

Abstract
Afferent discharge generated ectopically in the cell soma of dorsal root ganglion (DRG) neurons may play a role in normal sensation, and it contributes to paraesthesias and pain after nerve trauma. This activity is critically dependent on subthreshold membrane potential oscillations; oscillatory sinusoids that reach threshold trigger low-frequency trains of intermittent spikes. Ectopic firing may also enter a high-frequency bursting mode, however, particularly in the event of neuropathy. Bursting greatly amplifies the overall ectopic barrage. In the present report we show that subthreshold oscillations and burst discharge occur in vivo, as they do in vitro. We then show that although the first spike in each burst is triggered by an oscillatory sinusoid, firing within bursts is maintained by brief regenerative post-spike depolarizing afterpotentials (DAPs). Numerical simulations were used to identify the cellular process underlying rebound DAPs, and hence the mechanism of the spike bursts. Finally, we show that slow ramp and hold (tonic) depolarizations of the sort that occur in DRG neurons during physiologically relevant events are capable of triggering sustained ectopic bursting, but only in cells with subthreshold oscillatory behavior. Oscillations and DAPs are an essential substrate of ectopic burst discharge. Therefore, any consideration of the ways in which cellular regulation of ion channel synthesis and trafficking implement normal sensation and, when disrupted, bring about neuropathic pain must take into account the effects of this regulation on oscillations and bursting.
Keywords: depolarizing afterpotential, dorsal root ganglion, ectopic firing, neuropathic pain, pain, paresthesia, subthreshold oscillations
Sensory signals normally originate at axonal transducer endings in skin, muscle, and other peripheral tissues. The somata of sensory neurons in segmental dorsal root ganglia (DRGs) also generate afferent activity, but this is sparse in intact animals and its sensory consequences are likely to be minor (Wall and Devor, 1983; Devor, 1999). After nerve injury, however, discharge originating ectopically within DRGs is greatly augmented and can be a major contributor to neuropathic dysesthesias and chronic pain (Kirk, 1974; Wall and Devor, 1983; Burchiel, 1984; Nordin et al., 1984;Kuslich et al., 1991; Kajander et al., 1992; Sheen and Chung, 1993; Na et al., 1995; Blenk et al., 1997; Devor and Seltzer, 1999; C.-N. Liu et al., 2000a,b; Lyu et al., 2000; Zhang et al., 2000; Boucher and McMahon, 2001) (but see Li et al., 2000). The underlying process is therefore of considerable practical interest.
The process is also of theoretical interest. DRGs are located within a bony foramen, protected from external stimuli. Moreover, even when artificially depolarized, few DRG neurons are capable of firing more than a brief spike burst. The slow, tonic depolarizations normally caused by physiological stimuli rarely evoke any spikes at all (Villiere and McLachlan, 1996; Amir et al., 1999; C.-N. Liu et al., 2000a). How, then, does ectopic afferent discharge occur? We showed recently in vitro that DRG neurons with repetitive firing capability have a unique property. Rather than their discharge being triggered by the classical rhythmogenic process whereby a sustained depolarizing current repeatedly draws the membrane potential toward spike threshold, firing in these cells is caused by sinusoidal oscillations in the membrane potential. Spikes arise when peaks of normally subthreshold sinusoids reach threshold (Amir et al., 1999; C.-N. Liu et al., 2000a). We now confirm that this also appliesin vivo.
In vivo and in vitro, oscillation sinusoids normally reach threshold only intermittently, yielding slow, irregular firing (Wall and Devor, 1983). Axotomy enhances firing by increasing the number of oscillating neurons and shifting the membrane potential at which oscillations occur closer to rest (Vr) (Amir et al., 1999; C.-N.Liu et al., 2000a). Additional amplification results from replacement of the usual slow intermittent firing pattern with high-frequency bursts. In principle, bursting could simply result from a higher proportion of oscillation sinusoids reaching threshold as proposed recently by Wu et al. (2001) on the basis of recordings from the mesencephalic nucleus of the trigeminal nerve (MesV), but this is not the case. Rather, we show that bursting in DRG neurons reflects recurrent spike triggering by post-spike depolarizing afterpotentials (DAPs). The first spike in each burst is triggered by an oscillation sinusoid; the burst itself is maintained by DAPs.
In addition to augmenting spontaneous ectopic discharge, oscillations and DAP-triggered bursts render somata of sensory neurons responsive to physiologically relevant stimuli that were previously inert. We illustrate this by demonstrating sustained burst discharge in response to artificially generated slow ramp and hold depolarizations. Finally, we document sustained ectopic bursting in response to natural stimuli to which DRGs are subject in situ despite their privileged anatomical location.
MATERIALS AND METHODS
Animals and surgery
Experiments were performed using male and female rats of the Wistar-derived Sabra strain, both mature animals (>300 gm) and animals 2–5 weeks of age (20–145 gm). All protocols were in accordance with national and University regulations for the humane care and use of laboratory animals and followed the ethical guidelines of the International Association for the Study of Pain (Zimmermann, 1983).
Animals used for in vivo recording, and most of those used for in vitro recording, were intact (nonoperated). However, for the in vitro experiment on adrenergic stimulation (see Results), we used eight rats that had undergone unilateral neurectomy 2–10 d before electrophysiological evaluation. Briefly, under pentobarbital anesthesia (Nembutal, 50 mg/kg, i.p.), the sciatic nerve was exposed in the lower part of the popliteal fossa, tightly ligated with 5-0 silk, and cut just distal to the ligature (six rats). Alternatively, the L4 or L5 spinal nerve was exposed 5–10 mm distal to the ganglion, tightly ligated with 5-0 silk, and cut just distal to the ligature (two rats). In both cases, ∼5 mm of the distal nerve stump was excised. Surgical wounds were closed in layers, and the animals were treated with a topical bacteriostatic powder and penicillin (50,000 U/kg, i.m.). Recovery was uneventful. All animals were maintained under standard colony conditions, two to three per cage, in clear plastic shoebox cages bedded with pine shavings. Food and water was available ad libitum. The light/dark cycle was 12 hr.
Electrophysiological preparations
In vivo preparation. Intact rats (n = 10) were anesthetized with Nembutal (50 mg/kg, i.p., followed by ∼20 mg · kg−1 · hr−1as needed). A tracheotomy was performed, one carotid artery was cannulated for monitoring arterial pressure and for administration of fluids, and the animal was mounted prone in a spinal frame with legs in extension. Electrocardiogram and heart rate were monitored, and rectal temperature was maintained at 37.5°C using a feedback-controlled radiant heater. Dorsal roots (DRs) L3–6 and either the L4 or L5 DRG were exposed in a lower lumbar laminectomy and covered with warmed paraffin oil (34°C) in a pool formed of muscle and skin edges. To reduce movement artifacts, in most experiments the animal was paralyzed with gallamine triiodoethylate (Flaxedil, Specia, ∼10 mg/kg) or pancuronium bromide (∼2.0 mg/kg) and artificially respirated. End tidal PCO2 in the trachea was recorded using a Beckman LB-2 CO2 monitor.
In vitro preparation. Rats were overdosed with Nembutal (∼80 mg/kg, i.p.) and killed by carotid exsanguination. DRGs L4 or L5 were excised with the DR, spinal nerve, and a variable length of the sciatic nerve still attached. After ∼1 hr recovery in a modified Krebs' solution containing (in mm): NaCl 124, NaHCO3 26, KCl 3, NaH2PO4 1.3, MgCl 2, dextrose 10, and saturated with 95% O2 and 5% CO2 (pH 7.4, 290–300 mOsm, room temperature), the ganglia were mounted in a recording chamber and superfused with the Krebs' solution (1–2 ml/min, room temperature or 37°C) to which 2 mm CaCl2 was added. In some experiments, as noted, adrenaline (Teva) was added to the superfusion medium at a concentration of 10 μm, 100 μm,or 1 mm for periods of 1–2 min. We recognize that the 1 mm concentration is probably well above physiological levels.
Recording. Sharp glass microelectrodes were used for intracellular recording and stimulation (20–40 MΩ filled with 3 M KCl). Stimuli delivered through the recording microelectrode were either brief pulses (1 msec), prolonged steps (80 msec, 100 msec, or >2 sec), or slow ramp and hold stimuli (see below). Signals were bandpass filtered at DC-10 kHz and recorded digitally on magnetic videotape (Neurodata DR-484) for off-line analysis. We categorized DRG neurons as A- or C-neurons by axon conduction velocity (CV) and the waveform of the intracellularly recorded spike as described elsewhere (Koerber and Mendell, 1992; Amir and Devor, 1996). A-neurons were further categorized: Ainf neurons, which have an inflection on the falling phase of the action potential as assessed by analog differentiation, and relatively low CV (Amir and Devor, 1996), include many Aδ afferents and probably most of the myelinated nociceptors. A0 neurons, neurons without an inflection and with faster CV, are predominantly Aβ afferents, most of which are low-threshold mechanoreceptors (Koerber and Mendell, 1992; Amir and Devor, 1996). Some neurons showed a post-spike DAP. DAP amplitude was measured from the pre-spike baseline to the DAP peak. DAP latency was measured from the DAP peak to the peak of the preceding spike.
