

Abstract
In most postmitotic neurons, expression or activation of proteins that stimulate cell cycle progression or DNA replication results in apoptosis. One potential exception to this generalization is neuroblastoma (NB), a tumor derived from the sympathoadrenal lineage. NBs often express high levels of N-myc, a proto-oncogene that can potently activate key components of the cell cycle machinery. Here, we show that in postmitotic sympathetic neurons, N-myc can induce S-phase entry while protecting neurons from death caused by aberrant cell cycle reentry. Specifically, these experiments demonstrate that expression of N-myc at levels similar to those in NBs caused sympathetic neurons to reenter S-phase, as monitored by 5-bromo-2-deoxyuridine incorporation and expression of cell cycle regulatory proteins, and rescued them from apoptosis induced by withdrawal of their obligate survival factor, nerve growth factor. The N-myc-induced cell cycle entry, but not enhanced survival, was inhibited by coexpression of a constitutively hypophosphorylated form of the retinoblastoma tumor suppressor protein, suggesting that these two effects of N-myc are mediated by separate pathways. In contrast, N-myc did not cause S-phase entry in postmitotic cortical neurons. Thus, N-myc both selectively causes sympathetic neurons to reenter the cell cycle and protects them from apoptosis, potentially contributing to their transformation to NBs.
Keywords: neuronal cell cycle, neuronal apoptosis, NGF, pRb, neuroblastoma, cortical neurons, sympathetic neurons, N-myc, S-phase
Neuroblastoma (NB), a malignant tumor of children, is derived from the neural crest and arises in the sympathoadrenal system. Because NB is the most common extracranial tumor of children but is very rare in adults, it is likely that malignant transformation occurs at some point during the development of the sympathetic nervous system, perhaps during the transition from a neural crest precursor to a postmitotic sympathetic neuron. In this regard, two key questions involve the mechanisms that (1) maintain sympathetic precursor and/or postmitotic neurons in the cell cycle and (2) allow these transformed sympathetic cells to escape the default apoptotic pathways that are normally triggered when progenitors fail to appropriately exit the cell cycle (for review, see Miller et al., 2000).
Most previous work examining the cell cycle machinery involved in neuronal terminal mitosis has focused on CNS neurons. These studies have demonstrated that the retinoblastoma tumor suppressor, pRb, plays an essential role in inducing terminal mitosis (Clarke et al., 1992;Jacks et al., 1992; Lee et al., 1992, 1994; Slack et al., 1998). pRb binds to the E2F family of transcription factors, blocking transcriptional activation by E2F and of cell cycle genes, thereby preventing entry into the S-phase of the cell cycle (for review, seeSlack and Miller, 1996). When this pRb-mediated cell cycle exit is perturbed, CNS neurons apoptose, likely by a p53-mediated default apoptotic pathway (Macleod et al., 1996; Slack et al., 1998). It is unclear whether similar mechanisms are essential for the transition from a cycling sympathetic neuroblast to a postmitotic sympathetic neuron. In fact, the mechanisms appear to be fundamentally different: sympathetic neuroblasts express a neuronal phenotype while still dividing (Rothman et al., 1978; Rohrer and Thoenen, 1987; Memberg and Hall, 1995), whereas most CNS progenitors express a neuronal phenotype only when they undergo terminal mitosis (Lauder and Bloom, 1974;Rothman et al., 1980; Koulakoff et al., 1983; Menezes and Luskin, 1994;Gloster et al., 1999).
Significantly more is known about the mechanisms regulating sympathetic neuron survival and apoptosis. These neurons require nerve growth factor (NGF) and activation of the TrkA/NGF receptor to survive and, in their absence, undergo apoptosis (for review, see Kaplan and Miller, 2000). Apoptosis can occur via two pathways: a p75NTR-Jun-N-terminal kinase (JNK)-dependent pathway (Majdan et al., 1997, 2001; Bamji et al., 1998) and a second pathway that involves the stimulation of the cell cycle machinery (Freeman et al., 1994; Farinelli and Greene, 1996;Park et al., 1996, 1997). These pathways use p53 and its family member p73 as checkpoints to regulate apoptosis (Aloyz et al., 1998; Pozniak et al., 2000a). Thus, both the survival and terminal mitosis of sympathetic neurons may rely on two well known tumor suppressors, p53 and pRb. It is therefore surprising that neither pRb nor p53 is mutated in NB, although p53 localization is sometimes altered (Vogan et al., 1993; Goldman et al., 1996). Instead, the N-myc proto-oncogene is often found to be amplified in NB, and this amplification is one of the most important molecular markers for poor prognosis in patients with this tumor (Brodeur et al., 1984).
N-myc is a transcription factor that regulates a number of genes involved in cell proliferation and apoptosis and directly binds to cell cycle regulators such as pRb (Rustgi et al., 1991). The N-myc homozygous null mouse dies in midgestation and has fewer neuronal cells in the peripheral ganglia (Stanton and Parada, 1992; Stanton et al., 1992; Sawai et al., 1993). In the developing avian andDrosophila PNS, overexpression of N-myc stimulates G1/S-phase progression, ventral migration, and neural differentiation (Wakamatsu et al., 1997). In mice, the sequestration in the cytoplasm and thus inactivation of N-myc may be required for the neurotrophin-induced cell cycle arrest of sensory precursor cells (ElShamy et al., 1998). Consistent with their role as cell cycle regulators during development, myc family members are highly expressed in the childhood tumors NB, medulloblastoma, and Wilms' tumor (Nisen et al., 1986). However, the precise mechanisms whereby N-myc mediates its normal physiological role or its pathological oncogenic role are not clear.
Here, we have asked whether N-myc is capable of inducing postmitotic sympathetic neurons to reenter the cell cycle. We show that N-myc overexpression stimulated both cell cycle progression and survival of sympathetic neurons but had no effect on S-phase progression in postmitotic cortical neurons. Thus, sympathetic neurons are not completely locked out of the cell cycle, and N-myc can protect cycling neurons from apoptosis, thereby potentially contributing to malignant transformation.
MATERIALS AND METHODS
Sympathetic neuron cultures. Primary neuronal cultures were prepared from postnatal day 1 Sprague Dawley rat sympathetic superior cervical ganglia essentially as described previously (Ma et al., 1992). For immunocytochemistry and nuclear staining, MTT assays, or Western blot analysis, 6,000–10,000 cells per well were plated in 8-well chamber slides, 3000–5000 cells per well were plated in 96-well chamber slides, or 0.5–1 × 105 cells per well were plated in 6-well plates, respectively. All the plates were rat tail collagen-coated, and the cells were maintained in Ultraculture medium containing 2 mm glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (all from BioWhittaker, Walkersville, MD), and 3% rat serum (Harlan Bioproducts, Madison, WI). Neurons were initially cultured for 5 d in the presence of 50 ng/ml NGF (Cedarlane, Hornby, Ontario, Canada) and 7 μm cytosine arabinoside (CA). LAN-1–15N human neuroblastoma cells were the kind gift of Dr. C. Thiele (National Institutes of Health, Bethesda, MD).
Cortical neuron cultures. Primary cortical neuron cultures were obtained from embryonic day (E) 16–E17 mice. The meninges were removed, and cortical tissue was transferred into Neurobasal media (Invitrogen) containing 500 μM glutamine, 2% B27 supplement, and 1% penicillin–streptomycin (Invitrogen). The tissue was triturated into a single-cell suspension, and cells were plated in four-well chamber slides precoated with laminin and poly-d-lysine (Collaborative Research) at a density of 105 per well. Three days after plating, half of the media was changed with fresh media supplemented with 7 μm CA (final concentration). Three days later, all media was replaced with fresh media without CA. Infection with recombinant adenovirus was at 6 d after plating.
