

Abstract
Introduction:
Altered cell cycle reentry has been observed in Alzheimer’s disease (AD). Denticle-less (DTL) was predicted as the top driver of a cell cycle subnetwork associated with AD.
Methods:
We systematically investigated DTL expression in AD and studied the molecular, cellular, and behavioral endophenotypes triggered by DTL overexpression.
Results:
We experimentally validated that CDT2, the protein encoded by DTL, activated cyclin-dependent kinases through downregulating P21, which induced tau hyperphosphorylation and Aβ toxicity, two hallmarks of AD. We demonstrated that cyclin-dependent kinases inhibition by rosco-vitine not only rescued CDT2-induced cognitive defects but also reversed expression changes induced by DTL overexpression. RNA-seq data from the DTL overexpression experiments revealed the molecular mechanisms underlying CDT2 controlled cell cycle reentry in AD.
Discussion:
These findings provide new insights into the molecular mechanisms of AD pathogenesis and thus pave a way for developing novel therapeutics for AD by targeting AD specific cell cycle networks and drivers.
Keywords: Alzheimer’s disease, DTL, CDT2, CDK, Cell cycle reentry
1. Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that appears multifactorially and clinically and histopathologically heterogeneous [1]. In less than 1% of cases, the disease is associated with three rare mutations in β-amyloid (Aβ) precursor protein, presenilin-1, or presenilin-2 genes [2]. Its causes in the remaining more than 99% sporadic cases are unclear. Independent of cause, AD is characterized clinically by progressive dementia and histopathologically by the presence of numerous neurofibrillary tangles consisting of hyper-phosphorylated tau and senile plaques composed of Aβ [3,4]. Because 99% of AD cases appear to represent sporadic disease, its multifactorial features and neurobiological complexity hamper early clinical diagnosis, determination of mechanisms, and drug development.
Genome-wide transcriptional profiling may shed new light on the molecular interactions of cellular pathways that account for the complexity of AD. This in turn will facilitate the development of truly novel anti-AD treatments that target defined molecular networks and go beyond partial palliation of symptoms. In 2013, we systematically identified key molecular networks and drivers (regulators) in AD through an integrative multiscale network analysis of 1647 brain tissues from patients with AD and nondementia subjects [5]. Those molecular networks underlying AD enable an objective and precise illustration of the molecular interactions in AD brains and can be used to predict molecular and phenotypic changes in response to perturbations of key network drivers. The 2013 study revealed that a cell cycle–enriched module/subnetwork (CCM) had not only a strong association with AD clinical and pathological traits but also the second largest loss of gene-gene interactions in AD with respect to normal controls. Denticleless (DTL) was predicted as the top key driver of CCM, suggesting that DTL and its coding protein, CDT2, may play a prominent role in the pathogenesis of AD. Because the focus of the 2013 study was the network modeling of molecular dys-regulations in AD, no effort was made to investigate the role of CCM or DTL in AD.
Aberrant cell cycle, especially neuronal cell cycle reentry, has been observed in AD [6–12], but the exact molecular mechanisms underlying such cell cycle dysregulation remain elusive. DTL/CDT2 is an essential component of the early, radiation-induced G2/M checkpoint [13,14]. Proper DNA replication is crucial for maintaining genome stability [14–16]. As the substrate recognition subunit of CUL4 CRL ubiquitin ligase (CRL4) complex, CDT2 promotes precise DNA replication by the temporal regulation of replication licensing, which is required for cell proliferation and development [15,17,18]. DTL/CDT2 mainly mediates the ubiquitination and subsequent degradation of CDT1, Set8, and p21. CDT1 degradation in response to DNA damage is necessary to ensure proper cell cycle regulation of DNA replication [19–21]. Set8 degradation induces cell arrest in G2 and their failure to condense their chromosomes [22]. P21 is a member of major cyclin-dependent kinase (CDK) inhibitors (CKIs), which is expressed in G1 and G2 [23,24]. However, p21 degradation during S phase or following UV irradiation is essential to control replication licensing [24,25]. Therefore, DTL/CDT2 regulates cell cycle via the degradation on its substrates, especially p21 degradation, which may relieve the inhibition of CDKs. Meanwhile, the activation of CKIs has been shown in AD and participates in AD pathological alterations [26,27].
Based on these observations, we hypothesized that upregulation of DTL/CDT2 increases p21 degradation, which in turn releases CDKs activity and triggers AD process. In the present study, we systematically studied the molecular, cellular, and behavioral endophenotypes triggered by DTL overexpression (OE). We experimentally validated that CDT2-activated CDKs through downregulating P21, which in turn induced tau hyperphosphorylation and Aβ toxicity, two hallmarks of AD. RNA-seq data from the DTL OE experiments revealed that DTL regulated a large number of genes including several AD GWAS risk genes such as apolipoprotein E (APOE), CLU, and SORL1.
2. Methods
2.1. Plasmids, viruses, chemicals, and antibodies
The plasmids encoding pAOV-CMV-bGlobin-Vector-3FLAG orpAOV-CMV-bGlobin-CDT2–3FLAG were generated in our laboratory. All plasmids were sequenced and prepared using an endotoxin-free plasmid extraction kit (Tiangen). AAV-pAOV-CMV-bGlobin-Vector-3FLAG and AAV-pAOV-CMV-bGlobin-CDT2–3FLAG were constructed and packaged by Obio Technology CO, Ltd (Shanghai, China). Lipofectamine 2000 transfection reagents were from Invitrogen. Roscovitine was purchased from Sigma (Ros, Saint Louis, MO, USA). The bicincho-ninic acid protein detection kit was from Pierce (Rockford, IL, USA). Reagents for cell culture were from Gibco BRL (Gaithersburg, MD, USA). Reagents for cell culture were from Gibco BRL (Gaithersburg, MD, USA). Antibodies used in this study are listed in the Supplementary Table S4.
2.2. Cell culture, transfection, and drug treatment
The human embryonic kidney 293 (HEK293) cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Gibco BRL, Gaithersburg, MD, USA), HEK293 cells stably transfected with the longest human tau (tau441) (HEK293/tau), and mouse neuroblastoma N2a cells with stable expression of APP (amyloid precursor protein) (N2A-APP) were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Invitrogen; Bleiswijk, Netherlands) in the presence of 200 mg/mL G418 containing 10% fetal bovine serum. The cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Cells were seeded into 6- or 12-well culture plates 1 day before transfection, performed using Lipofectamine 2000 (Invitrogen), transfected with the mixture containing a total of 1.8 μg plasmids and 4 μL Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocols.
To explore the role of CDK activity on Cdt2-induced AD pathology, the cells transfected Cdt2 48 hours later were treated with roscovitine (10 μM) for 4 hours.
2.3. RNA extraction, reverse transcription, quantitative real-time polymerase chain reaction
Total RNA of cells was extracted using TRIzol Reagent (Invitrogen, Waltham, MA, USA). First-strand complementary DNA (cDNA) was synthesized from 500 ng total. RNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). Quantitative polymerase chain reaction (PCR) was performed in a 10 μL standard PCR reaction mixture prepared in duplicate using an Applied Biosystems 7900 Prism Real-Time PCR system and SYBR Premix Ex Taq (TaKaRa, Dalian, Japan), in accordance to the manufacturer’s protocol. Quantitative PCR primers were as follows: BACE1, sense: 5′-gactagga-gaccagaagtgaatg-3′ and antisense: 5′-cttctgtccaccc-tattttctgg-3′. The β-actin primers were used as the internal control. β-actin, sense: 5′-cacagactacctcatgaagatcc-3′ and antisense: 5′-cagctcgtaactcttctccag −3′.
2.4. Mouse brain tissue preparation and protein extraction
Mice were deeply anesthetized with chloral hydrate and immediately perfused with saline, and the brains were rapidly removed and snap-frozen for biochemical analysis. For brain protein extraction, hemispheres were first extracted in RIPA buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS), centrifuged at 14,000g for 20 min.
2.5. Immunoprecipitation
Cells were quickly dissected out and homogenized on ice in modified RIPA containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1% (v/v) Triton X-100, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA, 50 mM N-ethylmaleimide, 1 mM NaF, 1 mM Na3VO4, and 1 μg/mL each of protinin, leupeptin, pepstatin, and 1 mM phenylmethane sulfonyl fluoride before immunoprecipitation. The extracts (about 200 μg total proteins) were incubated with antibodies at 4° C overnight, and then protein G agarose was added and incubated at 4°C for 2 h. The agarose beads were collected, washed and resuspended in 60 μL of sample buffer containing 50 mM Tris-HCl, pH 7.6,2% SDS, 10% glycerol, 10 mM DTT, and 0.2% bromophenol blue, and boiled for 5 min and analyzed by Western blotting.
