

Abstract
Recent evidence suggests that reactive oxygen species (ROS), including superoxide, are not only neurotoxic but function as small messenger molecules in normal neuronal processes such as synaptic plasticity. Consistent with this idea, we show that brief incubation of hippocampal slices with the superoxide-generating system xanthine/xanthine oxidase (X/XO) produces a long-lasting potentiation of synaptic transmission in area CA1. We found that X/XO-induced potentiation was associated with a persistent superoxide-dependent increase in autonomous PKC activity that could be isolated via DEAE column chromatography. The X/XO-induced potentiation was blocked by the inhibition of PKC, indicating that the superoxide-dependent increase in autonomous PKC activity was necessary for the potentiation. We also found that X/XO-induced potentiation and long-term potentiation (LTP) occluded one another, suggesting that these forms of plasticity share similar cellular mechanisms. In further support of this idea, we found that a persistent, superoxide-dependent increase in autonomous PKC activity isolated via DEAE column chromatography also was associated with LTP. Taken together, our findings indicate that X/XO-induced potentiation and LTP share similar cellular mechanisms, including superoxide-dependent increases in autonomous PKC activity. Finally, our findings suggest that superoxide, in addition to its well known role as a neurotoxin, also can be considered a small messenger molecule critical for normal neuronal signaling.
Keywords: superoxide, long-term potentiation, hippocampus, protein kinase C, reactive oxygen species, synaptic plasticity
Long-term potentiation (LTP) is a long-lasting form of synaptic enhancement that has been postulated to underlie learning and memory mechanisms in the mammalian hippocampus (Malenka and Nicoll, 1999). The most commonly studied forms of LTP are induced by high-frequency stimulation (HFS), typically one or more trains of 100 Hz stimulation, of the Schaffer collateral–commissural input to CA1 pyramidal neurons. These forms of LTP are dependent on the activation of the NMDA subtype of glutamate receptors (Collingridge et al., 1983), influx of Ca2+ into the postsynaptic pyramidal neuron (Lynch et al., 1983; Malenka et al., 1988), and the production of small messenger molecules that, along with Ca2+, trigger the activation of a number of protein kinase signaling cascades (Roberson et al., 1996; Malenka and Nicoll, 1999).
One of the small messenger molecules produced after high-frequency stimulation that appears to be critical for LTP is the reactive oxygen species (ROS) superoxide. Evidence consistent with this notion includes the following findings: (1) NMDA receptor activation results in the production of superoxide in hippocampal slices (Bindokas et al., 1996), (2) cell-permeable scavengers of superoxide block LTP (Klann, 1998), (3) cell-impermeable scavengers of superoxide strongly attenuate LTP (Klann et al., 1998), and (4) hippocampal slices from transgenic mice that overexpress either cytoplasmic superoxide dismutase (SOD-1) or extracellular superoxide dismutase (EC-SOD) exhibit impaired LTP (Gahtan et al., 1998; Thiels et al., 2000).
In addition, it has been shown that superoxide can interact directly with protein kinase C (PKC), resulting in a persistent increase in autonomous PKC activity (Knapp and Klann, 2000). This is notable because persistent activation of PKC has been shown to be associated with and necessary for the expression of LTP (Klann et al., 1991, 1993;Wang and Feng, 1992; Sacktor et al., 1993; Hrabetova and Sacktor, 1996).
If a molecule such as a superoxide is to be deemed critical for LTP, then it should fulfill certain criteria (Sweatt, 1999). These criteria include the ability of the molecule to produce a persistent potentiation in synaptic transmission when added to hippocampal slices (Sweatt, 1999). Other small messenger molecules that have been shown to meet these criteria are arachidonic acid (Williams et al., 1989), nitric oxide (Böhme et al., 1991; Zhuo et al., 1993), carbon monoxide (Zhuo et al., 1993), and cAMP (Frey et al., 1993).
In this manuscript we show that brief exposure of hippocampal slices to xanthine/xanthine oxidase (X/XO), a superoxide-generating system, results in a PKC-dependent, long-lasting potentiation. The X/XO-induced potentiation occludes LTP, which suggests that both types of potentiation share similar cellular signaling mechanisms. Finally, we show that both X/XO-induced potentiation and LTP are associated with a persistent, oxidative activation of PKC. Taken together, these data are consistent with the notion that superoxide can function as a physiological signaling molecule and that the oxidative activation of PKC by superoxide is critical for LTP.
MATERIALS AND METHODS
Materials. Xanthine (X), superoxide dismutase (SOD), catalase, and bisindolylmaleimide (Bis) were purchased from Calbiochem (La Jolla, CA). 2-Amino-5-phosphonovaleric acid (APV) and DEAE-cellulose were purchased from Sigma (St. Louis, MO). Xanthine oxidase (XO) was purchased from Roche Molecular Biochemicals (Indianapolis, IN).
Preparation of hippocampal slices and extracellular recordings.Hippocampi from male Sprague Dawley rats (100–150 μg) were removed, and 400 μm slices were prepared with a McIlwain tissue chopper. The slices were perfused for 1–2 hr with a standard saline solution (in mm: 124 NaCl, 4.4 KCl, 26 NaHCO3, 10 d-glucose, 2 CaCl2, and 2 MgCl2, gassed with 95% O2/5% CO2, pH 7.4) in an interface tissue slice chamber at 30–32°C. Responses to Schaffer collateral stimulation in area CA1 were monitored for a minimum of 20 min before either incubation of the slices with X/XO or delivery of LTP-inducing HFS. Test stimuli (50 μsec) were given at a current (30–50 μA) that produced 50% of the maximum initial slope of the extracellular field EPSP (fEPSP). Responses to test stimuli were measured every 2.5 min as an average of four individual traces (0.1 Hz).
Measurement of superoxide generated from X/XO. The concentrations of superoxide produced by X/XO were determined spectrophotometrically (570 λ) by measuring the SOD-inhibitable conversion of nitroblue tetrazolium to diformazan (Halliwell and Gutteridge, 1989). Similar concentrations of superoxide produced by X/XO were observed when X/XO was present in a test tube and when X/XO was added to slices in a recording chamber.
Application of X/XO to hippocampal slices. To determine whether X/XO exerted an effect on hippocampal synaptic transmission, we monitored baseline responses for 20 min to ensure a stable baseline. Responses then were monitored while the slices were perfused with X/XO, boiled X/XO, or X/XO in the presence of SOD and catalase, usually for 10 min. After washout of the compounds the responses were monitored for an additional 45 min. In one set of experiments the NMDA receptor antagonist APV was present in the perfusate before, during, and after the application of X/XO. In another set of experiments paired pulse facilitation (PPF) was measured at several time points before and after perfusion with X/XO.
Induction of LTP. LTP-inducing HFS consisted of three 1 sec trains of stimuli (100 Hz) given 20 sec apart with the use of a current (60–100 μA) that elicited the maximum fEPSP. Responses to test stimuli were measured every 2.5 min as an average of four individual traces (0.1 Hz) for 45 min after delivery of the final train of HFS. Post-HFS fEPSPs were elicited by the same test stimulation intensity as before delivery of the HFS.
DEAE column chromatography of PKC. Hippocampal slices were frozen on dry ice 2 and 45 min after the washout of either X/XO or LTP-inducing HFS. For slices that were treated with X/XO, the entire hippocampal slice was homogenized in buffer as described previously (Knapp and Klann, 2000). The homogenates were separated into soluble and pellet fractions via centrifugation at 45,000 × gat 4°C for 45 min. For homogenates from X/XO-treated slices, the soluble fraction of hippocampal homogenates was applied to a 1 ml DEAE-cellulose column. The column was washed with 3 ml of column buffer (20 mm Tris-HCl, 1 mm EDTA, 100 ng/ml leupeptin, 100 ng/ml aprotinin, and 10 μg/ml benzamidine). PKC was eluted in 500 μl fractions by the stepwise addition of column buffer supplemented with 0.1 m NaCl, followed by column buffer supplemented with 0.25 m NaCl. The fractions were concentrated and desalted with the use of Microcon-3 ultrafugation devices (Millipore, Bedford, MA) for 2.5 hr at 4°C.