To test for oscillations, each cell was examined atVr (resting membrane potential) and then the membrane potential was depolarized in a slow ramp (∼20 mV/sec) and hold (>2 sec) until oscillations occurred or until more than or equal to −20 mV. Potentials obtained were further bandpass filtered at 1 Hz-1 kHz and digitized at 5 kHz (pClamp 6.0.3, Axon Instruments). Frequency components were quantified as power spectral density using a Fast Fourier Transform (FFT) routine (CP Analysis 5.1, Datawave). This analysis was applied to 1–4 sec epochs that did not contain spikes. Oscillations were usually obvious, but when necessary we used as a formal criterion that amplitude peaks be at least 1.5× the amplitude of the background noise level present during brief pauses in the oscillations or that there be a distinct peak in the FFT plot at the frequency expected from visual inspection of the voltage trace, or both.
Cross-excitation. Most DRG neurons show transient depolarization during tetanic stimulation of neurons that share the same DRG (Utzschneider et al., 1992; Amir and Devor, 1996). Here, two alternative stimulation protocols were used to induce this “DRG cross-excitation.” In the first, pulses were delivered through an Ag/AgCl electrode pair placed across the sciatic nerve. By adjusting stimulation intensity and polarity, a substantial proportion of the myelinated axons in the nerve could be activated without stimulating the axon of the impaled neuron itself. The proportion of neighboring neurons activated was assessed from the proportion of the maximal compound action potential (CAP) evoked by the stimulus as monitored through a recording suction electrode placed on the DR. In the second protocol, pulses were delivered to the DR while the CAP was monitored from the sciatic nerve. Stimulus pulses were monophasic 0.1- to 0.2-msec square-waves of ≤7mA, delivered in 10 sec tetani at 50 or 100 Hz. The stimuli were not sufficient to activate unmyelinated axons, as judged from both the latency of CAP components and the failure to record evoked spikes in intracellular recordings from C-neurons without an additional substantial increase in stimulation strength.
Computational model and statistics
Burst firing was simulated using a modified Hodgkin–Huxley (H–H) compartmental model. The simulations modeled a 50-μm-diameter isopotential cell using NEURON software [version 4.2.1; (Hines, 1989),www.neuron.yale.edu]. Specific membrane capacitance was 1 μF/cm2, specific longitudinal resistance was 110 Ω-cm, and temperature was 20°C. Membrane electrical properties, including the various kinetic terms, were based on values from the reconstructed action potential of the squid giant axon (Hodgkin and Huxley, 1952). However, some values were modified to more closely resemble mammalian DRG neurons as follows. (1) The maximal sodium conductance (gNa+max) was decreased to 50.0 mS/cm2 (Caffrey et al., 1992). (2) The leak conductance was increased to 0.7 mS/cm2 (Scroggs et al., 1994). (3) The reversal potential of the leak was set at −77.5 mV to approximate the K+ battery. (4) The maximal voltage-sensitive potassium conductance (gK+max) was decreased to 2.6 mS/cm2. Prolonged depolarizing steps (usually 600 msec) were given under current-clamp conditions. We used the Crank-Nicholson second-order accuracy method for integration (dt = 0.01 msec). For accuracy of calculations we followed the rule of thumb that the number of segments (computational compartments) should be more than or equal to section length/0.05 λ (Luscher et al., 1994; Segev and Burke, 1998). Our cell model was thus a single computational compartment.
Statistical comparisons are based on two-tailed t, χ2, and Fisher exact probabilities tests using a significance criterion of p = 0.05. Values are given ± the SD unless noted. We focused on A-neurons because these have been shown to be the main component of neuropathic discharge originating in the DRG (C.-N. Liu et al., 2000b; X. Liu et al. 2000;Boucher and McMahon, 2001).
RESULTS
Subthreshold oscillations and repetitive firing recordedin vivo
We have reported previously that in vitro, 5.5% of A-neurons (17 of 308; A0 + Ainf) from intact DRGs exhibit high-frequency subthreshold oscillations at Vr(∼100 Hz), with an additional 9.4% (29 of 308) beginning to oscillate on depolarization. Oscillation sinusoids typically remained phase-locked with a pure sine wave for only a few tens of cycles, before drifting by up to several Hertz. Defining a stable mean frequency requires sampling over epochs of several seconds (Amir et al., 1999; C.-N. Liu et al., 2000a). Oscillation amplitude was voltage sensitive, with a peak of 3–6 mV at membrane potentials of −55 to −30 mV. Most cells with oscillations fired long trains when depolarized by 2–3 mV from the potential at which they first showed oscillations. In contrast, cells without oscillations never fired repetitively, no matter how strongly they were depolarized.
We now document similar oscillatory behavior in vivo. Intracellular recordings were made from 16 neurons (14 A0, 2 Ainf) in DRGs from 10 intact adult rats. Two of these neurons (13%; one A0, one Ainf) exhibited oscillations of 50–55 Hz and repetitive firing atVr (Fig.1). The Ainf neuron fired singlet spikes at irregular intervals averaging 0.4 Hz, and the A0 neuron fired long bursts, typically lasting several seconds, with a fixed interspike interval (ISI) of 15 msec (67 Hz). Bursts were interrupted by brief pauses of 50–200 msec. The singlet action potentials and the spike bursts were always triggered by the rising (depolarizing) limb of oscillation sinusoids (Fig.1B). As in vitro, hyperpolarization fromVr decreased overall firing frequency by reducing oscillation amplitude, and hence reducing burst duration and lengthening the periods of spike-free oscillations between bursts (Fig. 1). Firing frequency within bursts also decreased (ISI = 15 msec at −43 mV, 20 msec at −50 mV).Vr of the cell with burst firing at rest was −43.0 mV. Vr of the other neurons was −46.6 ± 5.7 mV.
Fig. 1.

Subthreshold oscillations trigger sustained spike discharge in vivo. A, Recording setup showing dorsal (DR) and ventral (VR) roots and the sciatic nerve (SCN) innervating the hindpaw. The intracellular micropipette is used for recording and stimulation (R&Sic). Spontaneous burst discharge atVr is shown together with a dot display of the corresponding interspike intervals.B, Oscillation amplitude and the prevalence of evoked spikes decrease with a negative shift in the holding potential. Two epochs are shown at −50 mV, one in which spikes were present, and one without spikes. The neuron is an A0 neuron; the sciatic nerve was cut acutely.
An additional four A0 neurons had spike discharge originating at peripheral receptor endings (probably muscle proprioceptors; firing frequency = 0.25, 5, 18, and 22 Hz). This activity was easily distinguished from the activity of somatic origin because no oscillations were present, and neither depolarization nor hyperpolarization affected firing frequency. The remainder of this report is based on recordings in vitro.
Burst discharge and depolarizing afterpotentials
Singlet versus burst discharge pattern
From previous teased fiber recordings in vivo we know that ∼5% of DRG A-neurons in intact rats discharge spontaneously (Wall and Devor, 1983; Burchiel, 1984; Michaelis et al., 2000). Most fire slow irregular trains of singlet spikes (75%; 0.5–5 Hz), whereas others fire in a bursty (19%) or tonic (6%) pattern (ISI = 20–100 msec) (Wall and Devor, 1983). In vitro recordings (DRGs from intact rats) in the present study yielded the same overall level of activity (5%; 12 of 236 units fired spontaneously atVr). However, firing patterns were somewhat different in vitro: 42% of the cells had slow irregular discharge (5 of 12; 0.1–2.5 Hz; four A0 and one Ainf), 58% fired in on-off bursts (7 of 12; all A0); and none fired tonically.
In cells with slow irregular firing, individual spikes were triggered by individual oscillation sinusoids that reached threshold (Figs. 1,2). Mean firing rate mostly reflected the fraction of sinusoids that were suprathreshold, a value that depended on membrane potential and the distribution of sinusoid amplitudes. When cells were depolarized, sinusoid amplitude increased, and peaks came closer to threshold (Amir et al., 1999), increasing the rate of firing.
Fig. 2.

Slow irregular firing and burst discharge recorded from DRGs of intact (unoperated) rats in vitro.A, Irregular discharge triggered by subthreshold oscillations at Vr (A0 neuron;Vr = −47 mV). B,C, Spikes evoked in the same cell by an intracellular stimulus pulse and by axonal stimulation on the sciatic nerve (asterisk). Three superimposed traces are shown in each panel. This cell did not generate a DAP sufficient to trigger a second spike; subsequent spikes in the train are generated by subthreshold oscillations. D, Spontaneous burst firing in another cell (at Vr = −45 mV). Each burst is triggered by an oscillatory sinusoid that reached threshold.
In cells with burst firing, the first spike in each burst was likewise triggered by an oscillatory sinusoid (Figs. 1, 2D). However, subsequent spikes in the burst were not generated by this mechanism because the ISI within bursts did not match the period of the subthreshold oscillations seen just before the burst (Fig.3). It was invariably shorter; hence, instantaneous firing frequency early in each burst was always higher than the oscillation frequency.
Fig. 3.

The interspike interval within bursts matches the DAP latency, not the oscillation period. The two top panels show spike bursts from two different A0neurons (both at Vr = −45 mV). In each panel, two bursts are superimposed. The shorter of the two bursts ends in a subthreshold DAP (asterisk). The two top panels also show a run of subthreshold oscillations taken from just before the burst, showing that the oscillation period is greater than the ISI. The graph below plots corresponding data for 21 cells.