2.6. Western blot analysis
The mouse brain tissue or cells were lysed on ice (lysis buffer: 50 mM Tris, pH 7.4, 40 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1.5 mM Na3VO4, 50 mM NaF, 10 mM sodium pyrophosphate, 10 mM sodium β-glycero-phosphate, a complete protease inhibitors cocktail) and centrifuged for 20 min at 16,000 ×g. The supernatant was boiled in SDS loading buffer. Then, proteins were separated by 10% SDS-polyacrylamide gel electrophoresis gel and then transferred to nitrocellulose membrane. After blocking in 3% nonfat milk for 1 h at 25°C, the membranes were then incubated with primary antibodies at 4° C over-night. Finally, the membranes were incubated with anti-rabbit or anti-mouse IgG conjugated to IRDye (800CW) for 1 h at 25°C and visualized using the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA).
2.7. ELISA quantification of Aβ42
For measurement of Aβ42 species in cell medium, conditioned media of cells were recovered and debris was removed by centrifugation and applied to ELISA plates after 48 h of treatment. To detect the concentration of Aβ42 in total brain lysates, the mouse brains were homogenized in buffer (PBS with 5% BSA and 0.03% Tween-20, supplemented with protease inhibitor cocktail) and centrifuged at 16,000 × g for 20 min. Aβ42 was quantified using the Human/ Mouse Aβ1–42 ELISA Kit (Elabscience, Wuhan, China) in accordance with the manufacturer’s instructions. The Aβ42 concentrations were determined by comparison with the standard curve.
2.8. CDK activity assay
Cells were quickly dissected out and homogenized on ice in modified RIPA containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1% (v/v) Triton X-100, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 1 mMEDTA, 50 mM N-ethylmaleimide, and 1 mM phenylmethane sulfonyl fluoride before immunoprecipitation. The extracts (about 200 μg total proteins) were incubated with CDK antibodies at 4°C overnight, and then protein G agarose was added and incubated at 4°C for 2 h. The agarose beads were collected and washed with 1 mL cold PBS for three times; the purified CDK (20 μg) complex-bound beads (20 μL) were incubated with Histone1 in the presence of 2 μL of ATP (10 nM) in kinase assay buffer (20 mM Tris-HCl pH 7.6, 20 mM MgCl2, 1 mM EDTA, and 0.1 mM DTT) at 30°C for 30 minutes, washed and resuspended in 60 μL of sample buffer containing 50 mM Tris-HCl, pH 7.6,2% SDS, 10% glycerol, 10 mM DTT, and 0.2% bromophenol blue, and boiled for 5 min and analyzed the phosphorylation level of Histone1 at threonine sites by Western blotting.
2.9. Open field (OF) tests
The animals were individually placed at the center of a 45 × 45 × 45 cm3 white acrylic box and left to freely move within it for 10 min. For all behavioral tests, data were gathered and analyzed with a video tracking system (HVS Imagen, UK). Room temperature was maintained at 20° C.
2.10. Novel object recognition (NOR) tests
Novel object recognition (NOR) tests were performed a day after the OF test in a 45 × 45 × 45 cm3 white acrylic box. Animals were pretrained to habituate to the box for two consecutive days, without objects. For testing, animals were placed individually at the center of the box in the presence of two identical objects (old objects) for 10 min. After that period, the box and objects were cleaned with 50% methanol solution. The animal was later (after 2 h) exposed to one of the old objects and a new object of a different shape and color, and the box and objects were cleaned again to continue with the next animal. The recognition index was calculated as the time spent exploring the new object divided by the time exploring both objects.
2.11. Morris water maze tests
The Morris water maze (MWM) tests were performed a day after the NOR tests. The standard MWM procedure was used with minor modifications as described previously. The water maze test contains acquisition training and probe trial. During the acquisition training, the mice were trained to find a submerged platform hidden 1 cm under water by using constant cues outside the pool. During each trial, mice have up to 60 s to find the hidden platform; otherwise, it would be guided to the platform and forced to stay on it for 20 s. Acquisition training contained four trials a day, for six consecutive days. The probe trial is used to test the memory of animals. In the seventh day, the hidden platform was removed and each mouse was allowed to swim freely for 60 s. The swimming pathway, escape latency of mice to find the hidden platform, and the time spent in the target quadrant were recorded by a digital device connected to a computer.
2.12. Golgi staining
Mice were perfused with approximately 200 mL of normal saline containing 0.5% sodium nitrite, followed by 200 mL of 4% formaldehyde solution and 200 mL Golgi fixative (5% chloral hydrate, 4% formaldehyde and 5% potassium dichromate) for 4 h in the dark. The brains were incubated in the same Golgi fixative for 3 days and transferred to a silver solution containing 1% silver nitrate for 7 days in the dark. The solution was changed each day. Coronal brain sections of hippocampus tissue were cut into 85 mm sections using a vibrating microtome (Leica VT1000S; Leica, Germany).
2.13. Long-term potentiation
Mice (3 months old) were used for all our electrophysiology experiments. Mice were deeply anesthetized as mentioned previously. When all pedal reflexes were abolished, brains were removed and placed in ice-cold oxygenated slicing solution containing the following: 225 mM sucrose, 3 mM KCl, 1.25 mM NaH2PO4, 24 mM NaHCO3, 6 mM MgSO4, 0.5 mM CaCl2, and 10 mM D-glucose. Coronal slices (350 μm thick) containing the dorsal hippocampus were cut at 4–5 °C in the slicing solution using a Leica VT1000S vibratome and then transferred to an incubation chamber filled with oxygenated slicing solution in a 30°C water bath for 1 h before being recorded. Slices were laid down in a chamber with an 8 × 8 microelectrode array in the bottom planar (each 50 × 50 mm in size, with an interpolar distance of 150 μm) and kept submerged in artificial cerebrospinal fluid (1–2 mL/min) with a platinum ring glued by nylon silk. Signals were acquired using the MED64 System (Alpha MED Sciences, Panasonic). The field excitatory postsynaptic potentials in CA1 neurons were recorded by stimulating the Schaeffer fibers from dentate gyrus. Long-term potentiation was induced by applying three trains of high-frequency stimulation (100 Hz, 1-second duration).
2.14. Statistical analyses
Data were expressed as mean ± standard error of the mean and analyzed using commercial software (GraphPad Prism; GraphPad Software, Inc, La Jolla, CA). The two-way analysis of variance or one-way analysis of variance, or a Student’s t-test, was used to determine the different means among the groups, and differences with P < .05 were considered as statistically significant.
2.15. Human AD transcriptomic data set from the MS-NBTR cohort
To examine DTL expression in AD, we used a large-scale human AD postmortem brain gene expression data set from the Mount Sinai NIH NeuroBioBank (MS-NBTR) AD cohort [28,29]. In this data set, RNA-seq profiles were generated from brain tissues in four brain regions, including parahippo-campal gyrus (PHG), inferior frontal gyrus, superior temporal gyrus, and frontal pole. For a subset of 125 brains, microarray gene expression profiles from 19 cortical regions had been also generated in our recent study. A number of cognitive/neuropathological traits were characterized for the MS-NBTR cohort, including Clinical Dementia Rating, Braak neurofibrillary tangle score [30,31], CERAD (Consortium to Establish a Registry for Alzheimer’s Disease diagnosis) diagnoses and ratings of pathology, and mean neuritic plaque density in cortical regions (PlaqueMean). Following Wang et al. [29], samples were classified into 2 or 3 groups of different severity stages for each trait and differential expression was conducted between groups through a moderate t-test by using the limma package [32].
2.16. DTL OE RNA sequencing and differential expression analysis
2.16.1. Cell culture and RNA isolation protocol
HEK293 cells were seeded into six-well culture plates 1 day before transfection, performed using Lipofectamine 2000 (Invitrogen), and transfected with the mixture containing a total of 1.8 μg plasmids and 4 μL Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocols. The cells transfected Cdt2 for 48 hours, and total RNA of cells was extracted using TRIzol Reagent (Invitrogen, Waltham, MA, USA) according to the RNA isolation protocol. Cells were levitated and centrifuged to collect the cell pellet, and 1.5 mL TRIzol was added to the cell pellet (about 1 × 107 cells per 1.5 mL). The homogenate was centrifuged at 12,000 ×g for 5 minutes at 4° C, and the supernatant was transferred to a new 2.0 mL tube, added 0.3 mL of chloroform/isoamyl alcohol (24:1) per 1.5 mL of TRIzol reagent. The tubes were shaken vigorously for 15 seconds and centrifuged at 12,000 ×g for 10 minutes at 4°C. After that, the mixture was separated into three layers: the lower phenol-chloroform phase, an interphase, and an upper aqueous phase. RNA remained in the aqueous phase, and the aqueous phase was transferred to a new 1.5 mL tube, added equal volume of supernatant of isopropyl alcohol. Mixed well and placed at −20°C for 2 hours for precipitation. Centrifuged at 13600 rpm for 20 minutes at 4°C and removed the supernatant. The RNA pellet was washed with 1 mL of 75% ethanol, resuspended, and centrifuged at 13600 rpm for 3 minutes at 4°C. This step was repeated again, to completely remove the ethanol without disturbing the pellet. The RNA pellet was got air-dry in the biosafety cabinet and added 25 μL~100 μL of diethyl pyrocarbonate-treated water to dissolve the RNA pellet.