For the slices that were given LTP-inducing HFS, the CA1 region between the stimulating and recording electrodes was dissected and homogenized in buffer as described previously (Knapp and Klann, 2000). The homogenates were separated into soluble and pellet fractions as described above, and the soluble fraction of hippocampal homogenates was applied to a 100 μl DEAE-cellulose column. The column was washed with 300 μl of column buffer, and PKC was eluted in 50 μl fractions by the stepwise addition of column buffer supplemented with 0.1m NaCl, followed by column buffer supplemented with 0.25m NaCl. The fractions were concentrated and desalted via Microcon-3 ultrafugation devices for 30 min at 4°C.
PKC activity assays. Autonomous and cofactor-dependent PKC activity in each preparation was determined by using the selective PKC substrate NG(28-43) as described previously (Knapp and Klann, 2000).
RESULTS
Xanthine/xanthine oxidase induces a superoxide-dependent, long-lasting potentiation of hippocampal synaptic transmission
To determine whether superoxide could induce a long-lasting potentiation of hippocampal synaptic transmission, we incubated rat hippocampal slices with X/XO, a superoxide-generating system (Hille and Nishino, 1995). We found that a brief 10 min incubation of hippocampal slices with concentrations of X/XO that produced 1–5 μmsuperoxide resulted in a slight but significant depression of synaptic transmission while X/XO was present in the perfusate (fEPSP slope = 85 ± 9% of control, n = 10) (Fig.1A). However, this depression was transient and was followed by a slowly rising potentiation in the slope of the fEPSP, which reached a maximum 35–45 min after the washout of X/XO (fEPSP slope = 159 ± 7% of control, n = 10) (Fig. 1A,B). The potentiation of the fEPSP slope that was induced by X/XO persisted for at least 60 min after reaching the maximum potentiation (data not shown). We observed a similar potentiation in the slope of the fEPSP when hippocampal slices were incubated with concentrations of X/XO that produced 10 μm superoxide (fEPSP slope = 170 ± 5% of control, n = 8) (Fig.1A). Incubation of hippocampal slices with higher concentrations of X/XO that produced 50 μmsuperoxide resulted in a more robust transient depression of synaptic transmission while X/XO was in the perfusate (51 ± 7% of control, n = 6) (Fig. 1A). After the transient depression the slope of the fEPSP recovered to baseline, but we observed no long-lasting potentiation 45 min after the washout of X/XO (fEPSP slope = 97 ± 11% of control, n= 6) (Fig. 1A). Thus, potentiation of hippocampal synaptic transmission by X/XO is a concentration-dependent phenomenon. We used concentrations of X/XO that produce 1–5 μm superoxide for the remainder of our studies.
Fig. 1.

X/XO-induced potentiation is concentration- and time-dependent. A, X/XO-induced potentiation is dependent on the concentration of superoxide. Stable baseline responses of the fEPSP slope were recorded in hippocampal area CA1 for 20 min before the addition of X/XO (indicated by the bar) for 10 min. X concentrations were kept constant at 20 μg/ml for all experiments. XO concentrations were 2 μg/ml (open circles, n = 10), 15 μg/ml (open squares, n = 8), or 25 μg/ml (filled squares, n = 6). These X/XO concentrations produced superoxide concentrations of 1–5, 10, and 50 μm, respectively. Error bars indicate SEM for the indicated number of determinations. When we compared the fEPSP slope 45 min after the washout of X/XO with the fEPSP slope immediately before the addition of X/XO, statistically significant potentiation was observed for XO concentrations of 2 and 15 μg/ml (p < 0.001 by paired Student'st test). B, Representative fEPSPs before, 30 min after, and 45 min after treatment with X/XO (20 and 2 μg/ml). Calibration: 2 mV, 3 msec. C, X/XO-induced potentiation is dependent on the duration of X/XO incubation. Stable baseline responses were recorded for 20 min before the addition of X/XO (20 and 2 μg/ml) for 5 (open triangles, n= 4), 10 (open circles, n = 10), or 30 (filled triangles, n = 4) min as indicated by the bars. Error bars are SEM for the indicated number of determinations. When we compared the fEPSP slope 45 min after the washout of X/XO with the fEPSP slope immediately before the addition of X/XO, statistically significant potentiation was observed for the 5 and 10 min X/XO incubations (p < 0.01 and 0.001, respectively, by paired Student's t test).
We also determined whether the X/XO-induced potentiation in hippocampal synaptic transmission was time-dependent. A 5 min incubation of hippocampal slices with concentrations of X/XO that produced 1–5 μm superoxide did not result in a transient depression of synaptic transmission while X/XO was in the perfusate (fEPSP slope = 101 ± 8% of control, n = 4) (Fig.1C). However, similar to experiments with 10 min incubations of X/XO, we did observe a slowly rising potentiation in the slope of the fEPSP that peaked 35–45 min after the washout of X/XO (fEPSP slope = 136 ± 9% of control, n = 4) (Fig.1C). In contrast, a 30 min incubation of hippocampal slices with the same concentration of X/XO produced a transient depression of synaptic transmission while X/XO was in the perfusate (fEPSP slope = 89 ± 6% of control, n = 4) (Fig.1B), followed by a slowly rising potentiation that peaked 35–45 min after the washout of X/XO (fEPSP slope = 131 ± 9% of control, n = 4) (Fig.1C). These results indicate that X/XO-induced potentiation of synaptic transmission can be induced with incubations of X/XO in a range of 5–30 min. We used 10 min incubations for the remainder of our studies.
The X/XO superoxide-generating system also produces hydrogen peroxide. Therefore, we determined whether either superoxide or hydrogen peroxide was responsible for the X/XO-induced potentiation of synaptic transmission. In a subset of experiments in which hippocampal slices were incubated with X/XO, we added either SOD or catalase to remove superoxide and hydrogen peroxide, respectively. As illustrated in Figure 2A, hippocampal slices incubated with X/XO in the presence of SOD did not exhibit a long-lasting potentiation of the fEPSP 45 min after the washout of these compounds (fEPSP slope = 107 ± 5% of control,n = 8). Hippocampal slices incubated with X/XO in the presence of catalase (Fig. 2A) did exhibit a long-lasting potentiation of the fEPSP 45 min after the washout of these compounds (fEPSP slope = 136 ± 4% of control,n = 8). However, this increase was not as robust as that observed when slices were treated with X/XO alone, which suggests that hydrogen peroxide is necessary for the full X/XO-induced enhancement of synaptic transmission. Interestingly, the effect of catalase on X/XO-induced potentiation is very similar to the effect of catalase on LTP in mouse hippocampal slices (Thiels et al., 2000). To ensure that the X/XO-induced potentiation was not attributable to nonspecific effects of XO, we treated slices with X and XO that had been inactivated by boiling. As shown in Figure 2B, we observed neither a transient depression while X/boiled XO was in the perfusate (fEPSP slope = 96 ± 6% of control,n = 8) nor a slowly rising potentiation after the washout of X/boiled XO (fEPSP slope = 105 ± 5% of control,n = 8). Taken together, these data indicate that, whereas superoxide absolutely is required for X/XO-induced potentiation, hydrogen peroxide also contributes this type of potentiation in hippocampal slices.
Fig. 2.
