

Abstract
In the premature infant, hypoxic-ischemic damage to the cerebral white matter [periventricular leukomalacia (PVL)] is a common and leading cause of brain injury that often results in chronic neurologic disability from cerebral palsy. The cellular basis for the propensity of white matter injury to occur in the developing brain and the greater resistance of the adult white matter to similar injury remains unknown. By using a neonatal rat model of hypoxic-ischemic injury, we found that the mechanism of perinatal white matter injury involved maturation-dependent vulnerability in the oligodendroctye (OL) lineage. The timing of appearance of late OL progenitors was the major developmental factor that accounted for the susceptibility of the neonatal white matter to injury. Late OL progenitors were the major OL lineage stage killed by apoptosis, whereas early OL progenitors and more mature OLs were highly resistant. The density of pyknotic late OL progenitors was significantly increased in the ischemic hemisphere (67 ± 31 cells/mm2) versus the control hemisphere (2.2 ± 0.4 cells/mm2; mean ± SEM; p = 0.05), which resulted in the death of 72 ± 6% of this OL stage. Surviving late OL progenitors displayed a reactive response in which an increase in cell density was accompanied by accelerated maturation to a P27/kip1-positive oligodendrocyte. Because we showed recently that late OL progenitors populate human cerebral white matter during the high risk period for PVL (Back et al., 2001), maturation-dependent vulnerability of OL progenitors to hypoxia–ischemia may underlie the selective vulnerability to PVL of the white matter in the premature infant.
Keywords: hypoxia–ischemia, development, cell lineage, progenitor, cerebral white matter, cerebral cortex, O4 antibody, O1 antibody, NG2, immunohistochemistry, microglia, periventricular leukomalacia, prematurity, Ki-67, MIB-5, P27, actin, spectrin, cytochromec, caspase-3
During human brain development, between 23 and 32 weeks gestation, the periventricular cerebral white matter is at increased risk for injury from hypoxia–ischemia (H-I) [periventricular leukomalacia (PVL)]. Although PVL is the most common form of brain injury in premature infants and results in cerebral palsy in 5–15% of survivors, the cellular target in PVL is unknown (Volpe, 2000). Immaturity of the cerebral blood supply and a propensity for impaired cerebral autoregulation are the major vascular factors related to the susceptibility of the periventricular region to ischemia (Volpe, 1998). PVL results in a chronic disturbance of myelination, which suggests that oligodendrocyte (OL) progenitors are a major target cell of ischemic injury in PVL (Back and Volpe, 1997). However, the in vivo susceptibility of OL progenitors to H-I is unclear. The four successive stages of OL development can be defined by the presence of different cell type-specific surface antigens (see Fig.1A) (Pfeiffer et al., 1993). Use of these markers has demonstrated that late OL progenitors are the predominant OL stage in human cerebral white matter during the time of peak incidence of PVL (Back et al., 2001).
Fig. 1.

Susceptibility of late OL progenitors to H-I at P2. A, The four successive stages in the OL lineage are depicted at the top. Shown at the bottomare the markers that were applied to define each stage.B, The normal low-power distribution of O4-labeled cells in the corpus callosum in the control hemisphere. C, The ischemic corpus callosum contralateral to that in Bcontains numerous pyknotic O4-labeled cells (arrowheads). Adjacent to the infarct (bottom left) are numerous intensely labeled cells (arrows). These apparent reactive cells are discussed in the context of Figure 4. D, Pyknotic cells at various stages of degeneration. A halo of degenerating processes surrounds one pyknotic cell (arrow). Many pyknotic cells (arrowheads) at a more advanced stage of degeneration had no discernible processes. E, High-power detail of pyknotic OL progenitors (arrowheads). Typical morphology of a control cell (inset). F, The density (cells per square millimeter) of pyknotic late OL progenitors in the ischemic (Ipsilateral) hemisphere was significantly increased relative to the control (Contralateral) hemisphere (*p = 0.05; unpaired Student's ttest). G, One week after H-I at P2, the area of the ischemic (Ipsilateral) corpus callosum was significantly decreased by ∼20% (*p = 0.0004; unpaired Student's t test). Scale bars:B, C, 100 μm; D, 50 μm; E and inset, 25 μm.
In the neonatal rat, late OL progenitors are the predominant OL lineage stage in the cerebral hemispheres (corpus callosum and cortex) between postnatal day (P) 1 and P5 (Gard and Pfeiffer, 1989). We hypothesized that if late OL progenitors were subjected to a hypoxic-ischemic insult, they would be selectively vulnerable relative to more or less mature cells of the OL lineage. To test this hypothesis, we used a well established neonatal rat model of hypoxic-ischemic brain injury in which cerebral white matter injury increases closer to birth (Rice et al., 1981; Johnston, 1983; Sheldon et al., 1996; Vannucci et al., 1999). In this model, unilateral ligation of the carotid artery is followed by a variable period of exposure to hypoxia (Levine, 1960).
We found distinct differences in susceptibility to H-I across the OL lineage. We demonstrate enhanced susceptibility of late OL progenitors to death from H-I relative to earlier or later stages in the OL lineage. Late OL progenitors had two major responses to H-I: death that is associated with activation of caspase-3 or accelerated maturation to a post-mitotic P27/kip1-positive reactive OL. These studies show that predilection for injury to the cerebral white matter in the developing brain is related to maturation-dependent vulnerability of the OL lineage to H-I.
MATERIALS AND METHODS
Animal surgical procedures. The left common carotid artery was ligated in Sprague Dawley litters (12 pups per litter) at P2 or P7 as we described previously (Han et al., 2000). After a 2 h recovery period, pups were placed in containers (submerged in a 37°C water bath to maintain normothermia) through which humidified oxygen (6% for 4 h at P2 or 8% for 2.5 h at P7) and balanced nitrogen flowed. Thereafter, the pups were returned to their dams until they were killed. It should be noted that the durations of hypoxia studied were the maximum tolerated by the animals at each age.
