

Abstract
Activation of ERK (extracellular signal-regulated kinase) MAP (mitogen-activated protein) kinase in dorsal horn neurons of the spinal cord by peripheral noxious stimulation contributes to short-term pain hypersensitivity. We investigated ERK activation by peripheral inflammation and its involvement in regulating gene expression in the spinal cord and in contributing to inflammatory pain hypersensitivity. Injection of complete Freund's adjuvant (CFA) into a hindpaw produced a persistent inflammation and a sustained ERK activation in neurons in the superficial layers (laminae I–IIo) of the dorsal horn. CFA also induced an upregulation of prodynorphin and neurokinin-1 (NK-1) in dorsal horn neurons, which was suppressed by intrathecal delivery of the MEK (MAP kinase kinase) inhibitor U0126. CFA-induced phospho-ERK primarily colocalized with prodynorphin and NK-1 in superficial dorsal horn neurons. Although intrathecal injection of U0126 did not affect basal pain sensitivity, it did attenuate both the establishment and maintenance of persistent inflammatory heat and mechanical hypersensitivity. Activation of the ERK pathway in a subset of nociceptive spinal neurons contributes, therefore, to persistent pain hypersensitivity, possibly via transcriptional regulation of genes, such as prodynorphin and NK-1.
Keywords: ERK, MAP kinase, prodynorphin, neurokinin-1, spinal cord, inflammatory pain
Input to the spinal cord dorsal horn from high-threshold nociceptors induces central sensitization, a heterosynaptic facilitation that outlives the initiating stimulus for tens of minutes and that plays a major role in the generation of immediate postinjury pain hypersensitivity (Woolf, 1983). The increased neuronal excitability appears to result from post-translational regulation, such as phosphorylation, of key membrane receptors and channels (Woolf and Salter, 2000; Ji and Woolf, 2001).
Recently, we showed that ERK (extracellular signal-regulated kinase), a member of the MAPK (mitogen-activated protein kinase) family, is activated with a short latency (<1 min) in superficial dorsal horn neurons by noxious but not by innocuous stimuli. This activation contributes to short-term (<1 hr) pain hypersensitivity (Ji et al., 1999). Because the involvement of ERK activation in generating acute pain hypersensitivity can be detected well before any transcriptional change manifests, ERK activation in the spinal cord, as in the hippocampus (English and Sweatt, 1997; Impey et al., 1998, 1999; Winder et al., 1999), is likely to contribute to short-term changes in neuronal excitability by post-translational regulation.
Activated ERK is also, however, translocated to the nucleus in which it phosphorylates the transcription factor cAMP element-binding protein (CREB), via the CREB kinase Rsk2, subsequently activating cAMP response element (CRE)-mediated gene expression (Xing et al., 1996; Impey et al., 1998; Obrietan et al., 1999). ERK activation is required for long-term potentiation and long-term memory (English and Sweatt, 1997;Atkins et al., 1998; Impey et al., 1998, 1999). However, apart from immediate early genes such as c-fos, the specific target genes regulated by ERK responsible for long-lasting synaptic plasticity are primarily unknown (Xia et al., 1996; Sgambato et al., 1998).
Injection of irritant chemicals into a hindpaw of the rat produces a localized tissue inflammation and inflammatory pain hypersensitivity (Stein et al., 1988; Dubner and Ruda, 1992). Peripheral inflammation results in the transcriptional activation of many genes in dorsal horn neurons (Hunt et al., 1987; Wisden et al., 1990; Dubner and Ruda, 1992;Ji et al., 1994, 1995; Mannion et al., 1999; Woolf and Costigan, 1999,Samad et al., 2001), among which, prodynorphin and the substance P receptor neurokinin-1 (NK-1) have been intensively studied (Iadarola et al., 1988; Ruda et al., 1988; Schafer et al., 1993; McCarsson and Krause, 1994; Abbadie et al., 1996). Increased prodynorphin expression after inflammation has been suggested to be involved in the inflammation-induced enhanced excitability and subsequent development of expanded dorsal horn neuronal receptive fields (Hylden et al., 1991;Dubner and Ruda, 1992). Several lines of evidence suggest that NK-1 in the dorsal horn also plays an important role in inflammatory pain hypersensitivity (Traub, 1996; De Felipe et al., 1998; Ma et al., 1998;Woolf et al., 1998).
Noxious stimulation and inflammation induce CREB phosphorylation in dorsal horn neurons (Ji and Rupp, 1997; Messersmith et al., 1998), as well as ERK activation (Ji et al., 1999). CRE sites are present, moreover, in the promoter regions of both the prodynorphin and NK-1 genes (Hershey et al., 1991; Cole et al., 1995). This raises the possibility that the ERK pathway may play a role in regulating expression of CRE-containing genes, such as prodynorphin and NK-1, in the dorsal horn after inflammation and in this way contribute to altered pain sensitivity. We now investigated this.
MATERIALS AND METHODS
Animals and drugs. Adult male Sprague Dawley rats (230–300 gm) were used according to Massachusetts General Hospital Animal Care institutional guidelines. Animals were anesthetized with pentobarbital (50 mg/kg, i.p.). Complete Freund's adjuvant (CFA) (100 μl) was injected into the plantar surface of a hindpaw. For intrathecal drug delivery, a polyethylene-10 catheter was implanted into the intrathecal space of the spinal cord at the lumbar enlargement, and 10 μl of the MEK (MAP kinase kinase) inhibitor U0126 (1 μg; dissolved in 10% DMSO; Calbiochem, La Jolla, CA) was administered. DMSO (10%) was injected as vehicle control. For sustained drug delivery, an Alzet osmotic pump (3 d pump, 1 μl/hr) was filled with the MEK inhibitor U0126 (0.5 μg/ml) in 50% DMSO, and the catheter of the pump was implanted intrathecally at least 3 hr before CFA injection. DMSO (50%) was used as vehicle control.