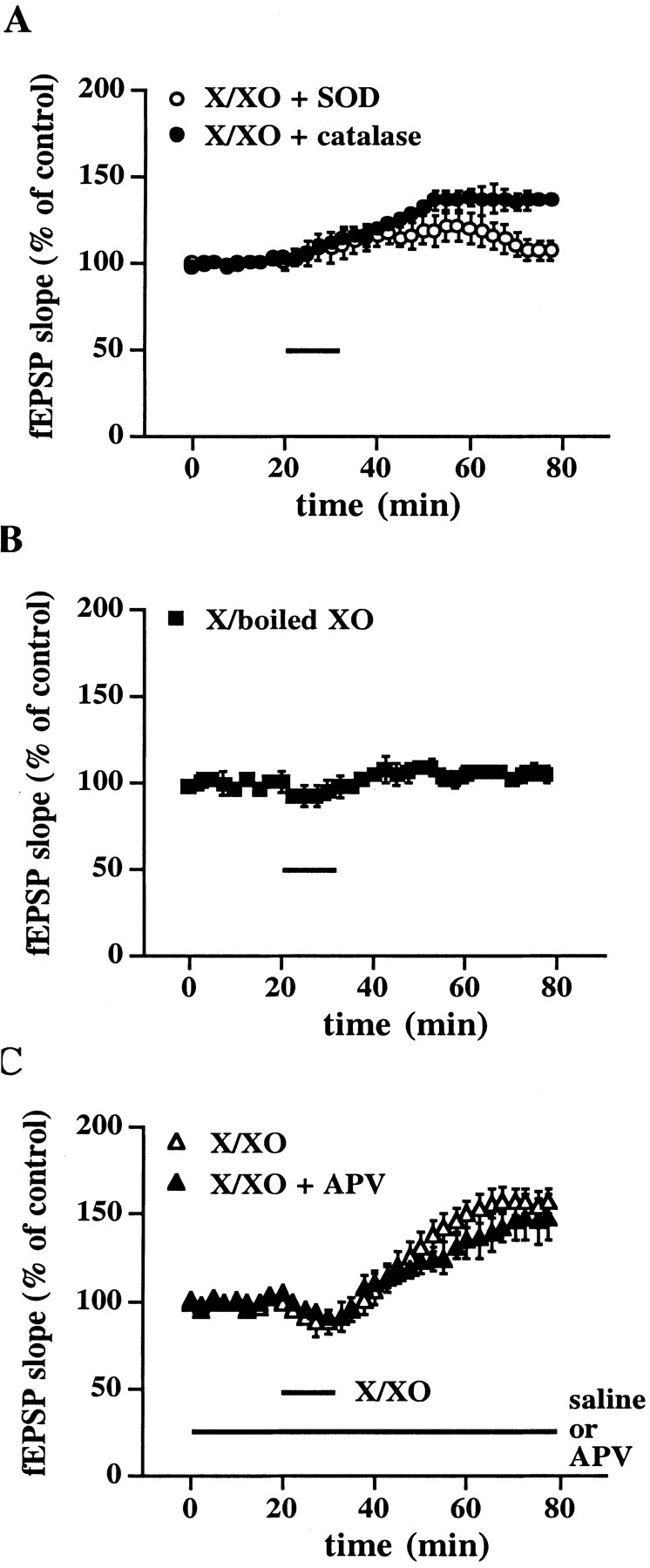
Characterization of X/XO-induced potentiation.A, X/XO-induced potentiation is superoxide-dependent. Stable baseline responses were recorded for 20 min before the slices were incubated with X/XO and antioxidant enzymes for 10 min (indicated by the bar). Open circles are ensemble averages from slices incubated with X/XO (20 and 2 μg/ml) and SOD (25 μg/ml). Filledcircles are ensemble averages from slices incubated with X/XO and catalase (25 μg/ml). Error bars are SEM for eight determinations. When we compared the fEPSP slope 45 min after the washout of X/XO with the fEPSP slope immediately before the addition of X/XO, statistically significant potentiation was observed in the presence of catalase (p < 0.001). B, Specificity of X/XO-induced potentiation.Filled squares are ensemble averages from slices incubated with X (20 μg/ml) and boiled XO (2 μg/ml). Error bars are SEM for eight determinations. C, X/XO-induced potentiation is downstream of NMDA receptor activation. Stable baseline responses were recorded for 20 min before the slices were incubated with X/XO (20 and 2 μg/ml) for 10 min either with (filled triangles) or without (open triangles) the NMDA receptor antagonist APV (50 μm) in the perfusate (indicated by thebars). Error bars are SEM for six determinations. There was no statistically significant difference in the potentiation between the groups 45 min after the washout of X/XO (p > 0.05 by paired Student'st test).
Interestingly, the transient depression in synaptic transmission that we observed when hippocampal slices were treated with X/XO alone was blocked in the presence of either SOD or catalase (Fig.2A). These data suggest that superoxide and hydrogen peroxide are necessary for the transient depression in synaptic transmission that is induced by X/XO.
If an ROS such as superoxide acts as a cellular messenger in LTP, then its actions should be independent of NMDA receptor activation because the application of exogenous superoxide would be downstream of this step in the pathway. Therefore, we examined the effect of APV, an NMDA receptor antagonist that blocks the induction of LTP (Collingridge et al., 1983), on X/XO-induced potentiation. When hippocampal slices perfused with 50 μm APV were incubated with X/XO for 10 min, we observed a transient depression of synaptic transmission while X/XO was in the perfusate (fEPSP slope = 92 ± 5% of control, n = 6) (Fig. 2C), followed by the slowly rising potentiation of synaptic transmission (fEPSP slope = 148 ± 13% of control, n = 6) (Fig.2C). The potentiation induced by X/XO in the presence of APV was not different from those slices treated with X/XO alone (fEPSP slope = 158 ± 7% of control, n = 6). These results are consistent with the notion that superoxide might act as a cellular messenger downstream of NMDA receptor activation in LTP.
Cell-impermeable scavengers of superoxide have been shown to attenuate LTP (Klann et al., 1998), suggesting that superoxide may need to enter the extracellular space after LTP-inducing stimulation. In addition, the incubation of hippocampal slices with X/XO has been shown to result in a superoxide-dependent increase in the release of glutamate (Pellegrini-Giampietro et al., 1988). Taken together, these data suggest the possibility that X/XO-induced potentiation is expressed presynaptically. Therefore, we examined the effects of X/XO on PPF, a well characterized presynaptic process in which facilitation is revealed when the second of two presynaptic action potentials results in increased neurotransmitter release relative to the response of the first action potential. Therefore, we measured PPF before and after slices were incubated with X/XO. We observed a small but significant increase in PPF (117 ± 6% of control, n = 6) immediately after the washout of X/XO (Fig.3, top) that coincided with the transient depression induced by X/XO (Fig. 3, bottom). Interestingly, we observed small but significant increases in PPF that coincided with the slowly rising potentiation both 20 (92 ± 3% of control, n = 6) and 30 min (90 ± 4% of control, n = 6) after the washout of X/XO (Fig. 3). However, PPF returned to control levels by the time that potentiation stabilized 40 min after the washout of X/XO (Fig. 3). Taken together, these results suggest that the cellular mechanisms underlying both the transient depression and the initial potentiation induced by X/XO are at least partially presynaptic.
Fig. 3.

Effect of X/XO on PPF. PPF was examined every 10 min with four sets of paired pulses, with an interpair interval of 20 sec and an interpulse interval of 50 msec. The change in PPF was expressed as the percentage of PPF at 0 min (the ratio of the second response to the first response). There was no alteration in PPF until immediately after the washout of X/XO (20 and 2 μg/ml), when we observed a increase in PPF that coincided with transient depression induced by X/XO. We also observed decreases in PPF 20 and 30 min after the washout of X/XO that coincided with the slowly rising potentiation induced by X/XO. Error bars are SEM for six determinations. *Statistical significance with a paired Student's ttest (p < 0.05).