Recombinant adenovirus infections. The adenovirus coding for human N-myc was constructed as described (He et al., 1998). Briefly, the N-myc cDNA was cloned into the pAdTrack shuttle vector, recombined with the pAdEasy adenoviral vector in bacteria, transfected, and amplified in human embryonic kidney (HEK)293 cells. The viruses were purified on CsCl gradients and titered in HEK293 cells, as we have described previously (Slack et al., 1996). The other adenoviruses have been described previously and encode a constitutively hypophosphorylated form of pRb (Chang et al., 1995; Toma et al., 2000), p53 (Slack et al., 1996; Pozniak et al., 2000a), kinase-inactive TrkA (Vaillant et al., 1999), green fluorescent protein (GFP) (Pozniak et al., 2000a; Toma et al., 2000) (Quantum Biotechnologies), andEscherichia coli β-galactosidase (Slack et al., 1996) (gift of Dr. Frank Graham, McMaster University, Canada). The adenoviruses were used to infect cultured sympathetic neurons at various concentrations of infective virus particles added per cell (multiplicity of infection; MOI). Infections of sympathetic neurons were performed by replacing the culture media with viruses diluted in Ultraculture containing no serum or CA and with glutamine, penicillin, streptomycin, and NGF as described above. Infections of cortical neurons were similar except that viruses were diluted in Neurobasal medium containing glutamine, B27, penicillin, and streptomycin. After 24 hr the media was replaced with the same media without virus.
Survival experiments. Neurons were cultured for 5 d and infected as described above. To measure potential virus-induced cell death, the neurons were maintained for an additional 4 d after infection and analyzed. The MTT assays, which measure mitochondrial function and reflect cell viability and survival, were performed in 96-well plates, each condition as a triplicate as described previously (Bamji et al., 1998). Data from these assays were expressed as a percentage of MTT signal obtained in 10 ng/ml NGF (100% survival) versus zero NGF (0% survival). For the neurotrophin withdrawal experiments, neurons were infected with recombinant adenovirus, and 2 d after infection, NGF was washed out by four 1 hr washes of Ultraculture alone. Neurons were then maintained in Ultraculture containing penicillin, streptomycin, and glutamine, with or without NGF, for 24–48 hr before analysis. MTT assays were performed 2 d after NGF withdrawal as indicated above. For the assessment of live/dead cells using Trypan blue, neurons were withdrawn from NGF for 2 d and stained with 20% Trypan blue, and the percentage of white (live) and blue (dead) of 300 or more cells in three random microscope fields was quantitated. For analysis of apoptotic nuclei with Hoechst, neurons were withdrawn from NGF for 48 hr, fixed with 4% paraformaldehyde (PFA), and then stained with Hoechst 33258 (Sigma, St. Louis, MO) diluted 1:3000 in PBS. Quantitation was as for Trypan blue analysis. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was performed at 24–48 hr after NGF withdrawal, essentially as described previously (Vaillant et al., 1999;Pozniak et al., 2000a). Briefly, neurons were fixed for 15 min at room temperature with 4% PFA, 0.25% glutaraldehyde (Fluka AG), and 0.2% Triton X-100 (Sigma) in 40 mm PIPES, 2.5 mm EGTA, 1 mmMgCl2, and washed with PBS. The cells were then incubated for 1 hr at 37°C in the TUNEL reaction mix of 20 μl TdT buffer/1.5 μl terminal deoxynucleotidyl transferase (TdT) enzyme (both from Promega Corporation) and 1 μl of biotin-16-dUTP (Boehringer Mannheim). After the TUNEL reaction, cells were washed, incubated for 1 hr with streptavidin-conjugated Cy3 secondary antibody (1:500; Jackson ImmunoResearch Laboratories) in PBS at room temperature, washed, Hoechst-labeled, and mounted. Digital image acquisition and analysis were performed with the Northern Eclipse software (Empix Inc.) using a Sony XC-75CE charge-coupled device video camera.
5-Bromo-2-deoxyuridine incorporation and immunocytochemistry. For immunocytochemistry and 5-bromo-2-deoxyuridine (BrdU) incorporation analysis, sympathetic neurons were plated in collagen-coated eight-well chamber slides and cultured for 5 d as described above, and cortical neurons were plated in four-well chamber slides and cultured as described. On the fifth and sixth days, for sympathetic and cortical neurons, respectively, cells were infected with the indicated virus or virus combination. For N-myc immunostaining, the cells were fixed in 4% paraformaldehyde, permeabilized in 0.5% Triton X-100 in PBS, rinsed, and incubated overnight with 1 μg/ml anti-human N-myc monoclonal antibody (Oncogene Research Products). After the fixed cells were washed with PBS, the secondary anti-mouse-Cy3 antibody was added for 1 hr, the cells were again washed, and the nuclei were labeled with Hoechst dye. For the analysis of DNA synthesis, 10 μm BrdU (Boehringer Mannheim) in fresh media was added for 24–48 hr. Neurons were fixed with ethanol, treated with 2N HCl for 10 min, 0.1 mNa2B4O7, pH 8.5, for 10 min, washed with PBS, and incubated either with monoclonal anti-BrdU (1:100; Chemicon) and polyclonal anti-neurofilament M (1:200; Chemicon) or with polyclonal sheep anti-BrdU (1:150; Research Diagnostics Inc.) and monoclonal anti-microtubule-associated protein 2 (MAP2) (1:400; Sigma) in PBS overnight at 4°C. After they were washed in PBS, the cells were incubated with secondary FITC or Cy3-conjugated anti-sheep antibody (1:300; Jackson ImmunoResearch) for 1 hr at room temperature, washed, and labeled with Hoechst (1:3000) to stain nuclei. In some experiments, 4, 8, or 16 μm cytosine arabinoside (a DNA polymerase inhibitor) was added at the same time as BrdU. Data acquisition and image analysis were performed as described above; for each treatment, at least 300 cells in each of three to six random fields were analyzed. For double labeling of neurons and astrocytes in cortical neuron cultures, cells were incubated with a monoclonal antibody to MAP2 (1:400; Sigma) and a polyclonal antibody to GFAP (1:800; Sigma). X-gal staining for β-galactosidase was performed essentially as described (Slack et al., 1996).
Immunoprecipitations and Western blot analysis. Cells were cultured and infected, and where indicated, NGF was withdrawn as described above. The cultured cells were rinsed briefly in cold TBS and then lysed in TBS lysis buffer (137 mm NaCl, 20 mm Tris, pH 8, 1% v/v NP-40, and 10% glycerol) or in urea extraction buffer (8 m urea, 4% 3-[(3-deoxycholic acid (cholamidopropyl)dimethylammonio]-1-propanesulfonate), 40 mm Tris, pH 8.0) supplemented with Mini Complete protease inhibitor mixture (Boehringer Mannheim) and 1.5 mm sodium vanadate. Lysates were scraped into Eppendorf tubes and rocked for 10 min at 4°C. Samples were cleared by centrifugation, and the protein concentration was determined by the BCA assay (Pierce Chemical) using BSA as a standard. For SDS-PAGE, samples were boiled in sample buffer, separated by 7.5–15% SDS-PAGE gradient gels, and transferred to nitrocellulose membranes. The filters were blocked with 5% skim milk powder in TBS-T (TBS + 1% Tween 20) for 1–2 hr and incubated with primary antibodies against p75NTR (1:1000;Promega), phospho-JNK (1:5000; Promega), MAPK (1:500; Santa Cruz Biotechnology), N-myc (1:1000; Oncogene Research Products), α-tubulin (1:5000; Oncogene Sciences), cyclin-dependent kinase 2 (cdk2) (1:1000, Santa Cruz Biotechnology), or cyclin E (2 μg/ml; Upstate Biotechnology).
RESULTS
Posmitotic sympathetic neurons enter S-phase of the cell cycle in response to overexpression of N-myc
To determine whether overexpression of N-myc is sufficient to perturb the cell cycle status and/or survival of postmitotic sympathetic neurons, we constructed a recombinant adenovirus expressing human N-myc and GFP under the control of separate but identical cytomegalovirus promoters (Fig.1A). We then used this virus to transduce neonatal, postmitotic sympathetic neurons that had been cultured for 5 d in the presence of 50 ng/ml NGF. A substantial number of neurons were transduced by infection with this virus at MOIs as low as 50, and this number increased with increasing viral MOIs so that as many as 95% of the neurons were detectably infected at an MOI of 300, as indicated by expression of GFP (Fig.1B). Confirmation that the GFP-positive neurons also expressed increased levels of N-myc was obtained by performing immunocytochemistry for N-myc on the infected neurons (Fig.1D). All cells that expressed GFP also expressed high levels of N-myc, and much of this N-myc was localized to the nucleus, as would be predicted. Western blot analysis for N-myc confirmed this morphological data and demonstrated that the virus expressed a recombinant protein of the appropriate molecular weight of 64 kDa (Fig.1C). Importantly, these Western blots also demonstrated that N-myc is endogenously expressed in cultured postmitotic sympathetic neurons. Moreover, the levels of human N-myc expression in cultures obtained at MOIs of 100–300 were similar to those seen in the human neuroblastoma cell line LAN-1–15N (Ciccarone et al., 1989), where the N-myc gene is amplified (Fig. 1C). No conclusions, however, can be drawn regarding the relative level of human N-myc overexpression in sympathetic neurons, because the antibody may not cross-react equally well with the human (adenoviral) and rodent (endogenous) proteins.
