

Abstract
Increasing evidence has suggested an important role for environmental factors such as exposure to pesticides in the pathogenesis of Parkinson's disease. In experimental animals the exposure to a common herbicide, rotenone, induces features of parkinsonism; mechanistically, rotenone-induced destruction of dopaminergic neurons has been attributed to its inhibition of the activity of neuronal mitochondrial complex I. However, the role of microglia, the resident brain immune cells in rotenone-induced neurodegeneration, has not been reported. Using primary neuron-enriched and neuron/glia cultures from the rat mesencephalon, we discovered an extraordinary feature for rotenone-induced degeneration of cultured dopaminergic neurons. Although little neurotoxicity was detected in neuron-enriched cultures after treatment for 8 d with up to 20 nm rotenone, significant and selective dopaminergic neurodegeneration was observed in neuron/glia cultures 2 d after treatment with 20 nm rotenone or 8 d after treatment with 1 nm rotenone. The greatly enhanced neurodegenerative ability of rotenone was attributed to the presence of glia, especially microglia, because the addition of microglia to neuron-enriched cultures markedly increased their susceptibility to rotenone. Mechanistically, rotenone stimulated the release of superoxide from microglia that was attenuated by inhibitors of NADPH oxidase. Furthermore, inhibition of NADPH oxidase or scavenging of superoxide significantly reduced the rotenone-induced neurotoxicity. This is the first report demonstrating that microglia play a pivotal role in rotenone-induced degeneration of dopaminergic neurons. The results of this study should advance our understanding of the mechanism of action for pesticides in the pathogenesis of Parkinson's disease.
Keywords: pesticides, microglia, superoxide, dopamine, Parkinson's disease, neurotoxicity
Parkinson's disease (PD) is characterized by a progressive and selective loss of dopaminergic neurons in the substantia nigra (SN), resulting in movement disorders (Olanow and Tatton, 1999). Sporadic PD accounts for >90% of the incidence of the disease, with the remaining fraction of mostly familial PD suspected to be linked to mutations in several recently identified genes, including α-synuclein and parkin (Langston, 1998;Polymeropoulos, 1998; de Silva et al., 2000). Unlike its familial counterpart, idiopathic PD usually begins in the fifth decade of life and progresses over a period of 10–20 years. Although the etiology of idiopathic PD remains unknown, epidemiological and case control studies have implicated rural living, well water consumption, and pesticide exposure as potential risk factors for PD (Priyadarshi et al., 2001). Among these factors, exposure to pesticides and insecticides appears to have one of the strongest correlations with an increased incidence of PD (Gorell et al., 1998; Ritz and Yu, 2000; Herishanu et al., 2001).
Recent success in demonstrating the degenerative effect of several pesticides on the nigrostriatal dopaminergic system in experimental animals has revitalized interest in identifying specific agents as possible causes of PD (Betarbet et al., 2000; Thiruchelvam et al., 2000). For example, Betarbet et al. (2000) reported that chronic administration of a common herbicide, rotenone, resulted in a selective destruction of the nigrostriatal dopaminergic system, formation of cytoplasmic inclusions in nigral neurons, and induction of hypokinesia and rigidity in rats, reproducing the key features of human PD. The selective toxicity for dopaminergic neurons in the SN bestowed on rotenone has been attributed to its inhibition of the activity of complex I of the mitochondrial respiratory chain and the unique vulnerability of dopaminergic neurons to oxidative damage as a result of mitochondrial complex I inhibition (Greenamyre et al., 1999; Jenner, 2001).
Microglia are the resident immune cells in the brain (Kreutzberg, 1996;Gonzalez-Scarano and Baltuch, 1999). Under normal conditions the microglia play a role in immune surveillance. Microglial activation has been associated with neurodegeneration via the production of a variety of proinflammatory and neurotoxic factors, including tumor necrosis factor-α (TNFα), interleukin-1β (IL-1β), eicosanoids, nitric oxide (NO), and superoxide (Chao et al., 1992; Cassarino et al., 1997;Liu et al., 2000, 2001a; McGuire et al., 2001). Of the various factors released by activated microglia, reactive oxygen species such as superoxide free radical appear to play a key role in the inflammation-mediated oxidative damage to neurons. One of the major sources of superoxide is the activity-dependent, multi-component, and plasma membrane-associated NADPH oxidase (Babior, 1999). Because the midbrain area that encompasses the SN has the highest density of microglia in the brain (Lawson et al., 1990; Kim et al., 2000), microglial activation may be an early event contributing to the degeneration of SN dopaminergic neurons as a consequence of a variety of oxidative insults. However, the role of microglia in rotenone-induced neurotoxicity has not been reported.
Using mesencephalic cultures, we report in this study that rotenone, at concentrations that were nontoxic to dopaminergic neurons in neuron-enriched cultures, induced selective degeneration of dopaminergic neurons in neuron/glia cultures. The rotenone-induced dopaminergic neurodegeneration was at least partially mediated by the activation of microglia and the NADPH oxidase-mediated release of superoxide free radical.
MATERIALS AND METHODS
Materials. Cell culture ingredients were obtained from Invitrogen (San Diego, CA). [3H]dopamine (DA; 30 Ci/mmol) and GABA (90 Ci/mmol) were from NEN (Boston, MA). Monoclonal antibodies against the CR3 complement receptor (OX-42) and microtubule-associated protein-2 (MAP-2) were from BD PharMingen (San Diego, CA) and Roche (Indianapolis, IN), respectively. The polyclonal anti-glial fibrillary acidic protein (GFAP) antiserum and antibody diluent were from Dako (Carpinteria, CA). The polyclonal anti-tyrosine hydroxylase (TH) antiserum was a gift from GlaxoWellcome (Research Triangle Park, NC). The monoclonal anti-neuron-specific nuclear protein (Neu-N) antibody was from Chemicon (Temecula, CA). The ED1 antibody and biotinylated isolectin were from Serotec (Raleigh, NC) and Sigma (St. Louis, MO), respectively. The Vectastain avidin–biotin complex (ABC) kit and biotinylated secondary antibodies were from Vector Laboratories (Burlingame, CA). Dextran and Ficoll-Hypaque were from Amersham Pharmacia (Piscataway, NJ). Apocynin (Fluka, Milwaukee, WI) and diphenylene iodonium (DPI; Molecular Probes, Eugene, OR) were dissolved in ethanol (200 mm) or dimethylsulfoxide (20 mm) as stock solutions, respectively. Rotenone was purchased from Calbiochem (San Diego, CA). For each experiment a fresh stock solution of rotenone (10 mm) was prepared in dimethylsulfoxide, which was diluted further to desired final concentrations in treatment medium before use. All other reagents were from Sigma.
Mesencephalic mixed neuron/glia cultures. Primary rat ventral mesencephalic neuron/glia cultures were prepared by following our previously published protocol with modifications (Liu et al., 2000). Briefly, ventral mesencephalic tissues were dissected from embryonic day 13/14 Fischer 344 rats and dissociated with a mild mechanical trituration. Cells were seeded to 24-well (5 × 105/well) culture plates precoated with poly-d-lysine (20 μg/ml) and maintained in 0.5 ml/well of MEM supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 10% heat-inactivated horse serum (HS), 1 gm/l glucose, 2 mml-glutamine, 1 mm sodium pyruvate, 100 μm nonessential amino acids, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2/95% air. Cultures were replenished with 0.5 ml/well fresh medium 3 d later and were used for treatment 7 d later. For treatment the cultures were maintained in 1 ml/well of MEM containing 2% FBS, 2% HS, 2 mml-glutamine, 1 mm sodium pyruvate, 50 U/ml penicillin, and 50 μg/ml streptomycin. Immunocytochemical analysis indicated that, at the time of treatment, the cultures were made up of ∼12% OX-42-immunoreactive (IR) microglia, 48% GFAP-IR astrocytes, and 40% Neu-N-IR neurons of which 2.8–3.8% were TH-IR neurons.