Primary antibodies. The O4 and O1 monoclonal antibodies were purified as described previously (Back et al., 2001). A rabbit polyclonal antibody against NG2 was generously provided by Dr. Joel Levine (State University of New York, Stony Brook, NY). The pan-axonal neurofilament marker SMI 312 (1:1000) was from Sternberger Monoclonals (Lutherville, MD). The microglial markerBandeiria griffonia isolectin B4, biotinylated (1:100; L2140) was from Sigma (St. Louis, MO). A rabbit anti-bovine glial fibrillary acidic protein (GFAP) antibody (Z-0334) was from Dako (Carpinteria, CA). Mouse monoclonal antibody MIB-5 (1:25; PNIM 2093, Beckman Coulter, Miami, FL) and rabbit polyclonal anti-P27 antisera (1:200; SC528, Santa Cruz Biotechnology, Santa Cruz, CA) were optimally visualized after antigen retrieval (30 min tissue incubation in 50 mm sodium citrate, pH 9.0, at 80°C). The rabbit polyclonal antibody against cytochrome c (1:100 with 0.1% Triton X-100; sc-7159) was from Santa Cruz Biotechnology. Dr. Anu Srinivasan generously provided the anti-activated caspase-3 antibody (Cm-1; Idun Pharmaceuticals, La Jolla, CA). Dr. Greg Cole (VA Medical Center UCLA, Sepulveda, CA) generously provided the rabbit polyclonal anti-fractin antisera (KYEAb4; 1:400). The α-spectrin rabbit polyclonal antibody used in this study specifically recognizes the caspase-3 cleavage product (p120) of α-spectrin (p120 antibody, 1:1000). The antibody was raised against a peptide corresponding to the N-terminal neoepitope generated by caspase-3 cleavage. The antibody was immunoaffinty purified using this N-terminal peptide and then cross-absorbed against a second peptide spanning the entire p120 cleavage site. Dr. Robert Siman (University of Pennsylvania, Philadelphia, PA) generously provided the rabbit polyclonal antibody Ab38 against calpain-cleaved spectrin.
All brain tissue was fixed for 24 h by immersion in ice-cold 4% paraformaldehyde in 0.1 m phosphate buffer and stored at 2–4°C in PBS. Free-floating sections (50 μm) were cut in ice-cold PBS on a Leica VTS-1000 vibrating microtome.
Quantification of the cell density of pyknotic OL lineage cells. A biotinylated O4 and an O1 monoclonal antibody were used to distinguish O4+O1− late OL progenitors from O4+O1+ immature OLs by immunofluorescence double labeling, and cells were counted as described previously (Back et al., 2001). Total OL lineage cells were defined as the total number of cells that labeled with the O4 monoclonal antibody (i.e., late OL progenitors plus immature OLs). Note that early OL progenitors were not quantified, because they showed no evidence of cell death in response to H-I (see Results). Briefly, cells were counted by selecting the nucleus as the smallest countable object. The nucleus was visualized with Hoechst 33324. Pyknotic and intact late OL progenitors were distinguished by a combination of nuclear and cellular morphology (see Results). A minimum of 20 high-power fields were counted with a 40× objective. Density was calculated from the total number of fields counted (0.0625 mm2 per field). Note that the O4- or O1-positive pyknotic cells did not label with the neuronal marker SMI 312, the astroglial marker GFAP, or the microglial marker isolectin B4.
Cells were counted in four adjacent regions of the forebrain in three serial adjacent coronal sections at the level of the mid-septal nuclei. Regions 1–3 were three adjacent areas of the corpus callosum from the midline (region 1) to its lateral extent (region 3). Region 1 comprised the parasagittal corpus callosum and in general contained the least number of pyknotic cells. Region 2 contained the supracallosal radiation and the underlying adjacent corpus callosum and was the most enriched in reactive OLs that were quantified in Figure4A. Region 3 comprised the lateral corpus callosum. Region 4 contained the cerebral cortex overlying the lateral corpus callosum (region 3). Regions 3 and 4 together were always the most enriched in pyknotic OL lineage cells. The cell counts reported in Figure 1F were from regions 3 and 4 and thus reflect injury to both the corpus callosum and the overlying cerebral cortex. In Figure 3C, the late OL progenitors were counted in region 4, and the immature OLs were counted in the adjacent white matter of region 3.
Fig. 4.

H-I triggers the proliferation of reactive OLs.A, By 24 h after H-I at P2 and P7, a significant increase in the density of O4+O1+ reactive OLs (*p= 0.04; **p = 0.02) occurred in the area adjacent to the ischemic lesion. B, Reactive OLs typically were visualized in clusters of two or more cells. C, A reactive OL in the apparent process of mitosis has a dividing nucleus (D) that was labeled with Hoechst 33324.E, Immature-appearing late OL progenitors in the ischemic penumbra at P8 (arrowheads) labeled for the O4 antibody (green) and the nuclear-associated cell proliferation marker MIB-5 (red). F, Mature-appearing reactive OLs (arrows) at P3 did not show nuclear-labeling with MIB-5 (arrowheads).G, Reactive OLs at P8 labeled with the O1 antibody (green, arrows) also labeled with an antibody against p27 that stained the nuclei of these and other cells (arrowheads). Scale bars:B–D, 10 μm; E,G, 15 μm; F, 20 μm.
Fig. 3.

