

Abstract
In microvessels of patients with coronary artery disease (CAD), flow-mediated dilation (FMD) is largely dependent upon the endothelium-derived hyperpolarizing factor H2O2. The goal of this study is to examine the influence of age and presence or absence of disease on the mechanism of FMD. Human coronary or adipose arterioles (~150 μm diameter) were prepared for videomicroscopy. The effect of inhibiting COX [indomethacin (Indo) or NOS (L-NAME), eliminating H2O2 (polyethylene glycol-cata-lase (PEG-CAT)] or targeting a reduction in mitochondrial ROS with scavengers/inhibitors [Vitamin E (mtVitamin E); phenylboronic acid (mtPBA)] was determined in children aged 0–18 years; young adults 19–55 years; older adults >55 years without CAD, and similarly aged adults with CAD. Indo eliminated FMD in children and reduced FMD in younger adults. This response was mediated mainly by PGI2, as the prostacyclin-synthase-inhibitor trans-2-phenyl cyclopropylamine reduced FMD in children and young adults. L-NAME attenuated dilation in children and younger adults and eliminated FMD in older adults without CAD, but had no effect on vessels from those with CAD, where mitochondria-derived H2O2 was the primary mediator. The magnitude of dilation was reduced in older compared to younger adults independent of CAD. Exogenous treatment with a sub-dilator dose of NO blocked FMD in vessels from subjects with CAD, while prolonged inhibition of NOS in young adults resulted in a phenotype similar to that observed in disease. The mediator of coronary arteriolar FMD evolves throughout life from prostacyclin in youth, to NO in adulthood. With the onset of CAD, NO-inhibitable release of H2O2 emerges as the exclusive mediator of FMD. These findings have implications for use of pharmacological agents, such as nonsteroidal anti-inflammatory agents in children and the role of microvascular endothelium in cardiovascular health.
Keywords: Vasodilation, Coronary artery disease, Flow-mediated dilation, Microvasculature
Introduction
The endothelium plays a critical role in vascular homeostasis by maintaining an anti-atherosclerotic and non-proliferative perivascular environment in part through shear-mediated release of nitric oxide (NO). Shear stress is one of the most physiologically important and widely conserved dilator stimuli in both conduit and resistance vessels, and across species.
In many cases, NO mediates flow-mediated dilation (FMD). Endothelial activation with inflammation and atherosclerosis develop at vascular sites associated with disturbances in normal laminar shear stress [41]. Natural aging and other cardiovascular risk factors can also augment vascular inflammation, reduce NO bioavailability, and decrease endothelial-dependent vasodilation. The mediator and magnitude of endothelium-dependent dilation is highly dependent upon age, species, vascular bed, and vessel size. Conduit vessels rely primarily on NO while the microcirculation utilizes a variety of mediators, including NO, prostacyclin (PGI2), and EDHF. Animal studies show that with normal aging, a transformation in the mediator of endothelium-dependent vasodilation can occur with vasodilator prostanoids contributing in infancy and NO in adulthood [18, 38]. However, it is never been determined if a similar transition occurs in humans, particularly in the coronary circulation. This could have important implications for use of COX-inhibitors in youth and in understanding microvascular function in health and disease.
Microvascular endothelial function is important for tissue perfusion, and its impairment is associated with poor prognosis and increased cardiovascular events [37,39] (reviewed by [1]). Microvascular dysfunction also predisposes to ischemia, even without conduit artery disease [8]. This issue becomes particularly important since we have previously demonstrated that the EDHF H2O2 mediates FMD in coronary arterioles from human subjects with coronary atherosclerosis (CAD) and that NO may play a role in similarly aged subjects without CAD [3, 16, 25]. As aging is an independent risk factor for atherosclerosis, it is important to understand its contribution to the regulation of vasodilation during different stages of life. Very little is known about how age affects the mechanism of endothelium dependent dilation in human resistance vessels, especially in the heart. This could be important in optimizing treatments to improve endothelial function and tissue perfusion [20]. The goal of this study is to determine the mechanisms underlying FMD in the human microcirculation throughout life, from early development through healthy adulthood, and with onset of disease. We hypothesize that shear elicits endothelium-dependent dilation throughout life. In the presence of disease where NO bioavailability is reduced, microvascular FMD is maintained by compensatory H2O2.
Materials and methods
Tissue acquisition and general protocol
Small coronary and adipose arterioles (maximal diameter under 300 μm; average = 115.6 ± 5.1 μm, n = 92, tissue acquired from 2011 to 2015) were isolated from de-identified, discarded surgical tissue collected on the day of the procedure. Additionally, rejected donor hearts were collected form the Wisconsin Donor Network organ procurement. Hearts and tissue were stored in cold cardiologic solution (4 °C) from removal until microvessels were isolated or tissue samples used for protein extraction. All procedures were approved by the MCW/Froedtert Hospital IRB. Consent was obtained from subjects at the VA medical center. For tissues received from Children’s Hospital of Wisconsin (CHW), subject assent was obtained by the Children’s Hospital tissue bank via a protocol approved by the CHW IRB. Tissue from all other sites was de-identified and considered surgical discard with no requirement for consent due to minimal risk, except for whole hearts obtained from the Wisconsin Organ Donor Network (otherwise unusable hearts) where historical clinical information was made available with PHI. IRB approval with waiver of consent was obtained from each hospital where surgical discard tissue was obtained in this manner.
Study groups
Four study groups were defined as follows: (A) children 0–18 years (mean age 5.1 ± 1, n = 28); (B) younger adults 19–55 years (mean age 40.3 ± 2, n = 25); (C) older adults >55 years without CAD (mean age 68.6 ± 1.4, n = 23), and (D) older adults diagnosed with CAD (mean age 62 ± 2.8, n = 46). No statistically significant difference in age was observed between CAD and non-CAD groups. Patient demographic are summarized in Table 1.
Table 1.
