

Abstract
Urocortin and urocortin II are members of the corticotropin-releasing hormone (CRH) family of neuropeptides that function to regulate stress responses. Two high-affinity G-protein-coupled receptors have been identified that bind CRH and/or urocortin I and II, designated CRHR1 and CRHR2, both of which are present in hippocampal regions of mammalian brain. The hippocampus plays an important role in regulating stress responses and is a brain region in which neurons are vulnerable during disease and stress conditions, including cerebral ischemia, Alzheimer's disease, and anxiety disorders. Here we report that urocortin exerts a potent protective action in cultured rat hippocampal neurons with concentrations in the range of 0.5–5.0 pm, increasing the resistance of the cells to oxidative (amyloid β-peptide, 4-hydroxynonenal, ferrous sulfate) and excitotoxic (glutamate) insults. We observed that urocortin is 10-fold more potent than CRH in protecting hippocampal neurons from insult, whereas urocortin II is ineffective. RT-PCR and sequencing analyses revealed the presence of both CRHR1 and CRHR2 in the hippocampal cultures, with CRHR1 being expressed at much higher levels than CRHR2. Using subtype-selective CRH receptor antagonists, we provide evidence that the neuroprotective effect of exogenously added urocortin is mediated by CRHR1. Furthermore, we provide evidence that the signaling pathway that mediates the neuroprotective effect of urocortin involves cAMP-dependent protein kinase, protein kinase C, and mitogen-activated protein kinase. This is the first demonstration of a biological activity of urocortin in hippocampal neurons, suggesting a role for the peptide in adaptive responses of hippocampal neurons to potentially lethal oxidative and excitotoxic insults.
Keywords: antalarmin, cAMP, excitotoxicity, lipid peroxidation, sauvagine, stress
Corticotropin-releasing hormone (CRH) and urocortin (Urc) are two members of a family of mammalian neuropeptides that function to regulate neuroendocrine, autonomic, and immunologic responses to stress (Owens and Nemeroff, 1991; Lovejoy and Balment, 1999). CRH is expressed widely in neurons in the brain, whereas urocortin exhibits a more restricted expression pattern (Morin et al., 1999). One of the few regions of mammalian brain that contains cells expressing both CRH and Urc is the hippocampus (Morin et al., 1999). The effects of CRH and Urc are mediated by two high-affinity receptors, CRHR1 and CRHR2, which are coupled to multiple G-proteins and which activate at least two intracellular signaling pathways (Perrin and Vale, 1999; Grammatopoulos et al., 2001). Both CRH and Urc are potent activators of CRHR1, although Urc binds CRHR2 with much higher affinity than CRH, and Urc therefore is believed to be the endogenous ligand for CRHR2 (De Souza, 1995; Perrin and Vale, 1999). The expression pattern of CRHR2 in brain is distinct and more restricted than that of CRHR1 (Van Pett et al., 2000). However, both receptors are expressed in several brain regions, such as the hippocampus (Van Pett et al., 2000). Although there is generally a poor correlation between the localization of Urc-containing projections and major sites of CRHR2 expression (Bittencourt et al., 1999), additional members of the CRH family that selectively bind CRHR2 were identified recently: UrcII (Reyes et al., 2001), UrcIII (Lewis et al., 2001), stresscopin, and stresscopin-related peptide (Hsu and Hsueh, 2001).
Studies with CRHR1 antagonists (Webster et al., 1996; Habib et al., 2000) and of mice lacking the CRHR1 gene (Smith et al., 1998; Timpl et al., 1998) suggest that this receptor is critical for activation of the hypothalamic–pituitary–adrenal (HPA) axis and stress responses. In contrast, studies of mice lacking the CRHR2 gene suggest that this receptor is responsible for the modulation of stress responses and maintenance of homeostasis after HPA axis activation (Coste et al., 2000; Kishimoto et al., 2000). The hippocampus plays a fundamental role in learning and memory processes (Martin et al., 2000) and in negative feedback regulation of the HPA axis during stress (McEwen, 1999). It is a major site of pathology in several disorders, including Alzheimer's disease (AD; Jellinger and Bancher, 1998) and ischemic stroke (Ott and Saver, 1993). Moreover, hippocampal atrophy and impairments in cognitive function occur in conditions characterized by hyperactivation of the HPA axis and excessive secretion of glucocorticoids, including AD, schizophrenia, depression, and anxiety disorders (Raber, 1998). Although CRH and Urc are critical components of the stress response, emerging evidence suggests that there are additional roles for these peptides. For instance, evidence has been provided that CRH can regulate learning and memory processes (Lee et al., 1993; Radulovic et al., 1999). Furthermore, CRH can protect against cell death caused by oxidative insults in tumor cell lines (Lezoualc'h et al., 2000) and in primary neuronal culture (Lezoualc'h et al., 2000; Pedersen et al., 2001), and Urc can protect primary cardiac myocytes from hypoxic/ischemic injury (Okosi et al., 1998; Brar et al., 1999, 2000). We now report that Urc exerts a potent cytoprotective activity toward cultured hippocampal neurons, suggesting a role for this peptide in adaptive responses of hippocampal neurons to insult under pathophysiological conditions.
MATERIALS AND METHODS
Primary neuronal cultures and experimental treatments.Cultures of dissociated cortical and hippocampal neurons were prepared from embryonic day 18 (E18) Sprague Dawley rats via methods described previously (Mattson et al., 1992). The cultures were maintained in Neurobasal medium with B27 supplements (Invitrogen, San Diego, CA), and all experiments were performed with 8-d-old cultures. Cell-signaling inhibitor studies and experiments with CRHR1/CRHR2 antagonists were performed in Neurobasal medium for 24 hr treatment periods to avoid toxic effects of the drugs (Pedersen et al., 2001). In all other experiments the treatments were performed in Locke's solution [containing (in mm) 154 NaCl, 5.6 KCl, 2.3 CaCl2, 1.0 MgCl2, 5 NaHCO3, 5 glucose, and 5 HEPES, plus 0.010 mg/ml phenol red, pH 7.2]. Stock solutions of human/rat CRH (Calbiochem, La Jolla, CA), rat Urc (Sigma, St. Louis, MO), and mouse UrcII (custom synthesis by Research Genetics, Huntsville, AL) were prepared in 5% glacial acetic acid at 100 μm, further diluted to 10 nm with dH2O, and stored at −20°C. The selective CRHR1 antagonist, antalarmin (provided by Dr. K. Habib, National Institute of Mental Health, Bethesda, MD), and the selective CRHR2 antagonist, antisauvagine-30 (provided by Dr. J. Rivier, The Salk Institute, La Jolla, CA), were prepared in dimethylsulfoxide (DMSO) to a final concentration of 10 μm and stored at −20°C. The compounds NF 449, PD 98059, bisindolylmaleimide I (BIM I), and H-89 (Calbiochem) were prepared at 200 μm in dH2O, at 4 mm in DMSO, at 10 μm in DMSO, and at 100 μm in DMSO, respectively, and stored at −20°C. A stock solution of 3-isobutyl-1-methylxanthine (IBMX; Sigma) was prepared at 50 mm in DMSO and stored at −20°C. The sources of and preparation of stock solutions of synthetic human amyloid β-peptide 25-35, 4-hydroxynonenal, ferrous sulfate, and l-glutamic acid were as described in our previous studies (Pedersen et al., 2000,2001).
Quantification of neuronal survival. Two different methods were used to quantify cell survival. The first method involved counting the number of viable hippocampal neurons, judged by morphological criteria, in premarked microscope fields before and at indicated time points after exposure to the experimental treatments as described previously (Mattson et al., 1992). The second method involved an assessment of nuclear morphology. After treatment the cultures were fixed with 4% paraformaldehyde (in 1× PBS) and stained with the fluorescent DNA-binding dye Hoechst 33342 (1 μg/ml in 1× PBS for 30 min). Cells were visualized under epifluorescence illumination (340 nm excitation; 510 nm barrier filter), using a 40× oil immersion objective and a Nikon Eclipse TE300 inverted microscope (300–500 cells/culture were counted in four separate cultures per treatment condition). The percentage of cells with condensed and/or fragmented chromatin was determined in each culture.