Maturation-dependent differences in OL lineage susceptibility are seen at P2 and P7. A, Injury to the ischemic (I) cerebral cortex and hippocampus at P2 and P7 was compared with control (C) by assay of tissue homogenates for DEVD-amino methyl coumarin cleavage reflecting activated caspase-3-like activity. Animals exposed to 6% hypoxia for 4 h at P2 had a similar increase in enzyme activity in the ischemic hemisphere compared with animals exposed for 2.5 h to 8% hypoxia at P7.B, Numerous reactive NG2-positive early OL progenitors (red, arrowheads andinset) in an ischemic lesion from a P2 animal overlapped in distribution with pyknotic late OL progenitors (green, short arrows). Note the apparent reactive OL (long arrow) that was resistant to H-I. C, After H-I at P7, the density (cells per square millimeter) of pyknotic late OL progenitors in the cerebral cortex was significantly greater than that of pyknotic immature OLs in the underlying corpus callosum (*p = 0.04; unpaired Student's t test). D, An ischemic lesion from a P2 animal was double labeled with O4 (red) and O1 (green) antibodies. Most pyknotic cells were O4+O1− late OL progenitors (red,arrows), and few pyknotic immature OLs were visualized (yellow, arrowheads). Note the double-labeled reactive-appearing OLs (yellow,long arrows) that appeared resistant to H-I. Scale bars:B, D, 50 μm.
Assessment of brain damage caused by H-I. Histological assessment of the damage resulting at 1 week after H-I at P2 was determined by calculating the amount of surviving tissue in 50 μm serial coronal sections that were stained with Cresyl violet, as described previously (Cheng et al., 1998). Briefly, sections were assessed rostrocaudally in each of three equally spaced coronal reference planes. The cross-sectional area of the corpus callosum was determined with the NIH image analysis system (version 1.57) linked to a Nikon microscope. The sections corresponded approximately to plates 18, 22, and 26 in a rat brain atlas (Paxinos and Watson, 1986). All measurements were done by an investigator blinded to the location of the ischemic hemisphere.
Fluorescence in situ detection of DNA fragmentation in OL progenitors. At 24 h after H-I, animals were killed, and the brains were processed for in situ detection of DNA fragmentation in the cerebral white matter. Brain sections were mounted and air-dried on subbed slides, permeabilized for 5 min in 1:1 ethanol/acetic acid (−20°C), and washed with PBS. An ApopTag-fluorescein in situ DNA fragmentation detection kit (Intergen, Purchase, NY) was used to carry out terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling (TUNEL) staining. For double fluorescence visualization of the O4 antibody and TUNEL label, tissue sections were first incubated overnight at 2–4°C with the O4 antibody (1:1000). They were next washed in PBS and incubated for 5 min in ice-cold 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, and immediately washed 3× in PBS. After a 2 h incubation with μ-chain-specific Texas Red-conjugated goat anti-mouse IgM (1:100; TI-2020; Vector Laboratories, Burlingame, CA), the tissue sections were washed in PBS, mounted, and air dried on subbed slides and processed in the dark for fluorescein–TUNEL labeling, as described above. Note that the 5 min incubation in 4% paraformaldehyde was required to prevent the solubilization of the O4 antibody–antigen complex by the subsequent exposure to 1:1 ethanol/acetic acid that was necessary for optimal visualization of the TUNEL staining.
DEVD cleavage assay. Animals from single litters (n = 6) underwent unilateral carotid ligation at P2 or P7 followed by hypoxia. The animals survived for 24 h before brain tissue ipsilateral and contralateral to the ischemic hemisphere was collected to measure DEVD-cleaving activity that reflects total activated caspase-1 and caspase-3 activity (Han et al., 2000).
Confocal microscopy. Tissue sections were analyzed with a Bio-Rad MRC 1024ES laser scanning confocal microscope, with an argon-ion laser coupled to an inverted microscope (Nikon Diaphot 200) and equipped with a 40× oil-immersion objective [Nikon Plan Fluor, numerical aperture (NA) 1.30] and a 100× oil-immersion objective (Nikon Plan Fluor, NA 1.30).
RESULTS
H-I causes the acute degeneration of late OL progenitors
Because the P2 rat forebrain is enriched in late OL progenitors (Gard and Pfeiffer, 1989), we first tested whether the predilection of the P2 cerebral white matter to H-I might be explained by a susceptible population of late OL progenitors. Tissue sections collected 24 h after H-I were double labeled with biotinylated O4 and O1 antibodies specific for the OL lineage (Fig.1A). Numerous pyknotic late OL progenitors (O4+O1−) were visualized in the ischemic hemisphere (Fig. 1C). Degeneration of late OL progenitors was greatest in the lateral corpus callosum and the overlying parietal cortex. Pyknotic progenitors displayed features of acute degeneration that included apparent condensation of the cell body, fragmentation of the process arbor, and labeling of both the plasma membrane and the cytoplasm with the O4 antibody (Fig.1D,E). Because O4 selectively binds to cell-surface glycoconjugates, the cytoplasmic labeling is consistent with a loss of plasma membrane integrity. The density of pyknotic late OL progenitors was significantly increased in the ischemic hemisphere (Fig. 1F) (ischemic hemisphere, 67 ± 31 cells/mm2 vs control hemisphere, 2.2 ± 0.4 cells/mm2; mean ± SEM;n = 3; p = 0.05). At this time in development, late OL progenitors comprised 57 ± 6% of the total OL lineage cells counted in these brain areas (Materials and Methods), and 72 ± 6% of these late OL progenitors were pyknotic. Thus, a very large percentage of OL lineage cells were damaged by H-I.
We next evaluated the degree of white matter injury that resulted in our model by quantifying the atrophy of the subcortical white matter at P9, 1 week after H-I at P2 (Fig. 1G). There was clear loss of tissue in the corpus callosum in the injured hemisphere. The area of ischemic corpus callosum was 3.8 ± 0.4 mm2 and was significantly reduced by 23 ± 6% relative to the contralateral corpus callosum (4.8 ± 0.2 mm2; mean ± SEM;n = 16; p = 0.0004).