Mesencephalic neuron-enriched cultures. Dissociated rat ventral mesencephalic cells were seeded first at 5 × 105 cells/well into poly-d-lysine-coated 24-well culture plates as described above. Then 2 d after initial seeding, cytosine β-d-arabinofuranoside (5–10 μm) was added to suppress the proliferation of glial cells. At 2–3 d later the cultures were changed back to fresh medium. Those 7-d-old cultures that contained <0.1% OX-42-IR microglia and 8% GFAP-IR astrocytes were used for treatment. Of the Neu-N-IR neurons, 2.7–3.9% were TH-IR neurons. Alternatively, cells were plated at 5 × 105 cells/well into 24-well culture plates and maintained in DMEM/F12 supplemented with 10% FBS, 6 gm/l glucose, 1× B27, 50 U/ml penicillin, and 50 μg/ml streptomycin (Lotharius et al., 1999). Then 2 d later the cultures were changed to serum-free Neurobasal medium containing 1× B27, 0.5 mml-glutamine, 0.01 μg/ml streptomycin, and 100 U/ml penicillin. At 7 d after the initial plating the cultures consisted of 3% GFAP-IR astrocytes, <0.1% OX-42-IR microglia, and 96% Neu-N-IR neurons of which 2.9–3.8% were TH-IR neurons. Initial comparison of the two aforementioned neuron-enriched cultures for response to rotenone-induced neurotoxicity yielded identical results. Therefore, the neuron-enriched cultures prepared with the use of cytosine β-d-arabinofuranoside were used in subsequent experiments.
Microglia-enriched cultures. Microglia were prepared from whole brains of 1-d-old Fischer 344 rats as described previously (Liu et al., 2001b). Briefly, brain tissues, devoid of meninges and blood vessels, were dissociated by a mild mechanical trituration. The isolated cells (5 × 107) were seeded in 150 cm2 culture flasks in DMEM/F12 containing 10% FBS, 2 mml-glutamine, 1 mm sodium pyruvate, 100 μm nonessential amino acids, 50 U/ml penicillin, and 50 μg/ml streptomycin. The cultures were maintained at 37°C in a humidified atmosphere of 5% CO2/95% air, and the medium was changed 4 d later. On reaching confluence (12–14 d), the microglia were separated from astrocytes by shaking the flasks for 5 hr at 180 rpm. The enriched microglia were >98% pure as determined by OX-42-IR and GFAP-IR.
Cell lines. The human monocytic U937 cells were obtained from American Type Culture Collection (Manassas, VA) and maintained at 37°C in RPMI 1640 supplemented with 10% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin in a humidified incubator with 5% CO2/95% air.
Preparation of rat neutrophils. Fresh blood was collected from the abdominal aorta of adult Fischer 344 male rats, and neutrophils were purified by following the modified protocol of Wang et al. (1995). Briefly, heparinized blood was sedimented with dextran, and the neutrophils were isolated by centrifugation through the Ficoll-Hypaque density gradient, followed by a hypotonic lysis to remove residual erythrocytes. Purified neutrophils (>95% viable cells, trypan blue exclusion) were resuspended in phenol red-free HBSS and kept on ice until use.
Immunocytochemistry. Immunostaining was performed essentially as described previously (Kim et al., 2000; Liu et al., 2000). Neurons were stained with the anti-MAP-2 antibody to detect both perikarya and neurites or the anti-Neu-N antibody to detect perikarya only. Dopaminergic neurons were detected with the anti-TH antibody. Astrocytes were recognized with the anti-GFAP antibody. Microglia were detected with an anti-complement type 3 receptor antibody (OX-42), an anti-macrophage-specific glycoprotein antibody (ED1), or the biotinylated isolectin. Briefly, after blocking, formaldehyde-fixed cells were incubated overnight at 4°C with primary antibodies diluted in antibody diluent (anti-MAP-2, 1:400; anti-Neu-N, 1:2000; anti-TH, 1:20,000; OX-42, 5 μg/ml; ED-1, 1:250; biotinylated isolectin, 13 μg/ml; or anti-GFAP, 1:1000). Except for biotinylated biotin, the bound primary antibody was visualized by incubation with an appropriate biotinylated secondary antibody, followed by the Vectastain ABC reagents and color development with 3,3′-diaminobenzidine. For double immunostaining, cultures were first stained with the anti-TH antibody and then with the anti-neu-N antibody followed by intensification using nickel sulfate. Images were recorded with a CCD camera and the MetaMorph software (Universal Imaging Systems, West Chester, PA). For visual enumeration of the immunostained cells in cultures, 10 representative areas per well were counted. For dopaminergic neurons the overall dendrite length for individual TH-IR neurons was measured by following our previously published protocol (Liu et al., 2001c). Two to four wells of identical treatment from each experiment and 50 TH-IR neurons per well were selected for measurement. Results were obtained from two separate experiments and were expressed as a percentage of the control cultures.
Uptake assays for tritiated DA or GABA. Cultures were washed two times with Krebs–Ringer buffer [containing (in mm) 16 sodium phosphate, 119 NaCl, 4.7 KCl, 1.8 CaCl2, 1.2 MgSO4, 1.3 EDTA, and 5.6 glucose; pH 7.4]. For DA and GABA uptake the cultures were incubated for 15 min at 37°C with 1 μm [3H]DA and 5 μm [3H]GABA in Krebs–Ringer buffer, respectively. After being washed (three times) with ice-cold Krebs–Ringer buffer, the cells were collected in 1N NaOH, and radioactivity was counted with a liquid scintillation counter (Liu et al., 2000). Nonspecific uptake was determined in parallel wells that received both the tritiated tracer and 10 μmmazindol for DA uptake or 10 μm NO-711 for GABA uptake (Suzdak et al., 1992). In addition, for GABA uptake, comparable results were obtained with the inclusion of β-alanine (1 mm), which preferentially affects glial GABA uptake (Mabjeesh et al., 1992).
Nitrite, TNFα, and IL-1β assays. The production of NO was determined by measuring the accumulated level of its stable metabolite, nitrite, in the supernatant with the Griess reagent (Green et al., 1982). The assay had a detection limit of ∼0.5 μm. TNFα or IL-1β released into the culture medium was measured with respective rat enzyme-linked immunosorbent assay kits from R&D Systems (Minneapolis, MN), with detection limits of 5 pg/ml, as described previously (Liu et al., 2000).
Measurement of superoxide production. The release of superoxide was determined by measuring the superoxide dismutase (SOD)-inhibitable reduction of cytochrome c as described previously (Chao et al., 1992; Liu et al., 2000). Primary microglia (0.5–1 × 105/well) were grown overnight in 96-well plates in DMEM containing 10% FBS. For superoxide assay the cultures were washed twice with HBSS and then maintained in 100 μl/well of phenol red-free HBSS. U937 monocytes or neutrophils were washed twice with HBSS and seeded (0.5–1 × 105/well) in 100 μl/well in 96-well plates. Added to each well was 50 μl of HBSS containing desired concentrations of drugs and with or without 800 U/ml SOD, immediately followed by 50 μl of 160 μm ferricytochromec in HBSS. The cultures were incubated for 30 min at 37°C; the absorbance at 550 nm was read with a SpectraMax Plus microplate spectrophotometer equipped with a solid-state heated (37°C) chamber (Molecular Devices, Sunnyvale, CA). To determine the effect of NADPH oxidase inhibitors on superoxide release, we preincubated the cells for 5 min with the vehicle or the inhibitors before the addition of rotenone. The amount of SOD-inhibitable superoxide was calculated by using a molar extinction coefficient of 2.11 × 104/m/cm for cytochrome c at 550 nm (Massy, 1959).
Statistical analysis. Statistical significance was assessed with an ANOVA, followed by the Bonferroni's t test via the StatView program (Abacus Concepts, Berkeley, CA). A value ofp < 0.05 was considered statistically significant.