Reverse transcription-PCR and sequencing. Total RNA was extracted with the use of TRIzol reagent (Invitrogen) from dissected tissues of E18 rats and from 8-d-old cultures of dissociated cortical and hippocampal neurons prepared from E18 rats. First-strand cDNAs were prepared from 2 μg of total RNA, using the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen) and using oligo-dT according to the manufacturer's instructions. The cDNAs were amplified directly by using PCR and Platinum PCR SuperMix (Invitrogen) with 10 μl of first-strand reaction mix, 10 μm forward primer, and 10 μm reverse primer. The following sequences of the CRHR1 primers were used: forward, 5′-CAA CAC GAC AAA CAA TGG-3′, and reverse, 5′-GCA AGA AGA GGA CAA AGG-3′; the following sequences of the CRHR2 primers were used: forward, 5′-TCA TCA CCA CCT TCA TCC-3′, and reverse, 5′-CAG CCT TCC ACA AAC ATC-3′. Primers were designed on the basis of the published sequences for rat brain CRHR1 (Chang et al., 1993; Perrin et al., 1993) and CRHR2 (Lovenberg et al., 1995). The conditions for PCR were 94°C for 2 min, followed by 33 cycles of 94°C for 30 sec, 50°C for 30 sec, and 72°C for 45 sec, followed by 72°C for 10 min. The PCR products were separated by electrophoresis in 2% agarose gels, extracted with the QIAquick gel extraction kit (Qiagen, Chatsworth, CA), and subcloned into the pCR 4-TOPO vector (Invitrogen). Plasmid purification was performed with the Qiagen Plasmid Midi Kit, and plasmid DNA was sequenced by using T3 primer and an Applied Biosystems (ABI, Foster City, CA) Prism 3700 automated DNA sequencer.
Quantification of cAMP levels. The cells were pretreated for 1 hr with IBMX, an inhibitor of cAMP and cGMP phosphodiesterases, and then treated for 30 min with Urc, CRH, or UrcII in the continued presence of IBMX. After treatment the cells were washed once with 1× PBS, incubated for 10 min in 0.1 m HCl, and scraped into microcentrifuge tubes; homogenates were sonicated briefly and stored at −80°C until use. Direct cAMP measurements were performed with the nonacetylated version of a commercial kit (Assay Designs, Ann Arbor, MI). An aliquot of each homogenate was used for determination of protein content with the Pierce (Rockford, IL) bicinchoninic acid (BCA) kit. To determine the EC50 value of the Urc concentration–response, we fit a rectangular hyperbola to the data plus a constant to account for the levels of cAMP in untreated cells.
Immunoblot analysis. The cells were pretreated for 1 hr with PD 98059, BIM I, or H-89 and then treated with Urc for 15 min in the continued presence of each inhibitor. After treatment the cells were washed once with 1× PBS and lysed in the presence of 1× cell lysis buffer (Cell Signaling Technologies, Beverly, MA) supplemented with 1 mm phenylmethylsulfonyl fluoride. An aliquot of each extract equivalent to 40 μg of total protein was subjected to 15% SDS-PAGE. Proteins were transferred electrophoretically to a nitrocellulose sheet, which then was incubated overnight at 4°C in blocking solution (5% milk powder in 1× TTBS) and incubated for 2 hr in the presence of an antibody recognizing phosphorylated ERK1/2 (mouse monoclonal IgG2a at a final concentration of 200 ng/ml in blocking solution; Santa Cruz Biotechnology, Santa Cruz, CA). After washes in 1× TTBS, the membrane was incubated for 1 hr the presence of peroxidase-conjugated secondary antibody (goat anti-mouse IgG; Jackson ImmunoResearch, West Grove, PA) and washed further in 1× TTBS. Immunolabeled proteins were visualized by enhanced chemiluminescence (Amersham, Buckinghamshire, UK). Subsequently, the membrane was washed with 1× TTBS, incubated for 30 min in Ponceau S solution (1:10 dilution with dH2O; Sigma), washed again with 1× TTBS, and incubated for 1 hr in blocking solution. Then the membrane was reprobed with an antibody recognizing ERK 1/2 (rabbit polyclonal IgG at a final concentration of 200 ng/ml in blocking solution; Santa Cruz Biotechnology) and processed as above with a goat anti-rabbit IgG secondary antibody (Jackson ImmunoResearch). Band intensities were quantified by densitometry with NIH Image software.
RESULTS
Potencies and effectiveness of Urc, CRH, and UrcII in protecting cultured hippocampal neurons from oxidative and excitotoxic insults
We have demonstrated previously that treatment of primary rat hippocampal cultures with CRH can protect the cells from death induced by insults potentially relevant to the neurodegenerative process in AD (Pedersen et al., 2001). Specifically, we observed that treatment of the hippocampal cultures with CRH (≥5 pm) protected the cells from death induced by synthetic human amyloid β-peptide (Aβ) 25-35, the lipid peroxidation product 4-hydroxynonenal (HNE), ferrous sulfate (FeSO4), or l-glutamic acid (l-glut). Here, we compare the potencies and effectiveness of CRH with that of Urc and UrcII in protecting primary hippocampal neurons from death induced by these insults. Exposure of the cultures to aggregated Aβ25-35 at 1 μm resulted in neurite fragmentation and shrinkage of the cell body in many neurons during a 24 hr period, whereas the majority of neurons cotreated with Urc at 10 pm and Aβ25-35 at 1 μm had intact neurites and cell bodies with normal appearance (Fig.1A). Cotreatment with Urc decreased the extent of Aβ-induced cell death in a concentration-dependent manner (Fig. 1B). Significant protection of the neurons occurred with Urc at concentrations as low as 0.5 pm, and complete protection was afforded by Urc at concentrations in the range of 1–5 pm (Fig. 1B). As with CRH, we observed that Urc also protected against cell death that was induced by HNE, FeSO4, and l-glut (Fig. 2A). However, a 10-fold lower concentration of Urc achieved the same extent of protection when compared with CRH (Fig. 2B). For instance, complete protection against HNE-induced cell death occurred with CRH at 10 pm and with Urc at 1 pm. Half-maximal effects were observed with Urc at 0.5 pm and CRH at 5 pm, and a combined treatment of the cultures with Urc and CRH at these concentrations resulted in complete protection from HNE-induced cell death (Fig. 2B). In contrast to Urc and CRH, treatment of the cultures with UrcII at concentrations up to 100 pm was completely ineffective at preventing cell death induced by HNE (Fig. 2C).
Fig. 1.

Protective effects of Urc against Aβ-induced cell death in cultured rat hippocampal neurons. A, Cultures were exposed for 24 hr to Locke's solution alone (Control) or to Locke's solution containing Urc at 10 pm, Aβ25-35 at 1 μm, or the combination of Urc at 10 pm and Aβ25-35 at 1 μm; the cultures were photographed under phase-contrast optics with a 10× objective. Many of the neurons exposed to Aβ25-35 exhibited morphological signs of degeneration, i.e., cell body shrinkage and neurite fragmentation (arrows), whereas the majority of neurons treated with a combination of Urc and Aβ25-35 had neurites and cell bodies with normal appearance. B, The neuroprotective effect of Urc is concentration dependent. Cultures were exposed for 24 hr to Locke's solution alone (Control) or to Locke's solution containing Aβ25-35 at 1 μm, Urc at the indicated concentrations, or the combination of Aβ25-35 at 1 μm and Urc at the indicated concentrations. The cultures were photographed under phase-contrast optics with a 10× objective, and the percentage of remaining viable neurons was determined. Values are the mean and SD of determinations made in three cultures per treatment condition (*p = 0.0007 and #p < 0.0001 vs Aβ; one-way ANOVA and Fisher's PLSD).