Pyknotic late OL progenitors label for markers of cell death
To verify that pyknotic OL progenitors were killed by H-I, we immunostained and labeled cells for markers of cell death. By 24 h after H-I at P2, numerous nuclei in the lesion were visualized by the TUNEL method for in situ end-labeling of double-stranded DNA fragmentation (Gavrielli et al., 1992) (Fig.2A,B). Labeling for TUNEL and with the O4 antibody confirmed the oligodendroglial origin of many of the TUNEL-positive nuclei (Fig.2C). We did not detect TUNEL-positive nuclei that labeled with the astroglial marker GFAP or the microglial marker isolectin B4 (data not shown).
Fig. 2.
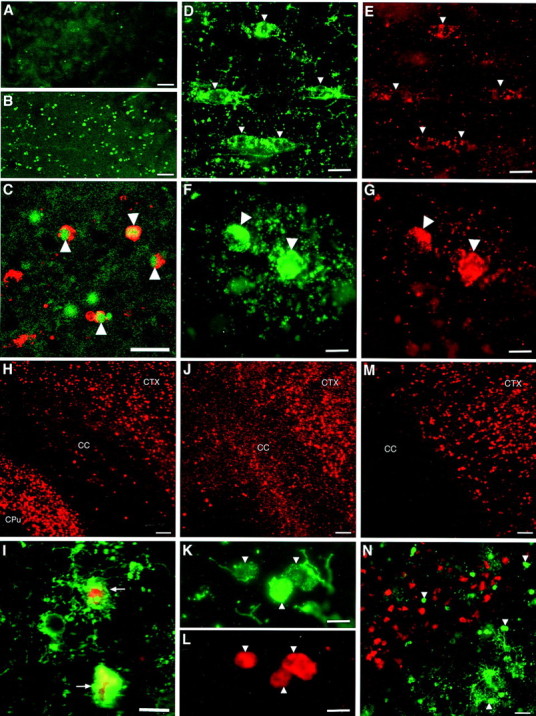
Pyknotic late OL progenitors label for markers of cell death. Numerous TUNEL-positive nuclei were detected in the ischemic corpus callosum (B) relative to control (A). C, Pyknotic O4 antibody-labeled cells (red, arrowheads) had TUNEL-positive nuclei (green).D, E, O4 antibody-labeled progenitors in the nonischemic hemisphere (D,arrowheads) displayed a punctate distribution of cytochrome c immunoreactivity in the soma and proximal processes (E). F,G, O4 antibody-labeled pyknotic OL progenitors (F, arrowheads) displayed a diffuse cytoplasmic distribution of cytochrome cimmunoreactivity (G). H, Numerous cells immunoreactive for CM1, an antibody against activated caspase-3, were visualized in the ischemic cerebral cortex (CTX) and caudate–putamen (CPu), but few were visualized in the corpus callosum (CC).I, Pyknotic O4-labeled cells at different stages of degeneration (green, arrows) were CM 1 immunoreactive. J, Numerous cells immunoreactive for fractin were visualized in the ischemic CTX and CC. Note that the CPu is not shown in this higher-power photomicrograph.K, L, Pyknotic O4-labeled cells (K, arrowheads) labeled for fractin (L). M, Numerous cells immunoreactive for the p120 antibody against a caspase cleavage product of spectrin were visualized in the ischemic CTX but were not detected in the corpus callosum. N, Higher-power detail of the junction between the cerebral cortex and the corpus callosum demonstrates that pyknotic O4 antibody-labeled cells (green, arrows) were not p120 antibody immunoreactive, in contrast to numerous apparent cortical neurons (red). Scale bars: A,B, 100 μm; C, 20 μm;D, E, 10 μm; F,G, 7 μm; H, 100 μm; I, 10 μm; J, 50 μm; K, L, 7 μm; M, 60 μm; N, 30 μm.
To further characterize the molecular events associated with death of the OL progenitors, we evaluated whether the pyknotic OL progenitors displayed cytoplasmic accumulation of cytochrome c, an upstream activator of neuronal apoptosis induced by H-I (Fujimura et al., 1998). Cytochrome c had a discrete punctate localization in the soma and processes of OL progenitors in the nonischemic hemisphere (Fig. 2D,E). By contrast, cytochrome c was diffusely distributed in the cytoplasm of pyknotic OL progenitors in the ischemic hemisphere (Fig.2F,G).
Numerous cells in the ischemic hemisphere were also labeled for activated caspase-3 (Fig. 2H), a major mediator of neuronal apoptosis (Schulz et al., 1999). We showed previously that neonatal neurons differ from adult neurons, in that caspase-dependent death is more prominent after H-I (Cheng et al., 1998; Han et al., 2000). Although many apparent neurons in the ischemic cerebral cortex and caudate–putamen were labeled for activated caspase-3, few cells of the OL lineage were labeled in the corpus callosum at 6, 12, 18, or 24 h after H-I at either P2 or P7. Staining for activated caspase-3 and with the O4 antibody showed that a small number of late OL progenitors labeled for activated caspase-3 at either age (Fig.2I).
To further evaluate the downstream mechanisms of death of late OL progenitors, we visualized two cytoskeletal proteins, actin and spectrin, that undergo caspase-3-dependent cleavage. The antibody fractin recognizes a cleavage product of actin that is specifically generated by caspase-1 or -3 (Yang et al., 1998). In previous studies, it was shown that fractin staining was markedly increased in frontotemporal cortex after H-I in the P7 rat (Pulera et al., 1998). Twenty-four hours after neonatal H-I at P2, the distribution of fractin staining (Fig. 2J) was similar to that of activated caspase-3 (Fig. 2D), but many more cells in the corpus callosum were labeled. Staining with the O4 antibody (Fig.2K) and for fractin (Fig. 2L) confirmed that numerous pyknotic late OL progenitors labeled strongly for fractin at 24 h after H-I. Because actin is a major cytoskeletal protein, the widespread accumulation of a caspase-cleaved actin fragment in ischemic late OL progenitors suggests that significant, deleterious caspase-dependent damage has occurred in the cell death pathway.
