981;7:23–25. doi: 10.1007/978-3-642-81553-9_7. [DOI] [PubMed] [Google Scholar]
- 23.Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139–144. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- 24.Giehl KM, Tetzlaff W. BDNF and NT-3, but not NGF, prevent axotomy-induced death of rat corticospinal neurons in vivo. Eur J Neurosci. 1996;8:1167–1175. doi: 10.1111/j.1460-9568.1996.tb01284.x. [DOI] [PubMed] [Google Scholar]
- 25.Koliatsos VE, Applegate MD, Kitt CA, Walker LC, DeLong MR, Price DL. Aberrant phosphorylation of neurofilaments accompanies transmitter-related changes in rat septal neurons following transection of the fimbria-fornix. Brain Res. 1989;482:205–218. doi: 10.1016/0006-8993(89)91183-9. [DOI] [PubMed] [Google Scholar]
- 26.Kreutzberg GW. Reaction of the neuronal cell body to axonal damage. In: Waxman SG, Kocsis JD, Stys PK, editors. The axon. Oxford UP; New York: 1995. pp. 355–374. [Google Scholar]
- 27.Lieberman AR. The axon reaction: a review of the principal features of perikaryal responses to axon injury. Int Rev Neurobiol. 1971;14:49–124. doi: 10.1016/s0074-7742(08)60183-x. [DOI] [PubMed] [Google Scholar]
- 28.Lighthall JW. Controlled cortical impact: a new experimental brain injury model. J Neurotrauma. 1988;5:1–15. doi: 10.1089/neu.1988.5.1. [DOI] [PubMed] [Google Scholar]
- 29.Martin LJ, Kaiser A, Price AC. Motor neuron degeneration after sciatic nerve avulsion in adult rat evolves with oxidative stress and is apoptosis. J Neurobiol. 1999;40:185–201. [PubMed] [Google Scholar]
- 30.Maxwell WL, Islam MN, Graham DI, Gennarelli TA. A qualitative and quantitative analysis of the response of the retinal ganglion cell soma after stretch injury to the adult guinea-pig optic nerve. J Neurocytol. 1994;23:379–392. doi: 10.1007/BF01666527. [DOI] [PubMed] [Google Scholar]
- 31.Maxwell WL, McCreath BJ, Graham DI, Gennarelli TA. Cytochemical evidence for redistribution of membrane pump calcium-ATPase and ecto-Ca-ATPase activity, and calcium influx in myelinated nerve fibres of the optic nerve after stretch injury. J Neurocytol. 1995;24:925–942. doi: 10.1007/BF01215643. [DOI] [PubMed] [Google Scholar]
- 32. Maxwell WL, Povlishock JT, Graham DL. A mechanistic analysis of nondisruptive axonal injury: a review. J Neurotrama 14 1997. 419 440 [Erratum (1997) 14:755] [DOI] [PubMed] [Google Scholar]
- 33.Maxwell WL, Kosanlavit R, McCreath BJ, Reid O, Graham DI. Freeze-fracture and cytochemical evidence for structural and functional alteration in the axolemma and myelin sheath of adult guinea pig optic nerve fibers after stretch injury. J Neurotrauma. 1999;16:273–284. doi: 10.1089/neu.1999.16.273. [DOI] [PubMed] [Google Scholar]
- 34.McBride RL, Feringa ER, Garver MK, Williams JK. Prelabeled red nucleus and sensorimotor cortex neurons of the rat survive 10 and 20 weeks after spinal cord transection. J Neuropathol Exp Neurol. 1989;48:568–576. doi: 10.1097/00005072-198909000-00007. [DOI] [PubMed] [Google Scholar]
- 35.McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AL. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- 36.Meaney DF, Thibault LE, Smith DH, Ross DT, Gennarelli TA. Diffuse axonal injury in the miniature pig: biomechanical development and injury threshold. Am Soc Mech Eng. 1993;25:169–175. [Google Scholar]
- 37.Merline M, Kalil K. Cell death of corticospinal neurons is induced by axotomy before but not after innervation of spinal targets. J Comp Neurol. 1990;296:506–516. doi: 10.1002/cne.902960313. [DOI] [PubMed] [Google Scholar]
- 38.Newcomb JK, Zhao X, Pike BR, Hayes RL. Temporal profile of apoptotic-like changes in neurons and astrocytes following controlled cortical impact injury in the rat. Exp Neurol. 1999;158:76–88. doi: 10.1006/exnr.1999.7071. [DOI] [PubMed] [Google Scholar]
- 39.Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab. 1999;19:443–451. doi: 10.1097/00004647-199904000-00010. [DOI] [PubMed] [Google Scholar]
- 40.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1986. [DOI] [PubMed] [Google Scholar]
- 41.Peters A, Palay S, Webster H. The fine structure of the nervous system. Oxford UP; New York: 1991. [Google Scholar]