Patient characteristics for microvessels used in studies
| Children | Younger adultsa | Older adults | CAD | |
|---|---|---|---|---|
| Characteristics | ||||
| Total samples | 28 | 25 | 23 | 46 |
| Sex, M/F | 15/13 | 15/10 | 14/9 | 29/17 |
| Age, years (average/median ± SD) | 5.1/3.0 ± 0.99 | 40.3/43.0 ± 2.01 | 68.6/68.5 ± 1.35 | 62.0/63.0 ± 2.78 |
| Race, AA/Cauc/Hisp/Asian | 4/19/2/2 | 7/17/1/0 | 4/19/0/0 | 11/17/0/0 |
| Underlying diseases/risk factors | ||||
| Coronary artery disease* | 0 | 0 | 0 | 46 |
| Tobacco | 0 | 7 | 4 | 17 |
| Hypertension* | 0 | 8 | 18 | 37 |
| Hypercholesterolemia* | 0 | 2 | 11 | 23 |
| Diabetes mellitus* | 0 | 1 | 7 | 14 |
| Congestive heart failure | 1 | 3 | 3 | 9 |
| MI | 0 | 0 | 1 | 6 |
| VD | 7 | 5 | 9 | 11 |
P < 0.05 vs. younger adults
Contains samples from donors of unknown ethnic background
Videomicroscopy
The microvascular preparation has been described previously [13,16]. In an organ chamber, both ends of a vessel are cannulated with glass micropipettes filled with PSS and pressurized to 60 mmHg. Vessels are constricted with endothelin-1 (0.1–1 nM) to achieve a 20–50% stable reduction in passive diameter. Flow is initiated using pipettes of identical impedance [23]. Changing the height of each reservoir in equal and opposite directions generates flow with no changes in luminal vessel pressure [22]. Data are reported as percent maximum diameter at a given pressure gradient. Two flow-response curves were generated in each vessel by varying reservoir pressure gradients between 5 and 100 cm H2O, producing shear stress of ~2–30 dynes/cm2. The first response was performed in the presence of vehicle and the second after adding inhibitor(s): Indo (10 μM); L-NAME (100 μM); polyethylene glycol-catalase (PEG-CAT, 500 U/ml); trans-2-phenyl cyclopropylamine (TPC, 100 μM); DETA (diethylenetriamine) NONOate (10 nM); and either of two mitochondrial-specific scavengers of ROS: triphenylphosphonium conjugated(mt-)Vitamin E (10 μM) and mtPBA (phenylboronic acid) (10 μM). Vasodilation was evaluated in response to flow or Iloprost, a synthetic PGI2 analogue (10 pM-1 μM). All pharmacological agents were added to the external bathing solution resulting in a volume increase of <1% of the circulating external bath solution. In each experiment, the endothelium-independent dilator papaverine (100 μM) was used to determine the maximal (passive) diameter at 60 mmHg and confirm integrity of smooth muscle relaxation.
Measurement of vascular reactive oxygen species (ROS)
The cell-permeable fluorescence sensor Mitochondria peroxy yellow 1 (MitoPYI; Tocris Bioscience, 10 μM) [12, 29] was used to selectively assess mitochondrial H2O2. To test specificity for H2O2, all experiments were repeated in the presence of PEG-CAT (500 U/mL). Vessels were prepared as for vasodilator studies, with MitoPY1 added to the buffer perfusing the lumen of the vessel, and maintained at 37 °C for the duration of the experiment [2, 16]. After allowing for cellular integration of the probe (30–45 min), the buffer in the organ chamber was exchanged, and vessel fluorescence intensity was measured before and after inducing a flow gradient of 100 cm H2O for 1–2 min. All vessel responses were compared with those obtained before flow, and data are expressed as percent change from baseline. MitoPY1 fluorescence was measured with a Nikon Eclipse TE200 microscope equipped with a 20× objective and a Hamamatsu C4742–95 camera, using an excitation wavelength of 489 nm and detection wavelength of 540 nm. Fluorescence data were analyzed using MetaMorph (Version NX) by outlining the vessel and after subtracting the fluorescence intensity of the background.
Materials
Endothelin-1 (ET-1) was obtained from Peninsula Laboratories and prepared in saline with 1% bovine serum albumin. mtPBA was prepared in the MCW free radical center and dissolved in DMSO. Other chemicals were obtained from Sigma-Aldrich. Agents were prepared in distilled water, PSS, or DMSO (DETANONOate, mitoPY1). All concentrations represent the final concentrations in the organ bath.
Immunohistochemistry (IHC)
Surgically discarded tissue was fixed in 10% zinc formalin, embedded in paraffin wax, cut into 4 μm sections, then rehydrated, followed by antigen retrieval and staining using a DAKO autostainer. Sections were incubated with secondary antibody or normal serum IgG as negative controls. The MCW/CHW Pathology Core facility was used for all IHC studies. Antibodies to both COX1 and COX2 (Santa Cruz Biotechnologies) were used.
Western blotting
30 μg protein was loaded onto a 10% Tris-HCl gel (Biorad) for SDS-PAGE western blot analysis. Transferred proteins on PVDF membrane were blocked with 5% nonfat milk in TBST and blotted overnight with antibodies against Cox-1 (1:500 dilution; sc-1752, Santa Cruz), Cox-2 (1:500 dilution; sc-1745, Santa Cruz) and GAPDH (1:5000; 6C5, abcam). After washing, the membranes were blotted with the appropriate HRP-conjugated secondary antibodies (Biorad) and protein signals were detected using chemiluminescence (SuperSignal West Pico, Thermo Scientific). Sample signals were then semi-quantified by densitometry and normalized to GAPDH. Data presented as fold change to control.
Statistical methods
Data are presented as mean ± SEM. For all concentration-response curves, differences between groups at each concentration were determined using a two-way (parameters—dose and treatment), repeated-measures analysis of variance (ANOVA). A post hoc Tukey’s test was used for comparison of individual differences. A probability value of P < 0.05 was considered to be statistically significant. For fluorescence studies and Western blot analysis, either a paired t test or one-way ANOVA with post hoc Tukey’s test was used, as indicated in the figure legends.
Results
Discarded human tissue was collected at the time of surgery from 122 patients. Detailed patient demographics are shown in Table 1. The maximal inner diameter of vessels from children (age 0–18 = 96.8 ± 7.5* μm, N = 34) was smaller than those from younger adults (age 18–55 = 136.0 ± 17.8 μm, N = 15). Vessels from older adults without (age >55 = 131 ± 8.9 μm, N = 11) and subjects with CAD (119 ± 7.5 μm, N = 34) were of similar maximal diameter (*P < 0.05 one-way ANOVA multiple comparisons versus control group (younger adults) (Holm-Sidak method). Patients were considered to have CAD if they had a history of CABG or PCI, >50% lesion of at least one coronary conduit vessel on cath, a standing clinical diagnosis of CAD, or via direct inspection of epicardial arteries (for explanted hearts).
Mechanism of FMD in coronary vessels changes throughout life and with onset of disease
To determine the mediator of FMD, we examined resistance vessels from patients in each of the four groups. There was no difference in the magnitude of dilation to flow between children (Fig. 1a) and young adults (Fig. 1b). Maximal FMD in older adults (Fig. 1c) was similar to that in adults with CAD (Fig. 1d) but in both groups was lower than in children and young adults.
Fig. 1.