We next visualized staining with the p120 antibody that recognizes a 120 kDa cleavage product of spectrin that is specifically generated by caspase-3. The distribution of p120 staining in the cerebral cortex was very similar to that of activated caspase-3 (Fig.2D), except that there was no staining detected in the corpus callosum (Fig. 2M). The distribution of p120-labeled cells did not overlap with that of O4 antibody-labeled pyknotic progenitors (Fig. 2N).
In support of the specificity of the fractin and p120 spectrin antibodies in our model for caspase-3 cleavage products, we found that neither antibody detected necrotic neurons in the forebrain of P7 rats that survived for 30 min after H-I. However, such neurons were readily detected with Ab38, an antibody directed against calpain-I-cleaved spectrin (data not shown) that also detects ischemic neurons in the adult brain (Roberts-Lewis et al., 1994). In addition we evaluated a novel small molecule caspase-3 inhibitor, MF-826, that when administered by intracerebroventricular injection, inhibited caspase-3 activation in a dose-dependent manner in the cortex and hippocampus 24 hr after H-I in the P7 rat (Han et al., 2001). In these same animals, MF-826 completely blocked staining for fractin and p120 in the ischemic hemisphere (data not shown).
Late OL progenitors are selectively vulnerable to H-I
We next evaluated whether OL maturation is associated with increased resistance to death from H-I. To determine the relative susceptibility of successive stages in the OL lineage to H-I, we first established conditions that produced a similar overall insult to the hemisphere at P2 and P7 (Fig.3A). We showed previously that the amount of activated caspase-3-like activity reflects the degree of neuronal injury after H-I in the P7 rat (Han et al., 2000), and we found similar levels of caspase-3-like activity in both the P2 and P7 cortex and hippocampus after H-I under the conditions used. Stages of OL lineage development proceed from the early OL progenitor to the late OL progenitor to the immature OL (Fig. 1A). Because these three stages are present in differing proportions at both P2 and P7, we next compared their relative susceptibility to H-I at these ages. As reported previously (Nishiyama et al., 1997; Reynolds and Hardy, 1997), numerous early OL progenitors, identified with the marker NG2, were present in similar numbers throughout the cerebral cortex and corpus callosum at both P2 and P7, and they overlapped in distribution with the other OL stages present at each age. The early OL progenitors were highly resistant to H-I, and no pyknotic cells were observed at either P2 or P7 at time periods up to 48 h after H-I. Rather, early OL progenitors appeared reactive and were characterized by a hypertrophic soma, thickened proximal processes, and a more elaborate process arbor (Fig. 3B, inset). The reactive early OL progenitors were distinct from other glial cell types and did not label for markers of microglia, astrocytes, or OLs. The differential susceptibility of early and late OL progenitors to H-I was indicated by the fact that numerous reactive early OL progenitors often overlapped in distribution with degenerating late OL progenitors (Fig.3B, short arrows).
We next compared the susceptibility of late OL progenitors with immature OLs. During normal OL lineage progression, late OL progenitors give rise to immature OLs (Fig. 1A). In the neonatal rat, late OL progenitors are the predominant OL lineage stage in the cerebral hemispheres (corpus callosum and cortex) between P1 and P5 (Gard and Pfeiffer, 1989). By P7 most of the late OL progenitors in the corpus callosum have matured to immature OLs. However, in the cerebral cortex at P7, maturation of late OL progenitors is delayed, and a minority of the cells are late OL progenitors (Reynolds and Hardy, 1997). We confirmed that in the nonischemic (control) cerebral cortex at P7, most of these two OL stages were late OL progenitors (72 ± 2.6%; mean ± SD). However, in the adjacent corpus callosum, 92 ± 6% were immature OLs. Hence, because H-I damaged both the cerebral cortex and the corpus callosum, we could directly compare the susceptibility of these two OL stages with H-I in the same area of injury (Fig. 3C). The density of pyknotic late OL progenitors was significantly increased in the ischemic cortex (40 ± 10 cells/mm2) as compared with the density of pyknotic immature OLs in the corpus callosum (13 ± 7 cells/mm2; mean ± SEM;n = 3; p = 0.04). The greater susceptibility of late OL progenitors to H-I at P7 was further supported by the fact that 49 ± 1% of the cortical late OL progenitors were pyknotic, whereas only 13 ± 8% of the callosal immature OLs were pyknotic. Hence, the markedly greater susceptibility of late OL progenitors to H-I was a stage-specific property that was independent of the age of the animal or the location of these cells in the forebrain.
H-I triggers reactive oligodendrogliosis
We next evaluated the observation that H-I appeared to promote accelerated OL maturation in the ischemic hemisphere. We found an increase in O1-labeled OLs, many of which displayed a reactive change in their morphology characterized by a hypertrophic soma and an elaborate arbor of thickened processes (Figs. 3B, long arrow,4F,G). These reactive OLs were also labeled more intensely in the ischemic hemisphere (Fig. 1C, arrows) compared with the control (Fig. 1B). We confirmed that an increase in O1-labeled OLs occurred in the ischemic hemisphere within 24 h after H-I at both P2 and P7 (Fig. 4A) and was greatest in a region likely to be the ischemic penumbra that was adjacent to the region of maximal death of late OL progenitors. At P3, the OL density was increased 1.6-fold (ischemic hemisphere, 89 ± 13 cells/mm2 versus control, 57 ± 5; mean ± SEM; n = 3, p = 0.02). The OL density at P8 was increased 2.1-fold (lesioned hemisphere, 234 ± 34 versus control, 112 ± 16, p = 0.04).
To account for the increased OL density after H-I at P2 and P7, we looked for evidence that late OL progenitors also increased after H-I at these ages. At both ages, the density of late OL progenitors increased in both the infarct and the penumbra. At P3, late OL progenitors in the entire ischemic region increased 1.6 ± 0.17-fold (mean ± SEM; n = 3) relative to control, which closely paralleled the 1.7 ± 0.15-fold increase in OLs. At P8, ischemic late OL progenitors were increased 3.2 ± 0.32-fold (mean ± SEM; n = 3) relative to control, and ischemic OLs were increased 2.1 ± 0.23-fold.