RESULTS
The presence of glia dramatically increases the sensitivity of dopaminergic neurons to rotenone-induced neurodegeneration
Primary mixed neuron/glia or neuron-enriched cultures were treated for 8 d with the vehicle control (control group) or 0.5–30 nm rotenone. [3H]DA uptake was measured to assess neurotoxicity to dopaminergic neurons. The treatment of neuron-enriched cultures with up to 20 nmrotenone did not have any significant effect on [3H]DA uptake (Fig.1A). Treatment of neuron-enriched cultures with higher concentrations of rotenone resulted in a sudden drop in [3H]DA uptake, and a near-complete loss of DA uptake was seen in cultures treated with 30 nm rotenone (Fig.1A). In contrast, in mixed neuron/glia cultures a significant reduction in [3H]DA uptake was observed after treatment with 1 nm rotenone (Fig. 1A). The decrease in DA uptake became more prominent with increasing concentrations of rotenone, and a 50% reduction was seen in cultures treated with 10 nmrotenone (Fig. 1A). Counts of TH-IR neurons indicated that no significant reduction in the number of TH-IR neurons was observed in neuron-enriched cultures after treatment for 8 d with up to 10 nm rotenone. However, a concentration-dependent decrease in the number of TH-IR neurons was seen in neuron/glia cultures, with a 50% decrease in the cultures treated for 8 d with 10 nm rotenone (Fig.1B). Analysis of the time dependence of rotenone-induced degeneration of dopaminergic neurons in neuron/glia cultures treated with 1–20 nm rotenone for 2–8 d demonstrated a time-dependent neurodegeneration (Fig. 1C). Although the reduction in DA uptake in cultures treated with 1 nm rotenone was only significant 8 d later, a significant reduction was seen in cultures treated with ≥20 nm rotenone as early as 2 d after treatment (Fig. 1C). The rapid onset of the rotenone-induced decrease in DA uptake at the higher concentrations (20 and 25 nm) was likely attributable to its direct neurotoxicity as a consequence of the inhibition of mitochondrial function.
Fig. 1.
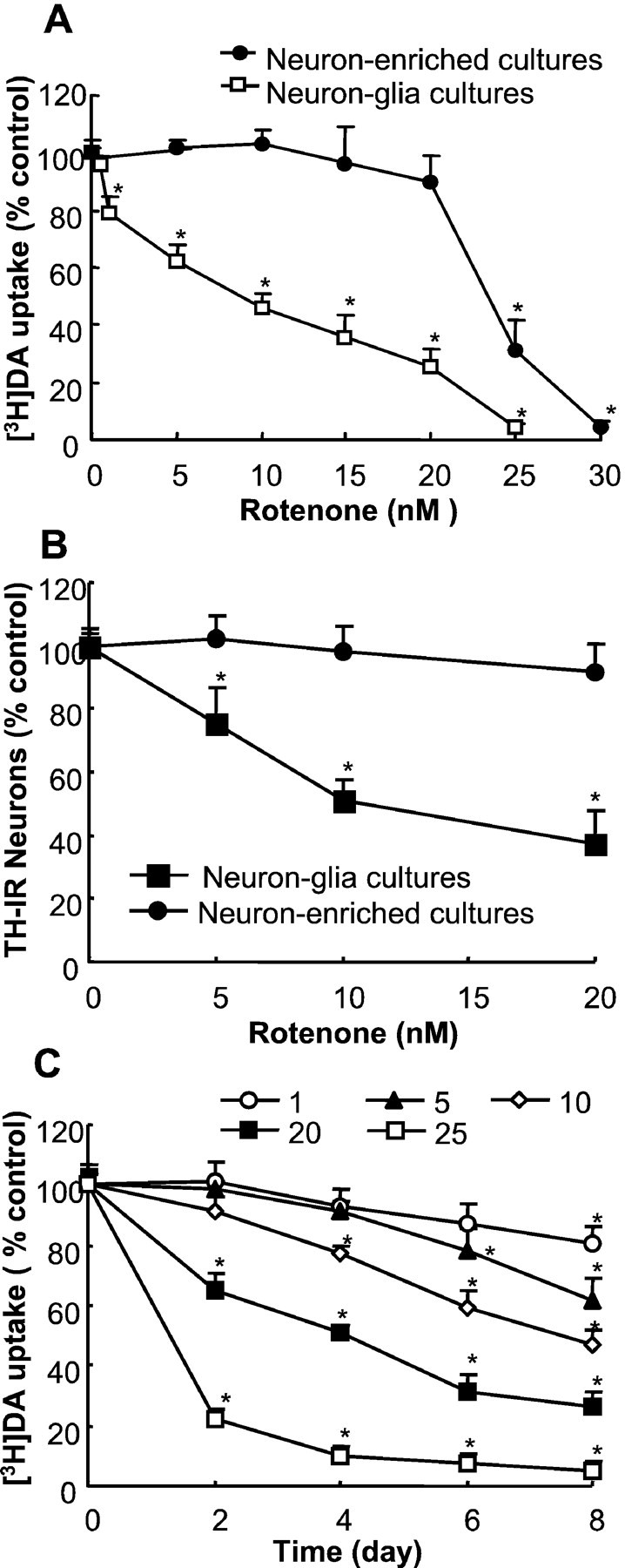
Differential sensitivity of dopaminergic neurons in neuron-enriched and mixed neuron/glia mesencephalic cultures to rotenone-induced degeneration. A, B, Primary neuron-enriched or neuron/glia cultures were treated for 8 d with the vehicle (control group) or 1–30 nm rotenone; degeneration of dopaminergic neurons was evaluated with the [3H]DA uptake assay (A) or quantification of TH-IR neurons in the cultures (B). C, Mixed neuron/glia cultures were treated for 2–8 d with the vehicle or 1–25 nmrotenone; the [3H]DA uptake was determined at the desired time points. The amount of DA uptake for the vehicle-treated neuron/glia cultures at the time of treatment and 8 d after treatment was 0.77 ± 0.04 and 0.65 ± 0.06 and for vehicle-treated neuron-enriched cultures was 0.81 ± 0.07 and 0.77 ± 0.08 pmol/min per well, respectively. The results for DA uptake (A, C) and cell counts (B) are expressed as a percentage of the control cultures and are the mean ± SEM of four experiments performed in triplicate. *p < 0.01, compared with the control.
Glia-enhanced neurodegeneration is selective for dopaminergic neurons
One of the hallmarks of Parkinson's disease is the selective loss of dopaminergic neurons in the SN. Therefore, it is of particular interest to determine whether the glia-enhanced neurodegeneration was selective for dopaminergic neurons among the various neuronal populations in the mixed neuron/glia culture. First, the cultures were treated with 1–25 nm rotenone for 8 d and then analyzed for the uptake of [3H]DA or [3H]GABA. As shown in Figure2A, although rotenone decreased [3H]DA uptake in a dose-dependent manner, [3H]GABA uptake in the cultures treated with up to 20 nm rotenone was not affected significantly. The difference in neurotransmitter uptake was reduced markedly in cultures treated with 25 nm rotenone (Fig. 2A). Consistent with the effect observed with the uptake assays, a quantification of immunostained neurons demonstrated that rotenone dose-dependently reduced the number of TH-IR neurons that accounted for ∼3.3% of the total neuronal population (Fig. 2B). In contrast, no significant reduction in Neu-N-IR or MAP-2-IR neurons, which represented the majority if not all of the neurons, was observed in cultures treated with 20 nm rotenone for 8 d (Fig. 2B). Again, a significant loss in Neu-N-IR or MAP-2-IR neurons was seen in cultures treated with 25 nm rotenone for 8 d (Fig.2B). In addition to a reduction in cell numbers, TH-IR neurons in rotenone-treated cultures displayed a less extensive dendritic network compared with those in control cultures, whereas the dendritic network of MAP-2-IR neurons, in general, was not affected significantly (Fig. 2C). Quantitation of the overall dendrite length of TH-IR neurons in the neuron/glia cultures demonstrated that, after treatment for 8 d with 10 and 20 nm rotenone, the average dendrite length reduced to 56.2 ± 6.6 and 30.0 ± 4.5% of that of the control cultures, respectively (p < 0.01 compared with control). In addition, double-label immunostaining for TH-IR and Neu-N-IR neurons further demonstrated the selective destruction of dopaminergic neurons in rotenone-treated neuron/glia cultures (Fig.2C).