- 42.Pettus EH, Povlishock JT. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 1996;722:1–11. doi: 10.1016/0006-8993(96)00113-8. [DOI] [PubMed] [Google Scholar]
- 43.Pettus EH, Christman CW, Giebel ML, Povlishock JT. Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. J Neurotrauma. 1994;11:507–522. doi: 10.1089/neu.1994.11.507. [DOI] [PubMed] [Google Scholar]
- 44.Povlishock JT, Erb DE, Astruc J. Axonal response to traumatic brain injury: reactive axonal change, deafferentation, and neuroplasticity. J Neurotrauma. 1992;9[Suppl 1]:S189–200. [PubMed] [Google Scholar]
- 45.Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenfeld J, Dorman ME, Griffin JW, Gold BG, Sternberger LA, Sternberger NH, Price DL. Distribution of neurofilament antigens after axonal injury. J Neuropathol Exp Neurol. 1987;46:269–282. doi: 10.1097/00005072-198705000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Shields DC, Schaecher KE, Hogan EL, Banik NL. Calpain activity and expression increased in activated glial and inflammatory cells in penumbra of spinal cord injury lesion. J Neurosci Res. 2000;61:146–150. doi: 10.1002/1097-4547(20000715)61:2<146::AID-JNR5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 48.Siegel SE, Agranoff BW, Albers RW, Fisher SK, Uhler MD. Basic neurochemistry: molecular, cellular and medical aspects. Lippincott-Raven; Philadelphia: 1999. [Google Scholar]
- 49.Singh LP, Aroor AR, Wahba AJ. Translational control of eukaryotic gene expression. Role of the guanine nucleotide exchange factor and chain initiation factor-2. Enzyme Protein. 1994;48:61–80. doi: 10.1159/000474972. [DOI] [PubMed] [Google Scholar]
- 50.Stone JR, Walker SA, Povlishock JT. The visualization of a new class of traumatically injured axons through the use of a modified method of microwave antigen retrieval. Acta Neuropathol (Berl) 1999;97:335–345. doi: 10.1007/s004010050996. [DOI] [PubMed] [Google Scholar]
- 51.Stone JR, Singleton RH, Povlishock JT. Antibodies to the C-terminus of the beta-amyloid precursor protein (APP): a site specific marker for the detection of traumatic axonal injury. Brain Res. 2000;871:288–302. doi: 10.1016/s0006-8993(00)02485-9. [DOI] [PubMed] [Google Scholar]
- 52.Stone JR, Singleton RH, Povlishock JT. Intra-axonal neurofilament compaction does not evoke local axonal swelling in all traumatically injured axons. Exp Neurol. 2001;172:320–331. doi: 10.1006/exnr.2001.7818. [DOI] [PubMed] [Google Scholar]
- 53.Sullivan HG, Martinez J, Becker DP, Miller JD, Griffith R, Wist AO. Fluid-percussion model of mechanical brain injury in the cat. J Neurosurg. 1976;45:521–534. [PubMed] [Google Scholar]
- 54.Teramoto N, Szekely L, Pokrovskaja K, Hu LF, Yoshino T, Akagi T, Klein G. Simultaneous detection of two independent antigens by double staining with two mouse monoclonal antibodies. J Virol Methods. 1998;73:89–97. doi: 10.1016/s0166-0934(98)00048-2. [DOI] [PubMed] [Google Scholar]
- 55.Van den Heuvel C, Lewis S, Wong M, Manavis J, Finnie J, Blumbergs P, Jones N, Reilly P. Diffuse neuronal perikaryon amyloid precursor protein immunoreactivity in a focal head impact model. Acta Neurochir Suppl (Wien) 1998;71:209–211. doi: 10.1007/978-3-7091-6475-4_60. [DOI] [PubMed] [Google Scholar]
- 56.Villegas-Perez MP, Vidal-Sanz M, Rasminsky M, Bray GM, Aguayo AJ. Rapid and protracted phases of retinal ganglion cell loss follow axotomy in the optic nerve of adult rats. J Neurobiol. 1993;24:23–36. doi: 10.1002/neu.480240103. [DOI] [PubMed] [Google Scholar]
- 57.Wang G, Achim CL, Hamilton RL, Wiley CA, Soontornniyomkij V. Tyramide signal amplification method in multiple-label immunofluorescence confocal microscopy. Methods. 1999;18:459–464. doi: 10.1006/meth.1999.0813. [DOI] [PubMed] [Google Scholar]
- 58.Wolf JA, Stys PK, Lusardi T, Meaney DF, Smith DH. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21:1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zipfel GJ, Babcock DJ, Lee JM, Choi DW. Neuronal apoptosis after CNS injury: the roles of glutamate and calcium. J Neurotrauma. 2000;17:857–869. doi: 10.1089/neu.2000.17.857. [DOI] [PubMed] [Google Scholar]