2.16.2. Differential expression analysis
Pair-ended RNA-seq data were generated with the Illumina HiSeq 2500 platform following the Illumina protocol. The raw sequencing reads were aligned to human hg19 genome using the STAR aligner (version 2.5.0b). Following read alignment, featureCounts [33] was used to quantify the gene expression at the gene level based on Ensembl gene model GRCh37.70. Genes with at least 1 count per million in at least one sample were considered expressed and hence retained for further analysis, otherwise removed. The gene-level read counts data were normalized using trimmed mean of the M-values normalization method [34] to adjust for sequencing library size difference. After normalization, transcriptome-wide differential gene expression analysis was performed between different treatment groups using R package limma [32]. Genes with fold change (FC) ≥ 1.2 and Benjamini-Hochberg false discovery rate (FDR)–adjusted P value ≤ .05 were considered differentially expressed genes (DEGs).
2.17. Construction of DTL-regulated signaling pathway network
To explore more broadly signaling pathways regulated by DTL, we used the human AD Bayesian network (BN) to rank and cluster the DEGs of DTL OE based on the network topology. Briefly, the DEGs were first projected onto the human AD BN to perform key driver analysis through testing the enrichment of each DEG’s network neighborhood for the rest of the DEGs. As a result, the DEGs were rank ordered by their enrichment significance. Then, the ranked DEGs were interlinked to form a network in such a way that the j-th ranked DEG was connected to one higher ranked DEG, say the k-th gene (k < j), if j was in the neighbor of the latter in the BN. When there were multiple possible k’s (i.e., multiple higher ranked DEGs), we kept only one edge between j and the smallest k (i.e., the top one) and trim the other edges to make the signaling network sparse. The edge direction was from a higher ranked node to a lower ranked node. With the network constructed, we further annotated each node by the functional pathways enriched in its downstream nodes. For presentation purpose, we removed from the final network those leaf nodes (i.e., without any downstream node) and nodes whose BN neighborhoods were not enriched for DEGs as they were less interesting in regard to interaction with other genes.
2.18. Set overlap analysis
Fisher’s exact test (FET) was employed to test for significant enrichment or depletion among different gene signatures. To control for multiple tests, Benjamini-Hochberg FDR approach was applied. When testing for functional enrichment, we overlapped the gene signatures with the gene ontology and canonical pathway annotations curated in the MSigDB [35] and FET was used to compute the P value significance.
3. Results
3.1. DTL is upregulated in human AD postmortem brain samples
We evaluated the gene expression of DTL at different stages of the AD progression in a large-scale postmortem brain transcriptomic data set from the MS-NBTR AD cohort [28,29]. Among the four brain regions profiled with RNA-seq from this cohort, DTL presented a 1.2-fold upregulation in demented subjects compared with nondemented normal controls in the PHG, a 1.5-fold upregulation in subjects with mild cognitive impairment compared with nondemented normal controls in the inferior frontal gyrus, and a 1.3-fold upregulation in subjects with severe plaque density compared with subjects with low plaque density in the superior temporal gyrus (Fig. 1A–C). When we combined the data from all four brain regions, a meta-analysis revealed that DTL was significantly upregulated in diseased brains for all the cognitive/neuropathological traits-Clinical Dementia Rating [36], Braak stage (a measure of hyperphosphorylated tau involvement) [37], CERAD score (a measure of AD diagnosis certainty) [38], and mean cortical neuritic plaque density [39] (2.3 ~ 3.5 fold, adjusted t-test P value ≤ .04), highlighting the increased power in the combined data. We confirmed the upregulated expression of DTL through a pan-cortical meta-analysis in a microarray gene expression data set spanning 19 cortical brain regions in a subset of the MS-NBTR cohort [28]. Compared with nondemented brains, DTL was upregulated by 1.6-fold (P value 1.4 × 10−3) in brains of persons with mild cognitive impairment and 1.6-fold (P value 1.2 × 10−3) in the brains of persons with frank dementia. In addition, we also examined DTL gene expression in an independent meta-analysis of postmortem brain samples curated in the AlzData database (http://www.AlzData.org). In this data set, DTL was upregulated in the entorhinal cortex region of the AD brains (1.2-fold, t-test P value 0.019) (Fig. 1D). Note that the analyses carried out here are novel and have not been reported elsewhere.
Fig. 1.
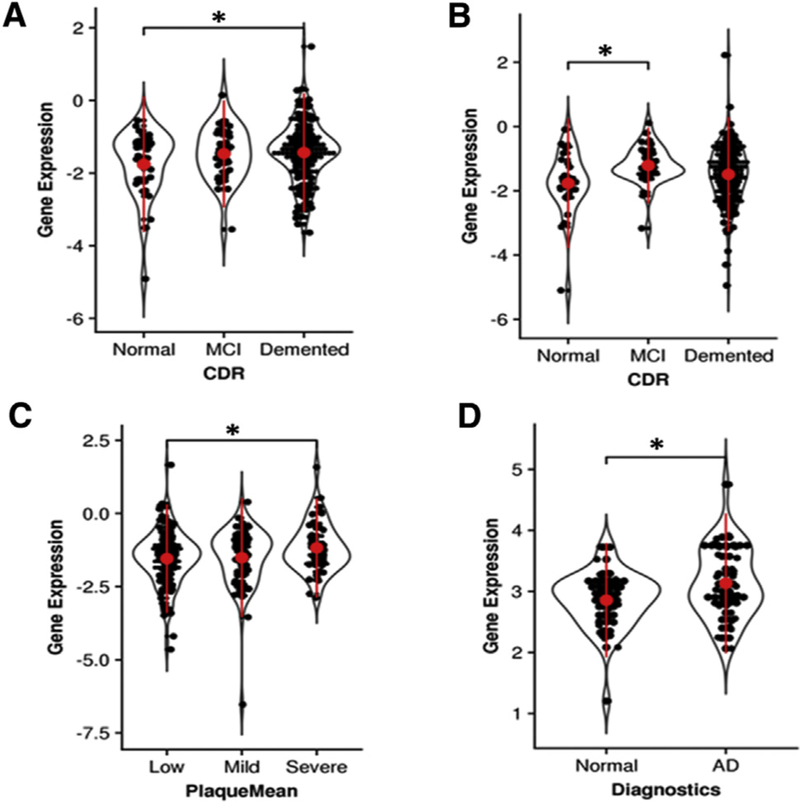
DTL is upregulated in human AD postmortem brains across different regions including (A) parahippocampus, (B) inferior frontal gyrus, (C) superior temporal gyrus, and (D) entorhinal cortex. P value significance is calculated from a one-tailed t-test. * P < .05. Abbreviations: AD, Alzheimer’s disease; CDR, Clinical Dementia Rating; MCI, mild cognitive impairment.
3.2. CDT2 downregulating P21 mediates the activation of CDKs
Because the aforementioned analysis of large-scale human brain transcriptomic data demonstrates that DTL is upregulated in AD, we investigate the mechanism underlying DTL and its coding protein CDT2’s participation in AD. We transfected HEK293 cells with flag-tagged CDT2 with special attention to the CDKs inhibitor, P21, the degradation of which is regulated by CDT2 [40]. We note that CDKs have been considered as very important protein kinases involved in AD [41–45]. We found that OE of CDT2 induced a marked decrease of P21 expression compared to controls (Fig. 2A and B). To validate that downregulation of P21 relieves the inhibition of CDKs and releases CDKs’ activities, a kinase activity assay was carried out. We found that CDT2 downregulation of P21 significantly increased the activities of CDK1, CDK2, CDK3, CDK4, and CDK5 compared to controls (Fig. 2C), whereas OE of CDT2 decreased the interaction of CDK5 with P21 (Supplementary Fig. S1A and S1B). Together, these findings indicate that CDT2 downregulation of P21 mediates the activation of CDKs.
Fig. 2.