Fig. 2.
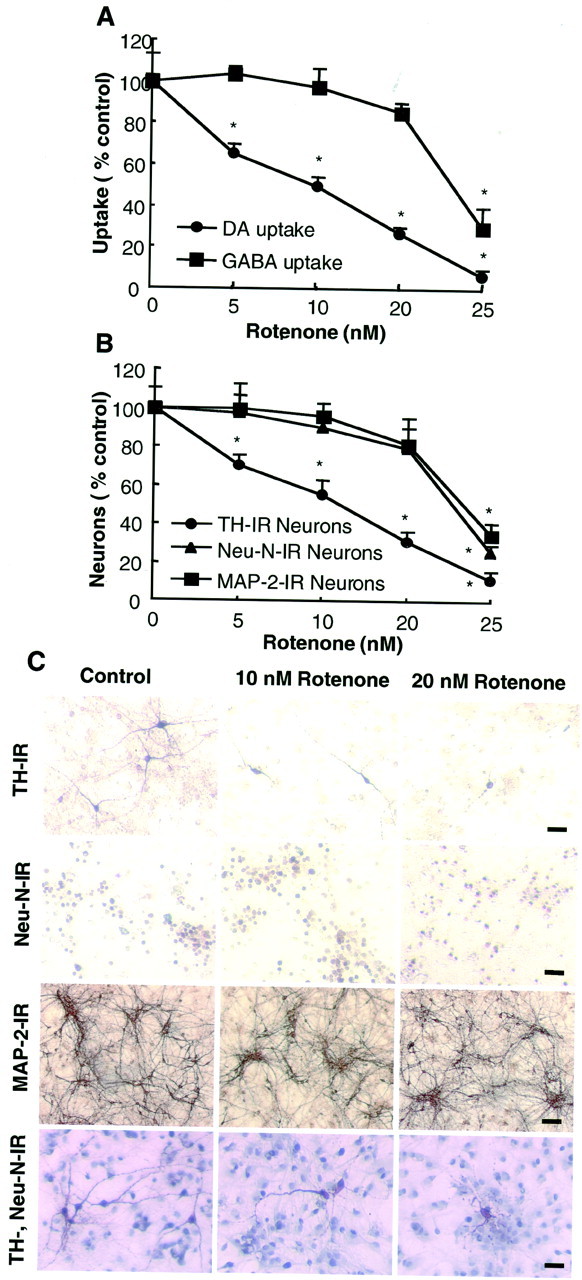
Selectivity of rotenone-induced neurodegeneration in mixed neuron/glia cultures. A, B, Mixed neuron/glia cultures were treated for 8 d with the vehicle or 5–25 nm rotenone. Afterward the cultures were assayed for uptake of [3H]DA or [3H]GABA (A) or were immunostained with anti-TH, Neu-N, or MAP-2 antibodies, followed by quantification of the positively stained cells (B). The amount of GABA uptake at the time of treatment and 8 d after treatment for the vehicle-treated neuron/glia cultures was 39.4 ± 4.2 and 35.2 ± 3.2 pmol/min per well, respectively. The number of Neu-N-IR, MAP-2-IR, and TH-IR neurons in the control cultures was ∼490, 460, and 14/mm2. The results are expressed as a percentage of the control cultures and are the mean ± SEM of three experiments performed in triplicate. *p < 0.01, compared with the control. C, Immunocytochemical analysis. Mixed neuron/glia cultures were treated for 8 d with the vehicle or 10 or 20 nm rotenone. Cultures then were immunostained for TH-IR, Neu-N-IR, or MAP-2-IR neurons or were double stained for TH-IR and Neu-N-IR neurons. Scale bars: TH-IR and Neu-N-IR neurons, 50 μm; MAP-IR neurons, 100 μm; neurons in the TH and Neu-N double-immunostained cultures, 75 μm.
The addition of microglia to neuron-enriched cultures significantly increases the sensitivity of dopaminergic neurons to rotenone-induced neurotoxicity
The vast difference in sensitivity of dopaminergic neurons to rotenone neurotoxicity between the mixed neuron/glia and neuron-enriched cultures prompted us to speculate that the neurotoxicity of rotenone was dependent on the presence of glia, especially that of microglia. To test this hypothesis directly, we added cells from primary microglia-enriched cultures to mesencephalic neuron-enriched cultures; DA uptake of neuronal cultures and microglia-supplemented neuronal cultures was compared after treatment with 10 nm rotenone for 8 d. As shown in Figure3, whereas the DA uptake of the neuron-enriched cultures was not affected by the addition of microglia (2.5–7.5 × 104/well), the addition of microglia significantly increased the sensitivity of dopaminergic neurons to rotenone-induced neurotoxicity. The increase in sensitivity to rotenone neurotoxicity positively correlated with the number of microglia added to the neuron-enriched cultures (Fig. 3). For example, the addition of 5 × 104microglia/well to neuron-enriched cultures, a level comparable with that observed in mixed neuron/glia cultures, reduced their DA uptake to 46% of the control cultures (Fig. 3), close to the degree of reduction observed in mixed neuron/glia cultures treated with 10 nmrotenone for 8 d (Fig. 1A). In contrast, after treatment for 8 d with 10 nm rotenone, no significant difference was detected in GABA uptake between the neuron-enriched cultures (96 ± 2% of control) and those with 7.5 × 104 microglia added per well (93% ± 4% of control, from two experiments performed in triplicate). These data further strengthen the notion that microglia play a pivotal role in dictating the sensitivity and selectivity of dopaminergic neurons to rotenone-induced degeneration.
Fig. 3.

Effect of the addition of microglia to neuron-enriched cultures on rotenone-induced degeneration of dopaminergic neurons. Neuron-enriched mesencephalic cultures were supplemented with 2.5–7.5 × 104microglia/well. At 24 hr later the cultures were treated with the vehicle or 10 nm rotenone, and DA uptake was determined 8 d after the treatment. The results are expressed as a percentage of the control cultures and are the mean ± SEM of three experiments performed in triplicate. *p < 0.01, compared with the control.
Rotenone-induced microglial activation precedes neurodegeneration
To study further the role of microglia in the rotenone-induced preferential and enhanced degeneration of dopaminergic neurons, we first examined whether rotenone could activate microglia in the mixed neuron/glia cultures. Microglial activation is characterized by dramatic changes in morphology and increased expression of surface antigens such as the complement type 3 receptor (Kreutzberg, 1996). Cultures were treated for 1 d with 10 nm rotenone, and microglia were detected with the antibody OX-42 that recognizes the complement receptor. Compared with control cultures, a significant portion of the OX-IR microglia in rotenone-treated cultures was larger in size than those in control cultures (Fig.4A). More prominently, the number of immunoreactive microglia in the rotenone-treated (1–10 nm) cultures increased significantly over that in the control cultures (Fig. 4B). A significant increase in OX-IR cells was seen in cultures that were treated for 1 d with 1 nm rotenone, and a 67% increase over the control cultures was observed with 10 nmrotenone (Fig. 4B). Similar results were obtained when the cultures were immunostained for other specific markers of microglia with isolectin and the antibody ED-1 (Fig.4B). These results demonstrated that rotenone was capable of inducing microglial activation, which preceded the degeneration of dopaminergic neurons in the mixed neuron/glia cultures.
Fig. 4.