Fig. 2.

Comparison of the potencies and effectiveness of Urc, CRH, and UrcII in protecting cultured rat hippocampal neurons from oxidative and excitotoxic insults. A, Cultures were exposed for 24 hr to Locke's solution alone (Control) or to Locke's solution containing Urc at 10 pm, Aβ25-35 at 1 μm, HNE at 1 μm, FeSO4 at 3 μm,l-glut at 20 μm, or the combinations as indicated. Values are the mean and SD of determinations made in four cultures per treatment condition. The extent of cell death caused by each insult alone was reduced in the presence of Urc (*p < 0.0001; one-way ANOVA and Fisher's PLSD).B, Cultures were exposed for 24 hr to Locke's solution alone (Control) or to Locke's solution containing HNE at 1 μm with or without the combinations as indicated. Values are the mean and SD of determinations made in four cultures per treatment condition. Submaximal and equally potent protective effects were observed with Urc alone at 0.5 pmand with CRH alone at 5 pm (*p < 0.0001 vs HNE; one-way ANOVA and Fisher's PLSD). Maximal protective effects were observed with Urc at 1 pm, CRH at 10 pm, or with the combination of Urc at 0.5 pm and CRH at 5 pm(*p < 0.0001 vs HNE;#p < 0.002 vs Urc, 0.5 pm+ HNE; +p = 0.0001 vs CRH, 5 pm + HNE; **p < 0.008 vs Urc, 0.5 pm + HNE; or CRH, 5 pm + HNE; one-way ANOVA and Fisher's PLSD). C, Cultures were exposed for 24 hr to Locke's solution alone (Control) or to Locke's solution containing HNE at 1 μm, UrcII at 1, 10, and 100 pm, or the combinations as indicated. Values are the mean and SD of determinations made in four cultures per treatment condition. UrcII was ineffective at protecting against HNE-induced cell death at any of the concentrations that were tested.
Evidence that the neuroprotective effects of Urc and CRH are mediated by CRHR1
Binding studies have demonstrated that Urc has a higher affinity for CRHR2 than does CRH (Perrin and Vale, 1999), whereas receptor expression studies have revealed that Urc potently can activate pathways downstream of CRHR1 and CRHR2 (Grammatopoulos et al., 2000). Because Urc exhibited a greater potency than CRH in protecting neurons from insult, we performed a series of studies aimed at determining which type of CRH receptor in fact was mediating these effects of the peptides. First, we performed RT-PCR analyses to establish whether both CRHR1 and CRHR2 were expressed in the cultured hippocampal neurons and to determine the relative abundance of CRHR1 and CRHR2 mRNAs within each sample. We observed expression of both CRHR1 and CRHR2 in untreated 8-d-old hippocampal and cortical cultures, with the levels of CRHR1 mRNA being several-fold higher than the levels of CRHR2 mRNA (Fig. 3). These results are consistent with the relative abundance of CRHR1 and CRHR2 mRNAs in the hippocampus, cortex, and midbrain of E18 rats (Fig. 3). We confirmed the presence of both CRHR1 and CRHR2 in the hippocampal cultures by cloning and sequencing the PCR products (Fig. 3).
Fig. 3.
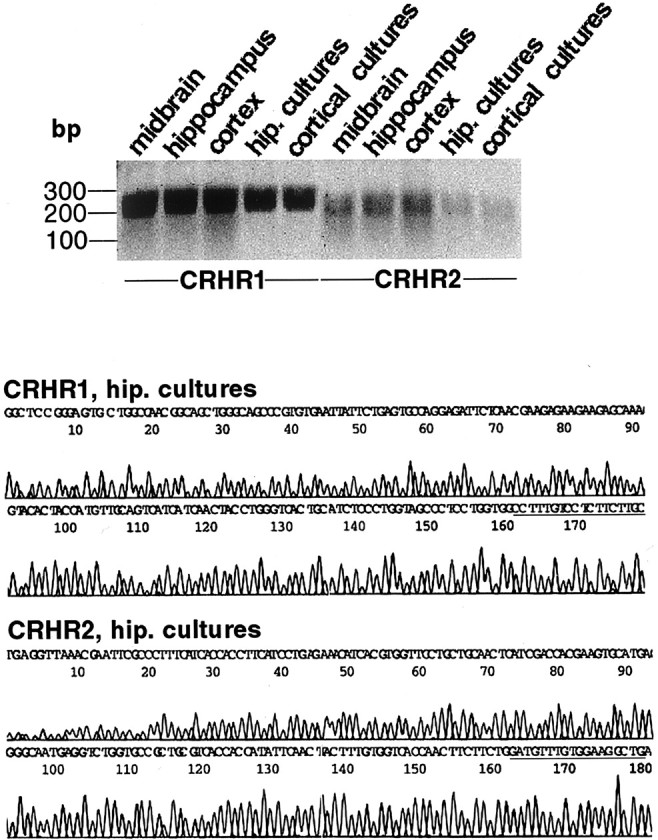
Expression of CRHR1 and CRHR2 in cultured rat hippocampal and cortical neurons. Top, Total RNA extracted from hippocampal and cortical cultures and from the midbrain, hippocampus, and cortex of E18 rat was subjected to first-strand reverse transcription, followed by PCR that used primers specific for either CRHR1 or CRHR2. The PCR products were separated by electrophoresis in a 2% agarose gel stained with ethidium bromide and were visualized with a FUJIFilm FLA-3000 fluorescent image analyzer and Image Gauge version 3.2 software. Equal loading of PCR products onto the gel was determined by using internal primers specific for GAPDH (data not shown). Similar results were obtained in three independent experiments. Bottom, Identity of the PCR products corresponding to the hippocampal cultures was confirmed by cloning and sequencing. Underlined sequences are those recognized by the reverse primers used for PCR amplification.
In a second set of studies we used highly selective antagonists for CRHR1 and CRHR2 to determine which of the two receptors mediated the protective effects of Urc and CRH in our cultures. In particular, we used the nonpeptide CRHR1 antagonist antalarmin (Ant; Webster et al., 1996; Chen et al., 1997) and the peptide CRHR2 antagonist antisauvagine-30 (aSVG-30; Rühmann et al., 1998; Higelin et al., 2001). As shown in Figure 4, pretreatment of the hippocampal cultures with Ant at 10 nm completely blocked the ability of Urc and CRH to protect against HNE-induced cell death. Interestingly, pretreatment of the cultures with Ant or aSVG-30 at 10 nm caused a slight potentiation in HNE-induced cell death (Fig. 5), suggesting that both CRHR1 and CRHR2 are occupied by ligand under basal conditions and serve a neuroprotective function when the cells are exposed to an insult. Significant protection by Urc against HNE-induced cell death was observed in cultures pretreated with aSVG-30, but not in cultures pretreated with Ant (Fig. 5). Collectively, the results of these studies suggest that the neuroprotective effects of exogenously added Urc and CRH are mediated exclusively by CRHR1, consistent with the much higher expression levels and therefore availability for ligand binding of CRHR1 versus CRHR2.
Fig. 4.

The neuroprotective effects of Urc and CRH are blocked by a CRHR1 antagonist. A, Cultures were exposed for 1 hr to fresh Neurobasal with or without antalarmin (Ant) at 10 nm and subsequently were exposed for 24 hr to Neurobasal alone (Control) or to Neurobasal containing Urc at 10 pm, HNE at 10 μm, Ant at 10 nm, or the combinations as indicated. Values are the mean and SD of determinations made in four cultures per treatment condition. The extent of cell death in cultures treated with HNE alone and in cultures treated with Ant followed by Urc and HNE was not statistically different. B, Cultures were exposed for 1 hr to fresh Neurobasal with or without Ant at 10 nm and subsequently were exposed for 24 hr to Neurobasal alone (Control) or to Neurobasal containing CRH at 10 pm, HNE at 10 μm, Ant at 10 nm, or the combinations as indicated. Values are the mean and SD of determinations made in four cultures per treatment condition. The extent of cell death in cultures treated with HNE alone and in cultures treated with Ant followed by CRH and HNE was not statistically different.