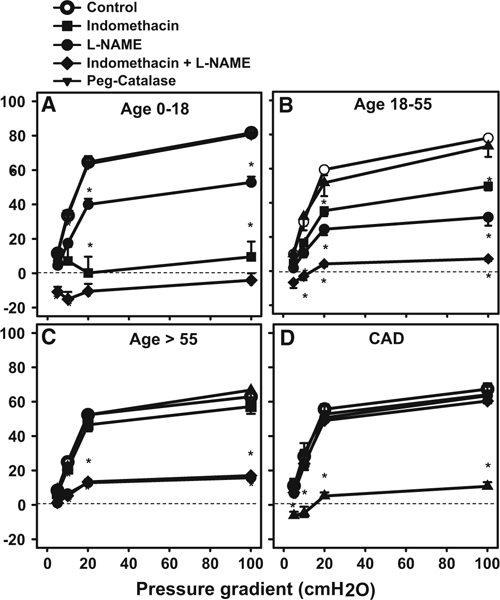
Mediator of flow-mediated dilation is changed throughout life and with onset of disease. Flow-mediated dilation (FMD) was compared in isolated atrial microvessels from a children age 0–18, b younger adults age 18–55, c older adults age >55 and d subjects with clinical diagnosed coronary artery disease independent of age. The mediator of FMD changed from PGI2/NO in children to NO/PGI2 in younger adults, to primarily NO in older adults, then to H2O2 in patients with CAD. #P < 0.05 vs. 18–55; *P < 0.05 two-way ANOVA RM Tukey’s post hoc analysis, N = 4–10
The mechanism of FMD varied in each group. In children, FMD was markedly attenuated by Indo but only partly reduced by L-NAME, indicating a primary role for prostaglandins, with a minor contribution from NO (Fig. 1a). L-NAME + Indo eliminated FMD in vessels from children. In younger adults, Indo modestly reduced FMD, with a much greater effect from L-NAME. The combination eliminated FMD (Fig. 1b). FMD in older adults was not altered by Indo but was eliminated by L-NAME (Fig. 1c). In vessels from subjects with CAD the mechanism of FMD shifted to H2O2 (inhibitable by PEG- CAT), with no effect of inhibiting prostaglandins and/or NO (Fig. 1d).
Prostacyclin-mediated dilation in vessels from children
Since products of cyclooxygenase (COX) contribute most to FMD in coronary arterioles from children, we determined the expression of COX1 and COX2 in human atrial tissue. Immunohistochemistry revealed prominent expression of COX1 in endothelial and medial vascular layers in both children and in adults with CAD, but with lower expression in young adults and older adults without CAD (Fig. 2a–h). COX2 expression was very low across all groups. Western blot analysis of whole heart tissue (left ventricle) indicated increased expression of both COX1 and COX2 in children’s tissue compared to cardiac tissue from older subjects without CAD (Fig. 2i, j).
Fig. 2.
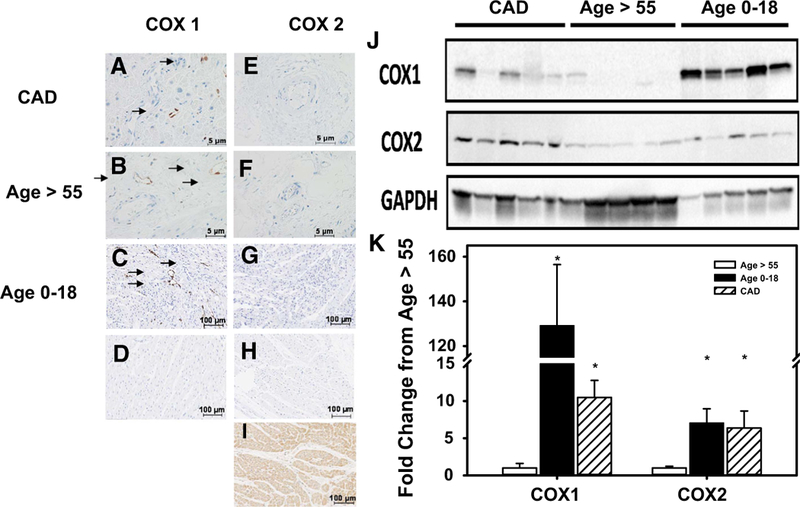
Cyclooxygenase expression in human atrial sections. Representative image of IHC in atrial sections for COX1 (a-d) and COX2 (e-i) and Western blot quantification of COX1 and 2 in left ventricle tissue (j, k). COX1 expression was significantly increased in vessels from atrial sections (IHC) and whole left ventricle (WB) tissue from pediatric subjects and patients with CAD compared to adult subjects without CAD. COX2 expression is elevated in vessels from atrial sections (IHC) and whole left ventricle (WB) of CAD or children’s tissue vs. non-CAD controls but not in microvessels of subjects with CAD. d, h Control sections without primary antibody staining. Positive control (i) for COX2 is derived from subjects with autoimmune disease *P < 0.05 two-way t test with Tukey’s post hoc analysis, IHC, N = 4; WB, N = 5
To determine whether the prototypical COX-derived dilator, PGI2 is responsible for FMD in arterioles from children, we examined dilation to the PGI2 analogue Ilo-prost in resistance vessels from children (age <18 years). Iloprost produced a dose-dependent dilation that was partly attenuated by L-NAME (Fig. 3a). PEG-CAT and DETA-NONOate had no effect on dilation. COX metabolizes arachidonic acid to form endoperoxide, which is converted to PGI2 by prostacyclin-synthase. To confirm the involvement prostacyclin-synthase, treatment with TPC (100 μM), a specific inhibitor of this enzyme, decreased flow-induced dilation (Fig. 3b) in coronary arterioles from children. Our data suggest a significant contribution of PGI2/PGI2 synthase in the mechanism of FMD in coronary vessels early in life.
Fig. 3.
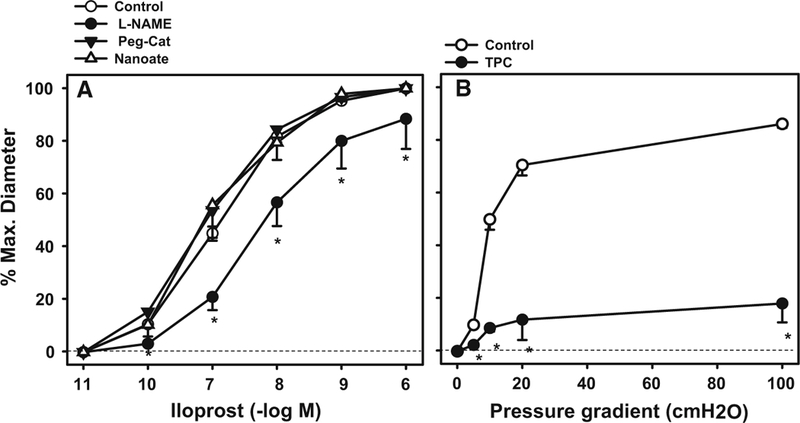
Prostacyclin receptor-and flow-mediated dilation in vessels from children age 0–18. Dilation was evaluated in isolated atrial microvessels in response to either the PGI2 mimic iloprost, or to shear. Inhibition of NOS with L-NAME partly attenuated iloprost mediated dilation. The PGI2 receptor antagonist TPC (100 μM) blocked FMD. *P < 0.05 two-way ANOVA RM Tukey’s post hoc analysis, N = 4–6
Origin and source of ROS as a vasodilator in disease
We have previously identified a role for mitochondrial H2O2 in mediating FMD in vessels from patients with CAD using inhibitors of the mitochondrial electron transport chain [2, 16]. In this study, we confirm this observation with two more direct methods. First, using mitoPYl we observed a robust increase in fluorescence during shear in adipose and atrial vessels from subjects with CAD (60 ± 5.2%). PEG-CAT reduced this response to 10 ± 5.7* % (*P < 0.05 vs. CAD) (Fig. 4a, b). We have previously demonstrated that no such increase is observed in vessels from subjects without CAD [3, 16].