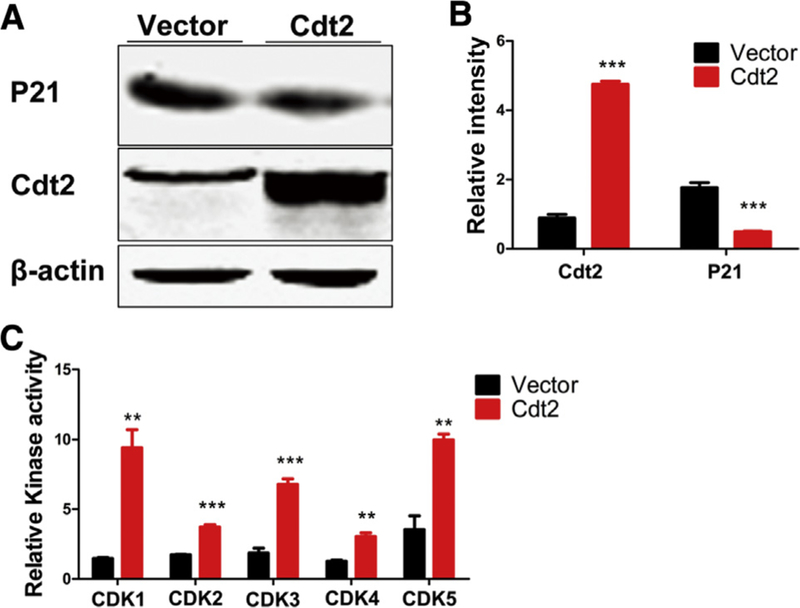
CDT2 activates CDKs through downregulating CDKs inhibitor P21. (A and B) Overexpression of CDT2 dramatically reduced P21 in HEK293 cells compared with controls. (C) Overexpression of CDT2 significantly increased the activities of CDK1, CDK2, CDK3, CDK4, and CDK5 compared with controls. All data represent mean ± standard error of the mean. ** P < .01; *** P < .001, versus vector control.
3.3. Identification of DTL/CDT2 OE signatures by RNA-seq
To study the transcriptional signature of DTL/CDT2 OE, we profiled transcriptomic profiles using RNA-seq in HEK293 cells transfected with plasmids of a human DTL gene construct. DTL/CDT2 OE signatures were identified using a moderated t-test comparing cells overexpressing DTL with control cells transfected with an empty vector; 2713 upregulated genes and 2760 downregulated genes were identified in DTL/CDT2 OE cells. The full list of DEGs is provided in Table S1. Among the upregulated DEGs, there were three CDK genes, including CDK11A (2.5-fold, FDR-adjusted P value 1.4 × 10−4), CDK16 (1.2-fold, FDR-adjusted P value 4.2 × 10−3), and CDK2 (1.22-fold, FDR-adjusted P value 0.03), whereas among the downregulated DEGs, there is one CDK gene CDK18 (0.79-fold, FDR-adjusted P value 0.03) and one CDK inhibitor CDKN1C (0.58-fold, FDR-adjusted P value 5.8 × 10−3) (Fig. 3A). A number of AD GWAS susceptibility genes were also among the DTL OE signatures, including 5 upregulated genes—APOE (2.4-fold, FDR adjusted P value 1.0 × 10 3), the ε4 variant of which is the most prominent risk factor for late-onset sporadic AD (LOAD), CASS4 (4.6-fold, FDR-adjusted P value 1.2 × 10−5), CELF1 (1.3-fold, FDR-adjusted P value 9.5 × 10−4), CLU (1.7-fold, FDR-adjusted P value 3.6 X 10_3), and ETFA (1.3, FDR-adjusted P value 4.1 × 10−3)—and 1 downregulated gene SORL1 (0.8-fold, FDR-adjusted P value 8.1 × 10−3) (Fig. 3B). In the present study, we found that the level of apoE 4 in DTL/CDT2 OE cells was markedly increased compared with controls (Supplementary Fig. S2). SORL1, also known as LR11, is an interesting target suppressed by DTL. The product of SORL1 is a neuronal APOE receptor that is also involved in APP processing and trafficking [46]. It has been reported that the level of SORL1 expression is reduced in AD brains and is positively correlated with Aβ accumulation [47].
Fig. 3.
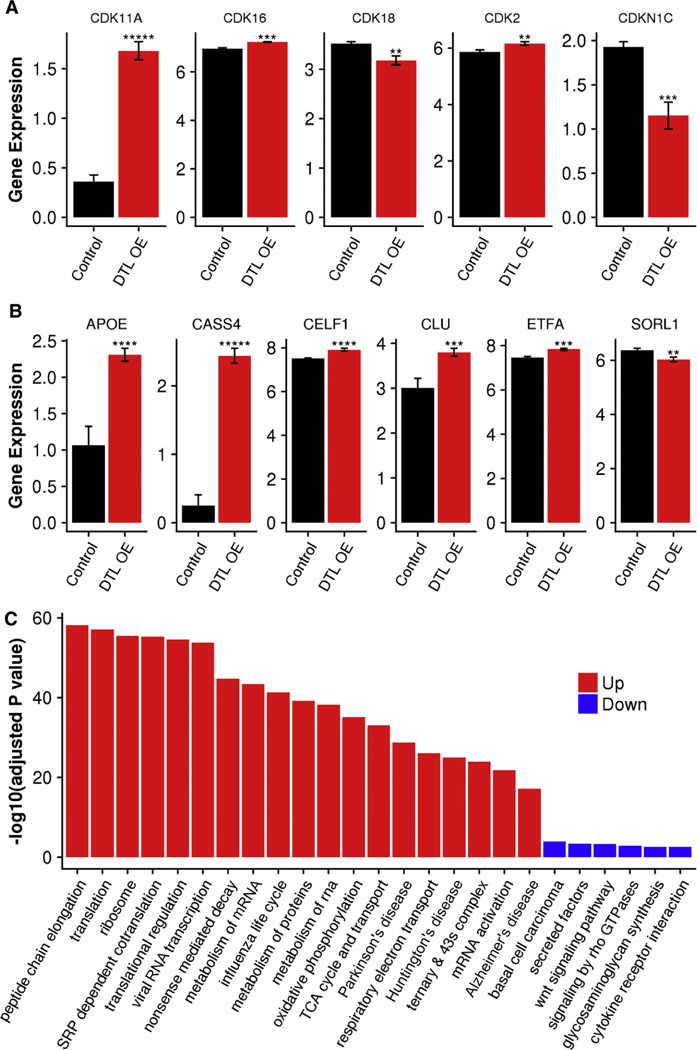
RNA-seq data from DTL overexpression experiments in HEK293 cells confirmed upregulation of several CDKs and further identified six AD GWAS risk genes as targets of DTL. (A) Differential expression of several CDK genes. (B) Differential expression of six AD GWAS susceptibility genes. (C) Functional pathways most enriched in the gene signatures upregulated (red bars) and downregulated (blue bars) by DTL overexpression. P value significance is calculated from a one-tailed t-test. ** P < .01; *** P < .001; **** P < .0001; ***** p < .00001. Abbreviations: AD, Alzheimer’s disease; APOE, apolipoprotein E; OE, overexpression.
As shown in Fig. 3C, the upregulated genes were enriched for transcription- and translation-related pathways, such as peptide chain elongation, translation, signal recognition particle-dependent co-translational protein targeting to membrane, 3’ UTR-mediated translational regulation, nonsense-mediated decay enhanced by the exon junction complex, and metabolism of mRNA. Influenza life cycle pathway was also enriched in the upregulated genes. This suggests that DTL plays an important role in modulating transcriptional and translational regulations in the cells. In contrast, downregulated genes were enriched for basal cell carcinoma, secreted factors, Wnt signaling pathway, signaling by Rho GTPases, glycosaminoglycan biosynthesis heparan sulfate, transmembrane receptor activity, and cytokine-cytokine receptor interaction.
We intersected the DTL OE signature with human AD gene expression signatures identified in the MS-NBTR cohort [29]. FET revealed a significant enrichment of the up-regulated genes induced by DTL OE within the upregulated genes detected in the MS-NBTR diseased brains in multiple regions including superior frontal gyrus, occipital visual cortex, and inferior frontal gyrus with respect to different cognitive/neuropathological traits (Table S2). For example, 65 up-regulated genes by DTL OE overlapped with the set of genes that were upregulated in the superior frontal gyrus in association with Braak stage, leading to a 1.7-fold enrichment with an adjusted P value of 1.1 × 10−3.
3.4. CDT2 upregulating CDKs induces tau hyperphosphorylation and Aβ toxicity
As protein kinases, CDK1 and CDK5 have been reported to be involved in tau phosphorylation during the progression of AD pathology [41−45,48]. Our present study shows that OE of CDT2 downregulates P21, which in turn releases CDKs’ activities. To assess whether CDT2 upregulation of CDK5 leads to AD pathological alterations, we overexpressed CDT2 in both in vivo C57BL/6J mice and in HEK293/tau cell lines via adeno-associated virus (AAV) injection and plasmid transfection, respectively. Western blotting showed the marked increase of tau phosphorylation at Ser396 in CDT2-treated C57BL/6J mice (Fig. 4A–B) and HEK293/tau cells (Supplementary Fig. S3A-B). The CDKs inhibitor roscovitine attenuated tau hyperphosphorylation in CDT2-treated mice (Fig. 4A–B) and cells (Supplementary Fig. S3A-B), while the level of total tau recognized by Tau5 remained unchanged across the three groups (Fig. 4A, C and Supplementary Fig. S3A, S3C). These data suggest that OE of CDT2 induces tau hyperphosphorylation via upregulation of CDKs.