Fig. 5.

The neuroprotective effects of Urc are not blocked by a CRHR2 antagonist. Cultures were exposed for 1 hr to fresh Neurobasal with or without Ant or antisauvagine-30 (aSVG-30) at 10 nm and subsequently were exposed for 24 hr to Neurobasal alone (Control) or to Neurobasal containing Urc at 10 pm, HNE at 10 μm, Ant at 10 nm, aSVG-30 at 10 nm, or the combinations as indicated. Values are the mean and SD of determinations made in four cultures per treatment condition. Pretreatment with Ant or aSVG-30 caused a slight potentiation of HNE-induced cell death (+p = 0.0001 and#p = 0.0074 vs HNE; one-way ANOVA and Fisher's PLSD). The protective effect of Urc was blocked in cultures pretreated with Ant, but not in cultures pretreated with aSVG-30 (*p < 0.0001 vs HNE + aSVG-30; one-way ANOVA and Fisher's PLSD).
The neuroprotective effect of Urc requires activation of cAMP-dependent protein kinase, protein kinase C, and mitogen-activated protein kinase
Although it is well established that increases in the levels of cAMP occur with the activation of CRHR1 and CRHR2, little is known regarding the signaling pathways that mediate responses to Urc and CRH. In addition to cAMP-dependent protein kinase (PKA), the results of recent studies suggest the involvement of protein kinase C (PKC; Chakravorty et al., 1999; Miyata et al., 1999) and mitogen-activated protein (MAP) kinase (Brar et al., 2000; Craighead et al., 2000; Grammatopoulos et al., 2000) in cellular responses to Urc and CRH. We have reported previously (Pedersen et al., 2001) that the protective effects of CRH in primary hippocampal cultures could be prevented by pretreatment with H-89, an inhibitor of PKA activity (O'Sullivan and Jamieson, 1992; Otmakhova et al., 2000). Thus we performed experiments to provide evidence for involvement of the cAMP–PKA pathway in the neuroprotective effect of Urc. Treatment of cultured hippocampal neurons with Urc or CRH caused an increase in cellular cAMP levels, an effect that could be blocked by pretreatment with Ant, whereas UrcII treatment did not alter the cellular levels of cAMP (Fig. 6A). The effect of Urc on cAMP levels was concentration-dependent, with an EC50 value of 0.58 pm (Fig.6B). Consistent with these results and our previously published observations with CRH (Pedersen et al., 2001), we observed that pretreatment of the cultures with H-89 blocked the ability of Urc to protect the neurons against HNE-induced cell death (Fig.7A).
Fig. 6.
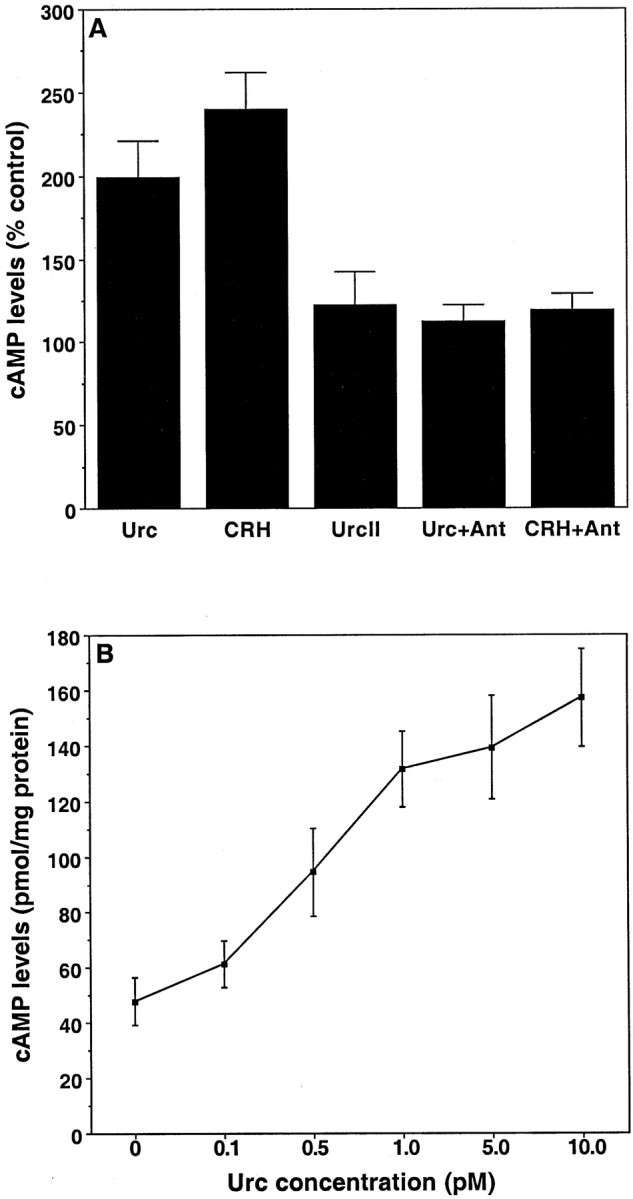
Elevation in cAMP concentrations by Urc and CRH in cultured rat hippocampal neurons. A, Cultures were exposed for 1 hr to Locke's solution with 50 μm IBMX and then were exposed for 30 min to Locke's solution alone (as the control) or to Locke's solution containing Urc at 1 pm, CRH at 10 pm, or UrcII at 10 pm in the continued presence of IBMX. Additional cultures were pretreated for 1 hr with IBMX at 50 μm and Ant at 10 nm and subsequently were treated for 30 min with Urc at 1 pm or CRH at 10 pm in the continued presence of both IBMX and Ant. Values are the mean and SD of determinations made in four cultures per treatment condition, expressed as a percentage of control cAMP levels (58.8 ± 8.7 pmol/mg protein). There was a statistically significant increase in the levels of cAMP in Urc- and CRH-treated groups (p < 0.0001 vs control; one-way ANOVA and Fisher's PLSD), but the levels of cAMP in control, UrcII, Urc + Ant, and CRH + Ant groups were not statistically different.B, Cultures were exposed for 1 hr to Locke's solution with 50 μm IBMX and then were exposed for 30 min to Locke's solution containing Urc at 0, 0.1, 0.5, 1.0, 5.0, or 10.0 pm in the continued presence of IBMX. Values are the mean and SD of determinations made in four cultures per treatment condition. There was a statistically significant increase in the levels of cAMP in cultures treated with Urc at concentrations ≥0.5 pm(p < 0.005 vs Urc at 0 or Urc at 0.1 pm; one-way ANOVA and Fisher's PLSD).
Fig. 7.

The neuroprotective effect of Urc requires G-protein-coupled receptor activation of PKA, PKC, and MAP kinase.A, Cultures were exposed for 1 hr to fresh Neurobasal with or without NF 449 at 200 nm, PD 98059 at 4 μm, BIM I at 10 nm, or H-89 at 100 nm; subsequently, the cultures were exposed for 24 hr to Neurobasal alone (Control) or to Neurobasal containing Urc at 10 pm, HNE at 10 μm, NF 449 at 200 nm, PD 98059 at 4 μm, BIM I at 10 nm, H-89 at 100 nm, or the combinations as indicated. Values are the mean and SD of determinations made in four cultures per treatment condition. Pretreatment with each of the inhibitors followed by treatment with Urc and HNE caused a statistically significant increase in the extent of cell death (p < 0.0001 vs Urc + HNE; one-way ANOVA and Fisher's PLSD). B, Immunoblot analysis demonstrating Urc activation of MAP kinases ERK1 and ERK2. Cultures were exposed for 1 hr to PD 98059 at 4 μm, BIM I at 10 nm, or H-89 at 100 nm and subsequently were exposed for 15 min to Urc with or without each of the inhibitors. The increased phosphorylation of ERK1 (p44) and ERK2 (p42) caused by Urc treatment is abolished in cultures pretreated with PD 98059 or BIM I and is attenuated in cultures pretreated with H-89. The levels of ERK1 and ERK2 in these samples are shown for comparison.C, Densitometric analysis of the immunoblots shown inB. The levels of phosphorylated ERK1 and ERK2 were normalized to the total levels of ERK1 and ERK2.