X/XO induces a persistent superoxide-dependent increase in autonomous PKC activity in hippocampal slices
Previously, we have observed a persistent increase in autonomous PKC activity in hippocampal slices exposed to X/XO, which can be isolated via DEAE-cellulose chromatography (Knapp and Klann, 2000). Once again, we treated hippocampal slices for 10 min with X/XO; 45 min after the washout of X/XO the slices were homogenized and subjected to DEAE-cellulose column chromatography. As has been reported previously, we observed two fractions of cofactor-dependent PKC activity that eluted from the DEAE columns with 0.1 and 0.25 m NaCl salt washes after an application of the soluble fraction of hippocampal homogenates in both control (X/boiled XO) and X/XO-treated slices (data not shown). In addition, we observed a unique peak of autonomous PKC activity that could be eluted with 0.25 m NaCl in X/XO-treated slices, but not in the control slices treated with X/boiled XO (Fig. 4). We did not observe this peak of autonomous PKC activity when X/XO-treated slices were incubated with SOD (Fig. 4). The unique peak of autonomous PKC activity induced by X/XO was not affected by incubation with catalase (Fig. 4). Taken together, these data indicate that X/XO induces a persistent increase in autonomous PKC activity in hippocampal slices that is dependent on superoxide but that is not dependent on hydrogen peroxide.
Fig. 4.

Effect of X/XO on autonomous PKC activity isolated from slices with DEAE column chromatography. Slices were incubated with X/boiled XO (open squares, n = 5), X/XO (open circles, n = 5), X/XO + SOD (filled squares, n = 5), or X/XO + catalase (filled circles,n = 5) for 10 min, and the incubations were followed by perfusion with normal saline solution for 45 min. Then the slices were frozen on dry ice and homogenized; the soluble fraction of the homogenates was applied to a DEAE column. PKC was eluted from the column with either 0.1 or 0.25 m NaCl (arrows), and autonomous PKC activity was measured in each fraction as described in Materials and Methods.
X/XO-induced potentiation in hippocampal slices is PKC-dependent
Because X/XO can induce a persistent, superoxide-dependent increase in autonomous PKC activity in hippocampal slices in a manner similar to that previously observed in electrically induced LTP (Klann et al., 1998), we hypothesized that the X/XO-induced potentiation observed in Figure 1 was PKC-dependent. To test this hypothesis, we added X/XO to hippocampal slices in the presence of 500 nmbisindolylmaleimide I (Bis), a highly selective PKC inhibitor that acts on the catalytic domain of PKC (Toullec et al., 1991). As shown in Figure 5A, we observed that Bis blocked the X/XO-induced potentiation in synaptic transmission (fEPSP slope = 111 ± 5% of control, n = 8). Therefore, activation of PKC is necessary for X/XO-induced potentiation. It is interesting to note that the transient depression induced by X/XO still was observed in the presence of Bis (Fig.5A), which suggests that this type of plasticity is not a manifestation of superoxide-induced stimulation of PKC.
Fig. 5.

Effect of the PKC inhibitor Bis on X/XO-induced potentiation and X/XO-induced increase in autonomous PKC activity.A, X/XO-induced potentiation is PKC-dependent. Stable baseline responses of the fEPSP slope were recorded for 20 min before the slices were incubated with X/XO (20 and 2 μg/ml) for 10 min in the presence of the PKC inhibitor Bis (500 nm) as indicated by the bars. Error bars are SEM for eight determinations. When we compared the fEPSP slope 45 min after the washout of X/XO with the fEPSP slope immediately before the addition of X/XO, no statistically significant potentiation was observed (p > 0.05 by paired Student'st test) B, X/XO-induced persistent increase in autonomous PKC activity is present after the washout of Bis. Slices were incubated with either normal saline (open circles, n = 5) or X/XO (filled circles, n = 5) in the presence of Bis as described in A. At the end of the experiment the slices were frozen and homogenized. The soluble fraction of the homogenates was applied to a DEAE column, and PKC was eluted from the column with either 0.1 or 0.25 m NaCl (arrows). Autonomous PKC activity was measured in each fraction as described in Materials and Methods.
Previously, we have investigated the modification of PKC induced by superoxide and determined that the regulatory domain of PKC is required because PKM, the free catalytic domain of PKC, was not stimulated by superoxide (Knapp and Klann, 2000). If superoxide stimulates PKC by acting on the regulatory domain, then we would expect to observe the superoxide-stimulated peak of autonomous PKC activity eluted from a DEAE column from slices exposed to X/XO in the presence of Bis. As shown in Figure 5B, we observed the expected unique peak of autonomous PKC activity from DEAE columns from slices treated with X/XO in the presence of Bis. This finding suggests that a block of the X/XO-induced potentiation by Bis cannot be attributable to interference with the superoxide-induced stimulation of PKC.
To ensure that Bis could inhibit oxidatively activated PKC, we performed in vitro assays to determine whether Bis could inhibit X/XO-induced increases in autonomous PKC activity. Purified PKC was treated with X/XO as described previously (Knapp and Klann, 2000) in the presence and absence of 500 nm Bis. Treatment of purified PKC with X/XO resulted in increased autonomous enzyme activity (control = 4.23 ± 0.34 pmol/min; X/XO = 18.34 ± 1.67 pmol/min; n = 4) that was inhibited completely by Bis (control plus Bis = 0.19 ± 0.02 pmol/min; X/XO = 0.25 ± 0.07 pmol/min; n = 4). Thus, Bis is able to inhibit oxidatively activated PKC.
X/XO-induced potentiation and LTP share similar cellular mechanisms
If the potentiation induced by superoxide contributes to LTP, then that potentiation should occlude LTP. To test this possibility, we treated slices with X/XO; 60 min after the washout of the X/XO the stimulation intensity was reduced to obtain a new baseline, and HFS was delivered to the slices. Figure6A shows that, after potentiation by X/XO, HFS did not result in a significant enhancement of synaptic transmission (fEPSP slope = 108 ± 9% of control, n = 6). However, if X/XO-induced potentiation was blocked by SOD, then HFS elicited LTP (fEPSP slope = 168 ± 8% of control, n = 6) (Fig. 6B). We also determined whether LTP could occlude X/XO-induced potentiation. LTP was induced with HFS; 30 min after the last train the stimulation intensity was reduced to obtain a new baseline. Figure 6Cshows that, after the establishment of LTP, the application of X/XO to the slices did not enhance synaptic transmission significantly (fEPSP slope = 110 ± 10% of control, n = 6). However, if LTP was blocked by APV, then the application of X/XO to the slices resulted in potentiation (fEPSP slope = 148 ± 8% of control, n = 6). Taken together, the findings in Figure6 suggest that the X/XO-induced potentiation and LTP share similar cellular mechanisms.
Fig. 6.

X/XO-induced potentiation and LTP occlude each other. A, X/XO-induced potentiation occludes LTP. Stable baseline responses of the fEPSP slope were recorded for 20 min before the slices were incubated with X/XO (20 and 2 μg/ml) as indicated by the bar. At 60 min after the washout of X/XO the stimulus intensity was reduced (indicated by thedown arrow) to match the fEPSP slope before exposure to X/XO. LTP-inducing HFS (indicated by the up arrow) then was delivered to the slices as described in Materials and Methods. Additional incubations of the slices with X/XO before HFS did not produce additional potentiation, indicating that the X/XO-induced potentiation was saturated (data not shown). Error bars are SEM for six determinations. B, LTP can be induced after a blockade of X/XO-induced potentiation by SOD. Slices were incubated with X/XO and SOD (25 μg/ml) as indicated by the bar. At 60 min after the washout of X/XO and SOD, LTP-inducing HFS (indicated by thearrow) was delivered to the slices. Error bars are SEM for six determinations. C, LTP occludes X/XO-induced potentiation. Stable baseline responses in hippocampal slices were recorded for 20 min before the delivery of LTP-inducing HFS as indicated by the up arrow. At 30 min after the last train of HFS the stimulus intensity was reduced (indicated by thedown arrow) to match the fEPSP slope before the HFS. Slices then were incubated with X/XO (20 and 2 μg/ml) as indicated by the bar. Additional delivery of HFS before incubation of the slices with X/XO did not produce additional potentiation, indicating that LTP was saturated (data not shown). Error bars are SEM for six determinations. D, X/XO can induce potentiation after a blockade of LTP by APV. HFS (indicated by thearrow) was delivered to the slices in the presence of APV (indicated by the bar). At 30 min after HFS the slices were incubated with X/XO as indicated by the bar. Error bars are SEM for six determinations.