Fig. 1.

Characterization of a recombinant adenovirus expressing human N-myc. A, Adenovirus construct encoding human N-myc and green fluorescent protein under separate but identical CMV promoters. B, Adenovirus N-myc (AdN)-myc is expressed in sympathetic neurons. Fluorescent micrograph of GFP expression in postmitotic sympathetic neurons 72 hr after infection with 300 MOI of AdN-myc. C, Western blot of equal amounts of protein isolated from LAN-1–15N neuroblastoma cells and from sympathetic neurons infected for 48 hr with 0–300 MOI of N-myc adenovirus and probed with an antibody for N-myc. Note that uninfected sympathetic neurons express N-myc and that LAN-1–15N cells express levels of N-myc similar to sympathetic neurons infected with 100–300 MOI of the N-myc adenovirus. D, Photomicrographs of sympathetic neurons infected with AdN-myc and then analyzed for expression of GFP and immunocytochemically for expression of N-myc (α-N-myc). Cells were also stained with the nuclear dye Hoechst 33258 to identify all of the cells in the field. Note that cells that are GFP positive are also overexpressing N-myc and that the overexpressed N-myc is primarily nuclear. Scale bar, 50 μm.
Using this recombinant adenovirus, we determined whether expression of N-myc at levels similar to those seen in NBs was sufficient to cause cell cycle entry in postmitotic sympathetic neurons. To ask this question, we infected neurons with the N-myc adenovirus and then added BrdU to the NGF-containing media 1 d later to label newly synthesized DNA. We then performed double-label immunocytochemistry 48 hr later for BrdU and for neurofilament-M, which is specific for neurons. As predicted, when neurons were mock infected or were infected with 200 MOI of control adenoviruses expressing β-galactosidase (Fig.2A) or GFP (data not shown), we detected no or very few neurons that were positive for BrdU, although some of the small percentage of non-neuronal cells present in these cultures were positive (Fig. 2A). Confirmation that most of the sympathetic neurons were transduced with 200 MOI of the β-galactosidase adenovirus, as we have reported previously (Slack et al., 1996), was obtained by staining the infected cultures with X-gal (Fig. 2E).
Fig. 2.
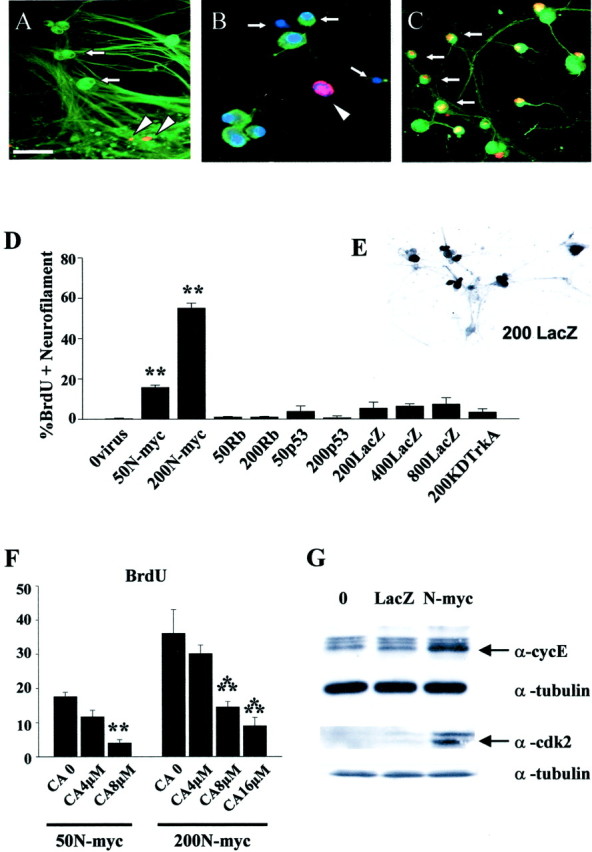
Overexpression of N-myc in postmitotic sympathetic neurons leads to S-phase entry.A–C, Photomicrographs of sympathetic neurons infected with recombinant adenovirus, pulse-labeled with BrdU, and then immunocytochemically analyzed for expression of neurofilament-M (green) and BrdU (red). In some cases, cells were also labeled with Hoechst to label all nuclei (blue). A, AdLacZ-expressing neurons do not incorporate BrdU. Neurons were infected with 200 MOI of a β-galactosidase adenovirus and maintained in 10 ng/ml NGF for 3 d. Note that none of the neurofilament-positive neurons are BrdU positive (arrows) and that the only BrdU-positive cells are neurofilament-negative non-neuronal cells (arrowheads).B, Uninfected neurons that were withdrawn from NGF in the presence of BrdU for 30 hr and then analyzed immunocytochemically. Note that a neurofilament-negative non-neuronal cell with an intact nucleus has incorporated BrdU (arrowhead) but that neurons in different phases of apoptosis are not BrdU positive (arrows). C, AdN-myc-expressing neurons incorporate BrdU. Neurons were infected with 200 MOI of N-myc adenovirus and maintained in the presence of NGF. One day later BrdU was added, and cells were analyzed immunocytochemically 48 hr later. Note that under these conditions, many of the neurofilament-positive neurons are BrdU positive (arrows). Scale bar:A, C, 100 μm; B, 50 μm. D, Quantitation of the percentage of cells positive for both BrdU and neurofilament after infection with various MOIs of recombinant adenovirus in the presence of NGF for 1 d, followed by the addition of BrdU for an additional 48 hr. Neurons were infected with recombinant adenoviruses expressing N-myc, constitutively hypophosphorylated pRb (Rb) (Toma et al., 2000), p53 (Slack et al., 1996), β-galactosidase (LacZ), or a kinase-inactive mutant of TrkA (KDTrkA) (Vaillant et al., 1999). Values derive from counts of at least 300 cells in four or more random microscope fields per condition, and error bars represent the SD of the mean. Similar results were obtained in four independent experiments. **p < 0.01; Student'st test. Note that only N-myc led to BrdU incorporation in sympathetic neurons. E, Staining for β-galactosidase (LacZ) in cultures of sympathetic neurons transduced with 200 MOI of the β-galactosidase adenovirus. Note that most of the sympathetic neurons are positively stained. F, Inhibition of N-myc stimulated cell cycle entry by DNA polymerase inhibition. Percentage of BrdU-positive neurons 48 hr after infection with 50 or 200 MOI AdN-myc, cultured in the presence of BrdU with or without 4, 8, or 16 μmcytosine arabinoside for the final 24 hr. Quantitation was performed as in D. Similar results were obtained in three independent experiments. Concentrations of CA shown here did not increase apoptotic cell morphology over the time of the experiment. **p < 0.05; ***p < 0.001.G, AdN-myc expression increases the levels of the S-phase markers cyclin E and cdk2. Western blot analysis of equal amounts of protein from sympathetic neurons that were uninfected (0) or infected with 200 MOI of adenoviruses expressing either β-galactosidase (LacZ) or N-myc for 2 d. Blots were reprobed for total α-tubulin as a loading control. Similar results were obtained in two independent experiments.
In contrast, when neurons cultured in the presence of NGF were infected with 200 MOI of the N-myc adenovirus, most of the neurofilament-positive neurons were also BrdU positive (Fig.2C). Quantitation of this result in four independent experiments revealed that up to 58% of the neurons were positive after a 48 hr pulse of BrdU (Fig. 2D). In contrast, no increase in neuronal BrdU incorporation was observed in neurons infected with up to 800 MOI of the β-galactosidase adenovirus (Fig.2D). To confirm that this enhanced BrdU incorporation was not caused simply by DNA damage or fragmentation, we performed similar experiments with sympathetic neurons that were withdrawn from NGF, a treatment that causes DNA fragmentation coincident with cellular apoptosis. NGF withdrawal never led to BrdU incorporation in sympathetic neurons (Fig. 2B). Similarly, no BrdU incorporation was observed after infection with adenoviruses expressing p53 (Aloyz et al., 1998) or a kinase-inactive form of TrkA (Vaillant et al., 1999) (Fig. 2D), both of which cause sympathetic neuron apoptosis in the presence of NGF.