Fig. 4.
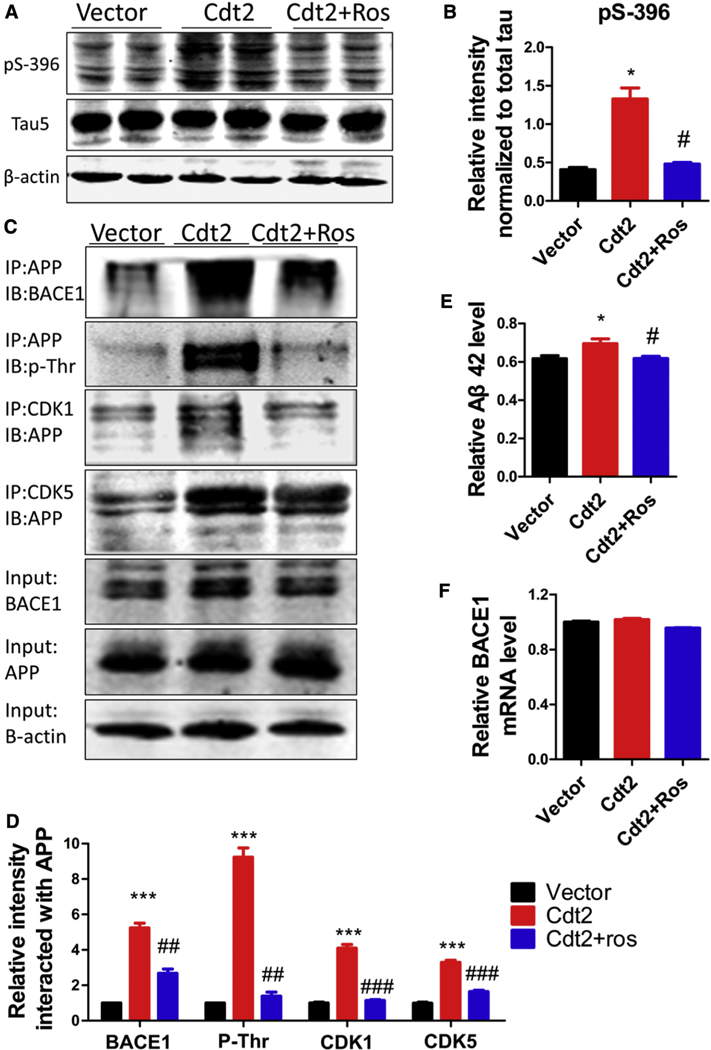
Overexpression of CDT2 induces tau hyperphosphorylation and Aβ overproduction. CDT2 was overexpressed in C57BL/6J mice in vivo via adeno-associated virus (AAV) injection. (A and B) Western blotting and statistical analysis showed the marked increase of tau phosphorylation at Ser396 in CDT2-treated C57BL/6J mice. The CDKs inhibitor roscovitine attenuated tau hyperphosphorylation in CDT2-treated mice, while the level of total tau recognized by Tau5 had no change among each group. (C and D) Transfection with CDT2 in N2a/APP cells induced a significant increase in the interaction of APP with BACE1, CDK1 and CDK5 and APP threonine phosphorylation levels compared with controls. Inhibition of CDKs by roscovitine markedly attenuated the interaction of APP with BACE1, CDK1 and CDK5 and APP threonine phosphorylation levels. (E) The level of Aβ42 was significantly increased in CDT2 mice compared with control animals, while CDKs inhibition by roscovitine restored Aβ42 level to normal. (F) BACE1 mRNA levels did not change among each group. All data represent mean 6 SEM. * P < .05; *** P < .001, versus vector control. # P < .05; ## P < .01; ### P < .001, versus Cdt2.
Besides tau hyperphosphorylation, Aβ-containing senile plaque is the other hallmark pathological change in AD. We therefore investigated the effect of CDT2 OE on Aβ toxicity. ELISA data showed that the level of Aβ42 was significantly increased in CDT2 mice compared with control animals, while CDKs inhibition by roscovitine restored the Aβ42 level to normal (Fig. 4D). A similar result was observed in N2a/APP cells transfected with CDT2 (Supplementary Fig. S3D). To further study the mechanism underlying Aβ increase under OE of CDT2, we detected BACE1, the rate-limiting enzyme in the process of Aβ generation from APP cleavage, and the interaction of BACE1 with APP [49,50]. Co-immunoprecipitation showed that the transfection with CDT2 in N2a/APP cells induced a significant increase in the interaction of BACE1 with APP compared with controls (Fig. 4E-F), whereas BACE1, APP, and BACE1 mRNA levels remained unchanged (Fig. 4G–I). However, inhibition of CDKs by roscovitine markedly attenuated the interaction of BACE1 with APP (Fig. 4E–F). This observation suggests that OE of CDT2 does not influence BACE1 expression and amount, yet CDT2 upregulation of CDKs may be associated with the interaction of BACE1 with APP, which then boosts Aβ production. We therefore hypothesize that activation of CDKs increases APP phosphorylation and augments phosphorylated APP interaction with BACE1. To test this hypothesis, we transfected N2a/APP cells with CDT2 and treated them with or without the CDKs inhibitor, roscovitine, and then performed co-immunoprecipitation. We found that transfection with CDT2 significantly increased the phosphorylation of APP and the interaction of APP with CDK1 or CDK5 compared with controls (Fig. 4J–K). These results imply that CDKs phosphorylating APP boosts its interaction with BACE1 and cleavage.
Taken together, OE of DTL and its coding protein CDT2 downregulates P21, whose deficiency relieves the inhibition of CDKs and in turn induces tau hyperphosphorylation and Aβ production by boosting phosphorylated APP interaction with BACE1.
3.5. CDK inhibition rescues CDT2-induced cognitive defects
Our data show that CDT2-induced upregulation of CDKs mediates two main neuropathological features of AD: tau hyperphosphorylation and Aβ toxicity. To further validate the role(s) of DTL in vivo and its coding protein CDT2 which regulates AD susceptibility and to confirm its significance on AD-associated behavior, synaptic regulation, and gene transcription, we injected AAV2-CDT2 into the entorhinal cortex of wild-type adult C57BL/6J mice. One month later, we first performed an OF test to assess anxiety and exploratory behavior that could have developed as a result of the AAV2-CDT2 injection. AAV2-CDT2 mice displayed similar patterns of anxiety and exploratory activity as control and AAV2-CDT2 + roscovitine animals (Supplementary Fig. S4). Next, an NOR test was administered by training on the first day and testing at the same time on the second day (Fig. 5A) to evaluate contextual memory. We observed a significant reduction in the NOR index (calculated as the time spent exploring the new object divided by the time exploring both objects) in AAV2-CDT2 mice compared with the control animals, while inhibition of CDKs by roscovitine restored these defects (Fig. 5B and C). Finally, we carried out the MWM test to assess learning and memory functions using this spatial reference memory task. AAV2-CDT2 mice revealed a marked deficit in finding a hidden platform compared with the control animals during the training days (Fig. 5D). For the probe trial, the latency (Fig. 5E) was markedly increased, and crossing numbers (Fig. 5F) and time spent in the target quadrant (Fig. 3G) were significantly decreased in AAV2-CDT2 mice compared with the control animals. Moreover, roscovitine treatment recovered these defects. The total swim distances remained comparable among the groups, indicating that CDT2 or roscovitine do not affect motor function (Fig. 5H–I). Together, these findings strongly support that CDK inhibition rescues CDT2-induced AD-like cognitive impairments.
Fig. 5.