Burst mechanism
We identified post-spike DAPs as the mechanism of burst discharge. In approximately one-third of cells checked, the action potential evoked by intracellular or axonal stimulation atVr was followed by a small DAP of 3.1 ± 2.4 mV (Fig. 4,arrows). A burst was triggered whenever an oscillatory sinusoid triggered a spike in a neuron that generated DAPs of sufficient amplitude. That is, if DAP amplitude was large enough to trigger a second spike, the DAP after the second spike triggered a third spike, the third a fourth, and so on for the duration of the burst. Within bursts the first ISI, i.e., the ISI between the first and second spikes in the train, exactly matched the latency of the DAP (6.4 ± 1.0 vs 6.5 ± 1.0 msec; n = 7;Vr, p > 0.2;r = 0.96; p < 0.005); it did not match the period of oscillations immediately preceding the burst (Fig. 3) (6.4 ± 1.0 vs 8.3 ± 1.0 msec; p < 0.05;r = 0.6; p > 0.2). Bursts always ended with a DAP that was just below spike threshold. From this we infer that what terminated bursts was a decline in DAP amplitude during the course of the burst (see below). Burst termination restored oscillations, setting the stage for triggering of the next burst. In all cells that did not fire bursts, the post-spike DAP was either small or nonexistent (Fig. 2A–C).
Fig. 4.

Afterdischarge bursts evoked by stimulus pulses are caused by DAPs. A, A brief (1 msec) depolarizing pulse was delivered intracellularly. In one trace, a single spike was evoked, followed by a post-spike DAP (arrow). In a second (superimposed) trace, the DAP was suprathreshold and triggered a second spike (Vr = −60 mV).B, Same cell but examined at several holding potentials. A brief (1 msec) hyperpolarizing pulse, which was subthreshold atVr, triggered a single anodal-break spike at −55 mV and a spike burst followed by a DAP and damped oscillations at −38 mV. Stimulus pulses are offset to aid visibility. The DAP (arrow) increased in amplitude with depolarization. C, Similar behavior after single-pulse (0.1 msec) axonal stimulation on the sciatic nerve (asterisk). D, Single-pulse axonal stimulation in this neuron produced a discharge burst (two sweeps superimposed). As during spontaneous bursts, spikes were synchronous with the DAP (arrow) but not with subthreshold oscillations (the epoch of oscillations shown was taken from just before the burst). All cells were A0; spikes inB–D are truncated.
The presence and amplitude of DAPs, and hence the likelihood of burst firing, correlated with other properties of the cell (Table1). These parameters were also sensitive to membrane potential. DAPs occurred preferentially in A0 neurons, in cells with a brief (<20 msec) rather than a more prolonged post-spike afterhyperpolarization (AHP), and in cells with subthreshold oscillations. DAP amplitude increased systematically with depolarization (0.37 ± 0.26 mV/mV;n = 24) to a peak at −35.8 ± 5.9 mV, and then it declined (Figs. 4, 5). As a consequence, slow ramp depolarization revealed DAPs in cells in which they were not initially visible. Thus, although atVr 31% of cells had DAPs, 96% had them on depolarization (8 of 26 vs 25 of 26; χ2, p < 0.001). Not surprisingly, cells with a DAP at Vrhad a more depolarized Vr than cells without (−52.8 ± 7.7 mV vs −58.8 ± 6.3 mV;p < 0.005). Correspondingly, many cells that fired singlet spikes at Vr became bursters on depolarization, with burst duration increasing with increasing depolarization (Fig. 1). Depolarization also decreased DAP latency (0.21 ± 0.17 msec/mV; n = 17), causing firing frequency within bursts to increase.
Table 1.
Characteristics of 18 DRG A-neurons that had a DAP atVr
| Cell type (% of cells sampled) | Presence of a brief or prolonged AHP (% cells sampled) | Presence of oscillations (atVr or depolarized) | |||
|---|---|---|---|---|---|
| A0 | Ainf | Brief (<20 msec) | Prolonged (≥20 msec) | Yes | No |
| 18 of 49 (37%) | 0 of 10 (0%) | 17 of 40 (43%) | 1 of 19 (5%) | 13 of 13 (100%) | 5 of 46 (11%) |
| p < 0.05* | p < 0.01* | p < 0.001* | |||
All cells were from intact rats, recorded in vitro.
p value based on the Fisher exact probabilities test.
Fig. 5.

DAP amplitude varies with membrane potential.Symbols represent data from 11 individual A0neurons.
Wu et al. (2001) argued recently that burst firing in MesV neurons arises directly from runs of oscillatory sinusoids that cross the spike threshold. We note, however, that in their results, as in ours, the period between spikes in a train is consistently shorter than the oscillation period (their Fig. 2, our Fig. 3). Rather, as we have shown, an ISI within bursts matches the DAP latency, and bursts are maintained by DAPs rather than by oscillations. This distinction is significant; oscillations and DAPs are not the same thing. By definition, a DAP is a brief depolarizing rebound potential that follows a spike. Subthreshold oscillations are ongoing trains of sinusoidal variation in the membrane potential, often completely dissociated from spike activity. On the other hand, these two waveforms appear to be reflections of the same underlying neuronal resonance characteristic [see below, and Amir et al. (2002)], and they sometimes appear in close association with one another (Table 1). For example, spikes are sometimes followed by a damped train of post-spike DAPs that appear to transition seamlessly into ongoing subthreshold oscillations (Figs. 1, 4B, D).
Cessation of bursts
What causes bursts to terminate? During the course of a burst, particularly a high-frequency burst, spike amplitude usually decreased and ISI increased, reducing instantaneous firing frequency (Fig.6A). The presence of spikes precluded measurement of DAP amplitude during bursts. However, each burst terminated in a DAP that was slightly below spike threshold. The failure of this final DAP to trigger a subsequent spike cut short the regenerative process that maintained the burst. Among the factors responsible for the apparent attenuation of DAP amplitude during bursts, and hence for shifting the peak of the DAP away from spike threshold, is a hyperpolarizing shift that developed during the course of the burst (Fig. 6). We have shown previously that this shift is caused primarily by a Ca2+-activated K+ conductance engaged during the burst (Amir and Devor, 1997).
Fig. 6.

Mechanism of repetitive burst firing in DRG neurons. A, This neuron was quiescent atVr, but subthreshold oscillations and burst discharge developed as a depolarizing ramp and hold current brought the cell toward −43 mV. Traces marked1-5 show at a higher sweep speed the parts of the main record that are marked with arrows. The first oscillation sinusoid to reach threshold triggered a prolonged spike burst (trace 2). During the course of the burst, ISI increased (spike rate accommodation). This can be seen by comparing ISI in traces 2 and 3, and also from the ISI dot display and the fast traces shown above the main record. As the burst progressed, membrane potential shifted several millivolts in the hyperpolarizing direction, leading to termination of the burst in a subthreshold DAP (trace 3). As the membrane subsequently drifted back toward the holding potential (−43 mV), the amplitude of oscillations increased, and ultimately a second burst was triggered (traces 4and 5). B, A similar process occurs when the initial depolarizing shift is caused by cross-depolarization. This cell did not fire when held at −45 mV. However, tetanic stimulation of the axons of neighboring neurons evoked cross-depolarization and repetitive burst firing (dashed line; 10 sec, 100 Hz pulses to the sciatic nerve at an intensity subthreshold for the axon of the impaled neuron but suprathreshold for the axons of neighbors).
After the cessation of each burst, the now hyperpolarized membrane revealed smaller oscillations than those that occurred immediately before the burst, or no oscillations at all. With time, membrane potential drifted back in the depolarizing direction, and as this occurred, oscillations reappeared and progressively grew in amplitude. The first oscillation sinusoid to reach spike threshold triggered a second DAP-driven burst, completing the burst cycle (Fig.6A).
Afterdischarge bursts
An additional consequence of DAPs is “afterdischarge.”In vivo, 2–5% of DRG neurons fire a burst of action potentials in response to single axonal stimulus pulses (intact and axotomized rats) (Devor and Wall, 1990; Delio et al., 1992). Such afterdischarge bursts were recorded here in vitro in a similar proportion of neurons [intact rats, 3% (8 of 236) neurons atVr; all A0] (Fig. 4). The bursts consisted of trains of 2 to >20 spikes after single brief axonal or intracellular stimulus pulses.
In vivo, afterdischarge is most common in neurons that already have ongoing burst discharge, and hence those that probably have subthreshold oscillations and DAPs (Devor and Wall, 1990). In such cells application of a stimulus pulse during a silent period triggers a burst, resetting the endogenous burst cycle and entraining it to the applied stimulus (Lisney and Devor, 1987). Afterdischarge in vitro was also associated with repetitive firing capability. Most neurons that showed afterdischarge atVr also fired spontaneously (5 of 8). In comparison, only 7 of the remaining 228 neurons, cells that did not show afterdischarge at Vr, fired spontaneously (p < 0.001). An additional 5 of the 228 neurons fired afterdischarge bursts when depolarized (by 6–26 mV from Vr; all A0 neurons), and 3 of them fired repetitively. All five developed subthreshold oscillations on depolarization.
All cells that generated afterdischarge bursts had DAPs. The stimulus pulse triggered the first spike in each burst; subsequent spikes were triggered by DAPs. As with spontaneous bursts (Fig. 3), the first ISI in afterdischarge bursts matched the latency of the DAP (both 7.2 ± 1.7 msec; n = 9; r = 1.0) (Fig.4D) more closely than they matched the period of oscillations (8.4 ± 1.2 msec; r = 0.67).