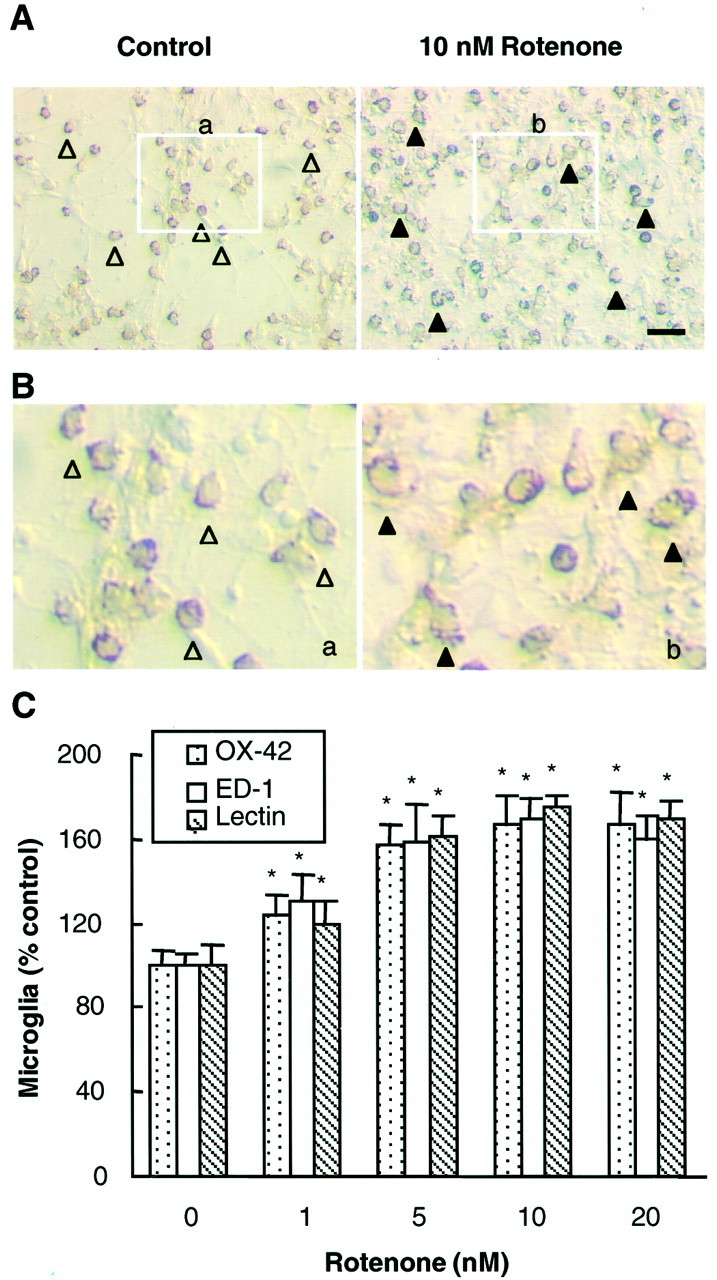
Effect of rotenone on microglial activation. Mixed neuron/glia cultures were treated for 1 d with the vehicle or 1–10 nm rotenone and then were immunostained with antibodies against specific markers of microglia. A, Immunocytochemical analysis with the OX-42 antibody of cultures treated for 1 d with the vehicle or 10 nm rotenone. In control cultures a significant portion of the OX-IR resting microglia was small and round (open arrowheads). After treatment with 10 nm rotenone, OX-42-IR microglia were enlarged significantly and were irregularly shaped (filled arrowheads), indicative of activation. Part of the images of OX-42-IR microglia (inclosed boxes, a and b) presented in A was cut out and presented inB for better representation of the differences.C, Quantification of microglial activation. Vehicle or rotenone-treated cultures were immunostained with OX-42, ED-1, or isolectin; positively stained microglia were counted. The number of OX-42-IR microglia in the control cultures was ∼120/mm2. The results are expressed as a percentage of the control cultures and are the mean ± SEM of three experiments performed in triplicate. *p< 0.01, compared with the control. Scale bar, 50 μm.
Rotenone induces NADPH oxidase-mediated release of superoxide from activated microglia
Activated microglia secrete a variety of proinflammatory and cytotoxic factors such as the cytokines TNFα and IL-1β and free radical NO and the reactive oxygen species (ROS), which work in concert to induce neurodegeneration. We first measured the accumulation of nitrite (an indicator of NO production), TNFα, or IL-1β in media from neuron/glia cultures after treatment with the vehicle or 1–20 nm rotenone for 1–8 d. However, the quantities of these factors in either vehicle- or rotenone-treated cultures were at or below the detection limits for the assays that were used (TNFα, 5 pg/ml; IL-1β, 5 pg/ml; NO, 0.5 μm).
Next, we examined the release of superoxide free radical from microglia by measuring the SOD-inhibitable reduction of cytochrome c. After incubation of microglia for 30 min with vehicle or 1–10 nm rotenone, a dose-dependent release of superoxide was observed (Fig.5A). Significant release of superoxide was detected in microglia treated with 5 or 10 nm rotenone. Similar profiles for superoxide release were observed in the human monocytic U937 cells that are of close lineage to macrophage and microglia (Fig. 5A). Because the main source of superoxide released is dependent on the activity of the membrane-associated NADPH oxidase that is expressed most abundantly in neutrophils (Griendling et al., 2000), we next tested the effect of rotenone on the release of superoxide from rat neutrophils. Indeed, significant release of superoxide was detected in neutrophils after treatment with concentrations of rotenone as low as 1 nm (Fig. 5A). The involvement of the activity of NADPH oxidase in rotenone-induced release of superoxide was determined by examining the effects of two mechanistically dissimilar inhibitors of NADPH oxidase, DPI and apocynin, with the former directly inhibiting the catalytic activity of the enzyme (Irani et al., 1997) and the latter preventing the assembly of the multi-subunit enzyme complex (Stolk et al., 1994). As shown in Figure 5B, both DPI (2.5–5 μm) and apocynin (0.25–0.5 mm) significantly inhibited the rotenone-induced release of superoxide from neutrophils. These results demonstrated that the rotenone-induced release of superoxide free radical involved the activity of NADPH oxidase.
Fig. 5.
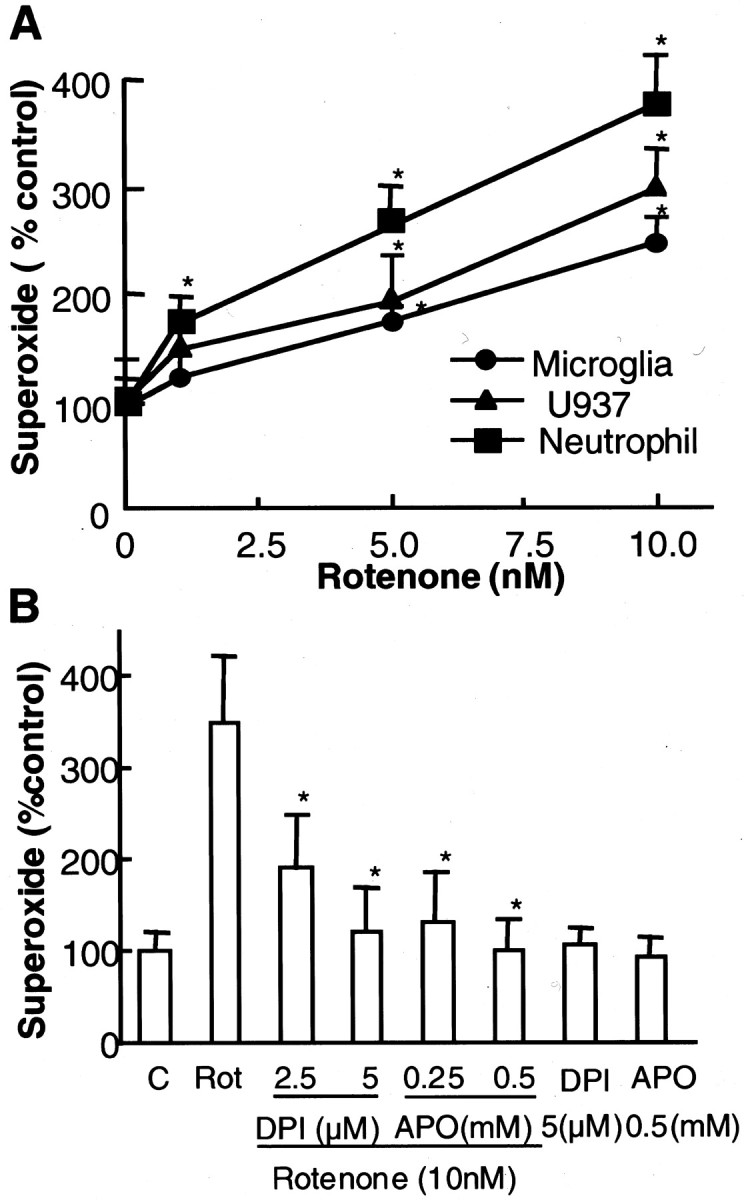
Rotenone stimulated the release of superoxide.A, Rat primary microglia, human monocytic U937 cells, or rat neutrophils were seeded to 96-well plates. Cells were treated with the vehicle or 1–10 nm rotenone. Superoxide production, measured as SOD-inhibitable cytochrome c reduction, was determined as described in Materials and Methods. The results are a percentage of the control cultures and are expressed as the mean ± SEM of three experiments performed in triplicate. *p < 0.01, compared with the control.B, Effect of NADPH oxidase inhibitors on rotenone-induced release of superoxide from neutrophils. Neutrophils were pretreated for 5 min with the vehicle or indicated concentrations of DPI or apocynin before treatment with 10 nm rotenone. Then the amount of superoxide released was determined as described in Materials and Methods. The results are a percentage of the control cultures and are expressed as the mean ± SEM of three experiments performed in triplicate. *p < 0.01, compared with rotenone-treated cells. C, Control; Rot, rotenone; APO, apocynin.