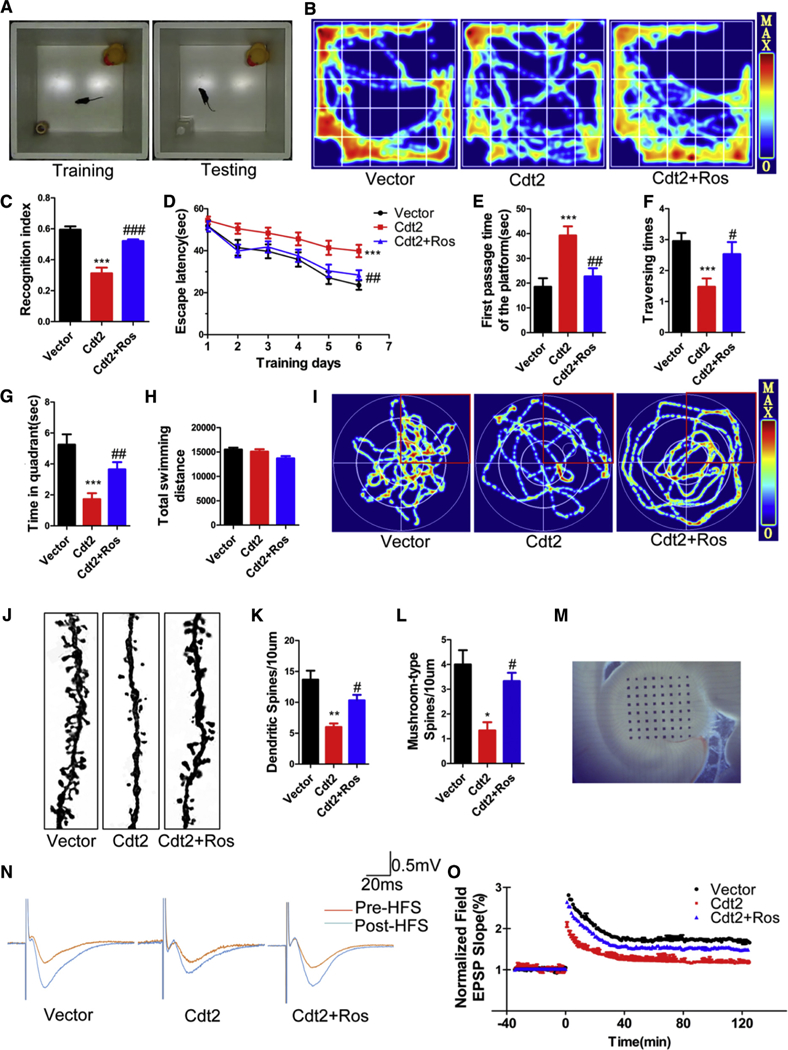
CDK inhibition rescues CDT2-induced AD-like cognitive impairments. Impact of overexpression of DTL and its coding protein CDT2 on AD susceptibility, AD behavior, and synaptic regulation was assessed through the injection of AAV2-CDT2 into the entorhinal cortex of wild type C57BL/6J mice. (A) The novel recognition index (calculated as the time spent exploring the new object divided by the time spent exploring both objects) in AAV2-CDT2 mice significantly decreased compared with the control animals. (B and C) Inhibition of CDKs by roscovitine recovered the defects induced by CDT2. (D) AAV2-CDT2 mice had a marked deficit in finding a hidden platform compared with the control animals during the training days using the Morris water maze test. (E) AAV2-CDT2 mice had significantly increased latency in the probe trial. (F) AAV2-CDT2 mice traversed less frequently. (G) AAV2-CDT2 mice spent less time in the target quadrant. (H and I) The total movement distances the three groups traveled were comparable. (J) Overexpressed CDT2 significantly reduced spine density and mushroom type, by contrast, inhibition of CDKs by roscovitine recovered these defects. (K) Quantification of spine density in (J). (L) Quantification of mushroom type in (J). (M) Overexpressed CDT2 substantially decreased the field excitatory postsynaptic potential slope. (N and O) Roscovitine rescued the damage or sabotage in the synaptic plasticity induced by overexpressed CDT2. All data represent mean 6 SEM. * P < .05; ** P < .01; ***, P < .001, versus vector control. # P < .05; ## P < .01, versus Cdt2; ###, P < .001.
To assess whether CDKs-triggered cognitive defect was associated with loss of the synaptic plasticity, we first performed Golgi staining and found that OE of CDT2 significantly reduced the density of mushroom type spine; by contrast, inhibition of CDKs by roscovitine recovered these defects (Fig. 5J). Spine density quantification is summarized in Fig. 5K-L. Next, we performed electrophysiology analysis (Fig. 5M). We found that OE of CDT2 substantially decreased the field excitatory postsynaptic potential slope, supporting that CDT2 amplification hampered synaptic plasticity, whereas roscovitine rescued these defects (Fig. 5N–O). These data strongly demonstrate that CDK inhibition reverses CDT2-induced synaptic plasticity deficits.
3.6. CDK inhibitor roscovitine reverses expression changes induced by DTL OE in HEK293 cells
We further explored whether inhibiting cyclin-dependent kinases can rescue the expression changes caused by DTL OE. For this purpose, we added roscovitine to the HEK293 cell cultures 48 hours after transfection with a DTL plasmid. After treating with roscovitine for 4 hours, RNAs were extracted from the cells and subjected to RNA-seq. We found that only five genes showed a significant change in the roscovitine-treated cells compared with the untreated control cells transfected with an empty vector. These included DTL, HBB, GH1, HSPA6, and DNAH17, all of which were upregulated with a similar FC as in the DTL OE cells (Table S1). The rest of the more than 5400 DEGs regulated by DTL OE were restored after roscovitine treatment to similar expression levels as in the control cells. As shown in Fig. 6A, most of the DTL OE signatures showed a reversed expression change with a similar absolute log2 FC value in the roscovitine-treated cells compared with the DTL OE-treated cells. This indicates that inhibiting CDK has no impact on the expression of DTL but will reverse nearly the entire compendium of transcriptomic changes induced by OE of DTL, further supporting the hypothesis that the DTL regulatory pathway is CDK dependent.
Fig. 6.
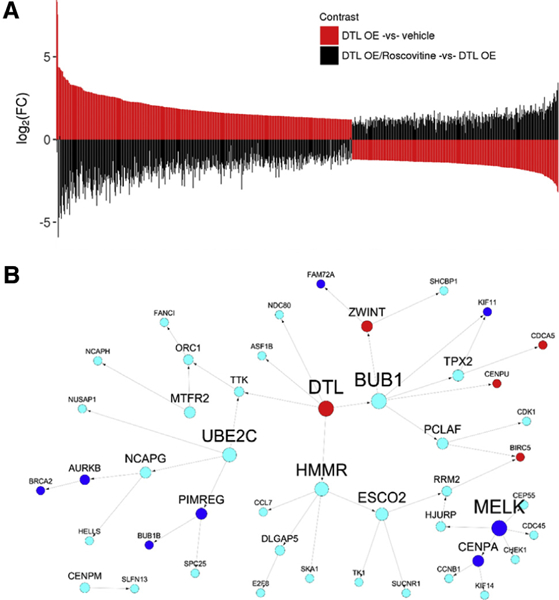
Gene expression changes in response to DTL overexpression are reversed by CDK inhibitor roscovitine and are enriched in the DTL centered causal network in AD. (A) CDK inhibitor roscovitine reverses the expression changes induced by DTL OE. The top panel shows the ordered log2 FC of the DTL OE signatures (DTL OE vs. vehicle control). The bottom panel shows the log2 FC between roscovitine-treated cells and DTL OE cells (DTL OE/roscovitine vs. DTL OE) for the DTL OE signatures, which are arranged in the same order as in the top panel. (B) DTL is a key driver in a cell cycle subnetwork in AD. Red and blue colors denote upregulated and downregulated genes induced by DTL OE in HEK293 cells, respectively. Cyan color represents the genes without significant differential expression in the same DTL OE experiment. Abbreviations: FC, fold change; OE, over-expression.
3.7. DTL regulated cell cycle signaling in AD
DTL was inferred as a key driver in a network module enriched with cell cycle genes identified in the Harvard Brain Tissue Resource Center expression data set [5]. We overlaid the DTL OE signature onto this cell cycle subnetwork and found a significant overlap (Fig. 6B). Of the 47 network genes neighboring DTL, 13 showed differential expression in the DTL OE assay, including 8 downregulated genes and 5 upregulated genes (2.6-fold, FET P value 2.4 × 10−3), suggesting that the human brain gene expression network is predictive of the DTL OE signature in the cell line under study.
We further extended the DTL network by incorporating genes significantly correlated with DTL in the prefrontal cortex region of the Harvard Brain Tissue Resource Center data set. Using Spearman’s correlation coefficient analysis and an FDR-adjusted P value threshold of 0.05, we identified 1729 positively correlated genes and 1454 negatively correlated genes in AD brains. A two-sided FET revealed that the genes upregulated by DTL OE were enriched for genes positively correlated with DTL in human AD brains (FET P = 3.04E-07, 1.4 FE), whereas the genes downregulated by DTL OE were enriched for genes negatively correlated with DTL in human AD brains (FET P = 1.2E-04, 1.3 FE) (Table S3). In summary, the present network analysis indicated a strong consistency between human brain tissue-based and cell line–based transcriptomic profiling regarding DTL signatures.
4. Limitation
In the present study, we detected cognition in CDT2 rats by the OF test to assay general locomotor activity levels and anxiety, NOR test to evaluate contextual memory, and MWM test to assess learning and memory functions. Actually, if AD-related depression needs to be detected, the forced swimming test can be used to evaluate “behavioral despair.” Because the MWM test per se may induce stress, Barnes maze, a dry-land maze, test can be performed to assess hippocampus-dependent spatial reference memory in the further experiment. We here used long-term potentiation as a persistent strengthening of synapses based on recent patterns of activity. The patch clamp technique can be especially useful in the future study of excitable neurons.