As an additional control for the specificity of BrdU incorporation, we determined whether we could rescue this effect by inhibiting DNA polymerase activity. To perform these experiments, neurons were infected with 50 or 200 MOI of N-myc adenovirus, and 4–16 μm DNA polymerase inhibitor CA was added at the same time as the BrdU. Twenty-four hours later, neurons were immunocytochemically double labeled for neurofilament and BrdU. Quantitation of these experiments (Fig. 2F) revealed that 50 and 200 MOI of N-myc caused ∼17 and 35%, respectively, of the neurons to incorporate BrdU and that CA inhibited this effect in a concentration-dependent manner.
To confirm that the enhanced BrdU incorporation reflected S-phase entry, we analyzed expression of two proteins known to be induced during S-phase of the cell cycle: cyclin-dependent kinase 2 (cdk2) and cyclin E (for review, see Ekholm and Reed, 2000). Specifically, sympathetic neurons were infected with 200 MOI of adenoviruses encoding either β-galactosidase or N-myc and were lysed 2 d later. Western blot analysis of equal amounts of protein revealed that levels of both of these proteins were increased in neurons overexpressing N-myc (Fig. 2G).
Finally, we determined whether N-myc overexpression caused sympathetic neurons to divide. Mitotic figures were never observed during extensive time-lapse analysis of multiple cultures, nor were dividing neuronal cell bodies observed. Because these analyses were performed at MOIs of N-myc adenovirus where >50% of neurons incorporated BrdU, we conclude that N-myc overexpression promotes S-phase entry but is insufficient to cause postmitotic neurons to proceed into mitosis itself.
Overexpression of N-myc inhibits sympathetic neuron apoptosis after NGF withdrawal
One hypothesis postulates that postmitotic neurons inevitably undergo apoptosis when they are forced to reenter the cell cycle. We therefore asked whether N-myc caused sympathetic neuron apoptosis coincident with cell cycle entry. Sympathetic neurons were infected with the N-myc adenovirus in the presence of NGF, and 4 d later we performed MTT assays, which measure mitochondrial function. Results of this experiment demonstrated that overexpression of N-myc had no adverse effects on sympathetic neuron survival (Fig.3A), despite the fact that it caused S-phase entry. Similar results were obtained when neurons were infected with a control GFP adenovirus at the same MOIs (Fig.3A). Thus, in sympathetic neurons, cell cycle reentry is not obligately coupled to apoptosis.
Fig. 3.

N-myc promotes sympathetic neuron survival after NGF withdrawal. A, AdN-myc does not decrease the survival of sympathetic neurons in the presence of NGF. MTT survival assays of sympathetic neurons selected in NGF, infected with 100–400 MOI of N-myc, GFP, or hypophosphorylated pRb adenovirus, maintained in NGF, and assayed 4 d after infection are shown. All conditions were performed in triplicate, and error bars represent the SD of the mean. Values derive from one representative experiment of three and are normalized to those obtained for uninfected neurons maintained in 10 ng/ml NGF, which are considered to be 100% survival. No significant alterations are seen in any of the conditions.B–D, Adenovirus-mediated expression of N-myc rescues sympathetic neurons from apoptosis induced by NGF withdrawal, as monitored by MTT assays (B), nuclear morphology (C), and Trypan blue exclusion (D). In all experiments, neurons were infected with adenovirus, withdrawn from NGF 2 d later, and analyzed after an additional 2 d. B, MTT assays of neurons infected with 80–320 MOI N-myc, GFP, or hypophosphorylated pRb adenovirus. Values were normalized to those obtained for uninfected neurons either maintained in NGF (100%) or withdrawn from NGF (1%). Values represent the average of three independent experiments, each of which was performed in triplicate. *p < 0.05; Student's t test comparing results obtained with the N-myc versus control GFP adenovirus. C,D, Percentage of living cells or nonapoptotic cells counted by Hoechst-labeled intact nuclei (C) or Trypan blue exclusion (D) after virus infection and NGF withdrawal. For both methods, values were obtained by counting three randomly selected microscope fields in each condition, and the error bars represent the SD of the mean. Values are representative results of one of two independent experiments. **p< 0.01; Student's t test comparing results obtained using the N-myc versus control β-galactosidase (LacZ) adenovirus.
We then asked whether overexpression of N-myc might provide a survival advantage to sympathetic neurons, as might be predicted by its role in promoting neuroblastoma. To perform these experiments, neurons were infected with 80–320 MOI of N-myc adenovirus; 2 d later they were withdrawn from NGF, and MTT assays were performed after an additional 2 d. This analysis revealed that N-myc significantly inhibited NGF withdrawal-induced apoptosis in a concentration-dependent manner (Fig.3B). Such a rescue was never observed with adenoviruses expressing either GFP (Fig. 3B) or β-galactosidase (data not shown). The rescue observed with N-myc overexpression was similar to or better than that observed with pro-survival proteins such as activated Ras (Mazzoni et al., 1999).
To confirm that the MTT assay was measuring neuronal survival and to determine the percentage of neurons that were rescued from apoptosis, we performed two additional assays. Neurons were infected with 50 or 200 MOI N-myc adenovirus, were withdrawn from NGF after 2 d, and then the percentage of live cells was determined 48 hr later by counting neurons with intact, Hoechst-positive nuclei (Fig.3C) or by Trypan blue exclusion (Fig. 3D). Both assays demonstrated a dose-dependent rescue of 65–85% of the neurons by overexpression of N-myc (Fig. 3C,D). As predicted, no such rescue was observed with similar MOIs of the β-galactosidase adenovirus. Finally, to demonstrate that N-myc was actually rescuing neurons from apoptosis, we performed TUNEL. Neurons were infected with adenoviruses expressing either N-myc and GFP or GFP alone, were withdrawn from NGF, and assayed 24 hr later. This analysis demonstrated that for neurons infected with the GFP virus, many of the GFP-positive cells were also TUNEL positive (Fig.4A–C). In contrast, for neurons infected with the N-myc/GFP virus, few of the GFP-positive cells were TUNEL positive (Fig.4D–F). Quantitation of the total number of TUNEL-positive cells confirmed that overexpression of N-myc significantly rescued sympathetic neurons from apoptosis after NGF withdrawal (Fig. 4G).
Fig. 4.

N-myc overexpression inhibits sympathetic neuron apoptosis after NGF withdrawal. A–F, TUNEL of sympathetic neurons infected with adenoviruses expressing GFP (AdGFP; A–C) or N-myc (this virus also expresses GFP) (AdN-myc;D–F) and withdrawn from NGF for 48 hr. Photomicrographs show TUNEL-positive nuclei (A, D), GFP-expressing cells (B, E), and Hoechst-positive nuclei (C, F) in the same fields (A–C andD–F). Arrowsindicate the same neurons in each field. Note that neurons infected with the adenovirus expressing only GFP (A–C) are TUNEL positive and display shrunken, apoptotic nuclei, but that those infected with the virus expressing both GFP and N-myc (D–F) are not TUNEL positive. Scale bar, 100 μm. G, Percentage of TUNEL-positive cells 24 hr after NGF withdrawal. Neurons were infected with various MOIs of the N-myc or GFP adenoviruses, and the total number of TUNEL-positive nuclei in three randomly selected fields was counted. The values represent the average of two independent experiments, and error bars represent the SD of the mean. **p < 0.01 in Student's t test comparing apoptotic (TUNEL-positive) cell percentage after infection with AdN-myc versus AdGFP.