The relevance of rotenone-stimulated superoxide release in mediating microglia-induced facilitation of dopaminergic neurodegeneration was established by the observation that neutralization of the reactivity of superoxide or inhibition of NADPH oxidase afforded neuroprotection. As shown in Figure6, the addition of a combination of SOD (50 U/ml) and catalase (50 U/ml) to the neuron/glia cultures right before and 24 hr after the addition of rotenone significantly reduced rotenone-induced damage to dopaminergic neurons. Neutralization of superoxide by SOD/catalase increased the DA uptake of cultures treated with 5 and 10 nm rotenone from 76 and 53% of the control cultures to 93 and 74% of the control cultures, respectively (Fig. 6). Pretreatment of neuron/glia cultures with 1 mmNG-nitro-l-arginine methyl ester, an inhibitor of nitric oxide synthase, did not afford any significant neuroprotection (data not shown). However, pretreatment of neuron/glia cultures with an inhibitor of NADPH oxidase, apocynin, before treatment with rotenone significantly attenuated the rotenone-induced degeneration of dopaminergic neurons measured either by [3H]DA uptake (Fig.7A) or counts of TH-IR neurons (Fig. 7B). The neuroprotective effect of apocynin was concentration-dependent over a range of 0.1–0.5 mm (Fig. 7C). Unexpectedly, treatment of cultures for 8 d with DPI (0.1–5 μm) resulted in significant damage to neurons in the culture (data not shown). Despite this, the results obtained with SOD/catalase and apocynin strongly suggested that NADPH oxidase-mediated production of superoxide in microglia underlies microglial facilitation of the rotenone-induced dopaminergic neurodegeneration.
Fig. 6.

Effect of SOD/catalase on rotenone-induced degeneration of dopaminergic neurons. SOD/catalase (50 U/ml each) were added to neuron/glia cultures right before and 24 hr after the addition of the indicated concentrations of rotenone. The cultures were assayed for [3H]DA uptake 8 d later. The results are the mean ± SEM of three experiments performed in triplicate. *p < 0.01, compared with the control.
Fig. 7.
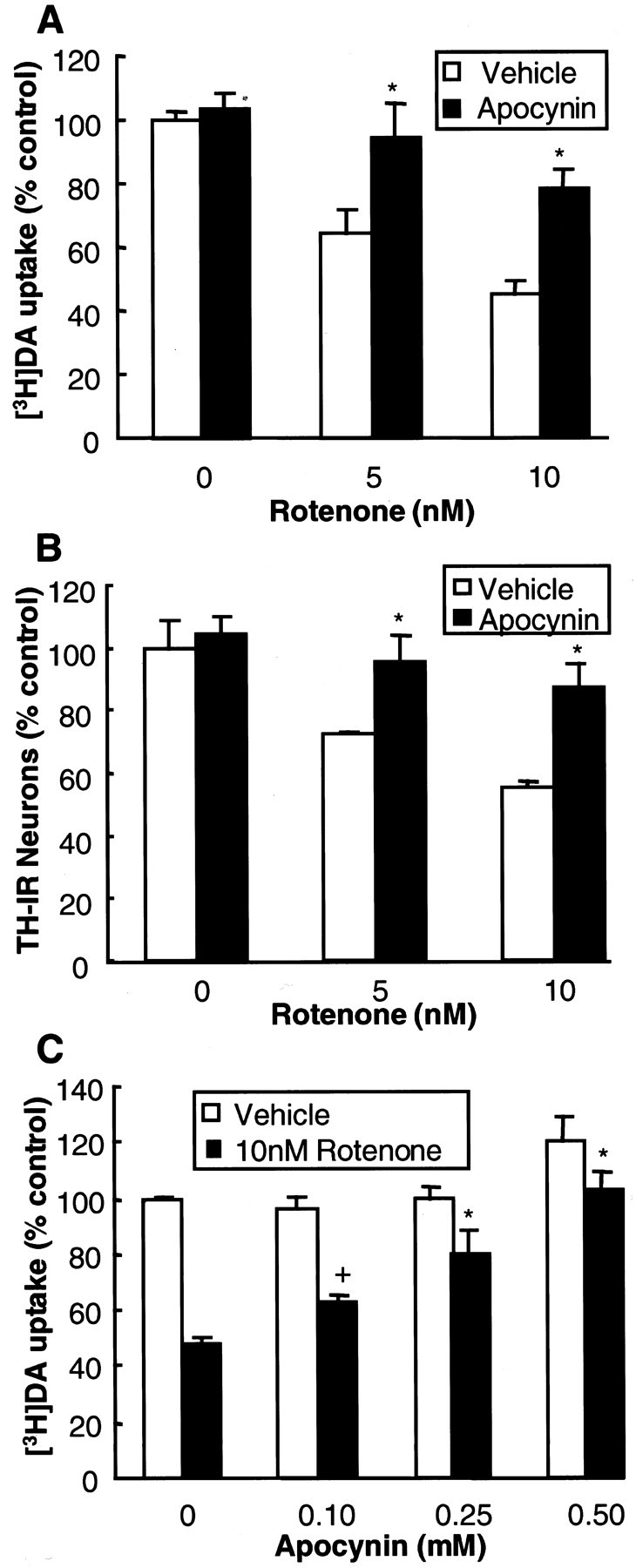
Effect of apocynin on rotenone-induced degeneration of dopaminergic neurons. A, B, Neuron/glia cultures were pretreated for 30 min with the vehicle or 0.25 mm apocynin before treatment with 5 or 10 nmrotenone. Then 8 d later the degeneration of dopaminergic neurons was assessed as [3H]DA uptake (A) and counts of TH-IR neurons (B). C, Neuron/glia cultures were pretreated for 30 min with the vehicle or the indicated concentration of apocynin before treatment with 10 nm rotenone. The cultures were assayed for [3H]DA uptake 8 d later. The results are the mean ± SEM of three experiments performed in triplicate. *p < 0.01 and+p < 0.05, compared with the control.
DISCUSSION
Using primary rat mesencephalic mixed neuron/glia and neuron-enriched cultures, this study for the first time demonstrates that microglia are actively involved in the rotenone-induced selective degeneration of dopaminergic neurons. At concentrations (1–15 nm) that are nontoxic to dopaminergic neurons in the neuron-enriched cultures, the presence of microglia in the mixed neuron/glia cultures enables rotenone to induce significant and selective degeneration of dopaminergic neurons. The dramatically enhanced dopaminergic neurodegeneration is primarily attributable to the rotenone-induced activation of microglia and their release of superoxide free radical as demonstrated by the measured SOD-inhibitable reduction of cytochrome c, the neuroprotective effect of SOD/catalase, and that of the NADPH oxidase inhibitor apocynin.