Computational model of the DAP
Both oscillatory behavior and associated spike bursts persist in the presence of wide-spectrum blockers of Ca2+ conductances but are eliminated by Na+ channel blockers (TTX, lidocaine) or reduced [Na+]o(Amir et al., 1999; Pedroarena et al., 1999; Wu et al., 2001). The DAPs responsible for bursting are therefore distinct from the prolonged Ca2+-dependent afterdepolarizations that sometimes occur in DRG neurons (see Discussion) (White et al., 1989). To gain insight into the membrane dynamics responsible for DAPs and bursting, we ran computational simulations of mammalian DRG neurons as described in Materials and Methods.
Just suprathreshold depolarizing steps triggered a single action potential followed by a DAP. The latency and amplitude of the DAP were like those observed in vitro (compare Fig.7, A and B, with Fig. 4A), and as in vitro, DAP amplitude was voltage sensitive. Increasing the amplitude of the depolarizing step increased the amplitude of the DAP, eventually triggering brief spike bursts. A further increase led to tonic discharge that persisted for as long as the stimulus was maintained (at least 2 sec) (Matzner and Devor, 1992).
Fig. 7.

Post-spike DAP simulated numerically using the H–H formalism. Cells held at Vr (−77.5 mV) were brought to near −55 mV at t = 0 using a 1 sec depolarizing current step. At t = 500 msec, a suprathreshold depolarizing pulse was given (0.2 msec, 10 nA) to trigger a spike. A, Two superimposed simulations using slightly different current steps. In one (1.030 nA), the post-spike DAP was subthreshold; in the second (1.033 nA), the DAP reached threshold and evoked a second spike. B, Enlargement of thetrace in A with the subthreshold DAP. Note the close resemblance to DAPs recorded from real DRG neurons (Fig.4A). C, Calculated transmembrane leak, Na+, and K+ currents underlying the voltage trace in B.
Plots of individual (calculated) transmembrane currents (Fig.7C) reveal the mechanism of the DAP in this model. After the spike, outward K+ and leak current induced a brief AHP during which Na+ current fell below its pre-spike value. This reprimed Na+ channels, making more of them available for opening. At the end of the AHP, when the membrane moved back in the depolarizing direction toward the holding potential, a small inward Na+ current was generated yielding the DAP. In cases where the DAP was relatively large, this process repeated itself, yielding damped oscillations (Matzner and Devor, 1992). Indeed, the H–H simulation can yield ongoing oscillations with frequency, amplitude, and voltage sensitivity similar to those recorded in vitro (Amir et al. 2002). The resonance characteristics underlying the DAP and subthreshold oscillations therefore appear to be related.
Repetitive firing capability: response to step depolarizations
The presence of subthreshold oscillations and DAPs critically determines whether a DRG neuron is capable of sustained firing in response to tonic artificial or physiological depolarizing stimuli.
All neurons tested fired a single spike at the beginning of a depolarizing step (n = 78; 48 A0, 30 Ainf; current at threshold 0.8 ± 0.3 nA generated a depolarization of 5.6 ± 2.0 mV), and about half of these fired a burst (i.e., more than or equal to two spikes) when current strength was increased (40 of 78; threshold 1.7 ± 1.1 nA; 11.9 ± 9.3 mV). The remainders never fired more than one spike, even using currents up to 10 nA, that generated depolarizations of 30–50 mV. Bursts occurred only in cells that generated a DAP. A0 neurons were more likely to fire a burst than Ainf neurons (33 of 48 vs 7 of 30;p < 0.001).
Burst duration and frequency were voltage sensitive. At threshold, bursts consisted of two or three spikes with a mean ISI of 7.2 ± 2.2 msec (equivalent to an instantaneous firing frequency of 138 ± 40 Hz). With increasing current strength, burst duration increased, persisting in some cells for the duration of the stimulus pulse (80 or 100 msec). However, using prolonged stimuli (>2 sec), bursts rarely lasted more than ∼200 msec (∼40 spikes) (Fig. 9A). Instantaneous firing frequency (reciprocal of the first ISI) increased with stimulation current along a continuous negatively accelerating frequency–current trajectory (Jack et al., 1983; Matzner and Devor, 1992). Bursts always ended in a subthreshold DAP, followed by damped oscillations in some cells (Fig. 8). Spike rate accommodation, i.e., the gradual increase in ISI during the course of a burst, occurred sometimes, especially during prolonged bursts. Although bursts occurred in cells without oscillations, longer burst durations were associated with oscillatory behavior. Thus, among cells with bursts of >15 spikes, 6 of 9 had oscillations. This compares with only 4 of 23 cells with bursts of fewer than 15 spikes (p < 0.005; all 32 neurons were A0).
Fig. 9.

Persistent firing during prolonged depolarizing steps depends on the presence of subthreshold oscillations and post-spike DAPs. A, Step depolarization (3 nA) triggered a single spike burst. Numbered traces show that oscillations present immediately after the burst damped out rapidly (traces 2–8). B, Under similar conditions, neurons with sustained subthreshold oscillations (traces 2–6) generate repeated spike bursts. Both neurons are A0; all spikes are truncated.
Fig. 8.
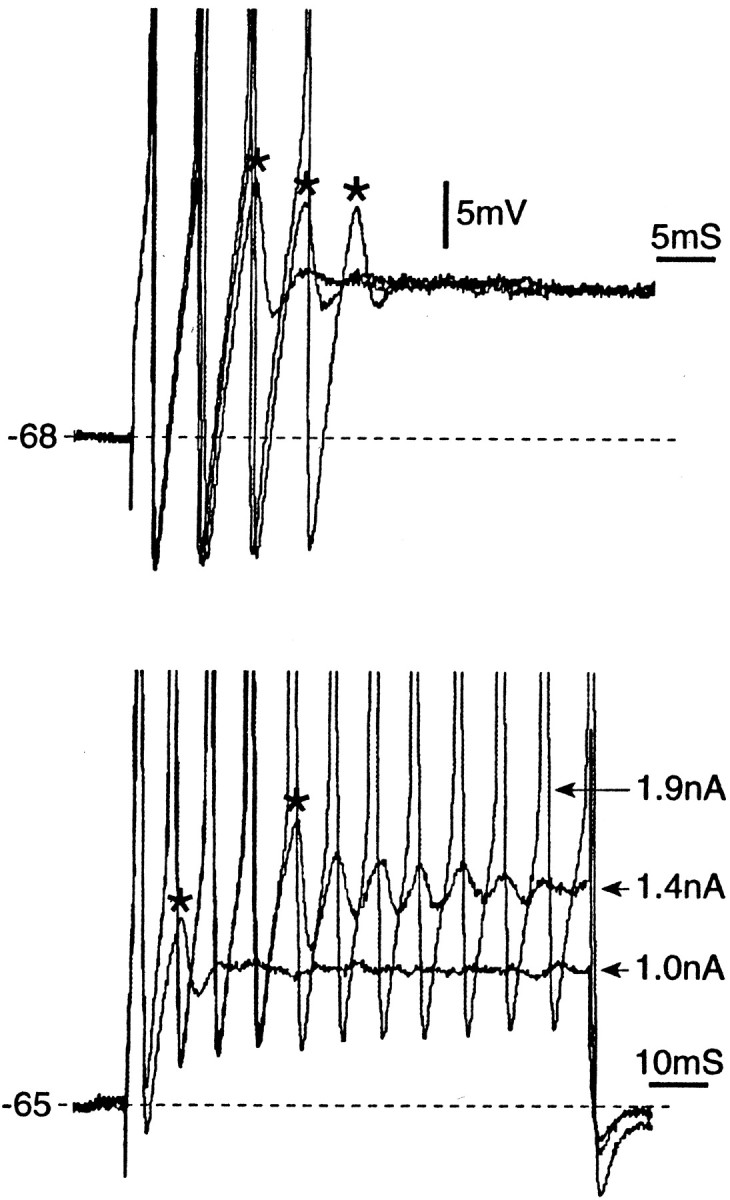
DAPs maintain spike bursts during depolarizing steps. The top panel shows three superimposed burst responses that contained two, three, and four spikes, respectively (80 msec, 1.4 nA depolarizing steps). Asterisks mark the subthreshold DAP that terminated the burst in each case. Thebottom panel is similar (data from a second neuron) except that current steps of 1.0, 1.4, and 1.9 nA were used. Burst duration increased with stimulus strength, persisting throughout the step in the case of the 1.9 nA stimulus. Again, bursts ended in a DAP. Both cells are A0; all spikes are truncated.
The ability of cells to fire in a sustained manner, for >200 msec, was critically dependent on the presence of subthreshold oscillations. For example, in the cell illustrated in Figure9B, an initial burst was followed by a series of shorter bursts that persisted indefinitely. Four such cells were encountered. In each case the recurrent bursts were a product of the oscillation/DAP process described above.
Physiologically relevant depolarizing stimuli
Under physiological and pathophysiological conditions in vivo, various processes are capable of depolarizing DRG somata despite their protected location (see Discussion). However, because all of these yield slow ramp and hold-like generator currents, without fast transients, it is likely that only oscillating cells respond with ectopic repetitive firing. We tested this prediction.