Fig. 4.
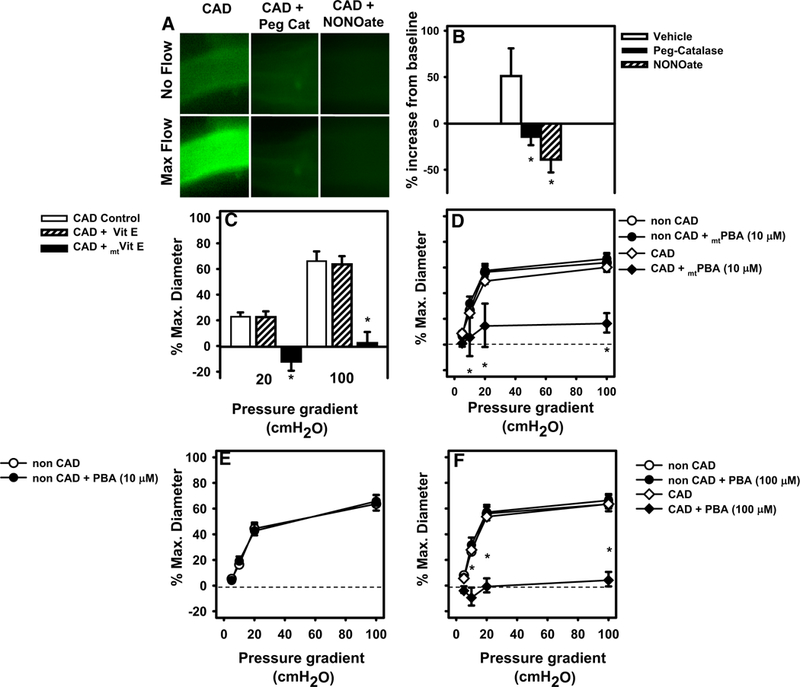
Mitochondrial H2O2 mediates FMD in vessels from subjects with CAD. a, b The H2O2 specific fluorescent probe PYI targeted to mitochondria (mitoPYI) was used to quantify H2O2 in isolated adipose and atrial vessels. Due to the volume and cleanness of vessels needed, both adipose vessels were used. Specificity of the probe for H2O2 was confirmed using Peg-catalase. c FMD in atrial microvessels was reduced in the presence of mitochondrial targeted antioxidant mtVitamine. e, d The mitochondrial targeted scavenger of H2O2 (mtPBA) reduced FMD in atrial arterioles from patients with CAD. e, f PBA (10 mM), the cellular scavenger of H2O2 had no effect on FMD but blocked FMD at a tenfold higher concentration. *P < 0.05 two-way ANOVA RM (dose response curve) or t test (single point dilation and fluorescence data) with Tukey’s post hoc, N = 4–5
Next, we tested the effect of novel antioxidant probes targeted to the mitochondria to more directly examine the contribution of mitoROS. Specifically, we used the mitochondrial targeted free radical scavenger mtVitamin E that eliminates superoxide and mtPBA which localizes in the mitochondrial matrix and eliminates H2O2 as well as per-oxynitrite with no effect on superoxide. FMD was reduced by mtVitamin E (10 μM, Fig. 4c) or mtPBA (10−5 M, Fig. 4d) in arterioles from patients with CAD. In contrast, mtPBA had no effect on FMD in arterioles from older adult patients without CAD (Fig. 4e; % max dilation with 10−5 mtPBA vehicle vs. scavenger: 64.2 ± 6.3 vs. 67.2 ± 4.1%). When similar concentrations of PBA or Vitamin E (not targeted to the mitochondria) were applied to vessels from CAD patients, FMD remained unchanged (Fig. 4c, e). However, tenfold higher doses of PBA (10−4 M) did reduce FMD. This higher concentration of PBA had no effect on FMD in arterioles from patients without CAD (Fig. 4f).
Role of NO in regulation of mtROS levels
NO, in doses subthreshold for direct vasodilation, is known to acutely inhibit formation of mtROS (reviewed by Larsen et al. [24]). Therefore, application of NO to vessels from patients with CAD might be expected to inhibit mitochondrial H2O2 generation, and therefore FMD. Treatment of vessels with the NO donor DETANONOate in a dose that does not produce dilation (10−8 M) reduced FMD in patients with CAD, but had no effect on dilation in non-CAD subjects (Fig. 5a). The same dose of DETANONOate completely eliminated flow-induced mitoPY 1 fluorescence (Fig. 4a, b).
Fig. 5.
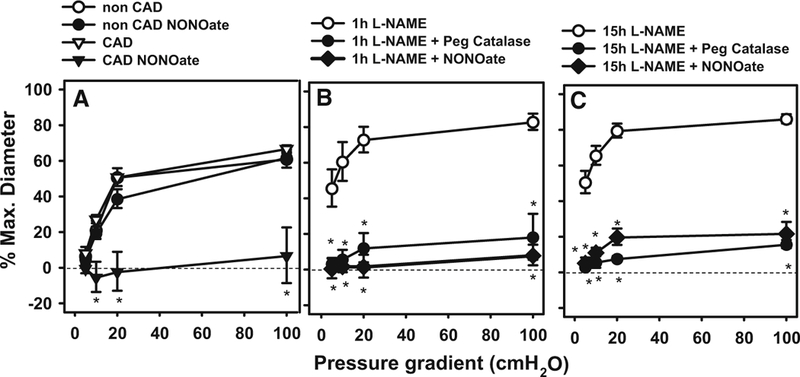
NO at a concentration subthreshold for dilation suppresses FMD in atrial arterioles from patients with and without CAD. a FMD was abolished by incubation with NONOate in atrial arterioles from patients with but not without CAD. b, c Treatment of adipose arterioles with L-NAME for either 1 or 18 h shifted the mechanism of dilation from NO to H2O2 (Peg-catalase inhibitable). The dilation was abolished by treatment with DETANONOate. *P < 0.05 two-way ANOVA RM Tukey’s post hoc analysis, N = 3–6
To assess the effect of prolonged NOS blockade in vessels form non-CAD subjects, we incubated vessels with L-NAME for either 1 or 15 h then tested dilation to graded levels of shear. Due to the greater length of these experiments, and our experience that viability of coronary vessels is significantly lower than for adipose after prolonged incubation, we performed this evaluation in adipose vessels. Prior experience indicates that adipose vessels exhibit the same basic phenotype as those isolated from atrial tissue. FMD was maintained in both cases but the mediator switched from NO to H2O2 (PEG-CAT inhibitable; Fig. 5b, c). Our previous studies indicate that prolonged incubation of vessels in media alone have no effect of mechanism of dilation [4, 14, 16].