Expression of constitutively hypophosphorylated pRb inhibits the S-phase entry caused by N-myc but has no effect on neuronal survival
One mechanism whereby N-myc is thought to regulate S-phase entry involves the retinoblastoma tumor suppressor protein (Rustgi et al., 1991; Lasorella et al., 2000). pRb is a key neuronal cell cycle regulator (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992,1994; Slack et al., 1998; Toma et al., 2000). Hypophosphorylated pRb binds to and inhibits proteins that promote S-phase entry, such as the E2F family (for review, see Slack and Miller, 1996). On this basis, we predicted that if in these experiments N-myc promotes S-phase entry via a mechanism involving pRb, then this S-phase entry should be inhibited by coincident overexpression of a mutant pRb that is constitutively hypophosphorylated. To test this prediction, sympathetic neurons were coinfected with the N-myc adenovirus and with increasing MOIs of a recombinant adenovirus expressing constitutively hypophosphorylated pRb (Chang et al., 1995; Toma et al., 2000).
To perform these experiments, we first asked whether expression of constitutively hypophosphorylated pRb on its own had any effect on sympathetic neuron S-phase entry. Neurons were infected with 50 or 200 MOI of pRb adenovirus, and then 1 d later BrdU was added for an additional 48 hr. Double-label immunocytochemistry revealed that few or none of the neurofilament-positive neurons were BrdU positive (Fig.2D). Having established that hypophosphorylated pRb had no effects on its own, we then determined whether it could rescue N-myc-mediated S-phase entry. Neurons were coinfected with 50 MOI N-myc and 200 MOI pRb adenoviruses for 1 d and then were incubated with BrdU for an additional 2 d. Double-label immunocytochemistry revealed that 50 MOI N-myc adenovirus caused ∼15% of sympathetic neurons to incorporate BrdU and that the pRb virus decreased this number in a concentration-dependent manner so that at 200 MOI pRb, <4% of neurons were BrdU positive (Fig.5A). In contrast, coinfection with 200 MOI of the β-galactosidase adenovirus had no significant effect (Fig. 5A). Thus, hypophosphorylated pRb rescued the N-myc-mediated S-phase entry.
Fig. 5.

Constitutively hypophosphorylated pRb inhibits the S-phase entry, but not the survival, caused by N-myc.A, Constitutively hypophosphorylated pRb rescues the N-myc-induced BrdU incorporation. Neurons were coinfected with 50 MOI N-myc adenovirus and 50–200 MOI pRb adenovirus, incubated with BrdU 1 d later, and then analyzed immunocytochemically for BrdU and neurofilament-M after an additional 2 d. As a control, neurons were coinfected with 200 MOI of a β-galactosidase adenovirus (LacZ). Quantitation was performed as described in Figure 2D. *p < 0.05; Student'st test, comparing N-myc plus pRb versus N-myc plus β-galactosidase. B, pRb has no effect on N-myc-induced survival after NGF withdrawal. Neurons were infected with 200 MOI N-myc in the presence or absence of 100 or 200 MOI hypophosphorylated pRb adenovirus, and 2 d later NGF was withdrawn. Neurons were stained with Hoechst, and intact, nonapoptotic nuclei were quantitated by counting at least 300 cells in each of three random fields.C, AdN-myc decreases p75NTR expression and inhibits the activation of JNK. Immunoblots of sympathetic neurons infected with 100 or 400 MOI of N-myc adenovirus in the presence (right panel) or absence (left panel) of NGF. Left panel, Neurons were infected with N-myc adenovirus for 48 hr and then withdrawn from NGF for an additional 24 hr. Equal amounts of protein were analyzed for expression of the p75NTR (α-p75), the activated, phosphorylated form of JNK (α-P-JNK), or for total MAP kinase protein levels (α-MAPK). Note that although p75NTR and phospho-JNK levels were reduced on infection with N-myc, total MAP kinase levels remained the same. Right panel, Neurons were infected with N-myc adenovirus and maintained in NGF for 72 hr before Western blot analysis. Immunoblots were first probed with an antibody to p75NTR and then reprobed with an antibody for total α-tubulin to demonstrate that similar amounts of protein were present in each lane. Note the concentration-dependent decrease in p75NTR levels in neurons infected with the N-myc adenovirus. Similar results were obtained in four independent experiments.
We then asked whether the survival effects seen with N-myc overexpression also involved pRb. Initially, we determined whether hypophosphorylated pRb itself had effects on sympathetic neuron survival. Neurons were infected with MOIs of the pRb adenovirus ranging from 80 to 400 and were then either maintained in NGF or withdrawn from NGF for 2 d before MTT assays. These experiments demonstrated that expression of hypophosphorylated pRb on its own had no effect on neuronal survival in either the presence or absence of NGF (Fig.3A,B). We then asked whether the N-myc-mediated survival was affected by constitutively hypophosphorylated pRb. To perform these experiments, sympathetic neurons were coinfected with 200 MOI N-myc and 100 or 200 MOI constitutively hypophosphorylated pRb virus for 2 d and were then withdrawn from NGF for an additional 24–48 hr. Staining of the neurons with Hoechst and quantitation of apoptotic nuclei revealed that coexpression of hypophosphorylated pRb had no effect on the decrease in apoptosis observed when N-myc was overexpressed (Fig. 5B). Thus, at a ratio of N-myc to pRb virus of 1:1, hypophosphorylated pRb rescued the N-myc-mediated S-phase entry but not its effects on neuronal survival, suggesting that the pathways used by N-myc to promote S-phase entry versus enhanced survival are distinct.
The apoptotic death of sympathetic neurons after NGF withdrawal requires the p75 neurotrophin receptor (Bamji et al., 1998), which directly activates a JNK-p53 apoptotic pathway (Aloyz et al., 1998). Moreover, the absolute cellular levels of p75NTR are a key determinant of its ability to cause neuronal apoptosis (Barrett et al., 1998;Majdan et al., 2001). Because N-myc is a transcription factor, it could potentially regulate sympathetic neuron apoptosis by regulating levels of this apoptotic receptor. To test this possibility, sympathetic neurons were cultured in 50 ng/ml NGF and then infected with 100 or 400 MOI of the N-myc adenovirus for 2 d. Western blot analysis revealed that N-myc overexpression led to a substantial decrease in the levels of p75NTR (Fig. 5C). Reprobing of the same blots for proteins such as α-tubulin or the MAP kinases revealed that this decrease was specific to p75NTR (Fig. 5C). Similar results were obtained for neurons that were withdrawn from NGF for 24 hr. p75NTR levels were significantly lower in neurons overexpressing N-myc, whereas levels of the MAP kinases were unchanged (Fig. 5C). Interestingly, concomitant with this decrease in p75NTR, levels of phosphorylated JNK, a key mediator of sympathetic neuron apoptosis after NGF withdrawal (Pozniak et al., 2000b; Harding et al., 2001), were also reduced (Fig. 5C). In contrast, levels of p75NTR or phospho-JNK were unaltered by infection with a control, GFP-expressing adenovirus (data not shown). Thus, one pRb-independent mechanism that could account for the enhanced survival observed when N-myc was overexpressed involves downregulation of the p75NTR–JNK apoptotic pathway.
N-myc does not cause S-phase entry in postmitotic cortical neurons
We next focused on postmitotic cortical neurons to ask whether N-myc could induce S-phase entry in CNS neurons. To perform these experiments, we used cultures of cortical neurons that were primarily free of glia. Specifically, cortical neurons were cultured at E16–17 and then maintained for 6 d to ensure that all neurons in the culture were postmitotic. To characterize the cultures, we immunostained them for MAP2, a neuron-specific protein, and for GFAP, an astrocyte-specific protein. This analysis revealed that >95% of the cells in these cultures were neurons, with only a small number of astrocytes present (Fig.6A–C). To ensure that the neurons were truly postmitotic, we also pulsed the cultures with BrdU for 48 hr; virtually none of the MAP2-positive neurons were BrdU positive (data not shown).
Fig. 6.