Because X/XO-induced potentiation and LTP are likely to share similar cellular mechanisms, we determined whether LTP was associated with superoxide-stimulated PKC. We delivered HFS to the Schaffer collateral input to area CA1, which resulted in LTP (for 2 min experiments, fEPSP slope = 181 ± 5% of control, n = 5; for 45 min experiments, fEPSP slope = 158 ± 7% of control,n = 5) (Fig.7A). We observed a typical elution profile of cofactor-dependent PKC activity from DEAE columns from LTP slices at both 2 and 45 min after the last train of HFS that was not different from control slices (Fig. 7B,C). However, we detected a unique peak of autonomous PKC activity in LTP slices both 2 and 45 min after the last train of HFS that was not detected in control slices (Fig.8A,B). These data are consistent with the idea that superoxide-induced stimulation of autonomous PKC activity is a biochemical mechanism involved in the induction and expression of both X/XO-induced potentiation and LTP.
Fig. 7.
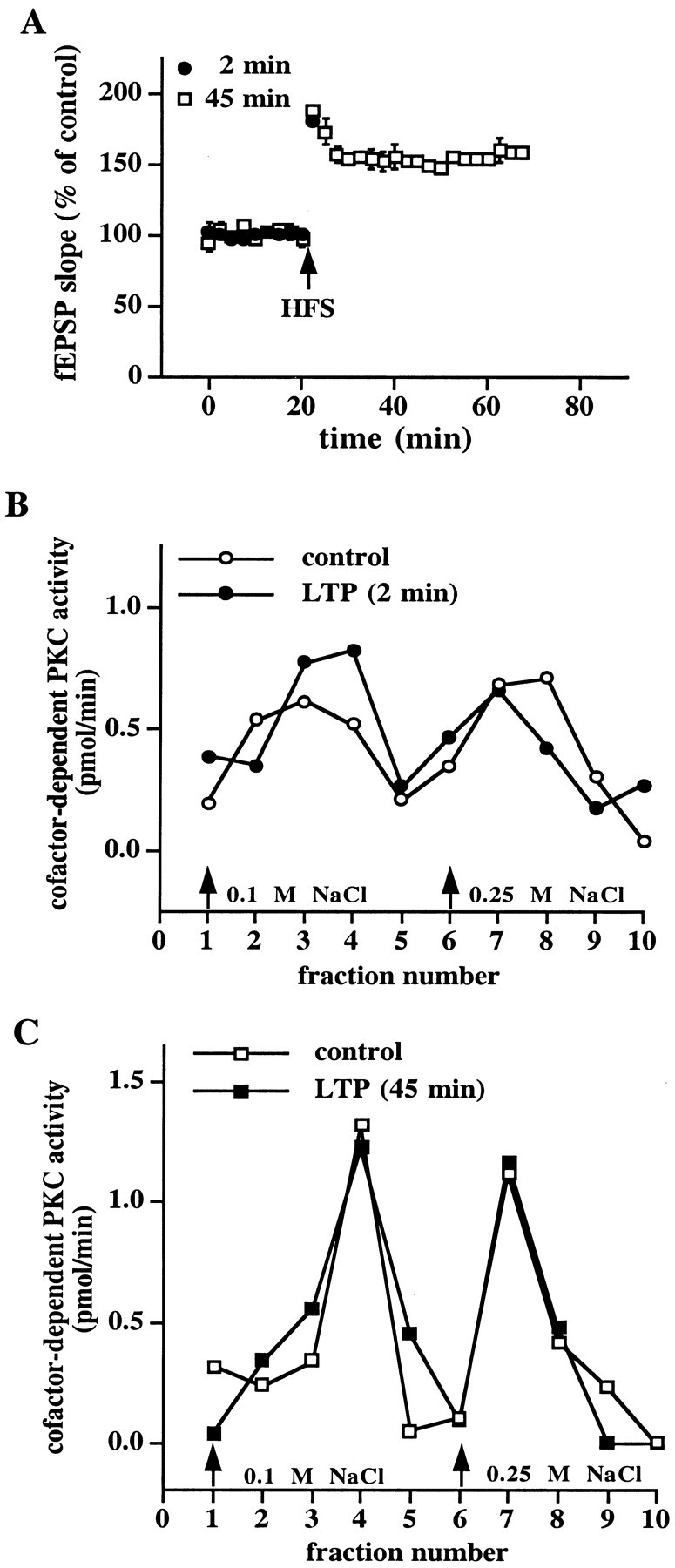
No alteration in the DEAE column elution profile of cofactor-dependent PKC activity after the induction of LTP.A, Induction of LTP. Stable baseline responses of the fEPSP slope were recorded in hippocampal slices for 20 min before the delivery of HFS as indicated by the arrow. Responses were recorded for either 2 min (filled circles,n = 5) or 45 min (open squares,n = 5) after the final train of HFS. Error bars are SEM for the indicated number of determinations. B, C, Elution profile of cofactor-dependent PKC activity from either control slices (open circles in B, open squares in C) or slices taken 2 min (filled circles in B) and 45 min (filled squares in C) after the final train of LTP-inducing HFS. Either 2 or 45 min after the final train of HFS the slices were frozen, and the area between the stimulating and recording electrodes was dissected and homogenized. The soluble fraction of the homogenates was applied to mini-DEAE columns, and PKC was eluted with either 0.1 or 0.25 m NaCl (arrows). Cofactor-dependent PKC activity was measured as described in Materials and Methods.
Fig. 8.

LTP is associated with a unique peak of autonomous PKC activity that elutes from DEAE columns. Shown are elution profiles of autonomous PKC activity from either control slices (open circles in A; open squares inB) or slices taken 2 min (filled circles in A) and 45 min (filled squares in B) after the final train of LTP-inducing HFS. Either 2 min (n = 5) or 45 min (n = 5) after the final train of HFS the slices were frozen, and the area between the stimulating and recording electrodes was dissected and homogenized. The soluble fraction of the homogenates was applied to mini-DEAE columns, and PKC was eluted with either 0.1 or 0.25 m NaCl (arrows). Autonomous PKC activity was measured as described in Materials and Methods.
We performed control experiments to ensure that the unique peak of superoxide-stimulated autonomous PKC activity was specific to LTP. First, we delivered test stimulation to slices with the same number of stimuli as given during HFS, but at low frequency (0.1 Hz). When the soluble fraction of homogenates from these slices was applied to DEAE columns, we did not observe the unique peak of autonomous PKC activity (Fig. 9A). In addition, we delivered HFS to slices in the presence of either the NMDA receptor antagonist APV or SOD and applied the soluble fraction of homogenates from these slices to DEAE columns. Once again, we did not observe the unique peak of autonomous PKC activity (Fig. 9B,C). These results indicate that the LTP-associated appearance of superoxide-stimulated PKC that eluted from the DEAE columns cannot be elicited by either low-frequency stimulation or HFS in the absence of LTP. The results of the APV and SOD experiments also indicate that NMDA receptor activation and the subsequent production of superoxide are necessary to trigger the oxidative activation of PKC.