Ramp and hold depolarization
In response to ramp and hold depolarization, most neurons did not fire even a single spike. This includes cells that did fire an initial burst on step depolarization (Fig.10A). Spiking was initiated, however, in a subset of 14 of the 61 cells tested (23%; all A0). Three cells fired a single burst during the ramp and then fell silent. The remaining 11 fired repetitively during both ramp and hold phases, with either irregularly spaced single spikes (n = 1) or repeated bursts (n = 10) (Fig. 6A). Firing threshold was 9.4 ± 6.6 mV positive to Vr (range 3–28 mV;n = 14). In all 14 cells, repetitive firing was a product of subthreshold oscillations and DAPs.
Fig. 10.

Burst firing during step versus ramp and hold depolarization. A, This neuron consistently fired a brief discharge burst in response to depolarizing steps (left). No spikes were ever evoked by the equivalent depolarization delivered as a slow ramp and hold (right). B, Likewise, neurons that fired in response to depolarizing steps (left) failed to respond to slow physiological depolarizations, evoked in this case by tetanic activation of neighboring neurons (cross-depolarization,right). The exception is neurons with subthreshold oscillations (Fig. 6B). Both neurons were A0.
DRG cross-depolarization
In vivo, ectopic discharge originating in DRG neurons is facilitated by stimulation of the axons of adjacent neurons that share the same ganglion or by stimulation of the skin innervated by the adjacent neurons (Devor and Wall, 1990). In vitro as well, most DRG neurons (∼90%) are transiently depolarized by spike activity in neighbors (Utzschneider et al., 1992; Amir and Devor, 1996,2000). However, in the previous in vitro studies of this “cross-depolarization” phenomenon, we failed to observe repetitive firing. In retrospect, this failure may have been attributable to our lack of appreciation at the time of the essential role of subthreshold oscillations, and hence the use of inadequate stimulation protocols. For example, we typically used 10 sec tetani of 50–100 Hz, which produced peak cross-depolarizations of ∼4–5 mV (Utzschneider et al., 1992; Amir and Devor, 1996). This is considerably less than the 9.4 ± 6.6 mV threshold for repetitive firing using ramp-like stimuli (above), and in any event would have been ineffective unless oscillating cells had been chosen specifically for study.
To test this explanation we selected eight A0neurons that showed subthreshold oscillations atVr (n = 1) or on depolarization (n = 7; 14.9 ± 16.5 mV positive toVr). Each cell was depolarized to its oscillatory threshold. Tetanic stimulation was then applied to the axons of neighboring neurons to evoke cross-depolarization (which averaged 3.7 ± 2.1 mV). In 5 of the 8 cells tested (63%), repetitive burst firing was evoked (Fig. 6B). One of the five cells was further depolarized until it began to fire, and in this case cross-depolarization accelerated the firing (Fig.11). In each case, firing consisted of spike bursts triggered by subthreshold oscillations and maintained by DAPs (as in Fig. 6A). For comparison, 13 neurons (11 A0, 2 Ainf) were similarly depolarized (by 15.4 ± 7.7 mV), but without the appearance of subthreshold oscillations. None of these showed spike discharge (χ2; p < 0.005), although the cross-depolarization evoked was comparable in amplitude (4.4 ± 3.3 mV vs 3.7 ± 2.1 mV; p > 0.2).
Fig. 11.

Cross-excitation within the DRG. Held at −45 mV, this A0 neuron (Vr = −60 mV) fired occasional spike bursts. Cross-depolarization evoked by tetanic stimulation of the axons of neighboring neurons (dashed line) enhanced the amplitude of subthreshold oscillations and increased the prevalence of bursts. Firing rate during bursts was not affected (ISI dot display is shown above each burst). Sample traces 1–4 (bottom) are taken from the times indicated. Short vertical linesare stimulus shock artifacts from the nerve stimulation.
The tetani used in this exercise were intense (10 sec trains at 50–100 Hz). However, the activity evoked is not outside of the range that can be evoked by strong natural stimuli or that may arise spontaneously as sites of ectopic activity in injured nerves. Moreover, as noted, we have shown previously that cross-excitation among DRG neurons can be generated using considerably weaker tetani, including physiological stimuli such as brushing the skin (Devor and Wall, 1990; Utzschneider et al., 1992).
Adrenaline
Activation of sympathetic post-ganglionic efferents and direct application of α-adrenergic agonists have been reported to depolarize DRG neurons and evoke discharge, particularly in nerve-injured animals (Burchiel, 1984; Devor et al., 1994; Petersen et al., 1996). Do subthreshold oscillations play an essential role? We tested 24 DRG A0 neurons, 5 from intact adult rats and 19 from rats that had undergone previous nerve injury. Cutting of sciatic (16 cells) or spinal nerves (3 cells) had similar effects, so results were combined. In most cases, we confirmed that the cell under study had indeed been axotomized by observing spikes in response to electrical stimulus pulses delivered to the cut nerve end.
None of the neurons from intact rats oscillated or fired atVr, either before or after adrenaline application. However, they did show a small depolarization within a few seconds of the start of adrenaline flow (10 μm;3.0 ± 1.8 mV; n = 5). Depolarization did not evoke oscillations or sustained firing. In axotomized DRGs, 2 of the 19 neurons tested had oscillations at Vrin drug-free medium, and an additional 12 oscillated when depolarized. All 14 of the oscillating cells fired repetitively when depolarized still further, whereas none of the 5 without oscillations did so. Adrenaline induced depolarization fromVr in 15 of the 19 neurons (79%; 3.6 ± 3.0 mV; n = 15). These included cells that generated oscillations in drug-free medium (10 of 14) and those that did not (5 of 5). Of the remaining four cells, two were hyperpolarized and in two there was no change in membrane potential.
Adrenaline affected oscillatory behavior and firing in 5 of the 19 axotomized neurons (26%; 4 sciatic, 1 spinal nerve injured). Remarkably, however, its effects were not mediated by membrane depolarization. In four of the five neurons adrenaline evoked oscillations and repetitive firing despite minimal or no depolarization (3.0, 1.0, 0 mV) (Fig.12A), or even slight hyperpolarization in one case (−4.0 mV). In the fifth neuron, activity was suppressed by adrenaline. Before administration the cell oscillated and fired repetitively at Vr. Adrenaline induced depolarization (100 μm, 3 mV; 1 mm, 4 mV) but a decrease in ongoing firing frequency (Fig. 12B). In vivo too, adrenaline sometimes suppresses spike discharge in DRG neurons (38%) (Burchiel, 1984; Devor et al., 1994). Adrenaline can apparently affect membrane resonance and firing properties through a mechanism independent of depolarization.
Fig. 12.

Repetitive firing in response to adrenergic stimulation is mediated by subthreshold oscillations. A, Bath-applied adrenaline (10 μm, betweenarrows) evoked an intense and prolonged (>1 min) discharge burst in this cell. Note that the adrenaline facilitated subthreshold oscillations and triggered the burst, without causing membrane depolarization. The burst was maintained by DAPs.B, In this cell, adrenaline (1 mm, betweenarrows) induced depolarization but a decrease in firing frequency. Firing was in singlets or doublets rather than bursts and was driven by subthreshold oscillations. Both neurons are A0, 3 d (A) and 10 d (B) after sciatic nerve section.
Unusual oscillatory modes
Slow oscillations
We encountered 3 neurons of 93 checked for this property that developed “slow” oscillations on depolarization fromVr (3%; all A0;in vitro; oscillation thresholds = −38, −43, −48 mV). In addition to oscillation frequency being low (∼25 vs ∼100 Hz in the cells described above) (Fig.13), the frequency appeared to be unaffected by depolarization. Oscillation amplitude, on the other hand, was voltage sensitive as in typically oscillating neurons. Slow oscillations did not trigger spikes. DAPs were absent in these cells atVr but emerged on depolarization.
Fig. 13.

Some DRG neurons generate low-frequency subthreshold membrane potential oscillations. A, “Typical” high-frequency oscillations. B, Low-frequency (24 Hz) oscillations. In both types of neurons, oscillation amplitude is voltage sensitive.
Nonsinusoidal voltage fluctations
Irregular low-frequency membrane potential fluctuations were seen at Vr in 3 of the 93 neurons (two A0, one Ainf; 3%). However, most of the others, both A0 and Ainf, also developed fluctuations when they were strongly depolarized (>80%) (Fig.14A). This includes neurons that had sinusoidal oscillations at more negative membrane potentials. Nonsinusoidal fluctuations were easily distinguished from subthreshold oscillations: they did not show an amplitude peak in their Fourier spectrum, and their power was concentrated at lower frequencies (<25 Hz) (Fig. 14B,C). With depolarization, the amplitude and hence the power density of the fluctuations increased, with the overall effect of increasing membrane instability.
Fig. 14.