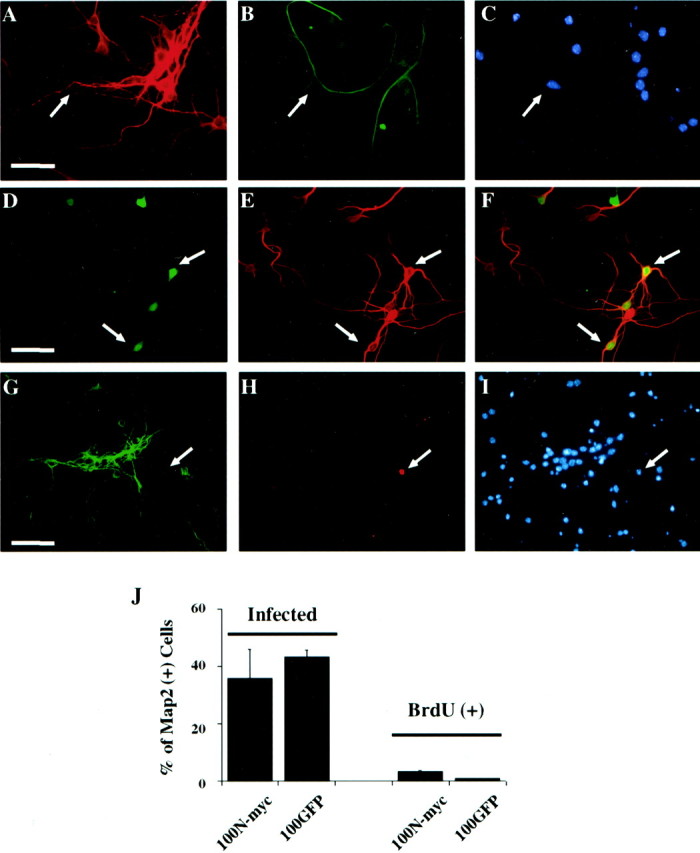
N-myc does not cause BrdU incorporation in postmitotic cortical neurons. A–C, Immunocytochemical analysis of postmitotic cortical neuron cultures. Neurons were cultured as described in Results and double labeled with antibodies for neuron-specific MAP2 (A) and astrocyte-specific GFAP (B). Cells were also stained with Hoechst to show cell nuclei (C). The same field is shown in all three panels, and arrowsindicate the occasional astrocyte present in the cultures. Note that most fields did not contain any GFAP-positive cells.D–F, Cortical neurons were infected with 100 MOI N-myc adenovirus (which also expresses GFP) and then immunostained for MAP2. Visualization of GFP (D) revealed those cells that were transduced with the N-myc adenovirus, whereas immunostaining for MAP2 (E) indicated that the infected cells were neurons. F is the merged image resulting from D and E.Arrows indicate cells that were positive for both GFP and MAP2. G–I, Cortical neurons were infected with 100 MOI N-myc adenovirus and then incubated in the presence of BrdU for 24 hr before double-label immunocytochemical analysis for MAP2 (G) and BrdU (H). The cells were also stained with Hoechst (I). The same field is shown in all three panels, and the arrow indicates a non-MAP2-positive non-neuronal cell that is BrdU positive.J, Left two bars, Quantitation of data showing that N-myc fails to promote S-phase entry in postmitotic cortical neurons. Quantitation is of data similar to those shown inD–F and indicates the percentage of MAP2-positive neurons that were GFP positive after infection with 100 MOI of N-Myc/GFP (100N-myc) or control GFP (100GFP) adenovirus. Right two bars, The percentage of MAP2-positive cells that were BrdU positive after infection with 100 MOI of either the N-myc or GFP adenoviruses. Similar results were obtained in three independent experiments.
We then used this culture system to determine whether overexpression of N-myc was sufficient to cause postmitotic cortical neurons to enter S-phase. Neurons were infected with 100 MOI N-myc or control, GFP adenovirus for 2 d and were then incubated with BrdU for 24 hr. Double-label immunocytochemical analysis for N-myc and MAP2 confirmed that ∼40–50% of the cortical neurons were infected under these conditions (Fig.6D–F,J). Similar numbers were infected with 100 MOI of the control GFP adenovirus (Fig. 6J). We then asked whether any of the cortical neurons incorporated BrdU by performing immunocytochemistry for MAP2 and BrdU (Fig.6G–I). Virtually none of the MAP2-positive neurons were positive for BrdU, although the occasional MAP2-negative non-neuronal cell did show BrdU immunoreactivity (Fig.6G–I). Quantitation of these data revealed that the percentage of BrdU-positive, MAP2-positive cells was similar in neurons infected with either the N-myc or control GFP adenoviruses (Fig. 6J). Thus, unlike sympathetic neurons, overexpression of N-myc did not cause postmitotic cortical neurons to enter S-phase, indicating that there is a fundamental difference in the control of S-phase entry in postmitotic cortical versus sympathetic neurons.
DISCUSSION
The experiments reported in this paper support a number of major conclusions, which together provide a molecular mechanism whereby the N-myc proto-oncogene could play a major role in the genesis and promotion of neuroblastoma. First, data presented here indicate that overexpression of N-myc at levels similar to those observed in NB promotes the survival and S-phase entry of postmitotic sympathetic neurons. These findings not only provide information about the mechanisms whereby N-myc might promote oncogenesis, but they also argue against the hypothesis that aberrant cell cycle entry inevitably causes neuronal apoptosis, at least in sympathetic neurons. Second, the N-myc-induced S-phase entry can be reversed by concomitant expression of constitutively hypophosphorylated pRb. In contrast, hypophosphorylated pRb had no effect on N-myc-mediated survival, indicating that these effects likely occur via separate downstream pathways, only one of which (the S-phase entry) involves pRb. Finally, our data demonstrate that N-myc does not cause S-phase entry in postmitotic cortical neurons, indicating that sympathetic and CNS neurons are “locked out” of the cell cycle by different mechanisms and suggesting that sympathetic neurons are preferentially susceptible to the oncogenic actions of N-myc. Thus, N-myc selectively causes postmitotic sympathetic neurons to reenter the cell cycle and allows them to survive under conditions in which they would normally undergo apoptosis, thereby potentially contributing to their transformation to NB.
Perhaps the most striking result reported here is the ability of N-myc to selectively cause postmitotic sympathetic neurons to reenter S-phase, as monitored by BrdU incorporation and increased expression of cyclin E and cdk2, both of which are normally increased during S-phase (Ekholm and Reed, 2000). However, although our data strongly support the conclusion that N-myc can cause S-phase entry, they also argue that N-myc is not sufficient to mediate further cell cycle progression, because we never observed mitotic figures or cell division. Similar results have been reported in transgenic skin cells that overexpressed c-myc: the cells entered S-phase but did not undergo cell division (Pelengaris et al., 1999). Why do only half of the neurons reenter S-phase in these experiments? One potential explanation involves neuronal maturity. Sympathetic neurons exit the cell cycle at time points ranging from E13 to postnatal day 1 (Hall and Landis, 1991), and hence after 5 d of culturing they range from being 4 to 12 d postmitotic. It may be that the more immature sympathetic neurons are preferentially sensitive to the cell cycle effects of N-myc. Alternatively, the explanation may be a technical one. BrdU labeling was performed for only 48 hr, and the length of time required for N-myc to induce a Go to S-phase transition may be variable.
How does N-myc mediate this S-phase entry? The results demonstrating that coexpression of hypophosphorylated pRb rescues the BrdU incorporation argues that N-myc mediates this effect via pRb. Such an effect could be mediated by direct interactions between N-myc and pRb (Rustgi et al., 1991), and it could also be indirectly mediated via an N-myc-induced increase in levels of the inhibitory basic helix-loop-helix protein, Id2, which binds to hypophosphorylated pRb and inhibits its ability to lock cells out of S-phase (Iavarone et al., 1994; Lasorella et al., 1996; 2000). An additional, potentially related mechanism involves N-myc-mediated downregulation of the cyclin-dependent kinase inhibitor p27 (Bouchard et al., 1999;Perez-Roger et al., 1999; Yang et al., 2001), which in fibroblasts is essential for induction of cyclin E–cdk2 kinase activity, but not for S-phase entry (Beier et al., 2000). Although the data presented here do not distinguish between these alternative explanations, we have observed that p27 levels are decreased and Id2 levels increased in sympathetic neurons overexpressing N-myc (our unpublished observations), suggesting that decreased p27 may collaborate with increased Id2 to trigger S-phase entry.