Discussion
There are four major new findings from this study. First, the primary mediator of FMD transitions with age from PGI2 in youth to NO in adulthood, and switches again with the onset of CAD to mitochondrial derived H2O2. The latter switch occurs more acutely and independent of age. Second, in older adults a small but significant reduction in FMD is observed when compared to younger individuals, independent of disease state. Third, blockade (1 h or more) of NOS evokes a compensatory shear-induced dilation mediated by H2O2. Finally, we show a critical role for NO in suppressing mitochondrial ROS and eliminating FMD in arterioles from subjects with CAD.
Vascular homeostasis is highly dependent upon factors released from the endothelium, the most prominent of which are NO, PGI2, and endothelium-derived hyperpolarization factors (EDHFs). Each factor produces vasodilation, and crosstalk between these pathways is well-established (NO inhibits ROS production in mitochondria, superoxide scavenges NO, and NO activates the COX enzymes to increase production of prostaglandins) [32]. We have demonstrated in children that COX metabolites are the primary mediators of dilation in FMD, but these metabolites play a diminishing role during the aging process and do not contribute to FMD in adults or in patients with CAD. We did not assess the specific contributions of COX1 vs. COX2 to FMD, but did find a significant increase of COX 1 expression (IHC Fig. 2a–d) in small arterioles of atrial sections. Furthermore, Western blot for COX1 confirmed a significant elevation in whole atrial tissue from children compared to younger adults. These data are consistent with an important role for PGI2 as a mediator of dilation to flow (Fig. 2j, k). Interestingly, in children’s samples as well as subjects with CAD levels of COX2 are elevated in whole heart samples but this was not evident in small arterioles by IHC. Due to small sample volumes we were not able to measure prostacyclin products, but with the use of the selective PGI2 synthase inhibitor, TPC, we demonstrate that prostacyclin-synthase is necessary for FMD in vessels from children. Interestingly, dilation to the PGI2 analogue iloprost was augmented by L-NAME confirming the well-established crosstalk between COX and NOS pathways in the vasculature [30, 35] reviewed by [21]. Our data are consistent previous work from Charpie et al. [6] comparing agonist- induced dilation in vertebral arteries from infants and adults, showing a change from PGI2 to NO mediated dilation to ACh. In their study, endothelium-dependent responses in arteries from infants increased prostaglandin activity compared to vessels from adult subjects, suggesting an age-dependent change in the primary mechanism responsible for acetylcholine-induced vasodilation [6]. Dilation in vessels from both age groups demonstrated an NO component; however, vessels from infants had an indomethacin-inhibitable component not observed in older adults. Together with our data a predominant role is suggested for PGI2 as a mediator of coronary dilation in infants.
An overall reduction in magnitude of FMD occurs in older age. It is intriguing to speculate that loss of the PGI2 component of FMD contributes to the age-related decline in endothelial-dependent dilation of coronary vessels. A similar change has been reported in both human [42] and animal models [10]. This process is accelerated by the development of atherosclerosis in human subjects, but due to the lack of development of age-dependent atherosclerosis in rodent models direct comparisons are difficult to make. Age-dependent increase in vascular stiffness or a contribution of clinical risk factors may also contribute to the change in dilator mechanism and decreased overall dilation. While the smooth muscle maximal dilation to papaverine was not impaired, arguing against such a possibility, we cannot exclude the effect of hypertension and other risk factors which are more prevalent in older individuals >55 years of age vs. those 18–55 years of age (Table 1).
The crosstalk between NO and mitochondrial ROS formation is complex. NO can directly decrease mitochondrial ROS formation by either inhibiting electron transport chain complexes [31] or through quenching superoxide resulting in formation of peroxynitrite [5]. In the human microcirculation, when NO bioavailability declines, mitochondrial H2O2 compensates as the mediator of FMD [3]. While NO is anti-inflammatory and generally thought to be protective against the development of atherosclerosis, H2O2 can stimulate smooth muscle cell proliferation and invoke endothelial inflammation, thereby increasing the risk for atherosclerosis [36]. Thus, reliance on H2O2 as the primary vasodilator in the coronary microcirculation instead of NO might have adverse consequences. The fact that mitochondrial ROS production is regulated by ambient NO and can be eliminated by small doses of exogenous NO as we demonstrate may explain why H2O2 does not contribute to FMD in otherwise healthy adults where NO predominates. However, sub-acute (at least 1 h) or chronic inhibition (15 h) of NOS allows for compensation by H2O2. We suspect that duration of exposure is critical since only 30 min of incubating non-CAD vessels with LNAME blocks FMD.
The physiological significance of shifting from NO to H2O2 as the mediator of microvascular FMD during heightened oxidative stress or in the presence of disease is not known. An intriguing observation is that the change from NO in health to H2O2 in CAD occurs independent of age. The age range for vessels from subjects with CAD in this study is 30–80 years. Of these six under the age of 55 fall into our “younger adult” group, but all show H2O2 as the dominant mechanism of FMD, whereas non-CAD vessels from this age group all had NO as the predominant mechanism of dilation. This indicates that onset of CAD and not age is regulating the change in dilator mechanism. An obvious advantage is that NO bioavailability is reduced during stress or cardiovascular disease, making H2O2 a more durable candidate for compensation. In conduit arteries such compensation does not occur; instead, loss of NO is typically manifest by attenuation of FMD [17]. Compensation is primarily seen in the microcirculation across species [27, 34]. Maintenance of vasodilator capacity is more important in the microcirculation, which compared to conduit arteries, contributes much more to tissue perfusion [7, 15]. However, it is well-established that both endothelial dysfunction in the microcirculation and reduced FMD in conduit vessels in patients with CAD directly correlate to clinical outcomes and can be an important prognostic tool [9, 28, 33]. Although H2O2 preserves FMD in CAD, there is a potential tradeoff since H2O2, unlike NO, is pro-inflammatory and pro-thrombotic. Since impaired microcirculatory function is a strong predictor of future cardiovascular events [20, 37] understanding the mechanisms that regulate the switch from NO to H2O2 could reveal novel approaches to preventing or reducing progression of vascular disease [3, 16].