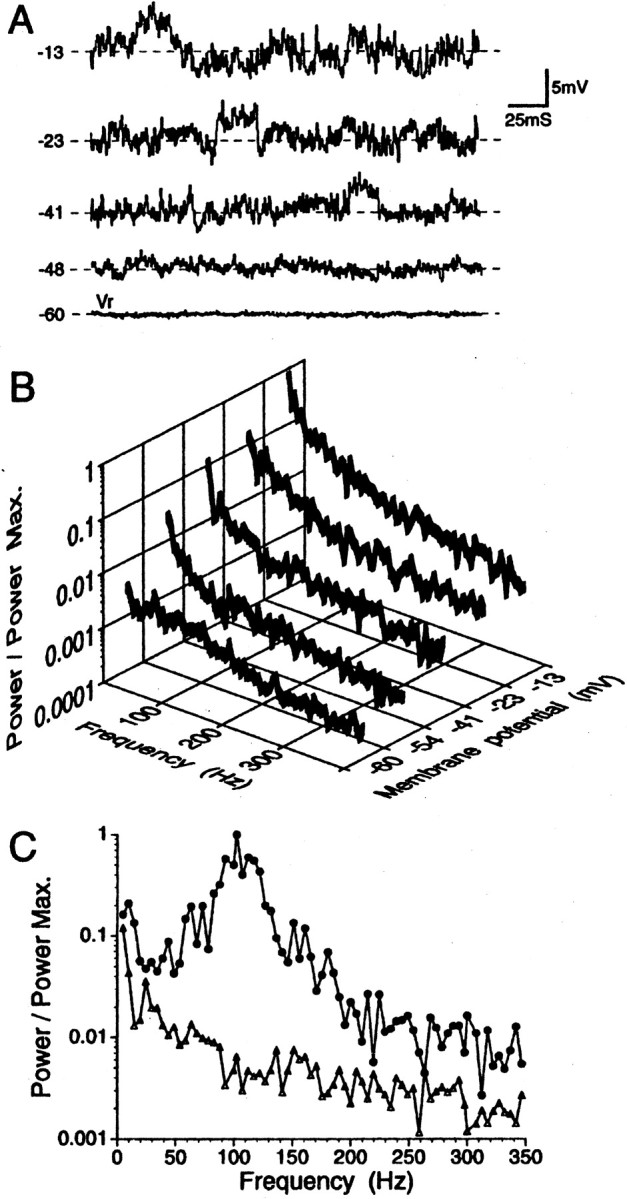
Nonsinusoidal fluctuations in the membrane potential increase in amplitude with depolarization. This is illustrated in original traces in A and in the corresponding FFT profile in B. C, Unlike neurons with typical high-frequency sinusoidal oscillations (filled circles) [data from Amir et al. (1999)], cells with nonsinusoidal fluctuations (triangles) did not show an amplitude peak in their FFT spectrum. In both A0 cells shown, FFT analysis was performed at the potential at which the oscillation/fluctuation amplitude was maximal (−35 mV, circles; −13 mV,triangles). FFT plots in B were individually normalized relative to the maximal power level observed in the particular cell. In C, normalization was relative to the cell indicated with filled circles.
Voltage fluctuations seen at Vr did not trigger spontaneous firing, and they rarely occasioned spikes on depolarization. There are two likely reasons. First, fluctuations tended to occur at membrane potentials sufficiently positive that most of the Na+ channels required for spike electrogenesis would have been inactivated. Second, the slope of the transient depolarization (dV/dt) at the leading edge of fluctuations was shallower than in the case of oscillations. Steep dV/dt aids in overcoming the prominent membrane accommodation characteristic of DRG A-neurons. This is a key attribute of high-frequency sinusoidal oscillations that permits them to facilitate repetitive firing (C.-N. Liu et al., 2000a). We note, however, that voltage fluctuations do trigger spikes in C-neurons (Amir et al., 1999). Nonsinusoidal fluctuations have been reported previously as a substrate for spiking in small-diameter DRG neurons in dissociated cultures (Mathers and Barker, 1984; Study and Kral, 1996).
DISCUSSION
Ectopic discharge generated in afferent A0neurons in the DRG is a major contributor to chronic dysesthesias and pain in neuropathy, and it may also play a small role in normal sensation (see introductory remarks). We have shown in vitro, and now also in vivo, that subthreshold membrane potential oscillations are a critical variable linking depolarization to ectopic firing. Only cells with oscillations are capable of firing in a sustained manner, spontaneously or in response to the sorts of tonic physiological stimuli that exacerbate neuropathic sensory symptoms in the clinical setting.
Oscillations alone sustain firing at low intensity. The full fury of ectopia is unleashed only when oscillations trigger afferent burst discharges (C.-N. Liu et al., 2000b). Our data indicate that bursting is a regenerative consequence of post-spike DAPs. This is so for spontaneous bursts, bursts generated at the onset of a depolarizing step, for afterdischarge bursts after brief stimulus pulses, and for bursting during tonic depolarization. Oscillations and DAPs act synergistically. The similarity of ectopic firing patterns in DRG somata and in neuromas and other sites of nerve trauma suggests that oscillations and DAPs may also underlie ectopic firing at these locations (Devor and Seltzer, 1999). Any consideration of the ways in which ion channel regulation and trafficking implement normal sensation and, when disrupted, bring about neuropathic pain (Devor and Seltzer, 1999; Waxman et al., 1999) must take into account the effects of this regulation on oscillations and burst discharge.
Depolarizing afterpotentials
Afterdepolarizations have been reported previously in afferent somata. Studying enzymatically dissociated rat DRG neurons with patch electrodes, White and Lovinger and colleagues (Lovinger and White, 1989; White et al., 1989) described a subpopulation of neurons with an afterdepolarization 70–200 msec in duration, and tens of millivolts in amplitude, generated by a transient (T-type) Ca2+ conductance. Short spike bursts, limited by the duration of the afterdepolarization, rode on this potential. A similar prolonged afterdepolarization was reported by Kocsis and coworkers in afferent axons bathed in 4-aminopyridine. This potential, which is not Ca2+ dependent, was attributed to a slow Na+ conductance (Honmou et al., 1994).
The brief, short-latency, rebound DAP that we have described here is clearly different. Although it appears frequently in published records, the only reports we are aware of that make mention of it are by Puil and coworkers in rat trigeminal ganglion slices (Puil et al., 1989) and by Pedroarena et al. (1999) in primary sensory neurons of MesV. In both studies, as in ours, fast rebound DAPs were associated with membrane resonance and subthreshold oscillations and were not blocked by Ca2+ channel antagonists or low [Ca2+]o. Rather, they appear to be caused entirely by inward Na+ and outward K+ (or leak) currents, and as we show here, they are accurately simulated by our modified H–H model.
The DAP as a signal amplifier
Oscillations per se drive ectopic discharge at low frequency (usually <5 Hz) and with an irregular ISI. This is the most common spontaneous firing pattern at Vr in vitro and hence in DRGs in vivo (Wall and Devor, 1983;Devor and Wall, 1990). The normally low level of electrogenesis in DRGs is amplified many-fold when irregular singlet spikes trigger high-frequency DAP-driven spike bursts (Amir et al., 1999; C.-N. Liu et al., 2000b). Within bursts, instantaneous firing rates often surpass 100 Hz. Bursting has at least two distinct amplifying effects: it augments the overall ectopic afferent barrage, and it powerfully reinforces marginally effective synaptic links (Lisman, 1997).
DAP-sustained bursts usually damp out after the first few tens of spikes. However, when damping is weak and the cell is depolarized to a level at which its DAP is prominent, spike-triggered bursts may be prolonged. Indeed, at least in vivo, DRG neurons sometimes continue firing for extended periods of time after a brief trigger pulse (Devor and Wall 1990; C.-N. Liu et al., 2000b; X. Liu, 2000). Entry into this self-sustained DAP-triggered firing mode may underlie common and previously unexplained sensory peculiarities in patients with peripheral neuropathies, such as touch-triggered pain paroxysms (in trigeminal neuralgia) and “hyperpathia” (Noordenbos, 1959;Rappaport and Devor, 1994).
Patients with hyperpathia often report sensations evoked by brief tactile stimuli that long outlast the stimulus itself. Likewise, response to repeated tapping on the skin may summate, sometimes building up to an intensely painful crescendo (Noordenbos, 1959). Such abnormalities, traditionally explained as CNS processes, are readily explained by endogenous rhythmogenesis in injured primary afferents, triggered by afferent spikes and sustained by DAPs. Although we by no means question the possible role of the CNS in hyperpathia, our results emphasize the contribution of PNS processes and specifically point to resonance mechanisms as promising targets for analgesic drug development.
DAPs and spontaneous repetitive firing in DRG neurons
In vitro, sustained firing atVr and on tonic depolarization (evoked firing) occurs only in neurons that generate subthreshold oscillations. Our recordings in vivo establish that repetitive spike discharge in DRGs, in the whole animal, also derive from subthreshold oscillations. Moreover, they confirm that oscillations are not an artifact of tissue excision or other aspects of the in vitropreparation.
The correspondence of this novel electrogenic process in vivo and in vitro rationalizes a large body of observations on ectopic firing made using teased fiber methodologies in whole animals (for review, see Devor and Seltzer, 1999). Qualitative and quantitative similarities include the magnitude of the ectopic barrage (percentage of neurons involved), potentiation of activity after axotomy, patterns of spike discharge, types of neurons that are active (primarily Aβ at early postoperative times), afterdischarge after single pulse stimuli, DRG cross-excitation, response to sympathetic stimulation, etc., but some parameters differ. These include the higher instantaneous frequency during bursts in vitro, the relatively high incidence in vivo of slow irregular firing versus bursting, and the rarity of tonic, as opposed to burst firing, in vitro. We suspect that these differences all derive from a single source: less prominent and more delayed DAPsin vivo. Small DAPs are less likely to trigger spike bursts and hence favor the slow irregular firing pattern most prevalentin vivo. Likewise, increased DAP latency reduces instantaneous firing frequency during bursts. Lower firing frequency, in turn, is expected to reduce the hyperpolarizing shift responsible for burst termination. This may explain why in vivo, ectopic firing sometimes persists indefinitely (ongoing, spontaneous discharge). Note that once initiated, tonic firing is maintained by the H–H process (i.e., by DAPs) and does not require subthreshold oscillations. Quantitative differences in DAP parameters in vitro versus in vivo could be caused by such factors as constituents of the extracellular medium, temperature, and effects of cell impalement that might alter Vrcompared with values in vivo. DAPs and bursting are enhanced at relatively depolarized potentials.