Results reported here also indicate that N-myc overexpression does not induce S-phase entry in cortical neurons, suggesting that sympathetic and cortical neurons are locked out of the cell cycle via distinct mechanisms. Such a difference could be predicted by considering the development of these two populations of neurons. Cortical neurons, like most CNS neurons, induce neuronal gene expression and undergo terminal mitosis at the same time (Gloster et al., 1999). Perturbation of this progenitor-to-postmitotic neuron transition, for example, via functional inhibition of the pRb family (Slack et al., 1998) or via overexpression of Id2 (Toma et al., 2000), leads to cellular apoptosis; in no conditions yet reported do cortical cells divide while expressing a neuronal phenotype. In contrast, sympathetic neuroblasts transition through a stage in which they express a neuronal phenotype while still dividing (Rothman et al., 1978; Rohrer and Thoenen, 1987; Memberg and Hall, 1995), suggesting that the nature of terminal mitosis differs in sympathetic versus CNS neurons. In that regard, our findings may indicate that the mechanisms locking most CNS neurons out of the cell cycle are much more stringent than for sympathetic neurons.
A somewhat surprising finding reported here is that, coincident with S-phase entry, N-myc promotes enhanced survival of sympathetic neurons in the absence of NGF. This is particularly surprising in light of findings indicating that aberrant cell cycle entry is one of the major mechanisms whereby NGF withdrawal causes sympathetic neuron apoptosis. In particular, NGF withdrawal causes increased cyclin D1 expression (Freeman et al., 1994), and inhibition of cdk4 and -6, both of which phosphorylate and activate pRb, is sufficient to delay NGF withdrawal-induced apoptosis (Park et al., 1997). However, in this regard, data presented here show that NGF withdrawal does not cause enhanced BrdU incorporation and that hypophosphorylated pRb is not, by itself, sufficient to rescue sympathetic neurons from apoptosis. Of themselves, our findings do not necessarily argue against a role for cell cycle dysregulation in NGF withdrawal-induced apoptosis, although they do demonstrate that this dysregulation does not actually lead to S-phase reentry. Instead, our data suggest that N-myc-induced survival mechanisms may be “dominant” to any apoptotic signals deriving from the coincident aberrant reentry into S-phase. Interestingly, data presented here suggest (but do not definitively establish) that one such N-myc-mediated mechanism may involve downregulation of p75NTR (Bamji et al., 1998).
N-myc is a true oncogene with overexpression in the sympathetic chain and adrenal medulla of transgenic mice that results, via unknown mechanisms, in malignant neuroblastoma (Weiss et al., 1997). The experimental and clinical data showing a strong correlation between N-myc gene amplification and poor outcome in neuroblastoma suggest that N-myc is involved in the malignant transformation of developing sympathetic precursors or neurons, or both. On the basis of our data showing that N-myc can promote S-phase entry and survival of “postmitotic” sympathetic neurons, we suggest a model in which N-myc contributes to malignant neuroblastoma by either stopping sympathetic neuroblasts from exiting the cell cycle or by collaborating with other risk factors to actually transform postmitotic neurons and cause them to reenter the cell cycle.
Footnotes
This work was supported by research grants from the National Cancer Institute of Canada and Canadian Institutes of Health Research (CIHR) to D.R.K. and F.D.M., respectively. D.R.K. is a recipient of the Harold Johns and Canadian Cancer Society Research Scientist Award, and F.D.M. is a CIHR Senior Scientist and a Killam Scholar. K.W. was supported by funds from the Finnish Academy, Yrjö Jahnsson Foundation, and the Finnish Medical Society Duodecim; F.B.H. was supported by a National Science and Engineering Research Council studentship.
Correspondence should be addressed to Dr. Freda Miller, Center for Neuronal Survival and Brain Tumor Research Center, Montreal Neurological Institute, McGill University, 3801 rue University, Montreal, Quebec, Canada H3A 2B4. E-mail:freda.miller@mcgill.ca.
REFERENCES
- 1.Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol. 1998;143:1691–1703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett GL, Georgious A, Reid K, Bartlett PF, Leung D. Rescue of dorsal root sensory neurons by nerve growth factor and neurotrophin-3, but not brain-derived neurotrophic factor or neurotrophin-4, is dependent on the level of the p75 neurotrophin receptor. Neuroscience. 1998;85:1321–1328. doi: 10.1016/s0306-4522(98)00006-2. [DOI] [PubMed] [Google Scholar]
- 4.Beier R, Burgin A, Kiermaier A, Fero M, Karsunky H, Saffrich R, Moroy T, Ansorge W, Roberts J, Eilers M. Induction of cyclin E-cdk2 kinase activity, E2F-dependent transcription and cell growth by Myc are genetically separable events. EMBO J. 2000;19:5813–5823. doi: 10.1093/emboj/19.21.5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, Reed S, Sicinski P, Bartek J, Eilers M. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999;18:5321–5333. doi: 10.1093/emboj/18.19.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- 7.Chang MW, Barr E, Seltzer J, Jiang YQ, Nabel GJ, Nabel EG, Parmacek MS, Leiden JM. Cytostatic gene therapy for vascular proliferative disorders with a constitutively active form of the retinoblastoma gene product. Science. 1995;267:518–522. doi: 10.1126/science.7824950. [DOI] [PubMed] [Google Scholar]
- 8.Ciccarone V, Spengler BA, Meyers MB, Biedler JL, Ross R. Phenotypic diversification in human neuroblastoma cells: expression of neural crest lineages. Cancer Res. 1989;49:219–225. [PubMed] [Google Scholar]
- 9.Clarke AR, Maandag ER, Van Roon M, Van der Lugt NMT, Van der Valk M, Hopper MI, Berns A, Te Riele H. Requirement for a functional Rb1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- 10.Ekholm SV, Reed SI. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol. 2000;12:676–684. doi: 10.1016/s0955-0674(00)00151-4. [DOI] [PubMed] [Google Scholar]
- 11.ElShamy WM, Fridvall LK, Ernfors P. Growth arrest failure, G1 restriction point override and S-phase death of sensory precursor cells in the absence of neurotrophin-3. Neuron. 1998;21:1003–1015. doi: 10.1016/s0896-6273(00)80619-4. [DOI] [PubMed] [Google Scholar]
- 12.Farinelli SE, Greene LA. Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. J Neurosci. 1996;16:1150–1162. doi: 10.1523/JNEUROSCI.16-03-01150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freeman RS, Estus S, Johnson EM., Jr Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 14.Gloster A, El-Bizri H, Bamji SX, Rogers D, Miller FD. Early induction of Ta1 α-tubulin transcription in neurons of the developing nervous system. J Comp Neurol. 1999;405:45–60. doi: 10.1002/(sici)1096-9861(19990301)405:1<45::aid-cne4>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 15.Goldman SC, Chen CY, Lansing TJ, Gilmer TM, Kastan MB. The p53 signal transduction pathway is intact in human neuroblastoma despite cytoplasmic localization. Am J Pathol. 1996;148:1381–1385. [PMC free article] [PubMed] [Google Scholar]
- 16.Hall AK, Landis SC. Early commitment of precursor cells from the rat superior cervical ganglion to neuronal or nonneuronal fates. Neuron. 1991;6:741–751. doi: 10.1016/0896-6273(91)90171-u. [DOI] [PubMed] [Google Scholar]
- 17.Harding TC, Xue L, Bienemann A, Haywood D, Dickens M, Tolkovsky AM, Uney JB. Inhibition of JNK by overexpression of the JNK binding domain of JIP-1 prevents apoptosis in sympathetic neurons. J Biol Chem. 2001;276:4531–4534. doi: 10.1074/jbc.C000815200. [DOI] [PubMed] [Google Scholar]
- 18.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iavarone A, Garg P, Lasorella A, Hsu J, Israel MA. The helix-loop-helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes Dev. 1994;8:1270–1284. doi: 10.1101/gad.8.11.1270. [DOI] [PubMed] [Google Scholar]
- 20.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 21.Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 22.Koulakoff A, Bizzini B, Berwald-Netter Y. Neuronal acquisition of tetanus toxin binding sites: relationship with the last mitotic cycle. Dev Biol. 1983;100:350–357. doi: 10.1016/0012-1606(83)90229-4. [DOI] [PubMed] [Google Scholar]