Fig. 9.
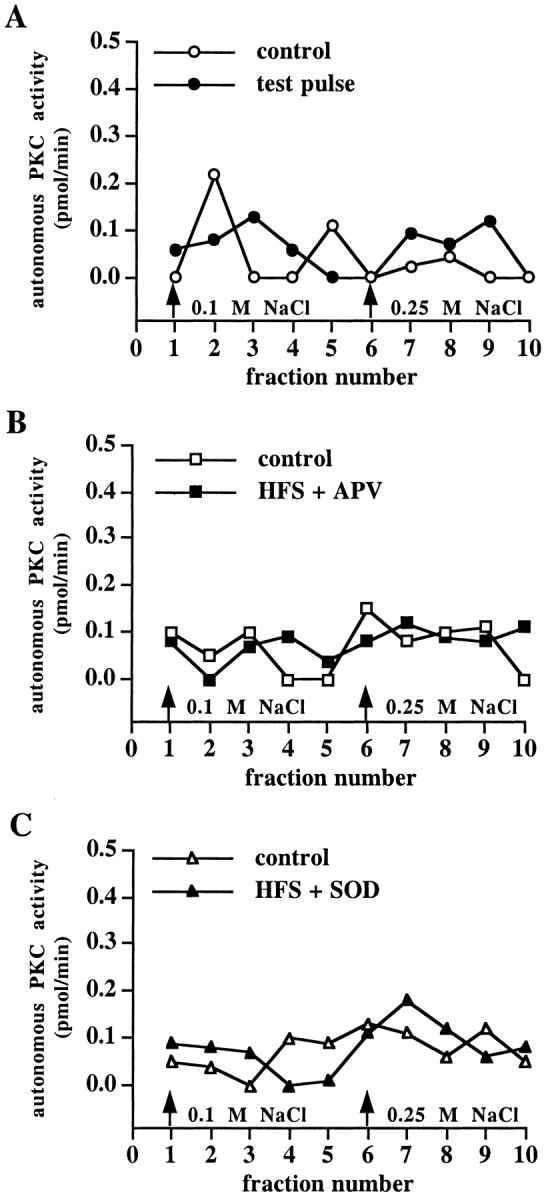
The LTP-associated unique peak of autonomous PKC activity is NMDA- and superoxide-dependent. Shown are elution profiles of autonomous PKC activity from control slices and slices that were given test pulses (A), slices in which HFS was delivered in the presence of APV (B), and slices in which HFS was delivered in the presence of SOD (C). In each panel, n = 5.
Because the PKC inhibitor Bis was able to block X/XO-induced potentiation (Fig. 5A), we asked whether this inhibitor was able to block LTP. As shown in Figure10, delivery of HFS in the presence of 500 nm Bis significantly attenuated LTP (fEPSP slope = 122 ± 6% of control, n = 6) compared with control slices from the same animal in which HFS was delivered in the presence of saline (fEPSP slope = 167 ± 5% of control, n = 6). These results are consistent with the idea that the oxidative activation of PKC plays a role in both forms of potentiation.
Fig. 10.

The PKC inhibitor Bis attenuates LTP. Open circles are ensemble averages from control LTP experiments. Filled circles are ensemble averages from slices given LTP-inducing HFS with 500 nm Bis in the perfusing solution (indicated by the bar). Responses recorded from slices given LTP-inducing HFS in the presence of Bis were compared with responses recorded from a control slice from the same animal (in an adjacent recording chamber) that received HFS in the absence of the inhibitor. Error bars are SEM for six determinations. When we compared the fEPSP slope 45 min after HFS with the fEPSP slope immediately before HFS, small but statistically significant potentiation was observed in the presence of Bis (p < 0.05 by paired Student'st test).
DISCUSSION
The results presented herein demonstrate that superoxide is necessary for X/XO-induced potentiation in area CA1 of rat hippocampal slices and that this type of potentiation requires superoxide-induced stimulation of autonomous PKC activity. In addition, we have shown that we can isolate superoxide-stimulated PKC not only from slices that were incubated with X/XO but also from slices in which LTP was induced. Finally, we have shown that X/XO-induced potentiation and LTP occlude one another. Taken together, these data suggest that X/XO-induced potentiation and LTP share similar cellular mechanisms, including the oxidative activation of PKC.
Previous results from studies on the effects of ROS on synaptic transmission indicate that the role of ROS in modulating synaptic transmission is complex, depending on numerous factors that contribute to the stringency of oxidation. Various ROS with divergent oxidative strengths have been shown to affect synaptic transmission. For example, hydroxyl radical is a stronger oxidant than hydrogen peroxide, which, in turn, is stronger than superoxide (Rice-Evans and Burdon, 1994). In previous studies strong oxidizing conditions that promote hydroxyl radical production were found to inhibit synaptic transmission (Pellmar, 1987; Auerbach and Segal, 1997). Another factor that influences the stringency of oxidation is the length of oxidant exposure. For example, prolonged (>55 min) exposure of hippocampal slices to superoxide inhibits synaptic transmission (Pellmar and Lepinski, 1992). In the studies presented herein, slices were exposed briefly (10 min) to low concentrations (Knapp and Klann, 2000) of ROS. Thus, we have characterized a physiological role for ROS in promoting a long-lasting enhancement in synaptic transmission.
We recently showed that superoxide-dependent stimulation of autonomous PKC activity is achieved via interaction of superoxide with the cysteine-rich zinc finger motif in the regulatory domain of the enzyme (Knapp and Klann, 2000). In a series of biochemical studies we showed that superoxide stimulates autonomous PKC activity via thiol oxidation and the release of zinc from the cysteine-rich zinc finger motifs. Based on the elution profiles of autonomous PKC activity from LTP samples (Fig. 8), it is reasonable to propose that LTP-inducing HFS induces a similar release of zinc from PKC. An obvious question arises: Which PKC isoform is stimulated by superoxide during LTP? PKC isoforms from each PKC subfamily (classical, novel, and atypical) can be stimulated by superoxide (Knapp and Klann, 2000), and numerous PKC isoforms are present in the superoxide-stimulated DEAE column fraction that elutes with 0.25 m NaCl (our unpublished observations). Thus, the identity of the PKC isoform(s) stimulated by superoxide after the induction of LTP will need to be determined in the future by other means.
In the current studies we focused on the participation of superoxide-stimulated PKC in the potentiation of hippocampal synaptic transmission. However, it is not our intention to imply that PKC is the only signaling enzyme that is affected by ROS such as superoxide during either X/XO-induced potentiation or LTP. There are a number of other effector molecules that are known to be important for synaptic plasticity and that are likely to be modulated by superoxide. For instance, activation of extracellular signal-regulated kinase (ERK) is necessary for LTP (English and Sweatt, 1997), and ERK activity in hippocampal slices is stimulated by ROS, including superoxide (Kanterewicz et al., 1998). Tyrosine kinase activity has been shown to be necessary for LTP (O'Dell et al., 1991; Grant et al., 1992; Kang and Schuman, 1995; Lu et al., 1998), and many tyrosine kinases have been shown to exhibit increased activity in response to ROS (Chan et al., 1986; Scheiven et al., 1993; Guyton et al., 1996). In addition, superoxide inhibits the activity of the protein phosphatase calcineurinin vitro (Wang et al., 1996) and in cultured hippocampal neurons after electrical stimulation (Bito et al., 1996). Therefore, it is reasonable to propose that both X/XO-induced potentiation and LTP involve superoxide-dependent increases in ERK activity and/or inhibition of calcineurin in addition to stimulation of autonomous PKC activity.