To elucidate further the neuroprotective signaling mechanisms evoked by Urc treatment of cultured hippocampal neurons, we determined the effects of compounds that inhibit the activation of G-proteins, PKC, and MAP kinase on the ability of Urc to protect against HNE-induced cell death. Pretreatment of the cultures with NF449, a selective antagonist of Gsα (Hohenegger et al., 1998), the selective PKC inhibitor bisindolylmaleimide I (BIM I; GF109203X;Heikkila et al., 1993; Huang et al., 1999), or the MAP kinase kinase inhibitor PD 98059 (Dudley et al., 1995) prevented or attenuated the neuroprotective effect of Urc, although none of the compounds affected the basal level of neuron survival (Fig. 7A). Two prominent MAP kinases that are known to be inhibited by PD 98059 are ERK1 (44 kDa) and ERK2 (42 kDa). MAP kinases are activated by phosphorylation on serine residues and have been shown to be substrates of PKA (Park et al., 2000) and PKC (Schonwasser et al., 1998). We therefore determined whether Urc does, in fact, activate MAP kinase in cultured hippocampal neurons and whether this requires PKA and/or PKC activities. As shown in Figure 7, B and C, Urc treatment of the cultures caused an increase in the levels of phosphorylated ERK1 and ERK2. This effect was prevented by pretreatment with PD 98059 or BIM I and was attenuated by pretreatment with H-89, suggesting that the ability of Urc to activate MAP kinases in hippocampal cultures is dependent on PKC activity and, to a lesser extent, on PKA activity. Our results suggest that the neuroprotective effect of Urc is dependent on G-protein-coupled receptor activation of signaling pathways involving PKA, PKC, and MAP kinase.
DISCUSSION
We have demonstrated the ability of Urc to protect cultured hippocampal neurons against cell death induced by amyloid β-peptide, oxidative stress, a product of membrane lipid peroxidation, and the excitatory neurotransmitter glutamate. We have demonstrated previously that CRH can protect primary hippocampal and cortical neurons from cell death induced by each of these insults, although a 10-fold higher concentration of CRH was required to achieve the same extent of neuroprotection than with Urc (Pedersen et al., 2001; this study). Although the present findings are the first to document a cytoprotective action of Urc in neurons, it has been reported that Urc protects cardiac myocytes cultured from neonatal rat against lethal ischemic injury and reduces the infarct size of an intact rat heart when applied before and after an ischemic insult (Okosi et al., 1998;Brar et al., 1999, 2000). In these studies Urc was applied at concentrations in the nanomolar range, whereas in cultured hippocampal neurons we observed that Urc can protect against cell death at concentrations as low as 0.5 pm. There is a discrepancy between the reported binding affinities of Urc and CRH toward CRHR1/CRHR2 and the concentrations of the peptides used in our studies (De Souza, 1995; Perrin and Vale, 1999). For instance, the binding affinity of CRH is in the range of 200–400 pm for CRH receptors in adult brain homogenates across a variety of species, including rat (De Souza, 1995). In addition to our studies, it was shown that CRH at a concentration of 10 pm protects IMR-32 cells from an Aβ-induced decrease in MTT reduction, although nanomolar concentrations were required to achieve full protection (Lezoualc'h et al., 2000). Overall, these results demonstrate that Urc and CRH can promote survival in various cell types. The potency of Urc and CRH toward embryonic neurons may be attributable to higher CRH receptor densities in these cells, based on the fact that CRH receptor densities peak in the CNS during early development (De Souza, 1995).
The ability of a CRHR1 antagonist to block neuroprotection by Urc and CRH demonstrates that the effect is, in fact, receptor-mediated. The compound Ant is a highly selective and potent antagonist of CRHR1, displacing 125I-ovine-CRH binding from rat frontal cortex homogenates with a Kiof 1.4 nm (Webster et al., 1996). In rhesus monkeys Ant has been shown to attenuate neuroendocrine, autonomic, and behavioral stress responses (Habib et al., 2000). We determined that both CRHR1 and CRHR2 are expressed in our neuronal cultures. However, given that the neuroprotective effect of Urc and CRH was blocked completely by Ant and unaltered by the selective CRHR2 antagonist aSVG-30, our results indicate that only CRHR1 is required for these peptides to protect neurons from cell death. Furthermore, we observed that UrcII, which selectively binds CRHR2 (Reyes et al., 2001), does not protect cultured hippocampal neurons from cell death. Although not tested in our study, we would expect a similar lack of neuroprotective function for UrcIII and stresscopin on the basis of their highly selective binding for CRHR2 (Hsu and Hsueh, 2001; Lewis et al., 2001). Although Urc is believed to be the cognate ligand for CRHR2, both Urc and CRH can bind and potently activate CRHR1 (Perrin and Vale, 1999). Notably, mice lacking the CRH gene exhibit normal behavioral responses to stressors, which can be attenuated by a selective CRHR1 antagonist related to Ant (Weninger et al., 1999). In these mice the behavioral responses to stressors may be attributable to actions of Urc. It is possible, therefore, that some of the effects of Urc in vivoare mediated by CRHR1, and the involvement of CRHR1 versus CRHR2 in mediating the various effects of Urc may be determined by the relative abundance of the receptors at sites of action. The lack of involvement of CRHR2 in the protective effects of Urc in cultured hippocampal neurons is likely attributable to a much greater availability of CRHR1 for binding compared with CRHR2.
All of the known effects of Urc and CRH involve receptor-coupled activation of adenylate cyclase and an increase in cellular levels of cAMP, and our data are consistent with this signaling pathway in the neuroprotective effect of these peptides. Furthermore, it is well documented in vitro that activation of the cAMP signaling pathway protects against neuronal death caused by a variety of insults, including FeSO4 and HNE in rat hippocampal cultures (Keller et al., 1998) and glutamate in rat cortical cultures (Morio et al., 1996). In addition to Gsα and the cAMP signaling pathway, CRH receptors are coupled to several other G-proteins, including Gq/11 linked to phospholipase C activation, IP3 production, and PKC activation (Grammatopoulos et al., 2000, 2001). Downstream targets of PKC include the MAP kinases ERK1 and ERK2. Indeed, Urc and CRH were reported to activate ERK1/2 in primary rat cardiac myocytes (Brar et al., 2000) and cortical neurons (Craighead et al., 2000), respectively. Our observations that neuroprotection by Urc could be prevented or attenuated by inhibitors of Gsα, PKA, PKC, or MAP kinase indicate that the effect is dependent on both Gsα- and Gq/11-activated signaling pathways. Note that Urc is 10-fold more potent in activating Gq/11 in cells expressing only CRHR1 (Grammatopoulos et al., 2000), which may explain the differences in potencies of Urc and CRH observed in our study. Similar to our findings, it was shown that CRH-induced cell shape changes in mollusk immunocytes require PKA and PKC, as well as PKB/Akt (Malagoli et al., 2000). Substrates of PKA (Park et al., 2000; Cavanaugh et al., 2001) and PKC (Schonwasser et al., 1998) include ERK1/2, and our results with BIM I and H-89 suggest that both PKA and PKC are responsible for Urc-induced phosphorylation of ERK1/2 in primary rat hippocampal neurons. Thus, emerging evidence supports a scenario in which at least two interdependent signaling pathways mediate the effects of Urc and CRH.