5. Discussion
The pathogenesis of LOAD remains elusive, and the clinical trials targeting the removal of toxic Aβ plaques from the brains of patients with AD have not appeared to be beneficial. Over past decades, intensive studies have identified more than two dozen genetic susceptibility loci associated with the late-onset AD [51,52] emphasizing the potential etiopathogenic heterogeneity of the disease. There is an urgent need for novel hypotheses and treatment targets for the disease. Using an integrative network method, we had previously predicted DTL as a potential key regulator in a cell-cycle subnetwork implicated in the neuropathology of AD. In this article, we systematically studied the transcriptomic signatures of DTL and validated through both in vivo and in vitro assays that DTL upregulates CDKs which in turn induces tau hyperphosphorylation and Aβ toxicity, suggesting a potential pathogenic role of DTL in AD. Although DTL was expressed at relatively low levels in adult brains, we found the brains of patients with high DTL expression were associated with worst cognitive/neuropathological outcome in the MS-NBTR AD cohort. As shown in Table 1, the top 10% of the samples with the highest DTL in the PHG of the MS-NBTR AD data set were enriched for demented (χ2 = 6.2, P value = .013) and definite AD (χ2 = 13.7, P value = .001) patients. The PHG was previously found to be the transcriptomically most severely affected brain region in AD through a pan-cortical transcriptomic network analysis in the MS-NBTR AD cohort. These data suggest that there exists a possible subtype of AD with high DTL expression. Although the DTL-specific subtype warrants further investigation, the finding has the important implication that it would be useful to use transcriptomic signatures to identify molecular subtypes of AD, a polygenic heterogenous disease, and subsequently develop optimal treatment for different subtype groups of different causes. A successful drug development for AD will require understanding of different etiopathogenic mechanisms involved and stratification of patients with AD by different disease subgroups in clinical trials.
Table 1.
Samples with high DTL expression in the parahippocampal gyrus in the MS-NBTR-AD cohort were associated with worst cognitive/ neuropathological outcome
|
DTL expression in AD |
CDR |
CERAD |
|||
|---|---|---|---|---|---|
| Demented | Nondemented | AD | pAD | Normal | |
| DTL | |||||
| High | 20 | 1 | 15 | 6 | 1 |
| Low | 119 | 61 | 64 | 51 | 65 |
| χ2 | 6.2 | 13.7 | |||
| P value | 0.013 | 0.001 | |||
Abbreviations: CDR, Clinical Dementia Rating; CERAD, neuropathology diagnostic certainty according to the Consortium to Establish a Registry for Alzheimer’s Disease; pAD, probable or possible Alzheimer’s disease.
NOTE. The top 10% samples with the highest DTL expression were assigned to the high group, whereas the remaining samples were assigned to the low group.
This article represents the first biological validation of the DTL-centered gene regulatory network in LOAD. The DEGs in cells overexpressing DTL significantly overlapped with the gene networks identified in human postmortem LOAD brains derived from two distinctly separate collections and cohorts (Fig. 6B and Supplementary Table S3), supporting the consistency of DTL-driven gene coexpression patterns between cell lines and human brain tissues. Many AD GWAS susceptibility genes are among the DTL OE signatures, including upregulated genes APOE 4, CASS4, CELF1, and CLU, and 1 downregulated gene SORL1 (Fig. 3B). APOE 4 is the strongest genetic risk factor for late-onset AD [53]. A large amount of evidence has demonstrated that in addition to increasing disease risk, apoE 4 stimulating the transcription factor AP-1 enhances transcription of APP and Ab levels and thereby accelerates early seeding of amyloid pathology [54,55], while apoE 4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy [56]. Our data, showing that apoE 4 protein was significantly increased in DTL/CDT2 OE cells compared with control, strongly imply that CDT2 upregulates apoE 4, which is in turn associated with AD neuro-pathologic processes. We demonstrated that CDT2 regulates the pathogenesis of AD through the activation of CDKs, which then upregulates apoE 4 expression. CASS4 (Cas scaffolding protein family member 4) is also known as HEPL, which is associated with NEDD9 and PTK2B to be involved simultaneous subtle regulation of calcium signaling, inflammatory response, and microtubule dynamics: all phenotypes highly relevant to LOAD [57]. Clusterin coded by CLU, also known as apolipoprotein-J, is a multifunctional lipoprotein involved in Aβ fibrillation, clearance, and complement inhibition [57]. In patients with AD, clusterin co-localizes with dystrophic neurites and Aβ in amyloid plaques [58]. SORL1 prevents trafficking of APP from the Golgi to endosomal compartments, where Aβ secretases reside [59]. Downregulation of SORLA in cultured cells or in transgenic mice has been shown to enhance the amyloidogenic process [60]. In the present study, we further showed that a CDKs inhibitor roscovitine reversed both the transcriptomic changes and the deleterious effects on neuropathology induced by DTL/CDT2 OE. We here speculate that CDT2-controlled cell cycle reentry in AD might be associated with tau pathology, Aβ toxicity, calcium signaling, inflammatory response, and microtubule dynamics via its regulation of AD-related susceptibility genes, which further account for synaptic loss and cognitive impairments.
The strong molecular, cellular, and behavioral phenotypes from DTL/CDT2 OE in this study not only validate DTL/CDT2 as a key regulator of AD pathogenesis but also provide new mechanistic insights. Hence, this study paves a way for developing novel therapeutics via inhibition of CDKs for AD by targeting cell cycle networks and drivers underlying AD. Toward this ultimate goal, we envision the following directions for future research: (1) specificity of cell cycle and DTL regulation in various AD pathological groups and APOE genotypes; (2) cell type specificity of cell cycle and DTL regulation in AD; and (3) interactions of the cell cycle pathway/network with other AD pathways/networks such as immune response, synaptic transmission, and nerve ensheathment across different brain cell types.
Supplementary Material
RESEARCH IN CONTEXT.
Systematic review: Cell cycle dysregulation or reentry has been observed in Alzheimer’s disease (AD), but the exact molecular mechanisms underlying such cell cycle dysregulation remains elusive. A recent systems biology study of AD predicted DTL as a key driver of a cell cycle gene subnetwork that was associated with AD pathophysiological traits.
Interpretation: DTL was found to be overexpressed in AD brains. We show that DTL activated CDKs through downregulating P21, which further induced tau hyperphosphorylation and Aβ toxicity. We demonstrated that CDK inhibition by roscovitine not only rescued CDT2-induced cognitive defects but also reversed expression changes induced by DTL overexpression. Molecular signatures of DTL overexpression validated the structure of the predicted cell cycle network associated with AD.
Future directions: Given that DTL has been comprehensively validated as a key regulator of AD pathogenesis from the molecular, cellular, and behavioral perspectives, we envision the following directions for the future research: (1) specificity of cell cycle and DTL regulation in various AD pathological groups and APOE genotypes; (2) cell type specificity of cell cycle and DTL regulation in AD; (3) interactions of the cell cycle pathway/network with other AD pathways such as immune response, synaptic transmission, nerve ensheathment, etc.
Acknowledgments
Funding: This work was supported in parts by grants from National Natural Science Foundation of China (31528010, 81571255, and 31771114), grant from Innovative Research Groups of the National Natural Science Foundation of China (81721005), grant from the Ministry of Science and Technology of China (2016YFC1305800), the Academic Frontier Youth Team Project to X.W. from Huazhong University of Science and Technology, and grants from the National Institutes of Health/National Institute on Aging (R01AG046170, RF1AG054014, RF1AG057440, R01AG057907, HHSN 271201300031).
Footnotes
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/joalz.2018.08.013.
The authors of this article have no conflict of interest to declare.