Study limitations
Several factors should be considered when interpreting these data. Studies involving human specimens suffer from heterogeneity among subjects. Tissues were collected from patients with diverse genetic backgrounds and underlying diseases. Although the source of vessels varied (coronary and different fat depots), in this study and prior publications we have consistently observed similarities in the mediator of FMD in adults in both health and disease [3, 16]. Additionally, tissue from children’s vessels was mostly not collected form atrial appendages, which accounts more than 90% of the source of coronary vessels in adult tissue. This may contribute to the significantly smaller vessel size found in children’s samples. However, close investigation of the mechanism of dilation in the smallest vessels and largest vessels in each cohort did not reveal any observable changes in FMD (supplemental Table 1).
Tissues were obtained from de-identified discarded specimens; therefore we were unable to collect comprehensive clinical data (e.g., full medication lists, extensive laboratory data) and are unable to fully evaluate their potential roles as contributing factors. This limitation is common to most studies involving human subjects and must be balanced against the advantage of studying responses in human tissue. The gender distribution (average 55 ± 3% males) of our patient samples was fairly balanced. The racial/ethical distribution, while skewed towards subjects from northern European descent (Caucasian; 86% age <18; 68% age 18–55; 83% age >55; 71% CAD) was representative of the population in our catchment area. Despite the limited sample size, and similar to prior studies from our lab [2, 16], we have not observed effects of gender or race on vasodilation. Importantly, we did not observe any influence of key baseline parameters (risk factors of subjects in Table 1) on the dilator or fluorescence results. The consistency of observed mechanisms of dilation in heterogeneous populations, make the findings more robust.
To evaluate a role for ROS in FMD, we utilized a multipronged approach using antioxidants or a H2O2 specific probe we have used previously (mitoPYl) [3, 16]. This could raise questions about specificity. For example, Vitamin E can attack a variety of ROS, and boronate reduces not only H2O2, but also peroxynitrite and HOCl. To ensure specificity, we confirmed that ROS-mediated dilation or increase in mitoPYl signal (H2O2 production) was inhibited by pegylated catalase which is specific for H2O2. Pegylation improves protein stability and allows it to move intracellularly. We have consistently found that PEG-CAT, but not always CAT is able to inhibit mitochondrial H2O2 fluorescence and functional responses (dilation) due to mitochondrial H2O2.
The beneficial effects of Vitamin E and PBA were seen only when targeted to the mitochondria using triph-enylphosphonium suggesting a critical role for mitochondrial derived ROS in mediating FMD. A tenfold larger dose of boronate also reduced FMD. We speculate that at the higher concentration boronate either entered the mitochondria in sufficient concentration, or was able to quench cytosolic H2O2, thereby inhibiting dilation.
In this study, we did not test the specific impact of prolonged reliance on H2O2 as the primary mediator of FMD. DelloStritto et al. [11] suggest that prolonged H2O2 exposure impairs TRPV1-dependent coronary vascular signaling, which in turn may contribute to microvascular dysfunction and tissue perfusion deficits associated with cardiovascular disease. Guarini et al. [19] have identified ROS induced mtDNA damage as a critical and reversible contributor to microvascular dysfunction in the coronary circulation that promotes heart failure.
We tested only one endothelium-dependent dilator, thus the findings may be specific to FMD. The rationale for using shear is threefold. First FMD is the most physiological endothelium-dependent dilator mechanism; second acetylcholine, the prototypical endothelium-dependent dilator elicits constriction in atrial arterioles of patients with CAD [26]; and third, other endothelium-dependent dilators, such as bradykinin or serotonin signal through multiple receptor pathways and are less frequently studied, limiting opportunities for comparison among species and vessel types. It will be important to confirm these findings with other endothelium-dependent dilators.
Significance
Our manuscript is the first to report the changes in the arteriolar mediator of FMD in the human heart during aging and with disease. Figure 6 illustrates the proposed changes in vasodilator mechanisms thoughtout life and with onset of CAD. The nature of the dilator substances released from the endothelium [PGI2, NO, and hydrogen peroxide (H2O2)] may play an important role in vascular homeostasis, with implications for atherosclerosis and vascular inflammation. It is intriguing to consider why these changes occur. Previous studies have linked lower prostacyclin levels in early life to increased risk for the development of coronary heart disease [40]. Loss of NO is also associated with worse outcomes. The vasodilator mediator released during shear may impact the onset and consequences of atherosclerosis. The switch from vaso-protective agonists, such as PGI2 or NO to pro-atherogenic H2O2 is consistent with the well-established idea that atherosclerosis is accompanied by a pro-inflammatory oxidative environment, mechanistically linked to onset of disease. Our previous work shows that acute stress in the form of brief elevations in intraluminal pressure initiates the same switch in the mechanism of FMD as does onset of CAD [2, 13] suggesting that the switch is compensatory for the loss of NO mediated dilation. We speculate the observed changes with onset of CAD represent a reversible stress adaption (telomerase and ceramide papers referenced here), but further studies have to be performed to confirm this hypothesis. Detailed mechanistic studies are also needed to understand the signaling pathways involved in this switch. This study has implications for use of common pharmaceuticals (e.g., NSAIDS; antioxidants) in children and adults with CAD, respectively.
Fig. 6.
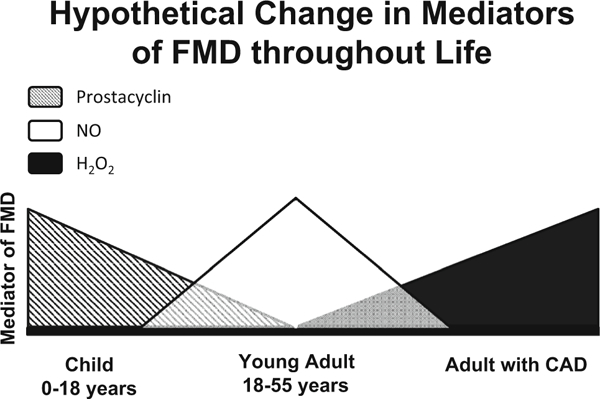
Proposed change in mediators of FMD throughout life. The mechanism of FMD in children is primarily mediated by PGI2 with a small contribution by NO. With healthy aging NO becomes the dominant mediator of dilation changes with a small PGI2 component. With onset of CAD NO mediated dilation is lost and H2O2 becomes the sole endothelial derived relaxing factor in response to shear
Supplementary Material
Acknowledgements
We thank the surgeons and nurses at Froedtert Hospital, the Division of Cardiothoracic Surgery at the Medical College of Wisconsin, the Cardiothoracic Surgery Division at the Zablocki Veterans Affairs Medical Center in Milwaukee, the Children’s Hospital of Wisconsin, the Aurora Medical Group Cardiovascular and Thoracic Surgery, Cardiothoracic Surgery Group of Milwaukee, the Wheaton Hospital Group including St. Joseph’s, The Wisconsin Heart Hospital, Elmbrook Memorial of Brookfield, and The Wisconsin Donor Network for providing tissue.