The insults used in the present study are relevant to the pathogenesis of several different neurodegenerative disorders. Mechanisms involving oxidative stress, lipid peroxidation, and/or excitotoxicity have been implicated in the degeneration of hippocampal and cortical neurons in AD, substantia nigra dopaminergic neurons in Parkinson's disease, spinal cord motor neurons in amyotrophic lateral sclerosis, and of the various neuronal populations affected in ischemic stroke (Olney et al., 1997; Mattson, 1998; Dirnagl et al., 1999; Ludolph et al., 2000; Sayre et al., 2001). We have provided evidence that Aβ causes apoptosis in neurons and increases their vulnerability to excitotoxicity by a mechanism involving membrane lipid peroxidation and the production of HNE (Mattson, 1997). Although speculative at present, Urc and/or CRH may serve a neuroprotective role analogous to neurotrophic factors that are upregulated in response to CNS injury (Mattson and Lindvall, 1997). Furthermore, upregulation in the levels of CRH and Urc during stress (Givalois et al., 2000; Weninger et al., 2000) may protect hippocampal neurons from the potentially damaging effects of glucocorticoids, released from the adrenal gland with the activation of the HPA axis (Raber, 1998; McEwen, 1999). This would provide a self-regulatory neuroprotective mechanism for the stress response axis. Thus, the functions of Urc and CRH in classical stress responses also may be extended to include the regulation of neuroprotective mechanisms.
The levels of CRH mRNA are increased markedly in the cerebral cortex and amygdala of rat brain in response to focal cerebral ischemia (Wong et al., 1995), and CRH immunoreactivity is increased in certain brain regions after kainic acid administration to rats (Piekut and Phipps, 1998). However, it has been shown that neuronal damage in these models can be attenuated by CRH receptor antagonists (Strijbos et al., 1994;Maecker et al., 1997). These results suggest that Urc and/or CRH promote neuronal death, yet evidence has been provided both in vitro and in vivo that CRH is not directly toxic to neurons (Craighead et al., 2000). One explanation for these observations is that Urc and/or CRH activate glial cells, which express CRH receptors (Bishop et al., 2000), causing them to release substances that are toxic to neurons. It also should be considered that the neuroprotective effects of Urc and CRH may be limited to only certain types of injury and modes of cell death. For instance, it has been reported that CRH is unable to protect primary rat cortical neurons from cell death induced by NMDA (Craighead et al., 2000). In this study, survival was assessed by measuring the release of lactate dehydrogenase, which is an indicator of necrotic cell death. The conditions under which the insults were used in our study would result in primarily an apoptotic form of cell death (Kruman et al., 1997; Guo et al., 1998). Interestingly, neuronal cell death by necrosis was shown to occur by a Gsα- and cAMP-mediated pathway inCaenorhabditis elegans (Berger et al., 1998). Thus, Urc and CRH may be able to protect neurons under conditions of acute injury, resulting in apoptosis, but actually promote neuronal death under conditions of chronic injury that result in necrosis. Although the exact roles of Urc and CRH in modulating neuronal injury in vivo remain to be determined, agonists of CRHR1 potentially may be used for the treatment of neurodegenerative conditions involving an apoptotic form of cell death.
Footnotes
This work was supported by the National Institute on Aging (NIA) and by a grant from the Alzheimer's Association. We thank Dr. Kamal Habib (Clinical Neuroendocrinology Branch, National Institute of Mental Health) for the gift of antalarmin, Dr. Jean Rivier (The Salk Institute for Biological Studies) for the gift of antisauvagine-30 peptide, and Wolfram Klapper (Christian-Albrechts-University, Kiel, Germany) for assistance with designing the primers for CRHR1 and CRHR2. We also thank S. Camandola and O. Milhavet (NIA) for helpful discussions.
Correspondence should be addressed to Mark P. Mattson, Gerontology Research Center, National Institute on Aging, 5600 Nathan Shock Drive-4F 02, Baltimore, MD 21224. E-mail: mattsonm@grc.nia.nih.gov.
REFERENCES
- 1.Berger AJ, Hart AC, Kaplan JM. Gαs-induced neurodegeneration in Caenorhabditis elegans. J Neurosci. 1998;18:2871–2880. doi: 10.1523/JNEUROSCI.18-08-02871.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bishop GA, Seelandt CM, King JS. Cellular localization of corticotropin-releasing factor receptors in the adult mouse cerebellum. Neuroscience. 2000;101:1083–1092. doi: 10.1016/s0306-4522(00)00413-9. [DOI] [PubMed] [Google Scholar]
- 3.Bittencourt JC, Vaughan J, Arias C, Rissman RA, Vale WW, Sawchenko PE. Urocortin expression in rat brain: evidence against a pervasive relationship of urocortin-containing projections with targets bearing type 2 CRF receptors. J Comp Neurol. 1999;415:285–312. [PubMed] [Google Scholar]
- 4.Brar BK, Stephanou A, Okosi A, Lawrence KM, Knight RA, Marber MS, Latchman DS. CRH-like peptides protect cardiac myocytes from lethal ischaemic injury. Mol Cell Endocrinol. 1999;158:55–63. doi: 10.1016/s0303-7207(99)00183-5. [DOI] [PubMed] [Google Scholar]
- 5.Brar BK, Jonassen AK, Stephanou A, Santilli G, Railson J, Knight RA, Yellon DM, Latchman DS. Urocortin protects against ischemic and reperfusion injury via a MAPK-dependent pathway. J Biol Chem. 2000;275:8508–8514. doi: 10.1074/jbc.275.12.8508. [DOI] [PubMed] [Google Scholar]
- 6.Cavanaugh JE, Ham J, Hetman M, Poser S, Yan C, Xia Z. Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J Neurosci. 2001;21:434–443. doi: 10.1523/JNEUROSCI.21-02-00434.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakravorty A, Mesiano S, Jaffe RB. Corticotropin-releasing hormone stimulates P450 17α-hydroxylase/17,20-lyase in human fetal adrenal cells via protein kinase C. J Clin Endocrinol Metab. 1999;84:3732–3738. doi: 10.1210/jcem.84.10.6053. [DOI] [PubMed] [Google Scholar]
- 8.Chang C-P, Pearse RV, II, O'Connell S, Rosenfeld MG. Identification of a seven transmembrane helix receptor for corticotropin-releasing factor and sauvagine in mammalian brain. Neuron. 1993;11:1187–1195. doi: 10.1016/0896-6273(93)90230-o. [DOI] [PubMed] [Google Scholar]
- 9.Chen YL, Mansbach RS, Winter SM, Brooks E, Collins J, Corman ML, Dunaiskis AR, Faraci WS, Gallaschun RJ, Schmidt A. Synthesis and oral efficacy of a 4(butylethylamino)pyrrolo [2,3-d]pyrimidine: a centrally active corticotropin-releasing factor1 receptor antagonist. J Med Chem. 1997;40:1749–1754. doi: 10.1021/jm960861b. [DOI] [PubMed] [Google Scholar]
- 10.Coste SC, Kesterson RA, Heldwein KA, Stevens SL, Heard AD, Hollis JH, Murray SE, Hill JK, Pantely GA, Hohimer AR. Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nat Genet. 2000;24:403–409. doi: 10.1038/74255. [DOI] [PubMed] [Google Scholar]
- 11.Craighead MW, Boutin H, Middlehurst KML, Allan SM, Brooks N, Kimber I, Rothwell NJ. Influence of corticotropin-releasing factor on neuronal cell death in vitro and in vivo. Brain Res. 2000;881:139–143. doi: 10.1016/s0006-8993(00)02759-1. [DOI] [PubMed] [Google Scholar]