References
- [1].Iqbal K, Grundke-Iqbal I. Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. J Cell Mol Med 2008;12:38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med 2016;18:421–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Malkki H Alzheimer disease: NGF gene therapy activates neurons in the AD patient brain. Nat Rev Neurol 2015;11:548. [DOI] [PubMed] [Google Scholar]
- [4].Vos SJ, Verhey F, Frölich L, Kornhuber J, Wiltfang J, Maier W, et al. Prevalence and prognosis of Alzheimer’s disease at the mild cognitive impairment stage. Brain 2015;138:1327–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J. Podtelezhnikov AA, et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell 2013;153:707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bialopiotrowicz E, Szybinska A, Kuzniewska B, Buizza L, Uberti D, Kuznicki J, et al. Highly pathogenic Alzheimer’s disease presenilin 1 P117R mutation causes a specific increase in p53 and p21 protein levels and cell cycle dysregulation in human lymphocytes. J Alzheimers Dis 2012;32:397–415. [DOI] [PubMed] [Google Scholar]
- [7].Hradek AC, Lee HP, Siedlak SL, Torres SL, Jung W, Han AH, et al. Distinct chronology of neuronal cell cycle re-entry and tau pathology in the 3xTg-AD mouse model and Alzheimer’s disease patients. J Alzheimers Dis 2015;43:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Norambuena A, Wallrabe H, McMahon L, Silva A, Swanson E, Khan SS, et al. mTOR and neuronal cell cycle reentry: How impaired brain insulin signaling promotes Alzheimer’s disease. Alzheimers Dement 2017;13:152–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wezyk M, Szybinska A, Wojsiat J, Szczerba M, Day K, Ronnholm H, et al. Overactive BRCA1 Affects Presenilin 1 in Induced Pluripotent Stem Cell-Derived Neurons in Alzheimer’s Disease. J Alzheimers Dis 2018;62:175–202. [DOI] [PubMed] [Google Scholar]
- [10].Wojsiat J, Prandelli C, Laskowska-Kaszub K, Martin-Requero A, Wojda U. Oxidative Stress and Aberrant Cell Cycle in Alzheimer’s Disease Lymphocytes: Diagnostic Prospects. J Alzheimers Dis 2015; 46:329–50. [DOI] [PubMed] [Google Scholar]
- [11].Herrup K The involvement of cell cycle events in the pathogenesis of Alzheimer’s disease. Alzheimers Res Ther 2010;2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Herrup K, Arendt T. Re-expression of cell cycle proteins induces neuronal cell death during Alzheimer’s disease. J Alzheimers Dis 2002;4:243–7. [DOI] [PubMed] [Google Scholar]
- [13].Sansam CL, Shepard JL, Lai K, Ianari A, Danielian PS, Amsterdam A, Hopkins N, et al. DTL/CDT2 is essential for both CDT1 regulation and the early G2/M checkpoint. Genes Dev 2006;20:3117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Abbas T, Dutta A. CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle 2011;10:241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev 2008;22:2507–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sehorn MG, Sigurdsson S, Bussen W, Unger VM, Sung P. Human meiotic recombinase Dmc1 promotes ATP-dependent homologous DNA strand exchange. Nature 2004;429:433–7. [DOI] [PubMed] [Google Scholar]
- [17].Beck DB, Burton A, Oda H, Ziegler-Birling C, Torres-Padilla ME, Reinberg D. The role of PR-Set7 in replication licensing depends on Suv4–20h. Genes Dev 2012;26:2580–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell 2010;40:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Benamar M, Guessous F, Du K, Corbett P, Obeid J, Gioeli D, et al. Inactivation of the CRL4-CDT2-SET8/p21 ubiquitylation and degradation axis underlies the therapeutic efficacy of pevonedistat in melanoma. EBioMedicine 2016;10:85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hu J, McCall CM, Ohta T, Xiong Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat Cell Biol 2004;6:1003–9. [DOI] [PubMed] [Google Scholar]
- [21].Ralph E, Boye E, Kearsey SE. DNA damage induces Cdt1 proteolysis in fission yeast through a pathway dependent on Cdt2 and Ddb1. EMBO Rep 2006;7:1134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, et al. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell 2010;40:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Balasubramanian S, Kim KH, Ahmad N, Mukhtar H. Activation of telomerase and its association with G1-phase of the cell cycle during UVB-induced skin tumorigenesis in SKH-1 hairless mouse. Oncogene 1999;18:1297–302. [DOI] [PubMed] [Google Scholar]
- [24].LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, et al. New functional activities for the p21 family of CDK inhibitors. Genes Dev 1997;11:847–62. [DOI] [PubMed] [Google Scholar]
- [25].Soria G, Speroni J, Podhajcer OL, Prives C, Gottifredi V. p21 differentially regulates DNA replication and DNA-repair-associated processes after UV irradiation. J Cell Sci 2008;121:3271–82. [DOI] [PubMed] [Google Scholar]
- [26].Copani A, Uberti D, Sortino MA, Bruno V, Nicoletti F, Memo M. Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci 2001;24:25–31. [DOI] [PubMed] [Google Scholar]
- [27].Chang KH, Vincent F, Shah K. Deregulated Cdk5 triggers aberrant activation of cell cycle kinases and phosphatases inducing neuronal death. J Cell Sci 2012;125:5124–37. [DOI] [PubMed] [Google Scholar]
- [28].Haroutunian V, Katsel P, Schmeidler J. Transcriptional vulnerability of brain regions in Alzheimer’s disease and dementia. Neurobiol Aging 2009;30:561–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang M, Roussos P, McKenzie A, Zhou X, Kajiwara Y, Brennand KJ, et al. Integrative Network Analysis of Nineteen Brain Regions Identifies Molecular Signatures and Networks Underlying Selective Regional Vulnerability to Alzheimer’s Disease. Genome Med 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathologica 2006;112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica 1991;82:239–59. [DOI] [PubMed] [Google Scholar]
- [32].Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- [34].Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–4. [DOI] [PubMed] [Google Scholar]
- [37].Braak H, Braak E. Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol 1996;92:197–201. [DOI] [PubMed] [Google Scholar]
- [38].Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991;41:479–86. [DOI] [PubMed] [Google Scholar]
- [39].Haroutunian V, Perl DP, Purohit DP, Marin D, Khan K, Lantz M, et al. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol 1998; 55:1185–91. [DOI] [PubMed] [Google Scholar]
- [40].Hall JR, Bereman MS, Nepomuceno AI, Thompson EA, Muddiman DC, Smart RC. C/EBPalpha regulates CRL4(Cdt2)-mediated degradation of p21 in response to UVB-induced DNA damage to control the G1/S checkpoint. Cell Cycle 2014;13:3602–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Smith PD, O’Hare MJ, Park DS. Emerging pathogenic role for cyclin dependent kinases in neurodegeneration. Cell Cycle 2004;3:289–91. [PubMed] [Google Scholar]
- [42].Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F x DP, in B-amyloid-induced neuronal death. J Biol Chem 1999;274:19011–6. [DOI] [PubMed] [Google Scholar]
- [43].Kimura T, Ishiguro K, Hisanaga S. Physiological and pathological phosphorylation of tau by Cdk5. Front Mol Neurosci 2014;7:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liu SL, Wang C, Jiang T, Tan L, Xing A, Yu JT. The Role of Cdk5 in Alzheimer’s Disease. Mol Neurobiol 2016;53:4328–42. [DOI] [PubMed] [Google Scholar]
- [45].Neve RL, McPhie DL. The cell cycle as a therapeutic target for Alzheimer’s disease. Pharmacol Ther 2006;111:99–113. [DOI] [PubMed] [Google Scholar]
- [46].Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A 2005;102:13461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Scherzer CR, Offe K, Gearing M, Rees HD, Fang G, Heilman CJ, et al. Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch Neurol 2004;61:1200–5. [DOI] [PubMed] [Google Scholar]
- [48].Illenberger S, Zheng-Fischhöfer Q, Preuss U, Stamer K, Baumann K, Trinczek B, et al. The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: implications for Alzheimer’s disease. Mol Biol Cell 1998;9:1495–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ghosh AK, Osswald HL. BACE1 (beta-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem Soc Rev 2014;43:6765–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Alcolea D, Martúnez-Lage P, Sanchez-Juan P, Olazaran J, Antúnez C, Izagirre A, et al. Amyloid precursor protein metabolism and inflammation markers in preclinical Alzheimer disease. Neurology 2015; 85:626–33. [DOI] [PubMed] [Google Scholar]
- [51].Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013; 45:1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sims R, vanderLee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 2017;49:1373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 1993;90:1977–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Huang YA, Zhou B, Wernig M, Sudhof TC. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 2017;168:427–441 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2012;2:a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017;549:523–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Beck TN, Nicolas E, Kopp MC, Golemis EA. Adaptors for disorders of the brain? The cancer signaling proteins NEDD9, CASS4, and PTK2B in Alzheimer’s disease. Oncoscience 2014;1:486–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nielsen HM, Mulder SD, Belien JA, Musters RJ, Eikelenboom P, Veerhuis R. Astrocytic A beta 1–42 uptake is determined by A beta-aggregation state and the presence of amyloid-associated proteins. Glia 2010;58:1235–46. [DOI] [PubMed] [Google Scholar]
- [59].Offe K, Dodson SE, Shoemaker JT, Fritz JJ, Gearing M, Levey AI, et al. The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J Neurosci 2006;26:1596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dodson SE, Andersen OM, Karmali V, Fritz JJ, Cheng D, Peng J, et al. Loss of LR11/SORLA enhances early pathology in a mouse model of amyloidosis: evidence for a proximal role in Alzheimer’s disease. J Neurosci 2008;28:12877–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.