The notion that multiple signaling cascades must be modulated by ROS for X/XO-induced potentiation is supported by the findings shown in Figure 5. We found that the PKC inhibitor Bis could block X/XO-induced potentiation (Fig. 5A). Because superoxide interacts with the regulatory domain of PKC (Knapp and Klann, 2000) and Bis inhibits PKC by interacting with the ATP-binding site in the catalytic domain (Toullec et al., 1991), we hypothesized that superoxide-stimulated PKC activity still should be present in hippocampal slices after the washout of Bis. This hypothesis was found to be correct (Fig.5B), indicating that Bis did not interfere with the interaction of superoxide with PKC. Interestingly, we observed no potentiation in synaptic transmission after the washout of Bis (Fig.5A). This result suggests that the superoxide-dependent stimulation of PKC activity alone is not sufficient for X/XO-induced potentiation. It is possible that X/XO-induced potentiation requires either the concomitant activation of PKC and ERK or the activation of PKC coupled with the inhibition of a protein phosphatase such as calcineurin.
We have shown that X/XO-induced potentiation is dependent on superoxide (Fig. 2A) and is occluded by LTP (Fig.6A). Taken together, these findings suggest that LTP, like X/XO-induced potentiation, is dependent on superoxide. This idea is supported further by previous findings showing that cell-permeable scavengers of superoxide block LTP (Klann, 1998), cell-impermeable scavengers of superoxide strongly attenuate LTP (Klann et al., 1998), and slices from mice that overexpress either SOD-1 (Gahtan et al., 1998) or EC-SOD (Thiels et al., 2000) exhibit deficient LTP. Thus, it is now critical to determine the source of superoxide production after LTP. Possible sources of LTP-dependent superoxide production that have been suggested include arachidonic acid metabolism, nitric oxide synthase, and the mitochondrial electron transport chain (Thiels et al., 2000). Another possibility is endogenous XO. There is evidence for XO activity being present in brain tissue (Markley et al., 1975) and from synaptosomal preparations (Deliconstantinos and Villiotou, 1996). However, we found that the incubation of hippocampal slices with X/boiled XO did not result in long-lasting potentiation, which suggests that endogenous XO is not capable of producing sufficient superoxide for LTP in hippocampal slice preparations. This finding also indicates that nonspecific effects of xanthine cannot account for X/XO-induced potentiation.
Another candidate enzyme for superoxide production during LTP is NADPH oxidase. This enzyme is a heterotetramer of cytosolic and membrane-associated proteins that can be activated by the small G-protein Rac1 (Shatwell and Segal, 1996). The expression of NADPH oxidase originally was characterized in phagocytic neutrophils of the immune system (Shatwell and Segal, 1996). However, there is evidence that NADPH oxidase and Rac1 also are expressed in the brain and, in particular, the hippocampus (Olenik et al., 1997; Mizuki et al., 1998). Interestingly, Rac1 is one of the proteins that has been shown to be associated with NMDA receptor multiprotein complexes (Husi et al., 2000), making it a candidate for regulation after the induction of LTP. It remains to be determined whether the cytosolic and/or membrane-associated NADPH oxidase proteins also are associated with NMDA receptor multiprotein complexes.
In addition to the role that superoxide plays in hippocampal LTP, superoxide also appears to be necessary for hippocampus-dependent learning and memory. For example, mice that overexpress EC-SOD were shown to have deficits in their ability to execute the eight-arm radial maze task (Levin et al., 1998) and in contextual fear conditioning (Thiels et al., 2000). Moreover, mice that overexpress SOD-1 were shown to have deficits in their ability to learn the spatial version of the Morris water maze task (Gahtan et al., 1998). Overall, these findings are consistent with superoxide signaling being a necessary component of hippocampus-dependent learning and memory. It will be of great interest to determine whether the superoxide-stimulated autonomous PKC activity associated with X/XO-induced potentiation and LTP also can be isolated in hippocampal lysates from rats that have acquired either contextual fear conditioning or various types of spatial tasks.
A number of our findings suggest that X/XO-induced potentiation and LTP share similar cellular mechanisms. This is supported by the following findings: (1) X/XO-induced potentiation and LTP occlude one another; (2) X/XO-induced potentiation and LTP are associated with superoxide-dependent increases in autonomous PKC activity; (3) the magnitudes of X/XO-induced potentiation and LTP are reduced by the PKC inhibitor Bis. However, several of our findings point to differences in these forms of potentiation. First, the onset of X/XO-induced potentiation is much slower than LTP (compare Figs.1A, 7A), similar to previous experiments with other small messenger molecules that can induce potentiation (Zhuo et al., 1993). Second, PPF is altered during the initial, slowly rising phase of X/XO-induced potentiation (Fig. 3), a result that is not observed consistently with LTP (for review, see Malenka and Nicoll, 1999). Finally, the PKC inhibitor Bis blocked X/XO-induced potentiation (Fig. 5A) but only attenuated LTP (Fig. 10). Taken together, these results suggest that cellular signaling mechanisms underlying X/XO-induced potentiation and LTP are similar, but not identical.
In closing, our studies demonstrate that superoxide can produce a persistent potentiation in synaptic transmission when added to hippocampal slices and that this potentiation shares similar cellular mechanisms with LTP. Thus, superoxide should be added to the list of small messenger molecules that are necessary for hippocampal LTP.
Footnotes
This work was supported by National Institutes of Health Grant NS34007 (E.K.) and National Research Service Award MH 18273 (L.T.K.). We thank Dr. Edda Thiels, Dr. Eric D. Norman, and Beatriz I. Kanterewicz for helpful comments throughout these studies and Dr. J. David Sweatt for critically reading this manuscript.
Correspondence should be addressed to Dr. Eric Klann, Department of Molecular Physiology and Biophysics, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030. E-mail: eklann@bcm.tmc.edu.