- 12.De Souza EB. Corticotropin-releasing factor receptors: physiology, pharmacology, biochemistry, and role in central nervous system and immune disorders. Psychoneuroendocrinology. 1995;20:789–819. doi: 10.1016/0306-4530(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 13.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 14.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel A. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Givalois L, Arancibia S, Tapia-Arancibia L. Concomitant changes in CRH mRNA levels in rat hippocampus and hypothalamus following immobilization stress. Brain Res Mol Brain Res. 2000;75:166–171. doi: 10.1016/s0169-328x(99)00290-9. [DOI] [PubMed] [Google Scholar]
- 16.Grammatopoulos DK, Randeva HS, Levine MA, Katsanou ES, Hillhouse EW. Urocortin, but not corticotropin-releasing hormone (CRH), activates the mitogen-activated protein kinase signal transduction pathway in human pregnant myometrium: an effect mediated via R1α, R2β CRH receptor subtypes, stimulation of Gq-proteins. Mol Endocrinol. 2000;14:2076–2091. doi: 10.1210/mend.14.12.0574. [DOI] [PubMed] [Google Scholar]
- 17.Grammatopoulos DK, Randeva HS, Levine MA, Kanellopoulou KA, Hillhouse EW. Rat cerebral cortex corticotropin-releasing hormone receptors: evidence for receptor coupling to multiple G-proteins. J Neurochem. 2001;76:509–519. doi: 10.1046/j.1471-4159.2001.00067.x. [DOI] [PubMed] [Google Scholar]
- 18.Guo Q, Fu W, Xie J, Luo H, Sells SF, Geddes JW, Bondada V, Rangnekar VM, Mattson MP. Par-4 is a mediator of neuronal degeneration associated with the pathogenesis of Alzheimer's disease. Nat Med. 1998;4:957–962. doi: 10.1038/nm0898-957. [DOI] [PubMed] [Google Scholar]
- 19.Habib KE, Weld KP, Rice KC, Pushkas J, Champoux M, Listwak S, Webster EL, Atkinson AJ, Schulkin J, Contoreggi C, Chrousos GP, McCann SM, Suomi SJ, Higley JD, Gold PW. Oral administration of a corticotropin-releasing hormone receptor antagonist significantly attenuates behavioral, neuroendocrine, and autonomic responses to stress in primates. Proc Natl Acad Sci USA. 2000;97:6079–6084. doi: 10.1073/pnas.97.11.6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heikkila J, Jalava A, Eriksson K. The selective protein kinase C inhibitor GF 109203X inhibits phorbol ester-induced morphological and functional differentiation of SH-SY5Y human neuroblastoma cells. Biochem Biophys Res Commun. 1993;197:1185–1193. doi: 10.1006/bbrc.1993.2602. [DOI] [PubMed] [Google Scholar]
- 21.Higelin J, Py-Lang G, Paternoster C, Ellis GJ, Patel A, Dautzenberg FM. 125I-antisauvagine-30: a novel, specific high-affinity radioligand for the characterization of corticotropin-releasing factor type 2 receptors. Neuropharmacology. 2001;40:114–122. doi: 10.1016/s0028-3908(00)00105-2. [DOI] [PubMed] [Google Scholar]
- 22.Hohenegger M, Waldhoer M, Beindl W, Boing B, Kreimeyer A, Nickel P, Nanoff C, Freissmuth M. Gsα-selective G-protein antagonists. Proc Natl Acad Sci USA. 1998;95:346–351. doi: 10.1073/pnas.95.1.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu SY, Hsueh AJW. Human stresscopin and stresscopin-related peptide are selective ligands for the type 2 corticotropin-releasing hormone receptor. Nat Med. 2001;7:605–611. doi: 10.1038/87936. [DOI] [PubMed] [Google Scholar]
- 24.Huang L, Killbride J, Rowan MJ, Anwyl R. Activation of mGluRII induces LTD via activation of protein kinase A and protein kinase C in the dentate gyrus of the hippocampus in vitro. Neuropharmacology. 1999;38:73–83. doi: 10.1016/s0028-3908(98)00168-3. [DOI] [PubMed] [Google Scholar]
- 25.Jellinger KA, Bancher C. Neuropathology of Alzheimer's disease: a critical update. J Neural Transm Suppl. 1998;54:77–95. doi: 10.1007/978-3-7091-7508-8_8. [DOI] [PubMed] [Google Scholar]
- 26.Keller JN, Hanni KB, Mattson MP, Markesbery WR. Cyclic nucleotides attenuate lipid peroxidation-mediated neuron toxicity. NeuroReport. 1998;9:3731–3734. doi: 10.1097/00001756-199811160-00029. [DOI] [PubMed] [Google Scholar]
- 27.Kishimoto T, Radulovic J, Radulovic M, Lin CR, Schrick C, Hooshmand F, Hermanson O, Rosenfeld MG, Spiess J. Deletion of Crhr2 reveals an anxiolytic role for corticotropin-releasing hormone receptor-2. Nat Genet. 2000;24:415–419. doi: 10.1038/74271. [DOI] [PubMed] [Google Scholar]
- 28.Kruman I, Bruce-Keller AJ, Bredesen DW, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee EH, Lee CP, Wang HI, Lin WR. Hippocampal CRF, NE, and NMDA system interactions in memory processing in the rat. Synapse. 1993;14:144–153. doi: 10.1002/syn.890140207. [DOI] [PubMed] [Google Scholar]
- 30.Lewis K, Li C, Perrin MH, Blount A, Kunitake K, Donaldson C, Vaughan J, Reyes TM, Gulyas J, Fischer W, Bilezikjian L, Rivier J, Sawchenko PE, Vale WW. Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF-2 receptor. Proc Natl Acad Sci USA. 2001;98:7570–7575. doi: 10.1073/pnas.121165198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lezoualc'h F, Engert S, Berning B, Behl C. Corticotropin-releasing hormone-mediated neuroprotection against oxidative stress is associated with the increased release of non-amyloidogenic amyloid β precursor protein and with suppression of nuclear factor-κB. Mol Endocrinol. 2000;14:147–159. doi: 10.1210/mend.14.1.0403. [DOI] [PubMed] [Google Scholar]
- 32.Lovejoy DA, Balment RJ. Evolution and physiology of the corticotropin-releasing factor (CRF) family of neuropeptides in vertebrates. Gen Comp Endocrinol. 1999;115:1–22. doi: 10.1006/gcen.1999.7298. [DOI] [PubMed] [Google Scholar]
- 33.Lovenberg TW, Liaw CW, Grigoriadis DE, Clevenger W, Chalmers DT, De Souza EB, Oltersdorf T. Cloning and characterization of a functionally distinct corticotropin-releasing factor receptor subtype from rat brain. Proc Natl Acad Sci USA. 1995;92:836–840. doi: 10.1073/pnas.92.3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ludolph AC, Meyer T, Riepe MW. The role of excitotoxicity in ALS—what is the evidence? J Neurol. 2000;247[Suppl 1]:I7–I16. doi: 10.1007/s004150050552. [DOI] [PubMed] [Google Scholar]
- 35.Maecker H, Desai A, Dash R, Rivier J, Vale W, Sapolsky R. Astressin, a novel and potent CRF antagonist, is neuroprotective in the hippocampus when administered after a seizure. Brain Res. 1997;744:166–170. doi: 10.1016/s0006-8993(96)01207-3. [DOI] [PubMed] [Google Scholar]
- 36.Malagoli D, Franchini A, Ottaviani E. Synergistic role of cAMP and IP3 in corticotropin-releasing hormone-induced cell shape changes in invertebrate immunocytes. Peptides. 2000;21:175–182. doi: 10.1016/s0196-9781(99)00203-x. [DOI] [PubMed] [Google Scholar]
- 37.Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 38.Mattson MP. Cellular actions of β-amyloid precursor protein, and its soluble and fibrillogenic peptide derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 39.Mattson MP. Modification of ion homeostasis by lipid peroxidation: roles in neuronal degeneration and adaptive plasticity. Trends Neurosci. 1998;21:53–57. doi: 10.1016/s0166-2236(97)01188-0. [DOI] [PubMed] [Google Scholar]