Funding This work was supported by National Institutes of Health Grants R01-HL-113612 (to D. D. Gutterman), R21-OD-018306 (to A. M. Beyer). We received support from the Clinical and Translational Science Award (CTSA) program of NCATS for writing assistance (Glenn Krakower Grant 8UL1TR000055).
Abbreviations
- BMI
Body mass index
- BSA
Bovine serum albumin
- CAD
Coronary artery disease
- COX
Cyclooxygenase
- eNOS
Endothelial nitric oxide synthase
- FMD
Flow-mediated dilation
- H2O2
Hydrogen peroxide
- L-NAME
NG-Nitro-L-arginine methyl ester
- mt
Mitochondrial
- MitoPY1
Mitochondria peroxy yellow 1
- NO
Nitric oxide
- PBA
Phenylboronate
- PGI2
Prostacyclin
- ROS
Reactive oxygen species
Footnotes
Compliance with ethical standards
Electronic supplementary material The online version of this article (doi:10.1007/s00395-016-0594-x) contains supplementary material, which is available to authorized users.
References
- 1.Bassenge E, Heusch G (1990) Endothelial and neuro-humoral control of coronary blood flow in health and disease. Rev Physiol Biochem Pharmacol 116:77–165. doi: 10.1007/3540528806_4 [DOI] [PubMed] [Google Scholar]
- 2.Beyer AM, Durand MJ, Hockenberry J, Gamblin TC, Phillips SA, Gutterman DD (2014) An acute rise in intraluminal pressure shifts the mediator of flow-mediated dilation from nitric oxide to hydrogen peroxide in human arterioles. Am J Physiol Heart Circ Physiol 307:H1587–H1593. doi: 10.1152/ajpheart.00557.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beyer AM, Freed JK, Durand MJ, Riedel M, Ait-Aissa K, Green P, Hockenberry JC, Morgan RG, Donato AJ, Peleg R, Gasparii M, Rokkas CK, Santos JH, Priel E, Gutterman DD (2015) Critical role for telomerase in the mechanism of flow mediated dilation in the human microcirculation. Circ Res. doi: 10.1161/circresaha.115.307918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beyer AM, Freed JK, Durand MJ, Riedel M, Ait-Aissa K, Green P, Hockenberry JC, Morgan RG, Donato AJ, Peleg R, Gasparri M, Rokkas CK, Santos JH, Priel E, Gutterman DD (2016) critical role for telomerase in the mechanism of flow-mediated dilation in the human microcirculation. Circ Res 118:856–866. doi: 10.1161/CIRCRESAHA.115.307918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cassina A, Radi R (1996) Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys 328:309–316. doi: 10.1006/abbi.1996.0178 [DOI] [PubMed] [Google Scholar]
- 6.Charpie JR, Schreur KD, Papadopoulos SM, Webb RC (1994) Endothelium dependency of contractile activity differs in infant and adult vertebral arteries. J Clin Invest 93:1339. doi: 10.1172/JCI117093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chilian WM, Eastham CL, Marcus ML (1986) Microvascular distribution of coronary vascular resistance in beating left ventricle. Am J Physiol Heart Circ Physiol 251:H779–H788 [DOI] [PubMed] [Google Scholar]
- 8.Cooper A, Heagerty AM (1998) Endothelial dysfunction in human intramyocardial small arteries in atherosclerosis and hypercholesterolemia. Am J Physiol Heart Circ Physiol 275:H1482–H1488 [DOI] [PubMed] [Google Scholar]
- 9.Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard-Herman M, Herrington D (2002) Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol 39:257–265. doi: 10.1016/S0735-1097(01)01746-6 [DOI] [PubMed] [Google Scholar]
- 10.Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, Kaley G (2002) Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res 90:1159–1166. doi: 10.1161/01.RES.0000020401.61826.EA [DOI] [PubMed] [Google Scholar]
- 11.DelloStritto DJ, Connell PJ, Dick GM, Fancher IS, Klarich B, Fahmy JN, Kang PT, Chen YR, Damron DS, Thodeti CK, Bratz IN (2016) Differential regulation of TRPV1 channels by H2O2: implications for diabetic microvascular dysfunction. Basic Res Cardiol 111:21. doi: 10.1007/s00395-016-0539-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dickinson BC, Chang CJ (2008) A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J Am Chem Soc 130:9638–9639. doi: 10.1021/ja802355u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durand MJ, Phillips SA, Widlansky ME, Otterson MF, Gutter-man DD (2014) The vascular renin-angiotensin system contributes to blunted vasodilation induced by transient high pressure in human adipose microvessels. Am J Physiol Heart Circ Physiol 307:H25–H32. doi: 10.1152/ajpheart.00055.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Durand MJ, Zinkevich NS, Riedel M, Gutterman DD, Nasci VL, Salato VK, Hijjawi JB, Reuben CF, North PE, Beyer AM (2016) Vascular actions of angiotensin 1–7 in the human microcirculation novel role for telomerase. Arterioscler Thromb Vasc Biol 36:1254–1262. doi: 10.1161/ATVBAHA.116.307518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feigl E (1983) Coronary physiology. Physiol Rev 63:1–205 [DOI] [PubMed] [Google Scholar]
- 16.Freed JK, Beyer AM, LoGiudice JA, Hockenberry JC, Gutterman DD (2014) Ceramide changes the mediator of flow-induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circ Res 115:525–532. doi: 10.1161/CIRCRESAHA.115.303881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerhard M, Roddy M-A, Creager SJ, Creager MA (1996) Aging progressively impairs endothelium-dependent vasodilation in forearm resistance vessels of humans. Hypertension 27:849–853. doi: 10.1161/01.HYP.27.4.849 [DOI] [PubMed] [Google Scholar]
- 18.Gray C, Harrison CJ, Segovia SA, Reynolds CM, Vickers MH (2015) Maternal salt and fat intake causes hypertension and sustained endothelial dysfunction in fetal, weanling and adult male resistance vessels. Sci Rep 5:9753–9761. doi: 10.1038/srep0975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guarini G, Kiyooka T, Ohanyan V, Pung YF, Marzilli M, Chen YR, Chen CL, Kang PT, Hardwick JP, Kolz CL, Yin L, Wilson GL, Shokolenko I, Dobson JG Jr, Fenton R, Chilian WM (2016) Impaired coronary metabolic dilation in the metabolic syndrome is linked to mitochondrial dysfunction and mitochondrial DNA damage. Basic Res Cardiol 111:29. doi: 10.1007/s00395-016-0547-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halcox JP, Schenke WH, Zalos G, Mincemoyer R, Prasad A, Waclawiw MA, Nour KR, Quyyumi AA (2002) Prognostic value of coronary vascular endothelial dysfunction. Circulation 106:653–658. doi: 10.1161/01.CIR.0000025404.78001.D8 [DOI] [PubMed] [Google Scholar]