- 23.Lasorella A, Iavarone A, Israel MA. Id2 specifically alters regulation of the cell cycle by tumor suppressor proteins. Mol Cell Biol. 1996;16:2570–2578. doi: 10.1128/mcb.16.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lasorella A, Noseda M, Beyna M, Iavarone A. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature. 2000;407:592–598. doi: 10.1038/35036504. [DOI] [PubMed] [Google Scholar]
- 25.Lauder JM, Bloom FE. Ontogeny of monoamine neurons in the locus coeruleus, raphe nuclei and substantia nigra of the rat. J Comp Neurol. 1974;155:469–482. doi: 10.1002/cne.901550407. [DOI] [PubMed] [Google Scholar]
- 26.Lee EHP, Chang CY, Hu N, Wang YCJ, Lai CC, Herrup K, Lee W, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 27.Lee EHP, Hu N, Yuan SSF, Cox LA, Bradley A, Lee W, Herrup K. Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev. 1994;8:2008–2021. doi: 10.1101/gad.8.17.2008. [DOI] [PubMed] [Google Scholar]
- 28.Ma Y, Campenot RB, Miller FD. Concentration-dependent regulation of neuronal gene expression by nerve growth factor. J Cell Biol. 1992;117:135–141. doi: 10.1083/jcb.117.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macleod KF, Hu Y, Jacks T. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 1996;15:6178–6188. [PMC free article] [PubMed] [Google Scholar]
- 30.Majdan M, Lachance C, Gloster A, Aloyz R, Zeindler C, Bamji S, Bhakar A, Belliveau D, Fawcett J, Miller FD, Barker PA. Transgenic mice expressing the intracellular domain of the p75 neurotrophin receptor undergo neuronal apoptosis. J Neurosci. 1997;17:6988–6998. doi: 10.1523/JNEUROSCI.17-18-06988.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Majdan M, Walsh GS, Aloyz R, Miller FD (2001) TrkA mediates developmental sympathetic neuron survival by silencing an ongoing p75NTR-mediated death signal. J Cell Biol, in press. [DOI] [PMC free article] [PubMed]
- 32.Mazzoni IE, Saïd FA, Aloyz R, Miller FD, Kaplan D. Ras regulates sympathetic neuron survival by suppressing the p53-mediated cell death pathway. J Neurosci. 1999;19:9716–9727. doi: 10.1523/JNEUROSCI.19-22-09716.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Memberg SP, Hall AK. Dividing neuron precursors express neuron-specific tubulin. J Neurobiol. 1995;27:26–43. doi: 10.1002/neu.480270104. [DOI] [PubMed] [Google Scholar]
- 34.Menezes JRL, Luskin MB. Expression of neuron-specific tubulin defines a novel population in the proliferative layers of the developing telencephalon. J Neurosci. 1994;14:5399–5416. doi: 10.1523/JNEUROSCI.14-09-05399.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller FD, Pozniak CD, Walsh GS. Neuronal life and death: an essential role for the p53 family. Cell Death Differ. 2000;7:880–888. doi: 10.1038/sj.cdd.4400736. [DOI] [PubMed] [Google Scholar]
- 36.Nisen PD, Zimmerman KA, Cotter SV, Gilbert F, Alt FW. Enhanced expression of the N-myc gene in Wilms' tumors. Cancer Res. 1986;46:6217–6222. [PubMed] [Google Scholar]
- 37.Park DS, Farinelli SE, Greene LA. Inhibitors of cyclin-dependent kinases promote survival of post-mitotic terminally differentiated PC12 cells and sympathetic neurons. J Biol Chem. 1996;271:8161–8169. doi: 10.1074/jbc.271.14.8161. [DOI] [PubMed] [Google Scholar]
- 38.Park DS, Levine B, Ferrari G, Greene LA. Cyclin-dependent kinase inhibitors and dominant negative cyclin-dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997;17:8975–8983. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pelengaris S, Littlewood T, Khan M, Elia G, Evan G. Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol Cell. 1999;3:565–577. doi: 10.1016/s1097-2765(00)80350-0. [DOI] [PubMed] [Google Scholar]
- 40.Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H. Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p27(Cip1). EMBO J. 1999;18:5310–5320. doi: 10.1093/emboj/18.19.5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000a;289:304–306. doi: 10.1126/science.289.5477.304. [DOI] [PubMed] [Google Scholar]
- 42.Pozniak CD, Walsh GS, Radinovic S, Kaplan DR, Miller FD. Evidence that p53 and p73 play a role in neuronal apoptosis. Soc Neurosci Abstr. 2000b;26:600. [Google Scholar]
- 43.Rohrer H, Thoenen H. Relationship between differentiation and terminal mitosis: chick sensory and ciliary neurons differentiate after terminal mitosis of precursor cells, whereas sympathetic neurons continue to divide after differentiation. J Neurosci. 1987;7:3739–3748. doi: 10.1523/JNEUROSCI.07-11-03739.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rothman TP, Gershon MD, Holtzer H. The relationship of cell division to the acquisition of adrenergic characteristics by developing sympathetic ganglion cell precursors. Dev Biol. 1978;65:322–341. doi: 10.1016/0012-1606(78)90030-1. [DOI] [PubMed] [Google Scholar]
- 45.Rothman TP, Specht LA, Gershon MD, Joh TH, Teitelman G, Pickel VM, Reis DJ. Catecholamine biosynthetic enzymes are expressed in replicating cells of the peripheral but not the central nervous system. Proc Natl Acad Sci USA. 1980;77:6221–6225. doi: 10.1073/pnas.77.10.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rustgi AK, Dyson N, Bernards R. Amino-terminal domains of c-myc and N-myc proteins mediate binding to the retinoblastoma gene product. Nature. 1991;352:541–544. doi: 10.1038/352541a0. [DOI] [PubMed] [Google Scholar]
- 47.Sawai S, Shimono A, Wakamatsu Y, Palmes C, Hanaoka K, Kondoh H. Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development. 1993;117:1445–1455. doi: 10.1242/dev.117.4.1445. [DOI] [PubMed] [Google Scholar]
- 48.Slack RS, Miller FD. Retinoblastoma in mouse neural development. Dev Genet. 1996;18:81–91. doi: 10.1002/(SICI)1520-6408(1996)18:1<81::AID-DVG9>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 49.Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Aloyz R, Lochmuller H, Hagighi A, Lach B, Seth P, Cooper E, Miller FD. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol. 1996;135:1085–1096. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Slack RS, El-Bizri H, Wong J, Belliveau DJ, Miller FD. A critical temporal requirement for the retinoblastoma protein family during neuronal determination. J Cell Biol. 1998;140:1497–1509. doi: 10.1083/jcb.140.6.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stanton BR, Parada LF. The N-myc proto-oncogene: developmental expression and in vivo site-directed mutagenesis. Brain Pathol. 1992;2:71–83. [PubMed] [Google Scholar]
- 52.Stanton BR, Perkins AS, Tessarollo L, Sassoon DA, Parada LF. Loss of N-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev. 1992;6:2235–2247. doi: 10.1101/gad.6.12a.2235. [DOI] [PubMed] [Google Scholar]
- 53.Toma JG, El-Bizri H, Barnabé-Heider F, Aloyz R, Miller FD. Evidence that helix-loop-helix proteins collaborate with retinoblastoma tumor suppressor protein to regulate cortical neurogenesis. J Neurosci. 2000;20:7648–7656. doi: 10.1523/JNEUROSCI.20-20-07648.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vaillant AR, Mazzoni I, Tudan C, Boudreau M, Kaplan DR, Miller FD. Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-Akt pathway to synergistically regulate neuronal survival. J Cell Biol. 1999;146:955–966. doi: 10.1083/jcb.146.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vogan K, Bernstein M, Leclerc JM, Brisson L, Brossard J, Brodeur GM, Pelletier J, Gros P. Absence of p53 gene mutations in primary neuroblastomas. Cancer Res. 1993;53:5269–5273. [PubMed] [Google Scholar]
- 56.Wakamatsu Y, Watanabe Y, Nakamura H, Kondoh H. Regulation of the neural crest cell fate by N-myc: promotion of ventral migration and neuronal differentiation. Development. 1997;124:1953–1962. doi: 10.1242/dev.124.10.1953. [DOI] [PubMed] [Google Scholar]
- 57.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16:2985–2995. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang W, Shen J, Wu M, Arsura M, FitzGerald M, Suldan Z, Kim DW, Hofmann CS, Pianetti S, Romieu-Mourez R, Freedman LP, Sonenshein GE. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene. 2001;20:1688–1702. doi: 10.1038/sj.onc.1204245. [DOI] [PubMed] [Google Scholar]