Immunohistochemistry. Rats were deeply anesthetized with pentobarbital (120 mg/kg, i.p.) and perfused through the ascending aorta with saline, followed by 4% paraformaldehyde with 1.5% picric acid. L4–L5 spinal cord segments were dissected and post-fixed for 2–4 hr. Transverse spinal cord sections (free floating, 30 μm) were cut and processed for immunohistochemistry using the ABC method as described previously (Ji et al., 1995, 1999). Briefly, sections were blocked with 2% goat serum in 0.3% Triton X-100 for 1 hr at room temperature (RT) and incubated overnight at 4°C with primary antibody. The sections were then incubated for 2 hr with biotinylated secondary antibody (1:200) and 1 hr with ABC complex (1:50; Vector Laboratories, Burlingame, CA) at RT. Finally, the reaction product was visualized with 0.05% DAB–0.002% hydrogen peroxide in 0.1m acetate buffer, pH 6.0, containing 2% ammonium nickel sulfate for 2–5 min. Some sections were processed with immunofluorescence by incubating overnight with primary antibody and 1 hr at RT with FITC-conjugated secondary antibody (1:300; Jackson ImmunoResearch, West Grove, PA). The following antibodies were used: anti-phospho-ERK (pERK) (also called anti-pMAPK; anti-rabbit, 1:500; New England Biolabs, Beverly, MA), anti-pERK (monoclonal; 1:300; New England Biolabs), anti-NK1 (anti-rabbit; 1:3000; Oncogene, Sciences, Uniondale, NY), and anti-prodynorphin (anti-guinea pig; 1:3000; kindly provided by Dr. R. Elde, University of Minnesota, Minneapolis, MN). Double immunofluorescence was performed by incubating a mixture of primary antibodies (mouse anti-pERK–rabbit anti-NK1 or rabbit anti-pERK–guinea pig anti-prodynorphin), followed by a mixture of corresponding secondary antibodies conjugated with either Cy3 or FITC.
In situ hybridization. Animals were rapidly sacrificed in a CO2 chamber, and L4–L5 spinal cord segments were removed and cut on a cryostat at a thickness of 20 μm. A vector (pSP65) with a 1.7 kb prodynorphin insert was kindly provided by Dr. Linda Kobierski (Harvard Medical School). Antisense RNA probe, and the corresponding sense control probe, were labeled by in vitro transcription using linearized DNA templates for prodynorphin and digoxigenin (DIG) labeling mixture for 2 hr at 37°C. Hybridization was processed as described previously (Ji et al., 1998). Tissue sections were air dried for 2 hr, fixed in 4% paraformaldehyde for 15 min, and acetylated in acetic anhydride (0.25%) for 10 min. Sections were prehybridized for 2 hr at RT and then incubated in hybridization buffer overnight at 60°C. After hybridization, sections were washed in decreasing concentrations of SSC (2×, 1×, and 0.2×) for 2 hr total. Sections were then blocked with 2% goat serum for 1 hr and incubated overnight at 4°C with alkaline phosphatase-conjugated anti-DIG antibody (1:5000; Boehringer Mannheim, Indianapolis, IN). Finally, the sections were visualized in 75 μg/ml nitroblue-tetrazolium-chloride, 50 μg/ml 5-bromo-4-chloro-3-indolyl-phosphate, and 0.24 mg/ml levamisole for 2–24 hr.
RNase protection. Dynorphin cDNA was generated by reverse transcription-PCR from rat DRG total RNA, using primers 5′-TGGAAAAGCCCAGCTCCTAGACCCT-3′ and 5′-TTCCTCGTGGGCTTGAAGTGTGAAA-3′ and cloned into pCRII (Invitrogen, San Diego, CA). The plasmid was linearized with EcoRV, and an antisense probe was synthesized using Sp6 RNA polymerase and labeled with [32P]UTP (800 Ci/mmol; NEN, Boston, MA). RNase protection assays (RPAs) were performed using the RPA III (Ambion, Austin, TX) protocol, as reported previously (Samad et al., 2001). Briefly, 10 μg of RNA samples were hybridized with labeled probe overnight at 42°C and then digested with RNase A/RNase T1 mix in RNase digestion buffer for 30 min at 37°C. Finally, samples were separated on denaturing acrylamide gel and exposed to x-ray films. A β-actin probe was used for each sample as loading controls.
Western blot. Animals were sacrified, and dorsal horns of the L4–L5 spinal segments were rapidly removed and homogenized with a hand-held pellet pestle in lysis buffer containing a cocktail of phosphatase inhibitors (100×) and proteinase inhibitors (25×; Sigma, St. Louis, MO). For NK-1 protein, the dorsal horns were directly homogenized in boiling SDS sample buffer (100 mmTris, pH6.8, 2% SDS, 20% glycerol, 10% β-mercaptoethanol, and 0.1% bromophenol blue). Protein samples were separated on SDS-PAGE gel (4–15% gradient gel; Bio-Rad, Hercules, CA) and transferred to polyvinylidene difluoride filters (Millipore, Bedford, MA). The filters were blocked with 3% milk and incubated overnight at 4°C with polyclonal anti-pERK (1:1000; New England Biolabs) or anti-NK-1 (1:5000; Oncogene Sciences) primary antibody. The blots were incubated for 1 hr at RT with HRP-conjugated secondary antibody (1:3000; Amersham Biosciences, Arlington Heights, IL) and visualized in ECL solution (NEN, Boston, MA) for 1 min and exposed onto hyperfilms (Amersham Biosciences) for 1–30 min. The blots were then incubated in stripping buffer (67.5 mm Tris, pH 6.8, 2% SDS, and 0.7% β-mercaptoethanol) for 30 min at 50°C and reprobed with polyclonal anti-ERK or anti-CREB antibody (1:3000; New England Biolabs) as loading controls.
Behavioral analysis. Animals were habituated to the testing environment daily for 2 d before baseline testing. Except for the heat test, all of the animals were placed on an elevated wire grid. For mechanical allodynia, the plantar surface of the hindpaw was stimulated with a series of von Frey hairs. The threshold was taken as the lowest force that evokes a brisk withdrawal response. For heat hyperalgesia, the plantar surface of a hindpaw was exposed to a beam of radiant heat through a transparent Perspex surface (Hargrevas et al., 1988). The withdrawal latency was recorded, with a maximum 15 sec as cutoff. The withdrawal latency was averaged over three trials.
Quantification and statistics. Eight nonadjacent sections from the L4–L5 lumbar spinal cord were randomly selected, and the numbers of immunoreactive or mRNA-positive neuronal profiles in the superficial laminae and/or deep laminae of the dorsal horn in each section were counted (under a 20× object field) by an observer blind to the treatment. The values from the eight sections were averaged for each animal. The data are represented as mean ± SEM. For RNase protection and Western blots, each experiment was repeated at least twice, and, in all cases, the same results were obtained. The density of specific bands was measured with a computer-assisted imaging analysis system (IP Lab software) and normalized against a loading control. Differences between groups were compared using Student'st test or ANOVA, followed by Fisher's PLSD. For nonparametric data, Mann–Whitney U test was applied. The criterion for statistical significance was p < 0.05.