- 40.Mattson MP, Lindvall O. Neurotrophic factors and the aging brain. In: Timiras PS, Bittar EE, editors. Advances in cell aging and gerontology, Vol 2. JAI; London: 1997. pp. 299–345. [Google Scholar]
- 41.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- 43.Miyata M, Okada D, Hashimoto K, Kano M, Ito M. Corticotropin-releasing factor plays a permissive role in cerebellar long-term depression. Neuron. 1999;22:763–775. doi: 10.1016/s0896-6273(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 44.Morin SM, Ling N, Liu X-J, Kahl SD, Gehlert DR. Differential distribution of urocortin- and corticotropin-releasing factor-like immunoreactivities in the rat brain. Neuroscience. 1999;92:281–291. doi: 10.1016/s0306-4522(98)00732-5. [DOI] [PubMed] [Google Scholar]
- 45.Morio H, Tatsuno I, Hirai A, Tamura Y, Saito Y. Pituitary adenylate cyclase-activating polypeptide protects rat-cultured cortical neurons from glutamate-induced cytotoxicity. Brain Res. 1996;741:82–88. doi: 10.1016/s0006-8993(96)00920-1. [DOI] [PubMed] [Google Scholar]
- 46.Okosi A, Brar BK, Chan M, D'Souza L, Smith E, Stephanou A, Latchman DS, Chowdrey HS, Knight RA. Expression and protective effects of urocortin in cardiac myocytes. Neuropeptides. 1998;32:167–171. doi: 10.1016/s0143-4179(98)90033-6. [DOI] [PubMed] [Google Scholar]
- 47.Olney JW, Wozniak DF, Farber NB. Excitotoxic neurodegeneration in Alzheimer disease. Arch Neurol. 1997;54:1234–1240. doi: 10.1001/archneur.1997.00550220042012. [DOI] [PubMed] [Google Scholar]
- 48.O'Sullivan AJ, Jamieson JD. Protein kinase A modulates Ca2+- and protein kinase C-dependent amylase release in permeabilized rat pancreatic acini. Biochem J. 1992;287:403–406. doi: 10.1042/bj2870403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otmakhova NA, Otmakhov N, Mortenson LH, Lisman JE. Inhibition of the cAMP pathway decreases early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 2000;20:4446–4451. doi: 10.1523/JNEUROSCI.20-12-04446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ott BR, Saver JL. Unilateral amnesic stroke. Six new cases and a review of the literature. Stroke. 1993;24:1033–1042. doi: 10.1161/01.str.24.7.1033. [DOI] [PubMed] [Google Scholar]
- 51.Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotropin-releasing factor. Pharmacol Rev. 1991;43:425–473. [PubMed] [Google Scholar]
- 52.Park JH, Park JK, Bae KW, Park HT. Protein kinase A activity is required for depolarization-induced proline-rich tyrosine kinase 2 and mitogen-activated protein kinase activation in PC12 cells. Neurosci Lett. 2000;290:25–28. doi: 10.1016/s0304-3940(00)01313-6. [DOI] [PubMed] [Google Scholar]
- 53.Pedersen WA, Luo H, Kruman I, Kasarskis E, Mattson MP. The prostate apoptosis response-4 protein participates in motor neuron degeneration in amyotrophic lateral sclerosis. FASEB J. 2000;14:913–924. doi: 10.1096/fasebj.14.7.913. [DOI] [PubMed] [Google Scholar]
- 54.Pedersen WA, McCullers D, Culmsee C, Haughey NJ, Herman JP, Mattson MP. Corticotropin-releasing hormone protects neurons against insults relevant to the pathogenesis of Alzheimer's disease. Neurobiol Dis. 2001;8:492–503. doi: 10.1006/nbdi.2001.0395. [DOI] [PubMed] [Google Scholar]
- 55.Perrin MH, Vale WW. Corticotropin-releasing factor receptors and their ligand family. Ann NY Acad Sci. 1999;885:312–328. doi: 10.1111/j.1749-6632.1999.tb08687.x. [DOI] [PubMed] [Google Scholar]
- 56.Perrin MH, Donaldson CJ, Chen R, Lewis KA, Vale WW. Cloning and functional expression of a rat brain corticotropin-releasing factor (CRF) receptor. Endocrinology. 1993;133:3058–3061. doi: 10.1210/endo.133.6.8243338. [DOI] [PubMed] [Google Scholar]
- 57.Piekut DT, Phipps B. Increased corticotropin-releasing factor immunoreactivity in select brain sites following kainate-elicited seizures. Brain Res. 1998;781:100–113. doi: 10.1016/s0006-8993(97)01219-5. [DOI] [PubMed] [Google Scholar]
- 58.Raber J. Detrimental effects of chronic hypothalamic–pituitary–adrenal axis activation. Mol Neurobiol. 1998;18:1–22. doi: 10.1007/BF02741457. [DOI] [PubMed] [Google Scholar]
- 59.Radulovic J, Ruhmann A, Liepold T, Spiess J. Modulation of learning and anxiety by corticotropin-releasing factor (CRF) and stress: differential roles of CRF receptors 1 and 2. J Neurosci. 1999;19:5016–5025. doi: 10.1523/JNEUROSCI.19-12-05016.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reyes TM, Lewis K, Perrin MH, Kunitake KS, Vaughan J, Arias CA, Hogenesch JB, Gulyas J, Rivier J, Vale WW, Sawchenko PE. Urocortin II: a member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc Natl Acad Sci USA. 2001;98:2843–2848. doi: 10.1073/pnas.051626398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rühmann A, Bonk I, Lin CR, Rosenfeld MG, Spiess J. Structural requirements for peptidic antagonists of the corticotropin-releasing factor receptor (CRFR): development of CRFR2β-selective antisauvagine-30. Proc Natl Acad Sci USA. 1998;95:15264–15269. doi: 10.1073/pnas.95.26.15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr Med Chem. 2001;8:721–738. doi: 10.2174/0929867013372922. [DOI] [PubMed] [Google Scholar]
- 63.Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith GW, Aubry J-M, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA. Corticotropin-releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–1102. doi: 10.1016/s0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- 65.Strijbos PJLM, Relton JK, Rothwell NJ. Corticotropin-releasing factor antagonist inhibits neuronal damage induced by focal cerebral ischemia or activation of NMDA receptors. Brain Res. 1994;656:405–408. doi: 10.1016/0006-8993(94)91485-0. [DOI] [PubMed] [Google Scholar]
- 66.Timpl P, Spanagel R, Sillaber I, Kresse A, Reul J, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet. 1998;19:162–166. doi: 10.1038/520. [DOI] [PubMed] [Google Scholar]
- 67.Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 68.Webster EL, Lewis DB, Torpy DJ, Zachman EK, Rice KC, Chrousos GP. In vivo and in vitro characterization of antalarmin, a nonpeptide corticotropin-releasing hormone (CRH) receptor antagonist: suppression of pituitary ACTH release and peripheral inflammation. Endocrinology. 1996;137:5747–5750. doi: 10.1210/endo.137.12.8940412. [DOI] [PubMed] [Google Scholar]
- 69.Weninger SC, Dunn AJ, Muglia LJ, Dikkes P, Miczek, Swiergiel AH, Berridge CW, Majzoub JA. Stress-induced behaviors require the corticotropin-releasing hormone (CRH) receptor, but not CRH. Proc Natl Acad Sci USA. 1999;96:8283–8288. doi: 10.1073/pnas.96.14.8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weninger SC, Peters LL, Majzoub JA. Urocortin expression in the Edinger–Westphal nucleus is up-regulated by stress and corticotropin-releasing hormone deficiency. Endocrinology. 2000;141:256–263. doi: 10.1210/endo.141.1.7277. [DOI] [PubMed] [Google Scholar]
- 71.Wong ML, Loddick SA, Bongiorno PB, Gold PW, Rothwell NJ, Licinio J. Focal cerebral ischemia induces CRH mRNA in rat cerebral cortex and amygdala. NeuroReport. 1995;6:1785–1788. doi: 10.1097/00001756-199509000-00019. [DOI] [PubMed] [Google Scholar]