Microglia, the resident immune cells in the brain, play a pivotal role in the detection of invading pathogens, host defense, and tissue repair (Kreutzberg, 1996; Aloisi, 1999). In response to injury or immunological challenges, microglia become readily activated and undergo dramatic changes in morphology, surface expression of molecules such as major histocompatibility complex, and the complement type 3 receptor, recognizable by the antibody OX-42 (Streit et al., 1988). Furthermore, activated microglia produce a wide array of immunomodulatory and cytotoxic factors, including TNFα, IL-1β, eicosanoids, NO, and superoxide free radical. Although a few of these factors are thought to contribute to tissue repair (Streit et al., 1988), a majority are believed to work via mechanisms not yet fully understood to induce neurodegeneration (McGeer et al., 1988; Chao et al., 1992; Jeohn et al., 1998; Minghetti and Levi, 1998; Liu et al., 2001a).
Numerous studies have suggested that, among the factors produced by activated microglia, oxygen free radicals play a prominent role in the degeneration of dopaminergic neurons. For example, stimulation of mesencephalic neuron/glia cultures with the bacterial endotoxin activated microglia to produce TNFα, IL-1β, NO, and superoxide (Kim et al., 2000; Liu et al., 2000). However, inhibition of microglial production of superoxide, but not the other factors, was most effective in protecting neurons, indicating that superoxide was a dominant degenerative factor for the dopaminergic neurons in the culture (Liu et al., 2000). In the current study the rotenone-induced degeneration of dopaminergic neurons in neuron/glia cultures may, at least in part, be attributable to the activation of microglia and NADPH oxidase-mediated release of superoxide free radical. This notion is supported by the following observations: (1) significant accumulation of NO was not observed, and inhibition of NO production did not afford neuroprotection; (2) although their participation in neurotoxicity could not be ruled out completely, the accumulated levels of IL-1β or TNFα were very low, and studies have shown that sufficient quantities of these factors, possibly even in combination with other factors (e.g., NO), are necessary to induce significant neurotoxicity (Jeohn et al., 1998); (3) significant release of superoxide, as measured by the SOD-inhibitable reduction of cytochrome c, was detected in rotenone-stimulated microglia as well as monocytes and neutrophils (Fig. 5A); (4) the rotenone-induced release of superoxide was sensitive to inhibitors of NADPH oxidase DPI and apocynin (Fig.5B); and (5) significant protection of dopaminergic neurons in the mixed neuron/glia cultures against rotenone-induced degeneration was achieved by the superoxide scavengers SOD and catalase and the NADPH oxidase inhibitor apocynin (Figs. 6, 7). NADPH oxidase is expressed widely in cells of the mesodermal lineage, including leukocytes and vascular cells (Babior, 1999). It is made up of at least seven subunits that are distributed between the cytosol and plasma membranes. In response to stimuli the cytosolic components, especially the p47 subunit, undergo extensive phosphorylation, and the entire group of cytosolic subunits (p40, p47, and p67) translocate to become associated with the membrane-bound heterodimeric cytochromeb558, composed of gp91 and p22. Two small G-proteins, Rac-1 and Rap1A, also are involved in the activation process of NADPH oxidase. The assembly of a fully functional NADPH oxidase catalyzes the transfer of electrons from NADPH to molecular oxygen, resulting in the generation of superoxide. It is believed that neutrophils and other immune cells generate superoxide as part of a repertoire to kill invading pathogens. In vascular and cardiac cells, NADPH oxidase-generated superoxide may be involved in cell signaling pathways (Griendling et al., 2000). Excessive and/or unintended activation of NADPH oxidase may result in tissue damage. It is, therefore, of tremendous significance to decipher the precise interaction between rotenone and members of the NADPH oxidase in relation to its stimulation of superoxide release from microglia.
It is worth noting that, although apocynin has been considered an effective inhibitor of the oxidative burst by blocking the assembly of the NADPH oxidase complex (Simons et al., 1990; Stolk et al., 1994), the neuroprotective effect of apocynin observed in this study (Fig. 7) may not be attributed exclusively to the inhibition of NADPH oxidase-mediated superoxide generation. The observation that SOD and catalase afforded only a partial neuroprotection also lends support to this notion. In other words, rotenone may be capable of inducing the release of additional neurotoxic factors for which the identities are not yet disclosed.
Oxygen free radicals are highly reactive and can interact with proteins, DNA, or RNA to alter their functions or induce lipid peroxidation, leading to eventual cell death (Facchinetti et al., 1998). Among the various populations of neurons in the brain, dopaminergic neurons in the SN are uniquely vulnerable to oxidative stress (Jenner and Olanow, 1998; Greenamyre et al., 1999). The elevated sensitivity of dopaminergic neurons to oxidative damage so far has been attributed in large part to their known reduced antioxidant capacity, increased accumulation of iron, and the concentration of neurochemicals such as dopamine that are prone to oxidative modification. However, of particular relevance to the current study are the reports that nonmitochondria-originated free radicals can, at least in vitro, impair complex I selectively and possibly trigger a self-amplifying cycle of additional free radical generation (Turrens and Boveris, 1980; Hasegawa et al., 1990). In others words the mixed neuron/glia culture system of the current study may be a reflection of the microenvironment in the brain: dopaminergic neurons may be under the dual assault of oxygen free radicals generated by rotenone-activated microglia and that generated as a consequence of the inhibition, by rotenone, of the mitochondrial complex I activity. Hence, it is plausible to speculate the existence of a potential amplification mechanism for superoxide generation in dopaminergic neurons as a consequence of the exposure of both microglia and dopaminergic neurons to rotenone. These observations, together with the fact that the nigral region has the highest concentration of microglia among the major brain regions that have been examined (Lawson et al., 1990; Kim et al., 2000), may be part of the mechanisms underlying the selective destruction of dopaminergic neurons in the SN after rotenone treatment (Betarbet et al., 2000; Foley and Riederer, 2000), although the exact mechanism remains to be understood fully.
It should be pointed out that, in this study, in neuron-enriched cultures treated with 20–30 nm rotenone a sudden drop in the viability of dopaminergic neurons was observed (Fig.1A). This reflects an important characteristic of rotenone working as a mitochondrial complex I inhibitor. Studies have shown that rotenone at between 25 and 30 nm will inhibit complex I activity significantly, which, in turn, will lead to a decrease in the mitochondrial membrane potential that is lethal to dopaminergic neurons (Greenamyre et al., 1999). Therefore, at these concentrations of rotenone, direct inhibition of neuronal mitochondrial complex I activity and the resultant decrease in mitochondrial membrane potential may have become the predominant force in determining the fate of dopaminergic neurons. However, exposure to concentrations of rotenone as low as 1 nm, which is at least 20 times lower than the critical concentration(s) needed to impair mitochondrial complex I activity directly, may be more relevant, at least in vitro, to the degeneration of dopaminergic neurons.
The present study using in vitro mesencephalic cultures provides important clues to our understanding of the rotenone-induced dopaminergic neurodegeneration. Continuing exploration of the role of microglia should further our quest to identify the environmental factors such as pesticides as potential factors involved in the pathogenesis of Parkinson's disease.
Footnotes
We thank Dr. Jerome L. Maderdrut for reading this manuscript.
Correspondence should be addressed to Bin Liu, F1-01, National Institute of Environmental Health Sciences, P.O. Box 12233, Research Triangle Park, NC 27709. E-mail: liu3@niehs.nih.gov.