RESULTS
ERK activation by peripheral inflammation
To investigate whether ERK is activated by peripheral inflammation, we injected 100 μl of CFA into the plantar surface of a hindpaw under pentobarbital anesthesia (50 mg/kg, i.p). This produced an area of localized swelling, erythema, and hypersensitivity to mechanical and thermal stimuli, which persisted for the duration of the experiment (48 hr). The inflammation induced by the CFA injection resulted in the induction of pERK in neurons in the medial superficial dorsal horn on the ipsilateral side of the lumbar enlargement (Fig.1a,b). No induction was found in the contralateral spinal cord (Fig. 1a). Intraplantar injection of saline (100 μl) only induced a very weak pERK induction (data not shown). The CFA-induced pERK was found only in neurons; all pERK cells expressed neuronal-specific nuclear protein, a marker for neuronal cells (data not shown). The pERK-labeled neurons were predominantly localized in laminae I–IIo, and the pERK was present in the nucleus, cytoplasm, and dendrites, as reported previously (Ji et al., 1999). The number of pERK neurons peaked at 10 min but remained elevated with a slow decline for 48 hr (Fig. 1c). This temporal pattern differs substantially from the transient (<2 hr) ERK activation evoked by intraplantar capsaicin (Ji et al., 1999). ERK activation by CFA was confirmed by Western blot analysis. The phosphorylation level of both ERK1 (44 kDa) and ERK2 (42 kDa) increased in the ipsilateral dorsal horn compared with the contralateral side at 30 min and 6 hr (Fig. 1d). Because ERK is only activated in a small subset of dorsal horn neurons, Western blot is less sensitive than immunohistochemistry in detecting ERK activation in the superficial dorsal horn.
Fig. 1.
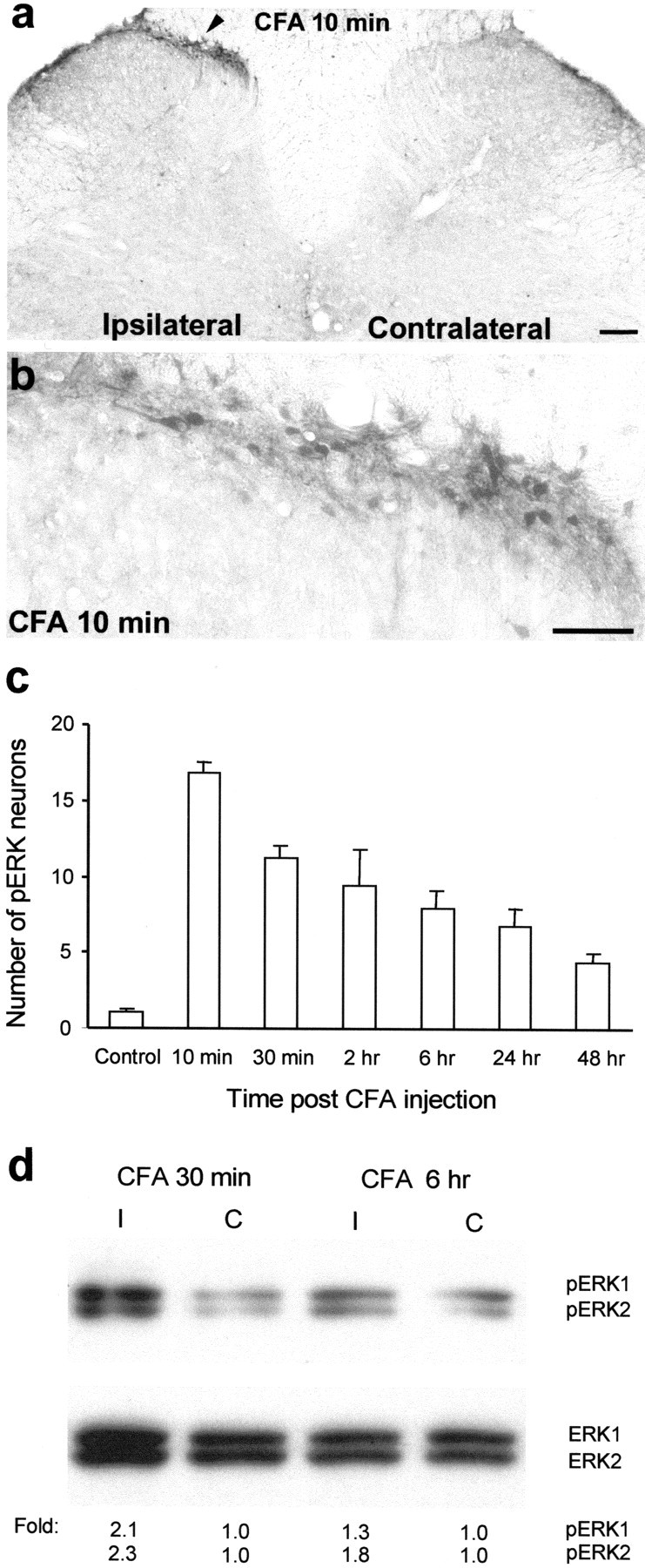
CFA induces a sustained activation of ERK.a, A low-magnification image showing induction of ERK phosphorylation in laminae I–IIo neurons of the ipsilateral spinal cord (indicated with an arrowhead) 10 min after CFA injection into a hindpaw. Scale bar, 200 μm. b, A high-magnification image of a, showing ERK activation in the medial superficial dorsal horn of the ipsilateral spinal cord 10 min after CFA injection. Scale bar, 50 μm. c, Time course of pERK induction after CFA administration measured by the number of pERK-positive neurons in the superficial (I–IIo) layers of the ipsilateral dorsal horn. Data are represented as mean ± SEM (n = 3). d, Western blot showing increased ERK phosphorylation of both ERK1 (44 kDa) and ERK2 (42 kDa) in the ipsilateral (I) dorsal horn compared with contralateral (C) side, 30 min and 6 hr after CFA injection. The bottom panel indicates levels of total ERK1 and ERK2, as loading controls. Foldrepresents comparative levels over the corresponding contralateral side after normalizing for loading.
Because pERK reached a peak level very rapidly (10 min) after the CFA injection, we tested whether the CFA also produced hyperalgesia at this time. CFA (100 μl) injected into the plantar surface of hindpaw in awake rats produced both immediate erythema and a rapid heat hyperalgesia. The paw-withdrawal latency (in seconds) decreased by 60% (from 10.8 ± 0.4 to 4.3 ± 0.7; p < 0.01; t test; n = 3) and 50% (from 9.7 ± 1.2 to 4.9 ± 1.3; p < 0.05) at 10 and 30 min, respectively. Saline-injected rats did not show any heat hypersensitivity.