In the area bordering the infarct, reactive OLs were frequently detected in clusters of two or more cells (Fig. 4B), and apparent dividing cells were detected (Fig.4C,D). We confirmed that the cell proliferation marker MIB-5 (Gerlach et al., 1997; Lui et al., 2000) labeled surviving late OL progenitors in the ischemic hemisphere (Fig.4E). However, the more mature-appearing reactive OLs did not label with MIB-5 (Fig. 4F), which indicates that they were post-mitotic. In fact, these reactive OLs also labeled for the p27Kip1 cyclin kinase inhibitor (Fig. 4G), which further supported their accelerated maturation and exit from the cell cycle (Casaccia-Bonnefil et al., 1997; Durand et al., 1997; Tikoo et al., 1998).
DISCUSSION
These findings demonstrate that during OL lineage maturationin vivo, the late OL progenitor is selectively vulnerable to H-I relative to earlier or later stages in the OL lineage. The data suggest that this may underlie the cellular basis for PVL, the most common cause of brain injury in the premature infant. Because of advances in neonatal intensive care, the number of surviving premature infants with PVL is increasing (Volpe, 1998). Thus, understanding the cellular basis for PVL is critical to developing preventive therapies.
Maturation-dependent vulnerability of the OL lineage to H-I
We evaluated the hypothesis that the increased predilection of cerebral white matter injury in the premature infant is related to maturation-dependent vulnerability of the OL lineage to H-I. We focused on the susceptibility of the late OL progenitor, because this OL stage predominates throughout the high-risk period for PVL (Back et al., 2001). We found distinct differences in susceptibility to H-I across the OL lineage. The most striking difference occurred between the early OL progenitor that was highly resistant to H-I and the late OL progenitor that was highly susceptible. This finding could not be explained by differences in the relative distribution or abundance of these two successive stages in the regions of ischemia. In agreement with previous studies (Nishiyama et al., 1996; Reynolds and Hardy, 1997), we also found that NG2+ early OL progenitors are similar to late OL progenitors in number and distribution in the P2 forebrain (data not shown). It also appears unlikely that the resistance of the early progenitors to H-I was related to the conditions used, because we found that a longer duration of hypoxia resulted in high mortality of the animals (data not shown).
That immature OLs were more resistant to H-I than late OL progenitors suggests that OL maturation is critical in determining resistance to H-I. This finding may be clinically significant, because the incidence of PVL declines around 30 weeks gestation, when late OL progenitors begin to differentiate and immature OLs increase threefold in the white matter (Back et al., 2001). Our data thus provide a cellular explanation for the susceptibility of preterm human cerebral white matter for PVL that relates to selective vulnerability of late OL progenitors to H-I. Our in vivo findings provide important biological relevance and validation for recent in vitroevidence that OL lineage maturation is accompanied by increased resistance to cell death from oxidative stress. We found previously that the late OL progenitor is markedly more susceptible than the mature OL to both endogenous and exogenous sources of oxidative stress (Back et al., 1998). More recent studies that used oxygen-glucose deprivation (OGD) as an in vitro model of H-I confirmed that resistance to OGD increased with OL maturation (Fern and Moller, 2000). The close agreement between our in vivo results and thesein vitro results thus indicates that in vitromodel systems offer powerful access to the molecular mechanisms that underlie the susceptibility of human cerebral white matter to injury.
Role of mitochondria in OL death
Although the basis for the selective vulnerability of the late OL progenitor to H-I remains unclear, the mechanism of death appears to involve a mitochondrial lesion, because the acute degeneration of late OL progenitors was accompanied by the apparent release of cytochromec from mitochondria to the cytoplasm. We showed previously that when late OL progenitors were killed in vitro by oxidative stress, these cells displayed ultrastructural features of apoptosis that included progressive swelling of mitochondria (Back et al., 1998). This disruption of mitochondrial integrity was accompanied by an intracellular rise in reactive oxygen species (ROS) production that contributed to cell death that was prevented by antioxidants. Because ROS production is a well established sequela of ischemia and reperfusion (Chan, 2001), one target of ROS toxicity in late OL progenitors in vivo may be the mitochondria. In fact, mitochondrial release of cytochrome c is a proximal event in neuronal apoptosis triggered by H-I in the adult brain (Fujimura et al., 1998) and is suppressed when the antioxidant enzyme Cu/Zn superoxide dismutase is overexpressed (Fujimura et al., 2000). Further studies are needed to determine whether mitochondrial dysfunction in late OL progenitors is also a consequence of ROS toxicity from H-I and whether mitochondrial injury may result in a feed-forward amplification of ROS production that is inevitably fatal to the cell. That OL precursors may be particularly susceptible to ROS toxicity is supported by the observation that the activity or expression of several antioxidant enzymes is reduced in OLs compared with other glia (for review, see Back and Volpe, 1999).
Molecular mechanisms of death of late OL progenitors
Our data indicate that H-I can trigger the death of late OL progenitors via a pathway that is similar to that of neonatal neurons. Distal to cytochrome c release, caspase-3 activation was detected in some late OL progenitors. Caspase-3 activation has also been detected in vitro in OLs treated with NGF or γ-radiation (Gu et al., 1999). Because the level of activated caspase-3 appears to be lower in late OL progenitors in vivothan in neonatal neurons, caspase-3 activation in late OL progenitors was supported by the detection of strong fractin immunoreactivity. Thus, the relative abundance of actin in the cytoskeleton of the late OL progenitor indirectly permitted the enhanced detection of activated caspase-3. Unexpectedly, we found that a caspase-3 cleavage product of spectrin was detectable in neurons but not in late OL progenitors, which suggests that spectrin is not a significant component of the late OL progenitor cytoskeleton. Although actin can be cleaved by either caspases or calpains (Villa et al., 1998; Yang et al., 1998), fractin immunoreactivity appears to be a reliable means to detect apoptotic late OL progenitors in the perinatal brain after H-I. This conclusion is supported by our finding that fractin staining was abolished in the ischemic hemisphere when animals were treated with a selective caspase-3 inhibitor MF-826 before H-I at P7.