- 21.Hink U, Münzel T (2006) COX-2, another important player in the nitric oxide-endothelin cross-talk good news for COX-2 inhibitors? Circ Res 98:1344–1346. doi: 10.1161/01.RES.0000228471.38761.93 [DOI] [PubMed] [Google Scholar]
- 22.Kuo L, Arko F, Chilian WM, Davis MJ (1993) Coronary venular responses to flow and pressure. Circ Res 72:607–615. doi: 10.1161/01.RES.72.3.607 [DOI] [PubMed] [Google Scholar]
- 23.Kuo L, Chilian WM, Davis MJ (1991) Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am J Physiol Heart Circ Physiol 261:H1706–H1715 [DOI] [PubMed] [Google Scholar]
- 24.Larsen FJ, Schiffer TA, Weitzberg E, Lundberg JO (2012) Regulation of mitochondrial function and energetics by reactive nitrogen oxides. Free Radic Biol Med 53:1919–1928. doi: 10.1016/j.freeradbiomed.2012.08.580 [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD (2011) H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res 108:566–573. doi: 10.1161/CIRCRESAHA.110.237636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller FJ, Dellsperger KC, Gutterman DD (1998) Pharmacologic activation of the human coronary microcirculation in vitro: endothelium-dependent dilation and differential responses to acetylcholine. Cardiovasc Res 38:744–750. doi: 10.1016/S0008-6363(98)00035-2 [DOI] [PubMed] [Google Scholar]
- 27.Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD (2003) Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res 92:e31–e40. doi: 10.1161/01.RES.0000054200.44505.AB [DOI] [PubMed] [Google Scholar]
- 28.Neunteufl T, Katzenschlager R, Hassan A, Klaar U, Schwarzacher S, Glogar D, Bauer P, Weidinger F (1997) Systemic endothelial dysfunction is related to the extent and severity of coronary artery disease. Atherosclerosis 129:111–118. doi: 10.1016/S0021-9150(96)06018-2 [DOI] [PubMed] [Google Scholar]
- 29.Ohsaki Y, O’Connor P, Mori T, Ryan RP, Dickinson BC, Chang CJ, Lu Y, Ito S, Cowley AW Jr (2012) Increase of sodium delivery stimulates the mitochondrial respiratory chain H2O2 production in rat renal medullary thick ascending limb. Am J Physiol Renal Physiol 302:F95–F102. doi: 10.1152/ajprenal.00469.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pinder AG, Pittaway E, Morris K, James PE (2009) Nitrite directly vasodilates hypoxic vasculature via nitric oxide-dependent and-independent pathways. Br J Pharmacol 157:1523–1530. doi: 10.1111/j.1476-5381.2009.00340.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poderoso JJ, Carreras MC, Lisdero C, Riobo N, Schopfer F, Boveris A (1996) Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys 328:85–92. doi: 10.1006/abbi.1996.0146 [DOI] [PubMed] [Google Scholar]
- 32.Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P (1993) Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci USA 90:7240–7244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shamim-Uzzaman QA, Pfenninger D, Kehrer C, Chakrabarti A, Kacirotti N, Rubenfire M, Brook R, Rajagopalan S (2002) Altered cutaneous microvascular responses to reactive hyperaemia in coronary artery disease: a comparative study with conduit vessel responses. Clin Sci 103:267–273. doi: 10.1042/cs1030267 [DOI] [PubMed] [Google Scholar]
- 34.Shimokawa H (2010) Hydrogen peroxide as an endothelium- derived hyperpolarizing factor. Pflugers Arch EJP 59:915–922. doi: 10.1007/s00424-010-0790-8 [DOI] [PubMed] [Google Scholar]
- 35.Sun D, Liu H, Yan C, Jacobson A, Ojaimi C, Huang A, Kaley G (2006) COX-2 contributes to the maintenance of flow-induced dilation in arterioles of eNOS-knockout mice. Am J Physiol Heart Circ Physiol 291:H1429–H1435. doi: 10.1152/ajpheart.01130.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taniyama Y, Griendling KK (2003) Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension 42:1075–1081. doi: 10.1161/01.HYP.0000100443.09293.4F [DOI] [PubMed] [Google Scholar]
- 37.Targonski PV, Bonetti PO, Pumper GM, Higano ST, Holmes DR Jr, Lerman A (2003) Coronary endothelial dysfunction is associated with an increased risk of cerebrovascular events. Circulation 107:2805. doi: 10.1161/01.CIR.0000072765.93106.EE [DOI] [PubMed] [Google Scholar]
- 38.Toda N (2012) Age-related changes in endothelial function and blood flow regulation. Pharmacol Ther 133:159–176. doi: 10.1016/j.pharmthera.2011.10.004 [DOI] [PubMed] [Google Scholar]
- 39.van de Hoef TP, van Lavieren MA, Damman P, Delewi R, Piek MA, Chamuleau SA, Voskuil M, Henriques JP, Koch KT, de Winter RJ, Spaan JA, Siebes M, Tijssen JG, Meuwissen M, Piek JJ (2014) Physiological basis and long-term clinical outcome of discordance between fractional flow reserve and coronary flow velocity reserve in coronary stenoses of intermediate severity. Circ Cardiovasc Interv 7:301–311. doi: 10.1161/CIRCINTERVENTIONS.113.001049 [DOI] [PubMed] [Google Scholar]
- 40.Wojakowski W, Gmński J (2001) Plasma levels of von Wille-brand factor, endothelin-1, prostacyclin and thromboxane in children from families with high risk of premature coronary artery disease. Scand J Clin Lab Invest 61:317–323. doi: 10.1080/00365510152379058 [DOI] [PubMed] [Google Scholar]
- 41.Wu C, Huang RT, Kuo CH, Kumar S, Kim CW, Lin YC, Chen YJ, Birukova A, Birukov KG, Dulin NO, Civelek M, Lusis AJ, Loyer X, Tedgui A, Dai G, Jo H, Fang Y (2015) Mechanosensitive PPAP2B regulates endothelial responses to atherorelevant hemodynamic forces. Circ Res 117:e41–e53. doi: 10.1161/CIRCRESAHA.117.306457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeiher AM, Drexler H, Saurbier B, Just H (1993) Endothelium-mediated coronary blood flow modulation in humans. Effects of age, atherosclerosis, hypercholesterolemia, and hypertension. J Clin Invest 92:652–662. doi: 10.1172/JCI116634 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.