Prodynorphin induction by inflammation
To investigate changes in the expression of prodynorphin in response to inflammation, we used RPA, in situhybridization, and immunohistochemistry. The peripheral inflammation resulted in a substantial upregulation of prodynorphin mRNA in the ipsilateral spinal dorsal horn 24 and 48 hr after CFA injection, as detected by the RPA (Fig. 2a). With the in situ hybridization, many strongly labeled prodynorphin mRNA-labeled neurons were found both in the superficial and deep layers of the ipsilateral dorsal horn 24 hr after CFA injection, whereas on the contralateral side, only a few weakly labeled neurons were detected (Fig. 2b). An increase in the number of prodynorphin peptide immunoreactive neurons was also found in the superficial and deep dorsal horn 48 hr after the CFA-induced inflammation (Fig. 2c).
Fig. 2.

CFA induces prodynorphin upregulation in the dorsal horn. a, An RNase protection assay reveals an increase in prodynorphin mRNA in the ipsilateral dorsal horn 24 and 48 hr after CFA injection. Fold represents comparative levels over control after normalizing for loading. b,In situ hybridization indicates an increased expression of prodynorphin mRNA in ipsilateral superficial and deep dorsal horn neurons 24 hr after CFA. Scale bar, 50 μm. c, Increased number of prodynorphin-immunoreactive neurons was induced in the ipsilateral superficial and deep dorsal horn by CFA injection at 48 hr. Scale bar, 50 μm.
ERK activation and prodynorphin expression
We then investigated whether prodynorphin mRNA expression in the dorsal horn is regulated by ERK activation. A specific and potent MEK inhibitor, U0126 (Favata et al., 1998), was intrathecally injected twice (1 μg), 30 min before and 6 hr after intraplantar CFA injection. This reduced the CFA-induced prodynorphin mRNA increase in the ipsilateral dorsal horn, as detected by the RPA (Fig.3a). The CFA-evoked increase in the number of prodynorphin mRNA-positive neurons in the superficial dorsal horn was also decreased by U0126 (2× 1 μg) without any effect on the number of labeled neurons in the deep laminae, as detected byin situ hybridization (Fig. 3b,c).
Fig. 3.

ERK activation regulates prodynorphin expression.a, Partial suppression of the CFA-induced increase in prodynorphin mRNA in the dorsal horn at 24 hr by U0126 (1 μg, intrathecally injected 30 min before and 6 hr after CFA).Fold represents comparative levels over control after normalizing for loading. b, Quantification of prodynorphin mRNA-positive neurons in laminae I–II and III–VI of the ipsilateral dorsal horn 24 hr after CFA injection. *p < 0.001, compared with control; +p < 0.001, compared with CFA (n = 4). c, In situhybridization showing an inhibition of the CFA-induced increase in prodynorphin mRNA-labeled neurons in the superficial dorsal horn by U0126 24 hr after CFA injection. Scale bar, 50 μm.
ERK activation and NK-1 expression
In agreement with previous studies (Abbadie et al., 1996, 1997), we observed increased NK-1 immunoreactivity in the superficial dorsal horn after CFA-induced inflammation using both immunohistochemistry and Western blot analysis (Fig.4a,c). However, in contrast to the previous studies (Abbadie et al., 1997; Honore et al., 1999), we found more NK-1-expressing cells after inflammation compared with our control (Fig. 4a,b). This discrepancy is attributable to the different detection thresholds for NK-1-positive neurons; our quantification, based on standard DAB staining, did not include weakly stained cells in control animals. These cells would be detected with confocal microscopy (Abbadie et al., 1997; Honore et al., 1999). The increase in NK-1-immunoreactive neurons we detected in lamina I (Fig. 4a) reflects the increase in staining intensity seen by others (Abbadie et al., 1997; Honore et al., 1999) in this lamina. To explore whether ERK activation is involved in the NK-1 upregulation, the MEK inhibitor U0126 was delivered intrathecally before the induction of inflammation via an osmotic pump (0.5 μg · μl−1 · hr−1for 2 d). MEK inhibition suppressed the CFA-induced elevation of NK-1-immunoreactive neurons in the superficial dorsal horn (Fig.4a,b). A Western blot analysis confirmed this (Fig. 4c). The NK-1 antibody recognized a single band of ∼46 kDa, which corresponds to the predicted molecular weight of cloned NK-1 receptor (Hershey and Krause, 1990).
Fig. 4.

ERK activation regulates NK-1 expression.a, Suppression of the CFA-induced increase in NK-1 immunoreactivity in the medial superficial dorsal horn at 48 hr by U0126 delivered via an osmotic pump. Scale bar, 50 μm.b, Quantification of the numbers of NK-1 neurons in laminae I–IIo of the ipsilateral dorsal horn 48 hr after CFA injection. *p < 0.001, compared with control; +p < 0.001, compared with CFA (n = 5). c, Western blot indicates that the CFA-induced NK-1 increase in the dorsal horn at 24 hr is inhibited by U0126 (1 μg, intrathecally injected 30 min before and 6 hr after CFA injection). CREB, a constitutively expressed protein, was used as a loading control. Fold represents comparative levels over control after normalizing for loading.
To test whether the pERK-positive neurons and prodynorphin—NK-1-expressing neurons belong to same subset of dorsal horn cells, we performed double immunofluorescence for pERK–prodynorphin and for pERK–NK-1. Almost all prodynorphin- and NK-1-positive neurons in the superficial dorsal horn also express pERK 24 hr after CFA injection (Fig. 5).
Fig. 5.

ERK is activated in a subset of prodynorphin- and NK-1-expressing neurons. pERK (red) is primarily colocalized with prodynorphin (green; left) and NK-1 (green; right) in the medial superficial dorsal horn 24 hr after CFA injection.Arrows indicate double-labeled neurons. Scale bar, 20 μm.
ERK activation and persistent inflammatory pain
To examine the functional consequences of ERK activation and its downstream effects on prodynorphin and NK-1 upregulation, we tested whether inhibition of ERK activation modified inflammatory pain hypersensitivity. Intrathecal administration of U0126 (1 μg) into non-inflamed animals, like another MEK inhibitor PD 98059 (Ji et al., 1999), produced no significant change in basal pain sensitivity measured in terms of mechanical withdrawal threshold (108% of the vehicle control) and heat withdrawal latency (113% of vehicle control), when tested 30 min after the administration. However, intrathecal administration of U0126 via an osmotic pump (0.5 μg · μl−1 · hr−1), started before the CFA injection and maintained for 48 hr, significantly reduced the inflammation-induced heat and mechanical hypersensitivity measured at 24 and 48 hr (Fig.6a,b).