Evoked repetitive firing in DRG neurons
Although the ability to generate subthreshold oscillations is a necessary condition for responsiveness to tonic stimuli in DRG A-neurons, it is not a sufficient condition. Except for cells that fire at Vr, there must also be a source of sustained depolarization, equivalent to the generator potential at sensory endings. Depolarization has two effects: it increases the amplitude of oscillatory sinusoids, and it brings them closer to the firing threshold of the neuron (Amir et al., 1999). Many conditionsin vivo are capable of inducing generator depolarizations in the DRG cell soma despite the protection of the bony intervertebral foramen. These include ischemia, hypoxia, systemic α-adrenergic agonists (e.g., serum adrenaline), sympathetic efferent discharge, elevated serum [K+], inflammatory mediators, spike activity in neighboring DRG neurons evoked by natural cutaneous stimuli or ectopia, and mechanical strain from relative movement of adjacent vertebrae during activity (Devor and Seltzer, 1999). Nonetheless, in healthy individuals, the DRG is only a minor source of afferent input. This is presumably because of the small number of intact neurons that have subthreshold oscillations atVr and the small amplitude of generator depolarizations produced in DRGs by physiological stimuli.
In the event of neuropathy, however, these relations can change radically. Axotomy greatly increases the population of neurons with oscillations at Vr, it shifts oscillation and firing threshold towardVr, and it enhances DAPs (Amir et al., 1999; C.-N. Liu et al., 2000a,b). The net effect is to augment spontaneous discharge and render neurons responsive to physiological stimuli that would otherwise have had no effect. There may also be an increase in the amplitude of generator depolarizations. For example, foramenal stenosis, disk herniation, or iatrogenically produced adhesions may increase the transmission of activity-generated strain forces from the vertebrae to the DRG and of traction forces transmitted by nerves or spinal roots, e.g., during straight leg lifting (Nordin et al., 1984; Kuslich et al., 1991; Nordin and Balagué, 1996;Nowicki et al., 1996; Zhang et al., 2000). Nerve injury is also known to trigger sprouting of postganglionic sympathetic endings within the DRG, a condition that may augment the local release of adrenergic agonists (McLachlan et al., 1993; Shinder et al., 1999). Finally, inflammation, either systemic or in local spinal tissues, may enhance ectopic firing originating in the DRG by increasing intraganglionic concentrations of an array of inflammatory mediators and by lowering tissue pH (Levine and Reichling, 1999). In this context, it is interesting to note that the DRG is uniquely devoid of a blood–nerve barrier (Allen and Kiernan, 1994; Devor, 1999). In light of its intrinsic oscillatory and burst-initiating properties, the DRG emerges as a novel and thus far unexploited target for pain control.
Footnotes
This work was supported by grants from the United States–Israel Binational Science Foundation and the German–Israel Foundation for Research and Development. M.M. received a Heisenberg fellowship from the Deutsche Forschungsgemeinschaft.
Correspondence should be addressed to Prof. Marshall Devor, Department of Cell and Animal Biology, Institute of Life Sciences, Hebrew University of Jerusalem, Jerusalem 91904, Israel. E-mail:marshlu@vms.huji.ac.il.
M. Michaelis's present address: Aventis Pharma Deutschland, Industriepark Hoechst, H821 65926 Frankfurt/Main, Germany.
REFERENCES
- 1.Allen DT, Kiernan JA. Permeation of proteins from the blood into peripheral nerves and ganglia. Neuroscience. 1994;59:755–764. doi: 10.1016/0306-4522(94)90192-9. [DOI] [PubMed] [Google Scholar]
- 2.Amir R, Devor M. Chemically mediated cross-excitation in rat dorsal root ganglia. J Neurosci. 1996;16:4733–4741. doi: 10.1523/JNEUROSCI.16-15-04733.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amir R, Devor M. Spike-evoked suppression and burst patterning in dorsal root ganglion neurons. J Physiol (London) 1997;501:183–196. doi: 10.1111/j.1469-7793.1997.183bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amir R, Devor M. Functional cross-excitation between afferent A- and C-neurons in dorsal root ganglia. Neuroscience. 2000;95:189–195. doi: 10.1016/s0306-4522(99)00388-7. [DOI] [PubMed] [Google Scholar]
- 5.Amir R, Michaelis M, Devor M. Membrane potential oscillations in dorsal root ganglion neurons: role in normal electrogenesis and in neuropathic pain. J Neurosci. 1999;19:8589–8596. doi: 10.1523/JNEUROSCI.19-19-08589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amir R, Liu C-N, Kocsis JD, Devor M (2002) Oscillatory mechanism in primary sensory neurons. Brain, in press. [DOI] [PubMed]
- 7.Blenk K, Habler H, Janig W. Neomycin and gadolinium applied to an L5 spinal nerve lesion prevent mechanical allodynia-like behaviour in rats. Pain. 1997;70:155–165. doi: 10.1016/s0304-3959(96)03314-3. [DOI] [PubMed] [Google Scholar]
- 8.Boucher TJ, McMahon SB. Neurotrophic factors and neuropathic pain. Curr Opinion Pharmacol. 2001;1:66–72. doi: 10.1016/s1471-4892(01)00010-8. [DOI] [PubMed] [Google Scholar]
- 9.Burchiel KJ. Spontaneous impulse generation in normal and denervated dorsal root ganglia: sensitivity to alpha-adrenergic stimulation and hypoxia. Exp Neurol. 1984;85:257–272. doi: 10.1016/0014-4886(84)90139-0. [DOI] [PubMed] [Google Scholar]
- 10.Caffrey JM, Eng DL, Black JA, Waxman SG, Kocsis JD. Three types of sodium channels in adult rat dorsal root ganglion neurons. Brain Res. 1992;592:283–297. doi: 10.1016/0006-8993(92)91687-a. [DOI] [PubMed] [Google Scholar]
- 11.Delio DA, Reuhl KR, Lowndes HE. Ectopic impulse generation in dorsal root ganglion neurons during methyl mercury intoxication: an electrophysiological and morphological study. Neurotoxicology. 1992;13:527–540. [PubMed] [Google Scholar]
- 12.Devor M. Unexplained peculiarities of the dorsal root ganglion. Pain [Suppl] 1999;6:S27–S35. doi: 10.1016/S0304-3959(99)00135-9. [DOI] [PubMed] [Google Scholar]
- 13.Devor M, Seltzer Z. Pathophysiology of damaged nerves in relation to chronic pain. In: Wall PD, Melzack R, editors. Textbook of pain, Ed 4. Churchill Livingstone; Edinburgh: 1999. pp. 129–164. [Google Scholar]
- 14.Devor M, Wall PD. Cross excitation among dorsal root ganglion neurons in nerve injured and intact rats. J Neurophysiol. 1990;64:1733–1746. doi: 10.1152/jn.1990.64.6.1733. [DOI] [PubMed] [Google Scholar]
- 15.Devor M, Janig W, Michaelis M. Modulation of activity in dorsal root ganglion (DRG) neurons by sympathetic activation in nerve-injured rats. J Neurophysiol. 1994;71:38–47. doi: 10.1152/jn.1994.71.1.38. [DOI] [PubMed] [Google Scholar]
- 16.Hines M. A program for simulation of nerve equations with branching geometry. Int J Biomed Comput. 1989;15:69–76. doi: 10.1016/0020-7101(89)90007-x. [DOI] [PubMed] [Google Scholar]
- 17.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol (Lond) 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honmou O, Utzschneider DA, Rizzo MA, Bowe CM, Waxman SG, Kocsis JD. Delayed depolarization and slow sodium currents in cutaneous afferents. J Neurophysiol. 1994;71:1627–1637. doi: 10.1152/jn.1994.71.5.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jack JJB, Noble D, Tsien RW. Electric current flow in excitable cells. Clarendon; Oxford: 1983. Repetitive activity in excitable cells. pp. 305–378. [Google Scholar]
- 20.Kajander KC, Wakisaka S, Bennett GJ. Spontaneous discharge originates in the dorsal root ganglion at the onset of a painful peripheral neuropathy in the rat. Neurosci Lett. 1992;138:225–228. doi: 10.1016/0304-3940(92)90920-3. [DOI] [PubMed] [Google Scholar]
- 21.Kirk E. Impulses in dorsal spinal nerve rootlets in cats and rabbits arising from dorsal root ganglia isolated from the periphery. J Comp Neurol. 1974;2:165–176. doi: 10.1002/cne.901550203. [DOI] [PubMed] [Google Scholar]
- 22.Koerber HR, Mendell LM. Functional heterogeneity of dorsal root ganglion cells. In: Scott SA, editor. Sensory neurons. Oxford UP; New York: 1992. pp. 77–96. [Google Scholar]