H-I can trigger accelerated maturation of late OL progenitors
We found that H-I can trigger a reactive response in both late OL progenitors and immature OLs in perinatal brain that is related to the OL stages present at the time of H-I. We confirmed that H-I, like other sources of brain injury, can trigger a reactive response in early OL progenitors (Nishiyama et al., 1997). Hence, multiple OL stages can mount a reactive response to brain injury. Several lines of evidence indicate that the late OL progenitor and immature OL undergo a reactive response to injury. First, both develop an altered morphology with an enlarged soma and elaborate thickened processes, which is atypical of normal late OL progenitors or immature OLs. Second, the distribution of reactive-appearing cells is restricted to regions of adjacent gray matter or white matter injury. Third, the distribution of these cells overlaps with that of reactive astrocytes and reactive microglia. Fourth, the reactive-appearing cells persist for at least 7 d after H-I at P2 (data not shown).
An unexpected finding is that the reactive response of the late OL progenitor was accompanied by accelerated OL lineage maturation. The 1.6-fold increase at P2 in O1 antibody-labeled OLs, which are normally a minor population of post-mitotic cells at this age, was paradoxical and indicated that H-I promoted accelerated maturation of the OL lineage. These cells did not label with markers for astrocytes or microglia and appear to derive from the major population of late OL progenitors that are mitotically active and directly precede immature OLs in the lineage (Fig. 1A). The accelerated maturation of the reactive late OL progenitors was supported by the detection of p27Kip1, a cyclin kinase inhibitor that is closely associated with OL lineage maturation and is expressed when late OL progenitors exit from the cell cycle to generate immature OLs (Casaccia-Bonnefil et al., 1997; Durand et al., 1997;Tikoo et al., 1998). We speculate that the accelerated maturation of reactive late OL progenitors may be a protective response to H-I, because we found that immature OLs were less susceptible to H-I than late OL progenitors. This reactive response may also be maladaptive if the reactive cells were to display arrested maturation. Although technically challenging, future studies with techniques to accurately measure rates of proliferation and death of specific OL lineage stages are needed to evaluate the relative contributions of progenitor death and reactive oligodendrogliosis to the myelination disturbances in PVL.
Clinical implications
Because we recently reported that late OL progenitors predominate throughout the high-risk period for PVL, our data suggest two potential mechanisms by which the late OL progenitor might contribute to the myelination disturbances of PVL. These mechanisms relate to our observation that late OL progenitors have two potential fates in response to H-I: death or accelerated maturation to a reactive OL. We speculate that death of late OL progenitors could disrupt myelination through a depletion of progenitors required to generate mature OLs. The reactive OLs may also contribute to myelination disturbances if these aberrant cells fail to differentiate to mature OLs with the normal potential to myelinate. Confirmation will require human clinicopathologic studies and the development of an animal model of selective white matter injury that reproduces the major features of PVL.
The response of the OL lineage to H-I also has clinical ramifications beyond perinatal brain injury. Early and late OL progenitors are present in the adult CNS (Scolding et al., 1995), where their roles in injury and repair are implicated in stroke and demyelinating and neurodegenerative disorders (Back and Volpe, 1999). Because OL progenitors were recently shown to be multipotent under some in vitro conditions (Kondo and Raff, 2000), the susceptibility of late OL progenitors to H-I may have important implications for brain injury and repair in both the developing and adult nervous system.
B.H.H. and N.L.L. contributed equally to this paper.
S.B. is supported by grants from the National Institutes of Health (NIH) (NS01855; NS41343; P30 HD 33703, Oregon Child Health Research Center, Training Program Award for Pediatric Physician-Scientists), March of Dimes Grant 6FY01-65, a Child Neurology Society Young Investigator Award, and the Medical Research Foundation of Oregon. D.M.H. is supported by NIH Grant NS35902. We are grateful to Dr. Greg M. Cole for generously providing the fractin antibody. We thank Douglas Beardsley for excellent technical assistance. We are grateful to Dr. Roger Simon for his invaluable advice.
Correspondence should be addressed to Dr. Stephen A. Back, Department of Pediatrics, NRC-5, Oregon Health Sciences University, 3181 S.W. Sam Jackson Park Road, Portland, OR 97201-3098. E-mail:backs@ohsu.edu.