Fig. 6.

Sustained infusion of an MEK inhibitor reduces CFA-induced inflammatory pain. The MEK inhibitor U0126 delivered by osmotic pump (0.5 μg · μl−1 · hr−1) before CFA injection reduces thermal hyperalgesia (a) and mechanical allodynia (b) 24 and 48 hr after CFA injection. These were measured by paw-withdrawal latency and paw-withdrawal threshold, respectively, and expressed as percentage of pre-CFA baseline measurements of vehicle control (50% DMSO). *p < 0.01, compared with corresponding vehicle control (n = 8).
Acute pain hypersensitivity (10–60 min after an intraplantar formalin injection) is reduced by inhibition of ERK activation, presumably by preventing post-translational changes (Ji et al., 1999). ERK activation by CFA could conceivably contribute to inflammatory pain hypersensitivity by either maintaining ongoing post-translational changes or inducing transcription of genes, such as prodynorphin and NK-1. In the former case, blocking ERK activation in established inflammation would be expected to reduce the pain hypersensitivity within tens of minutes as the substrates were dephosphorylated. If the contribution of ERK activation were through transcription, however, inhibiting ERK activation would be expected to have no immediate effect but rather a delayed effect. To test this, we intrathecally injected U0126 (1 μg) in rats with established inflammation (24 hr after CFA injection) and tested pain hypersensitivity 30 min, 6 hr, and 24 hr after the U0126 injection. Neither heat hyperalgesia nor mechanical allodynia was significantly affected by such post-treatment when tested at 30 min (Fig.7a,b). However, the post-treatment decreased heat hyperalgesia at 24 hr and mechanical allodynia at 6 hr (Fig. 7a,b), indicating a long-latency contribution of ERK activation to the maintenance of persistent inflammatory pain.
Fig. 7.

Post-treatment with an MEK inhibitor has a delayed effect on inflammatory pain. U0126 (1 μg) or vehicle (10% DMSO) was intrathecally administered 24 hr after CFA injection. Heat hyperalgesia (a) and mechanical allodynia (b) were tested 30 min, 6 hr, and 24 hr after the administration of the U0126. *p < 0.05, compared with corresponding vehicle control (n = 10). The data are expressed as percentage of pre-CFA baseline measurements of vehicle control.
DISCUSSION
ERK activation in nociceptive dorsal horn neurons
Peripheral inflammation induced, after a short latency, a persistent activation of ERK in laminae I–IIo neurons of the ipsilateral superficial dorsal horn. Inhibition of this activation, using an MEK inhibitor, blocked elevation of prodynorphin and NK-1 expression in this particular subset of dorsal horn neurons, as well as reduced inflammatory pain hypersensitivity. pERK was induced by CFA in the same subset of dorsal horn neurons that express prodynorphin and NK-1. Many NK-1- and dynorphin-expressing neurons in lamina I are projection neurons (Nahin et al., 1989; Marshall et al., 1996). Projection neurons in lamina I exhibit an enlargement of their receptive fields after CFA-induced inflammation (Dubner and Ruda, 1992). A targeted loss of NK-1-expressing neurons in lamina I has been shown to abolish inflammatory pain (Nichols et al., 1999). These studies indicate a critical role for the aforementioned superficial neurons in the reaction of the CNS to inflammation. A particular subset of C-nociceptor fibers, those that are NGF-responsive, and TrkA- and neuropeptide-expressing, terminate in laminae I and IIo, in an area overlapping the neurons that show ERK activation. Another subset of C-fibers, those that respond to the glial cell line-derived neurotrophic factor family of growth factors and are characterized by selective binding of the IB4 lectin, terminate in lamina IIi (Averill et al., 1995; Molliver et al., 1997). The neurons these fibers contact, many of which contain PKCγ (Malmberg et al., 1997), do not show ERK activation after capsaicin or CFA injection (R.-R. Ji et al., unpublished observations). The role of ERK in regulating pain hypersensitivity is, therefore, restricted to a particular subset of nociceptive dorsal horn neurons, only those located in laminae I–IIo, and this activation is likely to reflect activation only of TrkA-expressing C-fibers.
Transcriptional regulation in response to ERK activation
pERK was found in the nucleus of neurons after CFA injection (Fig.1a), pointing to a possible transcriptional role for the activated kinase. Unlike the transient activation (lasting <2 hr) induced after capsaicin injection (Ji et al., 1999), CFA produced persistent ERK activation (Fig. 1c). The sustained ERK activation after CFA injection is associated with persistent upregulation of prodynorphin mRNA (lasting >48 hr) (Fig.2a), whereas the transient pERK induced by capsaicin is associated with a shorter-lasting upregulation of prodynorphin mRNA (<6 hr; R.-R. Ji and C. J. Woolf, unpublished observation). ERK activation is likely to regulate the expression of prodynorphin and NK-1, both of which are CRE-containing genes, via CREB phosphorylation. CREB is required for dopamine-induced expression of prodynorphin in striatal neurons (Cole et al., 1995) and is phosphorylated in NK-1 receptor-expressing neurons after noxious stimulation (Anderson and Seybold, 2000). Interestingly, a CRE site has been shown to mediate a long-term sensitization of nociceptive neurons inAplysia (Lewin and Walters, 1999).