REFERENCES
- 1.Auerbach JM, Segal M. Peroxide modulation of slow onset potentiation in rat hippocampus. J Neurosci. 1997;17:8695–8701. doi: 10.1523/JNEUROSCI.17-22-08695.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 4.Böhme GA, Bon C, Stutzmann JM, Doble A, Blanchard JC. Possible involvement of nitric oxide in long-term potentiation. Eur J Pharmacol. 1991;199:379–381. doi: 10.1016/0014-2999(91)90505-k. [DOI] [PubMed] [Google Scholar]
- 5.Chan TM, Chen E, Tatoyan A, Shargill NS, Pleta M, Hochstein P. Stimulation of tyrosine-specific protein phosphorylation in the rat liver plasma membrane by oxygen radicals. Biochem Biophys Res Commun. 1986;139:439–445. doi: 10.1016/s0006-291x(86)80010-9. [DOI] [PubMed] [Google Scholar]
- 6.Collingridge GL, Kehl SJ, McClennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral–commissural pathway of the rat hippocampus. J Physiol (Lond) 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deliconstantinos G, Villiotou V. NO synthase and xanthine oxidase activities of rabbit brain synaptosomes: peroxynitrite formation as a causative factor of neurotoxicity. Neurochem Res. 1996;21:51–61. doi: 10.1007/BF02527672. [DOI] [PubMed] [Google Scholar]
- 8.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long-term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 9.Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- 10.Gahtan E, Auerbach JM, Groner Y, Segal M. Reversible impairment of long-term potentiation in transgenic Cu/Zn-SOD mice. Eur J Neurosci. 1998;10:538–544. doi: 10.1046/j.1460-9568.1998.00058.x. [DOI] [PubMed] [Google Scholar]
- 11.Grant SGN, O'Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- 12.Guyton KZ, Lui Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. J Biol Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- 13.Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Oxford UP; New York: 1989. [Google Scholar]
- 14.Hille R, Nishino T. Xanthine oxidase and xanthine dehydrogenase. FASEB J. 1995;9:995–1003. [PubMed] [Google Scholar]
- 15.Hrabetova S, Sacktor TC. Bidirectional regulation of protein kinase Mζ in the maintenance of long-term potentiation and long-term depression. J Neurosci. 1996;16:5324–5333. doi: 10.1523/JNEUROSCI.16-17-05324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SGN. Proteomic analysis of NMDA receptor–adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- 17.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 18.Kanterewicz BI, Knapp LT, Klann E. Stimulation of p42 and p44 mitogen-activated protein kinases by reactive oxygen species and nitric oxide in hippocampus. J Neurochem. 1998;70:1009–1016. doi: 10.1046/j.1471-4159.1998.70031009.x. [DOI] [PubMed] [Google Scholar]
- 19.Klann E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J Neurophysiol. 1998;80:452–457. doi: 10.1152/jn.1998.80.1.452. [DOI] [PubMed] [Google Scholar]
- 20.Klann E, Chen S-J, Sweatt JD. Persistent protein kinase activation in the maintenance phase of long-term potentiation. J Biol Chem. 1991;266:24253–24256. [PubMed] [Google Scholar]
- 21.Klann E, Chen S-J, Sweatt JD. Mechanism of protein kinase C activation during the induction and maintenance of long-term potentiation probed using a novel peptide substrate. Proc Natl Acad Sci USA. 1993;90:8337–8341. doi: 10.1073/pnas.90.18.8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klann E, Roberson ED, Knapp LT, Sweatt JD. A role for superoxide in protein kinase C activation and induction of long-term potentiation. J Biol Chem. 1998;273:4516–4522. doi: 10.1074/jbc.273.8.4516. [DOI] [PubMed] [Google Scholar]
- 23.Knapp LT, Klann E. Superoxide-induced stimulation of protein kinase C via thiol modification and modulation of zinc content. J Biol Chem. 2000;275:24136–24145. doi: 10.1074/jbc.M002043200. [DOI] [PubMed] [Google Scholar]
- 24.Levin ED, Brady TC, Hochrein EC, Oury TD, Jonsson LM, Marklund SL, Crapo JD. Molecular manipulations of extracellular superoxide dismutase: functional importance for learning. Behav Genet. 1998;28:381–390. doi: 10.1023/a:1021673703129. [DOI] [PubMed] [Google Scholar]
- 25.Lu YM, Roder JC, Davidow J, Salter MW. Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science. 1998;279:1363–1367. doi: 10.1126/science.279.5355.1363. [DOI] [PubMed] [Google Scholar]
- 26.Lynch G, Larson K, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- 27.Malenka RC, Nicoll RA. Long-term potentiation: a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 28.Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- 29.Markley HG, Faillance LA, Mezey E. Xanthine oxidase activity in rat brain. Biochim Biophys Acta. 1975;309:23–31. doi: 10.1016/0005-2744(73)90313-6. [DOI] [PubMed] [Google Scholar]
- 30.Mizuki K, Kadomatsu K, Hata K, Ito T, Fan Q-W, Kage Y, Fukumaki Y, Sakaki Y, Takeshige K, Sumimoto H. Functional modules and expression of p40phox and p67phox, SH3-domain-containing proteins involved in the phagocyte NADPH oxidase complex. Eur J Biochem. 1998;251:573–582. doi: 10.1046/j.1432-1327.1998.2510573.x. [DOI] [PubMed] [Google Scholar]
- 31.O'Dell TJ, Kandel ER, Grant SGN. Long-term potentiation in the hippocampus is blocked by tyrosine kinase inhibitors. Nature. 1991;353:558–560. doi: 10.1038/353558a0. [DOI] [PubMed] [Google Scholar]
- 32.Olenik C, Barth H, Just I, Aktories K, Meyer DK. Gene expression of small GTP-binding proteins RhoA, RhoB, Rac1, Cdc42 in adult rat brain. Brain Res Mol Brain Res. 1997;52:263–269. doi: 10.1016/s0169-328x(97)00270-2. [DOI] [PubMed] [Google Scholar]
- 33.Pellegrini-Giampietro DE, Cherici G, Alesiani M, Carla V, Moroni F. Excitatory amino acid release from rat hippocampal slices as a consequence of free-radical formation. J Neurochem. 1988;51:1960–1963. doi: 10.1111/j.1471-4159.1988.tb01187.x. [DOI] [PubMed] [Google Scholar]
- 34.Pellmar TC. Peroxide alters neuronal excitability in the CA1 region of guinea pig hippocampus in vitro. Neuroscience. 1987;23:447–456. doi: 10.1016/0306-4522(87)90068-6. [DOI] [PubMed] [Google Scholar]
- 35.Pellmar TC, Lepinski DL. Electrophysiological consequences of exposure of hippocampal slices to dihydroxyfumarate, a generator of superoxide radicals. Brain Res. 1992;569:189–198. doi: 10.1016/0006-8993(92)90630-r. [DOI] [PubMed] [Google Scholar]
- 36.Rice-Evans CA, Burdon RH. Free radical damage and its control. Elsevier; New York: 1994. [Google Scholar]
- 37.Roberson ED, English JD, Sweatt JD. A biochemist's view of long-term potentiation. Learn Mem. 1996;3:1–24. doi: 10.1101/lm.3.1.1. [DOI] [PubMed] [Google Scholar]
- 38.Sacktor TC, Osten P, Valsamis H, Jiang X, Naik MU, Sublette E. Persistent activation of the zeta isoform of protein kinase C in the maintenance of long-term potentiation. Proc Natl Acad Sci USA. 1993;90:8342–8346. doi: 10.1073/pnas.90.18.8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scheiven GL, Kirihara D, Burg DL, Geahlen RL, Ledbetter JA. p72syk tyrosine kinase is activated by oxidizing conditions that induce lymphocyte tyrosine phosphorylation and Ca2+ signals. J Biol Chem. 1993;268:16688–16692. [PubMed] [Google Scholar]
- 40.Shatwell KP, Segal AW. NADPH oxidase. Int J Biochem Cell Biol. 1996;28:1191–1195. doi: 10.1016/s1357-2725(96)00084-2. [DOI] [PubMed] [Google Scholar]
- 41.Sweatt JD. Toward a molecular explanation for long-term potentiation. Learn Mem. 1999;6:399–416. doi: 10.1101/lm.6.5.399. [DOI] [PubMed] [Google Scholar]
- 42.Thiels E, Urban NN, Gonzalez-Burgos GR, Kanterewicz BI, Barrionuevo G, Chu CT, Oury TD, Klann E. Impairment of long-term potentiation and associative memory in mice that overexpress extracellular superoxide dismutase. J Neurosci. 2000;20:7631–7639. doi: 10.1523/JNEUROSCI.20-20-07631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- 44.Wang JH, Feng DP. Postsynaptic protein kinase C essential to induction and maintenance of long-term potentiation in the hippocampal CA1 region. Proc Natl Acad Sci USA. 1992;89:2576–2580. doi: 10.1073/pnas.89.7.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Culotta VC, Klee CB. Superoxide dismutase protects calcineurin from inactivation. Nature. 1996;383:434–437. doi: 10.1038/383434a0. [DOI] [PubMed] [Google Scholar]
- 46.Williams JH, Errington ML, Lynch MA, Bliss TVP. Arachidonic acid induces long-term activity-dependent enhancement of synaptic transmission in the hippocampus. Nature. 1989;341:739–742. doi: 10.1038/341739a0. [DOI] [PubMed] [Google Scholar]
- 47.Zhuo M, Small SA, Kandel ER, Hawkins RD. Nitric oxide and carbon monoxide produce activity-dependent long-term synaptic enhancement in hippocampus. Science. 1993;260:1946–1950. doi: 10.1126/science.8100368. [DOI] [PubMed] [Google Scholar]