- 23.Kuslich S, Ulstro C, Michael C. The tissue origin of low back pain and sciatica. Orthop Clin North Am. 1991;22:181–187. [PubMed] [Google Scholar]
- 24.Levine J, Reichling D. Peripheral mechanisms of inflammatory pain. In: Wall P, Melzack R, editors. Textbook of pain, Ed 4. Churchill Livingstone; Edinburgh: 1999. pp. 59–84. [Google Scholar]
- 25.Li Y, Dorsi M, Meyer R, Belzberg A. Mechanical hyperalgesia after an L5 spinal nerve lesion in the rat is not dependent on input from injured nerve fibers. Pain. 2000;85:493–502. doi: 10.1016/S0304-3959(00)00250-5. [DOI] [PubMed] [Google Scholar]
- 26.Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- 27.Lisney SJW, Devor M. Afterdischarge and interactions among fibers in damaged peripheral nerve in the rat. Brain Res. 1987;415:122–136. doi: 10.1016/0006-8993(87)90275-7. [DOI] [PubMed] [Google Scholar]
- 28.Liu C-N, Michaelis M, Amir R, Devor M. Spinal nerve injury enhances subthreshold membrane potential oscillations in DRG neurons: relation to neuropathic pain. J Neurophysiol. 2000a;84:205–215. doi: 10.1152/jn.2000.84.1.205. [DOI] [PubMed] [Google Scholar]
- 29.Liu C-N, Wall PD, Ben-Dor E, Michaelis M, Amir R, Devor M. Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain. 2000b;85:503–521. doi: 10.1016/S0304-3959(00)00251-7. [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Eschenfelder S, Blenk K-H, Janig W, Habler H-J. Spontaneous activity of axotomized afferent neurons after L5 spinal nerve injury in rats. Pain. 2000;84:309–318. doi: 10.1016/s0304-3959(99)00211-0. [DOI] [PubMed] [Google Scholar]
- 31.Lovinger DM, White G. Post-natal development of burst firing behavior and the low-threshold transient calcium current examined using freshly isolated neurons from rat dorsal root ganglia. Neurosci Lett. 1989;102:50–57. doi: 10.1016/0304-3940(89)90306-6. [DOI] [PubMed] [Google Scholar]
- 32.Luscher C, Streit J, Lipp P, Luscher H-R. Action potential propagation through embryonic dorsal root ganglion cells in culture. II. Decrease of conduction reliability during repetitive stimulation. J Neurophysiol. 1994;72:634–643. doi: 10.1152/jn.1994.72.2.634. [DOI] [PubMed] [Google Scholar]
- 33.Lyu YS, Park SK, Chung K, Chung JM. Low dose of tetrodotoxin reduces neuropathic pain behaviors in an animal model. Brain Res. 2000;871:98–103. doi: 10.1016/s0006-8993(00)02451-3. [DOI] [PubMed] [Google Scholar]
- 34.Mathers DA, Barker JL. Spontaneous voltage and current fluctuations in tissue cultured mouse dorsal root ganglion cells. Brain Res. 1984;293:35–47. doi: 10.1016/0006-8993(84)91450-1. [DOI] [PubMed] [Google Scholar]
- 35.Matzner O, Devor M. Na+ conductance and the threshold for repetitive neuronal firing. Brain Res. 1992;597:92–98. doi: 10.1016/0006-8993(92)91509-d. [DOI] [PubMed] [Google Scholar]
- 36.McLachlan E, Janig W, Devor M, Michaelis M. Peripheral nerve injury triggers noradrenergic sprouting within dorsal root ganglia. Nature. 1993;363:543–546. doi: 10.1038/363543a0. [DOI] [PubMed] [Google Scholar]
- 37.Michaelis M, Liu X-G, Janig W. Axotomized and intact muscle afferents but no skin afferents develop ongoing discharges of dorsal root ganglion origin after peripheral nerve lesion. J Neurosci. 2000;20:2742–2748. doi: 10.1523/JNEUROSCI.20-07-02742.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Na HS, Ko KH, Back SK, Sung B, Yoo DJ, Hong SK. Role of signals from dorsal root ganglion in neuropathic pain in a rat model. Neurosci Lett. 1995;288:147–150. doi: 10.1016/s0304-3940(00)01231-3. [DOI] [PubMed] [Google Scholar]
- 39.Noordenbos W. Pain. Elsevier; Amsterdam: 1959. [Google Scholar]
- 40.Nordin M, Balagué F. Biomechanics and ergonomics in disk herniation accompanied by sciatica. In: Weinstein JN, Gordon SL, editors. Low back pain: a scientific and clinical overview. American Academy of Orthopedic Surgeons; Rosemont, IL: 1996. pp. 23–48. [Google Scholar]
- 41.Nordin M, Nystrom B, Wallin U, Hagbarth K-E. Ectopic sensory discharges and paresthesiae in patients with disorders of peripheral nerves, dorsal roots and dorsal columns. Pain. 1984;20:231–245. doi: 10.1016/0304-3959(84)90013-7. [DOI] [PubMed] [Google Scholar]
- 42.Nowicki B, Haughton V, Schmidt TA, Lim T-H, An H, Riley L, III, Yu L, Hong J-W. Occult lumbar lateral spinal stenosis in neural foramen subjected to physiologic loading. Am J Neuroradiol. 1996;17:1605–1614. [PMC free article] [PubMed] [Google Scholar]
- 43.Pedroarena CM, Pose IE, MH Y, Chase MH, Morales FR. Oscillatory membrane potential activity in the soma of a primary afferent neuron. J Neurophysiol. 1999;82:1465–1476. doi: 10.1152/jn.1999.82.3.1465. [DOI] [PubMed] [Google Scholar]
- 44.Petersen M, Zhang J, Zhang J-M, LaMotte RH. Abnormal spontaneous activity and responses to norepinephrine in dissociated dorsal root ganglion cells after chronic nerve constriction. Pain. 1996;67:391–397. doi: 10.1016/0304-3959(96)03146-6. [DOI] [PubMed] [Google Scholar]
- 45.Puil E, Miura R, Spigelman I. Consequences of 4-aminopyridine applications to trigeminal root ganglion neurons. J Neurophysiol. 1989;62:810–820. doi: 10.1152/jn.1989.62.3.810. [DOI] [PubMed] [Google Scholar]
- 46.Rappaport ZH, Devor M. Trigeminal neuralgia: the role of self sustaining discharge in the trigeminal ganglion. Pain. 1994;56:127–138. doi: 10.1016/0304-3959(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 47.Scroggs RS, Todorovic SM, Anderson EG, Fox AP. Variation in IH, IR and ILEAK between acutely isolated adult rat dorsal root ganglion neurons of different size. J Neurosci. 1994;14:271–279. doi: 10.1152/jn.1994.71.1.271. [DOI] [PubMed] [Google Scholar]
- 48.Segev I, Burke RE. Compartmental models of complex neurons. In: Koch C, Segev I, editors. Methods in neuronal modeling: from ions to networks, Ed 2. MIT; Cambridge, MA: 1998. pp. 93–136. [Google Scholar]
- 49.Sheen K, Chung JM. Signs of neuropathic pain depend on signals from injured nerve fibers in a rat model. Brain Res. 1993;610:62–68. doi: 10.1016/0006-8993(93)91217-g. [DOI] [PubMed] [Google Scholar]
- 50.Shinder V, Govrin-Lippmann R, Cohen S, Belenky M, Ilin P, Fried K, Wilkinson HA, Devor M. Structural basis of sympathetic-sensory coupling in rat and human dorsal root ganglia following peripheral nerve injury. J Neurocytol. 1999;28:743–761. doi: 10.1023/a:1007090105840. [DOI] [PubMed] [Google Scholar]
- 51.Study RE, Kral MG. Spontaneous action potential activity in isolated dorsal root ganglion neurons from rats with a painful neuropathy. Pain. 1996;65:235–242. doi: 10.1016/0304-3959(95)00216-2. [DOI] [PubMed] [Google Scholar]
- 52.Utzschneider D, Kocsis J, Devor M. Mutual excitation among dorsal root ganglion neurons in the rat. Neurosci Lett. 1992;146:53–56. doi: 10.1016/0304-3940(92)90170-c. [DOI] [PubMed] [Google Scholar]
- 53.Villiere V, McLachlan EM. Electrophysiological properties of neurons in intact rat dorsal root ganglia classified by conduction velocity and action potential duration. J Neurophysiol. 1996;76:1924–1941. doi: 10.1152/jn.1996.76.3.1924. [DOI] [PubMed] [Google Scholar]
- 54.Wall PD, Devor M. Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain. 1983;17:321–339. doi: 10.1016/0304-3959(83)90164-1. [DOI] [PubMed] [Google Scholar]
- 55.Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Sodium channels and pain. Proc Natl Acad Sci USA. 1999;96:7635–7639. doi: 10.1073/pnas.96.14.7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.White G, Lovinger DM, Weight FF. Transient low-threshold Ca2+ current triggers burst firing through an afterdepolarizing potential in an adult mammalian neuron. Proc Natl Acad Sci USA. 1989;86:6802–6806. doi: 10.1073/pnas.86.17.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu N, Hsiao C-F, Chandler S. Membrane resonance and subthreshold membrane oscillations in mesencephalic V neurons: participants in burst generation. J Neurosci. 2001;21:3729–3739. doi: 10.1523/JNEUROSCI.21-11-03729.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J-M, Li H, Brull SJ. Perfusion of the mechanically compressed lumbar ganglion with lidocaine reduces mechanical hyperalgesia and allodynia in the rat. J Neurophysiol. 2000;84:798–805. doi: 10.1152/jn.2000.84.2.798. [DOI] [PubMed] [Google Scholar]
- 59.Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]