ERK activation and inflammatory pain hypersensitivity
U0126 is a potent and selective MEK inhibitor (Favata et al., 1998), achieving inhibition of ERK activation even in the face of strong activators, such as phorbol esters, whereas other major signal transduction pathways are not affected (Roberson et al., 1999). This inhibitor has not only been used in in vitro studies (Roberson et al., 1999) but also in in vivo studies (Han and Holtzman, 2000; Kuroki et al., 2001). At the dose we used, we did not find obvious toxicity of this inhibitor, animals behaved normally, and locomotion was unaffected. Although basal pain sensitivity was not affected by the inhibitor, persistent inflammatory pain was reduced. This could conceivably result from either preventing some post-translational change mediated by the ERK signal transduction pathway, as for acute pain hypersensitivity (Ji et al., 1999), or a reduction in transcription of target genes, such as prodynorphin and NK-1. The involvement suggested by a number of different studies, of both the NK-1 receptor and prodynorphin in pain mechanisms, together with their regulation by ERK activation, is compatible with a hypothesis that ERK activation after inflammation contributes to pain hypersensitivity by regulating gene transcription. The temporal profile of the effect of blocking ERK activation represents additional support. The acute pain hypersensitivity established within minutes of intraplantar formalin can be reduced by preventing ERK activation (Ji et al., 1999), an effect that is too quick (<1 hr) to be mediated by an inhibition of transcription and is likely therefore to represent some post-translational change downstream of the activated ERK. At present, it is not clear what the substrate for such post-translational change is, but it may well be an ion channel or receptor, such as the NMDA or AMPA receptor (Woolf and Salter, 2000). Such post-translational changes underlie the induction and maintenance of central sensitization, a use-dependent plasticity that outlasts its initiating stimulus by tens of minutes (Woolf, 1983; Woolf and Wall, 1986). If inflammatory hypersensitivity were a manifestation only of a central sensitization maintained by ongoing afferent input from the inflamed tissue, then blocking the initiation of central sensitization, by inhibiting an ERK-mediated phosphorylation, should reduce the hypersensitivity over a periods of tens of minutes as the key proteins were dephosphorylated. The fact that MEK inhibition during established inflammation had no immediate effect, but rather only reduced mechanical and thermal hypersensitivity 6–24 hr later, argues that the role of ERK activation may well be via transcriptional regulation.
Dynorphin and NK-1 contribute to inflammatory pain hypersensitivity
A temporal correlation has been shown previously between the expression of prodynorphin and NK-1 and development of inflammatory pain hypersensitivity (Iadarola et al., 1988; Abbadie et al., 1996). Unlike other opioid peptides, intrathecal injection of dynorphin does not produce analgesia (Laughlin et al., 1997). Dynorphin has actually been found to be pronociceptive in some pathological pain states. For example, dynorphin A antiserum reduces the pain hypersensitivity after nerve injury (Nichols et al., 1997; Wegert et al., 1997; Malan et al., 2000), and neuropathic pain does not persist in prodynorphin knock-out mice (Wang et al., 2001). The pronociceptive action of dynorphin appears to be the result of its nonopioid actions (Laughlin et al., 1997), including an activation of NMDA receptors sufficient to induce excitotoxicity (Dubner and Ruda, 1992).
Inflammation induces NK-1 receptor upregulation in dorsal horn neurons and upregulation of its ligand, the neuropeptide substance P, in primary afferent neurons (Noguchi et al., 1988; Abbadie et al., 1996) (also see Woolf et al., 1998). NK-1 antagonists have been shown to reduce inflammatory pain (both hyperalgesia and mechanical allodynia) in several different animal models (Neumann et al., 1996; Ren et al., 1996; Traub, 1996; Ma et al., 1998; Woolf et al., 1998; Trafton and Basbaum, 2000), including NK-1 knock-out mice (De Felipe et al., 1998). The increased amount and internalization of the NK-1 receptor on the dendrites of dorsal horn neurons in response to noxious and innocuous stimuli after inflammation indicates that this receptor is activated by substance P in response to peripheral stimuli (Abbadie et al., 1997).
Conclusion
ERK activation has two roles in nociceptive plasticity in the dorsal horn: a short-latency contribution to acute noxious stimulus-induced central sensitization and an involvement in the induction and maintenance of inflammatory pain. The involvement of pERK in peripheral inflammatory pain hypersensitivity may be contributed to by its regulation of prodynorphin and NK-1 expression, as well as other target genes. ERK activation plays, therefore, a pivotal role in the functional plasticity and chemical phenotype of a group of neurons in the superficial dorsal horn, determining the activation of particular effector responses to divergent inputs, which in turn contribute to altered sensibility.
Footnotes
The work was supported by National Institutes of Health Grants RO1 NS38253 (C.J.W.) and RO1 NS40698 (R.R.J.). We thank Sara Billet for technical support, Dr. Linda Kobierski (Massachusetts General Hospital) for the prodynorphin cDNA vector, and Dr. Robert Elde (University of Minnesota) for prodynorphin antibody.
Correspondence should be addressed to Dr. Ru-Rong Ji, Neural Plasticity Research Group, Department of Anesthesia and Critical Care, Massachusetts General Hospital, Harvard Medical School, 149 13th Street, Room 4309, Charlestown, MA 02129. E-mail:ji@helix.mgh.harvard.edu.
REFERENCES
- 1.Abbadie C, Brown JL, Mantyh PW, Basbaum AI. Spinal cord substance P receptor immunoreactivity increases in both inflammatory and nerve injury models of persistent pain. Neuroscience. 1996;70:201–209. doi: 10.1016/0306-4522(95)00343-h. [DOI] [PubMed] [Google Scholar]
- 2.Abbadie C, Trafton J, Liu H, Mantyh PW, Basbaum AI. Inflammation increases the distribution of dorsal horn neurons that internalize the neurokinin-1 receptor in response to noxious and non-noxious stimulation. J Neurosci. 1997;17:8049–8060. doi: 10.1523/JNEUROSCI.17-20-08049.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson LE, Seybold VS. Phosphorylated cAMP response element binding protein increases in neurokinin-1 receptor-immunoreactive neurons in rat spinal cord in response to formalin-induced nociception. Neurosci Lett. 2000;283:29–32. doi: 10.1016/s0304-3940(00)00908-3. [DOI] [PubMed] [Google Scholar]