REFERENCES
- 1.Aloisi F. The role of microglia and astrocytes in CNS immune surveillance and immunopathology. Adv Exp Med Biol. 1999;468:123–133. doi: 10.1007/978-1-4615-4685-6_10. [DOI] [PubMed] [Google Scholar]
- 2.Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- 3.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 4.Cassarino DS, Fall CP, Swerdlow RH, Smith TS, Halvorsen EM, Miller SW, Parks JP, Parker WD, Jr, Bennett JP., Jr Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson's disease. Biochim Biophys Acta. 1997;1362:77–86. doi: 10.1016/s0925-4439(97)00070-7. [DOI] [PubMed] [Google Scholar]
- 5.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- 6.de Silva HR, Khan NL, Wood NW. The genetics of Parkinson's disease. Curr Opin Genet Dev. 2000;10:292–298. doi: 10.1016/s0959-437x(00)00082-4. [DOI] [PubMed] [Google Scholar]
- 7.Facchinetti F, Dawson VL, Dawson TM. Free radicals as mediators of neuronal injury. Cell Mol Neurobiol. 1998;18:667–682. doi: 10.1023/a:1020685903186. [DOI] [PubMed] [Google Scholar]
- 8.Foley P, Riederer P. Influence of neurotoxins and oxidative stress on the onset and progression of Parkinson's disease. J Neurol. 2000;247[Suppl 2]:II82–II94. doi: 10.1007/pl00007766. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 10.Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Richardson RJ. The risk of Parkinson's disease with exposure to pesticides, farming, well water, and rural living. Neurology. 1998;50:1346–1350. doi: 10.1212/wnl.50.5.1346. [DOI] [PubMed] [Google Scholar]
- 11.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 12.Greenamyre JT, MacKenzie GM, Peng T-I, Stephans SE. Mitochondrial dysfunction in Parkinson's disease. Biochem Soc Symp. 1999;66:85–97. doi: 10.1042/bss0660085. [DOI] [PubMed] [Google Scholar]
- 13.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 14.Hasegawa E, Takasgige K, Oishi T, Murai Y, Minikami S. 1-Methyl-4-phenylpyridinium (MPP+) induces NADH-dependent superoxide formation and enhances NADH-dependent lipid peroxidation in bovine heart submitochondrial particles. Biochem Biophys Res Commun. 1990;170:1049–1055. doi: 10.1016/0006-291x(90)90498-c. [DOI] [PubMed] [Google Scholar]
- 15.Herishanu YO, Medvedovski M, Goldsmith JR, Kordysh E. A case-control study of Parkinson's disease in an urban population of southern Israel. Can J Neurol Sci. 2001;28:144–147. doi: 10.1017/s0317167100052835. [DOI] [PubMed] [Google Scholar]
- 16.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 17.Jenner P. Parkinson's disease, pesticides, mitochondrial dysfunction. Trends Neurosci. 2001;24:245–247. doi: 10.1016/s0166-2236(00)01789-6. [DOI] [PubMed] [Google Scholar]
- 18.Jenner P, Olanow CW. Understanding cell death in Parkinson's disease. Ann Neurol. 1998;44:S72–S84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
- 19.Jeohn GH, Kong LY, Wilson B, Hudson P, Hong JS. Synergistic neurotoxic effects of combined treatments with cytokines in murine primary mixed neuron/glia cultures. J Neuroimmunol. 1998;85:1–10. doi: 10.1016/s0165-5728(97)00204-x. [DOI] [PubMed] [Google Scholar]
- 20.Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 22.Langston JW. Epidemiology versus genetics in Parkinson's disease: progress in resolving an age-old debate. Ann Neurol. 1998;44:S45–S52. doi: 10.1002/ana.410440707. [DOI] [PubMed] [Google Scholar]
- 23.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 24.Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther. 2000;293:607–617. [PubMed] [Google Scholar]
- 25.Liu B, Gao HM, Wang J-Y, Jeohn G-H, Cooper CL, Hong JS (2001a) Role of nitric oxide in inflammation-mediated neurodegeneration. Ann NY Acad Sci, in press. [DOI] [PubMed]
- 26.Liu B, Wang K, Gao HM, Mandavilli B, Wang JY, Hong JS. Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J Neurochem. 2001b;77:182–189. doi: 10.1046/j.1471-4159.2001.t01-1-00216.x. [DOI] [PubMed] [Google Scholar]
- 27.Liu B, Qin L, Yang SN, Wilson BC, Liu Y, Hong JS. Femtomolar concentrations of dynorphins protect rat mesencephalic dopaminergic neurons against inflammatory damage. J Pharmacol Exp Ther. 2001c;298:1133–1141. [PubMed] [Google Scholar]
- 28.Lotharius J, Dugan LL, O'Malley KL. Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J Neurosci. 1999;19:1284–1993. doi: 10.1523/JNEUROSCI.19-04-01284.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mabjeesh NJ, Frese M, Rauen T, Jeserich G, Kanner BI. Neuronal and glial γ-aminobutyric acid+ transporters are distinct proteins. FEBS Lett. 1992;299:99–102. doi: 10.1016/0014-5793(92)80109-t. [DOI] [PubMed] [Google Scholar]
- 30.Massy V. The microestimation of succinate and the extinction coefficient of cytochrome c. Biochim Biophys Acta. 1959;24:255–265. doi: 10.1016/0006-3002(59)90259-8. [DOI] [PubMed] [Google Scholar]
- 31.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 32.McGuire SO, Ling ZD, Lipton JW, Sortwell CE, Collier TJ, Carvey PM. Tumor necrosis factor alpha is toxic to embryonic mesencephalic dopamine neurons. Exp Neurol. 2001;169:219–230. doi: 10.1006/exnr.2001.7688. [DOI] [PubMed] [Google Scholar]
- 33.Minghetti L, Levi G. Microglia as effector cells in brain damage and repair: focus on prostanoids and nitric oxide. Prog Neurobiol. 1998;54:99–125. doi: 10.1016/s0301-0082(97)00052-x. [DOI] [PubMed] [Google Scholar]
- 34.Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- 35.Polymeropoulos MH. Autosomal dominant Parkinson's disease. J Neurol. 1998;245[Suppl 3]:P1–P3. doi: 10.1007/pl00007740. [DOI] [PubMed] [Google Scholar]
- 36.Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS. Environmental risk factors and Parkinson's disease: a metaanalysis. Environ Res. 2001;86:122–127. doi: 10.1006/enrs.2001.4264. [DOI] [PubMed] [Google Scholar]
- 37.Ritz B, Yu F. Parkinson's disease mortality and pesticide exposure in California, 1984–1994. Int J Epidemiol. 2000;29:323–329. doi: 10.1093/ije/29.2.323. [DOI] [PubMed] [Google Scholar]
- 38.Simons JM, Hart BA, Ip Vai Ching TR, Van Dijk H, Labadie RP. Metabolic activation of natural phenols into selective oxidative burst agonists by activated human neutrophils. Free Radic Biol Med. 1990;8:251–258. doi: 10.1016/0891-5849(90)90070-y. [DOI] [PubMed] [Google Scholar]
- 39.Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- 40.Streit WJ, Graeber MB, Kreutzberg GW. Functional plasticity of microglia: a review. Glia. 1988;1:301–307. doi: 10.1002/glia.440010502. [DOI] [PubMed] [Google Scholar]
- 41.Suzdak PD, Frederiksen K, Andersen KE, Sorensen PO, Knutsen LJ, Nielsen EB. NNC-711, a novel potent and selective γ-aminobutyric acid uptake inhibitor: pharmacological characterization. Eur J Pharmacol. 1992;224:189–198. doi: 10.1016/0014-2999(92)90804-d. [DOI] [PubMed] [Google Scholar]
- 42.Thiruchelvam M, Richfield EK, Baggs RB, Tank AW, Cory-Slechta DA. The nigrostriatal dopaminergic system as a preferential target of repeated exposures to combined paraquat and maneb: implications for Parkinson's disease. J Neurosci. 2000;20:9207–9214. doi: 10.1523/JNEUROSCI.20-24-09207.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang JP, Raung SL, Kuo YH, Teng CM. Daphnoretin-induced respiratory burst in rat neutrophils is, probably, mainly through protein kinase C activation. Eur J Pharmacol. 1995;288:341–348. doi: 10.1016/0922-4106(95)90047-0. [DOI] [PubMed] [Google Scholar]