- 4.Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- 5.Averill S, McMahon SB, Clary DO, Reichardt LF, Priestley JV. Localization of trkA receptors in chemically identified subgroups of adult rat sensory neurones. Eur J Neurosci. 1995;7:1484–1494. doi: 10.1111/j.1460-9568.1995.tb01143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole RL, Konradi C, Douglass J, Hyman SE. Neuronal adaptation to amphetamine and dopamine: molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron. 1995;14:813–823. doi: 10.1016/0896-6273(95)90225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Felipe C, Herrero JF, O'Brien JA, Palmer JA, Doyle CA, Smith AJ, Laird JM, Belmonte C, Cervero F, Hunt SP. Altered nociception, analgesia and aggression in mice lacking the receptor for substance P. Nature. 1998;392:394–397. doi: 10.1038/32904. [DOI] [PubMed] [Google Scholar]
- 8.Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci. 1992;15:96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- 9.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 10.Favata M, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 11.Han BH, Holtzman DM. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci. 2000;20:5775–5781. doi: 10.1523/JNEUROSCI.20-15-05775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hargrevas K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 13.Hershey AD, Krause JE. Molecular characterization of a functional cDNA encoding the rat substance P receptor. Science. 1990;247:958–962. doi: 10.1126/science.2154852. [DOI] [PubMed] [Google Scholar]
- 14.Hershey AD, Dykema PE, Krause JE. Organization, structure and expression of the gene encoding the rat substance P receptor. J Biol Chem. 1991;266:4366–4374. [PubMed] [Google Scholar]
- 15.Honore P, Menning PM, Rogers SD, Nichols ML, Basbaum AI, Besson JM, Mantyh PW. Spinal substance P receptor expression and internalization in acute, short-term, and long-term inflammatory pain states. J Neurosci. 1999;19:7670–7678. doi: 10.1523/JNEUROSCI.19-17-07670.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunt SP, Pini A, Evan G. Induction of c-fos-like protein in spinal cord neurons following sensory stimulation. Nature. 1987;328:632–634. doi: 10.1038/328632a0. [DOI] [PubMed] [Google Scholar]
- 17.Hylden JL, Nahin RL, Traub RJ, Dubner R. Effects of spinal kappa-opioid receptor agonists on the responsiveness of nociceptive superficial dorsal horn neurons. Pain. 1991;44:187–193. doi: 10.1016/0304-3959(91)90136-L. [DOI] [PubMed] [Google Scholar]
- 18.Iadarola MJ, Brady LS, Draisci G, Dubner R. Enhancement of dynorphin gene expression in spinal cord following experimental inflammation: stimulus specificity, behavioral parameters and opioid receptor binding. Pain. 1988;35:313–326. doi: 10.1016/0304-3959(88)90141-8. [DOI] [PubMed] [Google Scholar]
- 19.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between Erk and PKA is required for Ca2+ stimulation of CREB-dependent transcription and Erk nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 20.Impey S, Obrietan K, Storm DR. Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- 21.Ji RR, Rupp F. Phosphorylation of transcription factor CREB in rat spinal cord after formalin-induced hyperalgesia: relationship to c-fos induction. J Neurosci. 1997;17:1776–1785. doi: 10.1523/JNEUROSCI.17-05-01776.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ji RR, Woolf CJ. Neruonal plasticity and signal transduction in nociceptive neurons: Implications for the initiation and maintenance of pathological pain. Neurobiol Dis. 2001;8:1–10. doi: 10.1006/nbdi.2000.0360. [DOI] [PubMed] [Google Scholar]
- 23.Ji RR, Zhang X, Wiesenfeld-Hallin Z, Hokfelt T. Expression of neuropeptide Y and neuropeptide Y (Y1) receptor mRNA in rat spinal cord and dorsal root ganglia following peripheral tissue inflammation. J Neurosci. 1994;14:6423–6434. doi: 10.1523/JNEUROSCI.14-11-06423.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji RR, Zhang X, Nilsson S, Wiesenfeld-Hallin Z, Hokfelt T. Central and peripheral response to galanin induced by inflammation. Neuroscience. 1995;68:563–576. doi: 10.1016/0306-4522(95)94333-t. [DOI] [PubMed] [Google Scholar]
- 25.Ji RR, Schlaepfer TE, Aizenman CD, Qiu D, Hung JC, Rupp F. Repetitive transcranial magnetic stimulation activates specific neural regions. Proc Natl Acad Sci USA. 1998;95:15635–15640. doi: 10.1073/pnas.95.26.15635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji RR, Baba H, Brenner G, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- 27.Kuroki Y, Fukushima K, Kanda Y, Mizuno K, Watanabe Y. Neuroprotection by estrogen via extracellular signal-regulated kinase against quinolinic acid-induced cell death in the rat hippocampus. Eur J Neurosci. 2001;13:472–476. doi: 10.1046/j.0953-816x.2000.01409.x. [DOI] [PubMed] [Google Scholar]
- 28.Laughlin TM, Vanderah TW, Lashbrook J, Nichols ML, Ossipov M, Porreca F, Wilcox GL. Spinally administered dynorphin A produces long-lasting allodynia: involvement of NMDA but not opioid receptors. Pain. 1997;72:253–260. doi: 10.1016/s0304-3959(97)00046-8. [DOI] [PubMed] [Google Scholar]
- 29.Lewin MR, Walters ET. Cyclie GMP pathway is critical for inducing long-term sensitization of nociceptive sensory neurons. Nat Neurosci. 1999;2:18–23. doi: 10.1038/4520. [DOI] [PubMed] [Google Scholar]
- 30.Ma QP, Allochorne AJ, Woolf CJ. Morphine, the NMDA receptor antagonist MK801 and tachykinin NK1 receptor antagonist RP67580 attenuate the development of inflammation-induced progressive tactile hypersensitivity. Pain. 1998;77:49–57. doi: 10.1016/S0304-3959(98)00081-5. [DOI] [PubMed] [Google Scholar]
- 31.Malan TP, Ossipov MH, Gardell LR, Ibrahim M, Bian D, Lai J, Porreca F. Extraterritorial neuropathic pain correlates with multisegmental elevation of spinal dynorphin in nerve-injured rats. Pain. 2000;86:185–194. doi: 10.1016/s0304-3959(00)00243-8. [DOI] [PubMed] [Google Scholar]
- 32.Malmberg AB, Chen C, Susumu T, Basbaum AI. Preserved acute pain and reduced neuropathic pain in mice lacking PKC gamma. Science. 1997;278:279–283. doi: 10.1126/science.278.5336.279. [DOI] [PubMed] [Google Scholar]
- 33.Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, Holstege JC, Ji RR, Acheson A, Lindsay RM, Wilkinson GA, Woolf CJ. Brain-derived neurotrophic factor-a centrally acting modulator of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci USA. 1999;96:9385–9390. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marshall GE, Shehab SA, Spike RC, Todd AJ. Neurokinin-1 receptors on lumbar spinothalamic neurons in the rat. Neuroscience. 1996;72:255–263. doi: 10.1016/0306-4522(95)00558-7. [DOI] [PubMed] [Google Scholar]
- 35.McCarsson KE, Krause JE. NK-1 and NK-3 type tachykinin receptor mRNA expression in the rat spinal cord dorsal horn is increased during adjuvant or formalin-induced nociception. J Neurosci. 1994;14:712–720. doi: 10.1523/JNEUROSCI.14-02-00712.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Messersmith DJ, Kim DJ, Iadarola MJ. Transcription factor regulation of prodynorphin gene expression following rat hindpaw inflammation. Mol Brain Res. 1998;53:260–269. doi: 10.1016/s0169-328x(97)00308-2. [DOI] [PubMed] [Google Scholar]
- 37.Molliver DC, Wright DE, Leitner ML, Parsadanian AS, Doster K, Wen D, Yan Q, Snider WD. IB4-binding DRG neurons switch from NGF to GDNF dependence in early postnatal life. Neuron. 1997;19:849–861. doi: 10.1016/s0896-6273(00)80966-6. [DOI] [PubMed] [Google Scholar]
- 38.Nahin RL, Hylden JL, Iadarola MJ, Dubner R. Peripheral inflammation is associated with increased dynorphin immunoreactivity in both projection and local circuit neurons in the superficial dorsal horn of the rat lumbar spinal cord. Neurosci Lett. 1989;96:247–252. doi: 10.1016/0304-3940(89)90386-8. [DOI] [PubMed] [Google Scholar]
- 39.Neumann S, Doubell TP, Leslie T, Woolf CJ. Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature. 1996;384:360–364. doi: 10.1038/384360a0. [DOI] [PubMed] [Google Scholar]
- 40.Nichols ML, Lopez Y, Ossipov MH, Bian D, Porreca F. Enhancement of the antiallodynic and antinociceptive efficacy of spinal morphine by antisera to dynorphin A (1–13) or MK-801 in a nerve-ligation model of peripheral neuropathy. Pain. 1997;69:317–322. doi: 10.1016/S0304-3959(96)03282-4. [DOI] [PubMed] [Google Scholar]
- 41.Nichols ML, Allen BJ, Rogers SD, Ghilardi JR, Honore P, Luger NM, Finke MP, Li J, Lappi DA, Simone DA, Mantyh PW. Transmission of chronic nociception by spinal neurons expressing the substance P receptor. Science. 1999;286:1558–1561. doi: 10.1126/science.286.5444.1558. [DOI] [PubMed] [Google Scholar]
- 42.Noguchi K, Morita Y, Kiyama H, Ono K, Tohyama MA. Noxious stimulus induces the preprotachykinin-A gene expression in the rat dorsal root ganglion: a quantitative study using in situ hybridization histochemistry. Brain Res. 1988;464:31–35. doi: 10.1016/0169-328x(88)90015-0. [DOI] [PubMed] [Google Scholar]
- 43.Obrietan K, Impey S, Smith D, Athos J, Storm DR. Circadian regulation of cAMP response element-mediated gene expression in the suprachiasmatic nuclei. J Biol Chem. 1999;274:17748–17756. doi: 10.1074/jbc.274.25.17748. [DOI] [PubMed] [Google Scholar]
- 44.Ren K, Iadarola MJ, Dubner R. An isobolographic analysis of the effect of N-methyl-d-aspartate and NK1 tachykinin receptor antagonists on inflammatory hyperalgesia in the rat. Br J Pharmacol. 1996;117:196–202. doi: 10.1111/j.1476-5381.1996.tb15174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweat JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruda MA, Iadarola MJ, Cohen LV, Young WS. In situ hybridization histochemistry and immunohistochemistry reveal an increase in spinal dynorphin biosynthesis in rat model of peripheral inflammation and hyperalgesia. Proc Natl Acad Sci USA. 1988;85:622–626. doi: 10.1073/pnas.85.2.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samad A, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. An interleukin-1b-mediated induction of Cox-2 in the central nervous system contributes to inflammatory pain hypersensitivity. Nature. 2001;22:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 48.Schafer MK, Nohr D, Krause JE, Weihe E. Inflammation-induced upregulation of NK1 receptor mRNA in dorsal horn neurons. NeuroReport. 1993;4:1007–1010. doi: 10.1097/00001756-199308000-00003. [DOI] [PubMed] [Google Scholar]
- 49.Sgambato V, Pages C, Rogard M, Besson MJ, Caboche J. Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostrital stimulation. J Neurosci. 1998;18:8814–8825. doi: 10.1523/JNEUROSCI.18-21-08814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stein C, Millan MJ, Herz A. Unilateral inflammation of the hindpaw in rats as a model of prolonged noxious stimulation: alterations in behavior and nociceptive thresholds. Pharmacol Biochem Behav. 1988;31:445–451. doi: 10.1016/0091-3057(88)90372-3. [DOI] [PubMed] [Google Scholar]
- 51.Trafton JA, Basbaum AI. The contribution of spinal cord neurokinin-1 receptor signaling to pain. J Pain Suppl. 2000;1:57–65. doi: 10.1054/jpai.2000.9806. [DOI] [PubMed] [Google Scholar]
- 52.Traub RJ. The spinal contribution of substance P to the generation and maintenance of inflammatory hyperalgesia in the rat. Pain. 1996;67:151–161. doi: 10.1016/0304-3959(96)03076-X. [DOI] [PubMed] [Google Scholar]
- 53.Wang Z, Gardell LR, Ossipov MH, Vanderah TW, Brennan MB, Hochgeschwender U, Hruby VJ, Malan TP, Jr, Lai J, Porreca F. Pronociceptive actions of dynorphin maintain chronic neuropathic pain. J Neurosci. 2001;21:1779–1786. doi: 10.1523/JNEUROSCI.21-05-01779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wegert S, Ossipov MH, Nichols ML, Bian D, Vanderah TW, Malan TP, Porreca F., Jr Differential activities of intrathecal MK-801 or morphine to alter responses to thermal and mechanical stimuli in normal or nerve-injured rats. Pain. 1997;71:57–64. doi: 10.1016/s0304-3959(97)03337-x. [DOI] [PubMed] [Google Scholar]
- 55.Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by b-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- 56.Wisden W, Errington ML, Williams S, Dunnett SB, Waters C, Hitchcock D, Evan G, Bliss TVP, Hunt SP. Differential expression of immediate early genes in the hippocampus and spinal cord. Neuron. 1990;4:603–614. doi: 10.1016/0896-6273(90)90118-y. [DOI] [PubMed] [Google Scholar]
- 57.Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- 58.Woolf CJ, Costigan M. Transcriptional and post-translational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci USA. 1999;96:7723–7730. doi: 10.1073/pnas.96.14.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 60.Woolf CJ, Wall PD. Relative effectiveness of C primary afferent fibers of different origins in evoking a prolonged facilitation of the flexor reflex in the rat. J Neurosci. 1986;6:1433–1442. doi: 10.1523/JNEUROSCI.06-05-01433.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woolf CJ, Mannion RJ, Neumann S. Null mutations lacking substance: elucidating pain mechanisms by genetic pharmacology. Neuron. 1998;20:1063–1066. doi: 10.1016/s0896-6273(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 62.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAPK/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]