REFERENCES
- 1.Back SA, Volpe JJ. Cellular and molecular pathogenesis of periventricular white matter injury. MRDD Res Rev. 1997;3:96–107. [Google Scholar]
- 2.Back SA, Volpe JJ. Approaches to the study of diseases involving oligodendroglial death. In: Koliatsos V, Ratan R, editors. Cell death and diseases of the nervous system. Humana; Totowa, NJ: 1999. pp. 401–428. [Google Scholar]
- 3.Back SA, Gan X-D, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casaccia-Bonnefil P, Tikoo R, Kiyokawa H, Friedrich V, Jr, Chao MV, Koff A. Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27Kip1. Genes Dev. 1997;11:2335–2346. doi: 10.1101/gad.11.18.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Cheng Y, Deshmukh M, D'Costa A, Demaro J, Gidday J, Shah A, Sun Y, Jacquin M, Johnson E, Jr, Holtzman D. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J Clin Invest. 1998;101:1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durand B, Gao FB, Raff M. Accumulation of the cyclin-dependent kinase inhibitor p27/kip1 and the timing of oligodendrocyte differentiation. EMBO J. 1997;16:306–317. doi: 10.1093/emboj/16.2.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fern R, Moller T. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci. 2000;20:34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujimura M, Morita-Fujimura Y, Murakami K, Kawase M, Chan PH. Cytosolic redistribution of cytochrome c after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1998;18:1239–1247. doi: 10.1097/00004647-199811000-00010. [DOI] [PubMed] [Google Scholar]
- 11.Fujimura M, Morita-Fujimura Y, Noshita N, Sugawara T, Kawase M, Chan PH. The cytosolic antioxidant copper/zinc-superoxide dismutase prevents the early release of mitochondrial cytochrome c in ischemic brain transient focal cerebral ischemia in mice. J Neurosci. 2000;20:2817–2824. doi: 10.1523/JNEUROSCI.20-08-02817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gard AL, Pfeiffer SE. Oligodendrocyte progenitors isolated directly from developing telencephalon at a specific phenotypic stage: myelinogenic potential in a defined environment. Development. 1989;106:119–132. doi: 10.1242/dev.106.1.119. [DOI] [PubMed] [Google Scholar]
- 13.Gavrielli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerlach C, Golding M, Larue L, Alison MR, Gerdes J. Ki-67 immunoexpression is a robust marker of proliferative cells in the rat. Lab Invest. 1997;77:697–698. [PubMed] [Google Scholar]
- 15.Gu C, Casaccia-Bonnefil P, Srinivasan A, Chao MV. Oligodendrocyte apoptosis mediated by caspase activation. J Neurosci. 1999;19:3043–3049. doi: 10.1523/JNEUROSCI.19-08-03043.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han BH, D'Costa A, Back SA, Parsadian M, Patel S, Shah A, Gidday J, Srinvasan A, Deshmukh M, Holtzman D. BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol Dis. 2000;7:38–53. doi: 10.1006/nbdi.1999.0275. [DOI] [PubMed] [Google Scholar]
- 17.Han BH, Xu D, Choi J, Han Y, Xanthoudakis S, Roy S, Ng G, Tam J, Vaillancourt J, Collucci J, Giroux A, Robertson GS, Zamboni B, Nicholson DW, Holtzman DM. MF-826, a selective caspase-3 inhibitor attenuates apoptosis following neonatal hypoxia-ischemia. Soc Neurosci Abstr. 2001;27:763.2. [Google Scholar]
- 18.Johnston M. Neurotransmitter alterations in a model of perinatal hypoxic-ischemic brain injury. Ann Neurol. 1983;39:511–518. doi: 10.1002/ana.410130507. [DOI] [PubMed] [Google Scholar]
- 19.Kondo T, Raff M. Oligodendrocyte precursor cells reprogrammed to become multipotential stem cells. Science. 2000;289:1754–1757. doi: 10.1126/science.289.5485.1754. [DOI] [PubMed] [Google Scholar]
- 20.Levine S. Anoxic-ischemic encephalopathy in rats. Am J Pathol. 1960;36:1–17. [PMC free article] [PubMed] [Google Scholar]
- 21.Lui L, Rudin M, Kozlova EN. Glial cell proliferation in the spinal cord after dorsal rhizotomy or sciatic transection in the adult rat. Exp Brain Res. 2000;131:64–73. doi: 10.1007/s002219900273. [DOI] [PubMed] [Google Scholar]
- 22.Nishiyama A, Lin X-H, Giese N, Heldin C, Stallcup W. Co-localization of NG2 proteoglycan and PDGF α receptor on O2A progenitor cells in the developing rat brain. J Neurosci Res. 1996;42:299–314. doi: 10.1002/(SICI)1097-4547(19960201)43:3<299::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 23.Nishiyama A, Yu M, Drazba J, Tuohy V. Normal and reactive NG2+ glial cells are distinct from resting and activated microglia. J Neurosci Res. 1997;48:299–312. doi: 10.1002/(sici)1097-4547(19970515)48:4<299::aid-jnr2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 24.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1986. [DOI] [PubMed] [Google Scholar]
- 25.Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-k. [DOI] [PubMed] [Google Scholar]
- 26.Pulera MR, Adams LM, Lui H, Santos DG, Nishimura RN, Yang F, Cole GM, Wasterlain CG. Apoptosis in a neonatal rat model of cerebral hypoxia-ischemia. Stoke. 1998;29:2622–2630. doi: 10.1161/01.str.29.12.2622. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds R, Hardy R. Oligodendroglial progenitors labeled with the O4 antibody persist in the adult rat cerebral cortex in vivo. J Neurosci Res. 1997;47:455–470. doi: 10.1002/(sici)1097-4547(19970301)47:5<455::aid-jnr1>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 28.Rice JE, III, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- 29.Roberts-Lewis JM, Savage MJ, March VR, Pinsker LR, Siman R. Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J Neurosci. 1994;14:3934–3944. doi: 10.1523/JNEUROSCI.14-06-03934.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schulz J, Weller M, Moskowitz MA. Caspases as treatment targets in stroke and neurodegenerative diseases. Ann Neurol. 1999;45:421–429. doi: 10.1002/1531-8249(199904)45:4<421::aid-ana2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 31.Scolding N, Rayner P, Sussman J, Shaw C, Compston D. A proliferative adult human oligodendrocyte progenitor. NeuroReport. 1995;6:441–445. doi: 10.1097/00001756-199502000-00009. [DOI] [PubMed] [Google Scholar]
- 32.Sheldon RA, Chuai J, Ferreiro DM. A rat model for hypoxic-ischemic brain damage in very premature infants. Biol Neonate. 1996;69:327–341. doi: 10.1159/000244327. [DOI] [PubMed] [Google Scholar]
- 33.Tikoo R, Osterhout DJ, Casaccia-Bonnefil P, Seth P, Koff A, Chao MV. Ectopic expression of p27kip1 in oligodendrocyte progenitor cells results in cell-cycle growth arrest. J Neurobiol. 1998;36:431–440. [PubMed] [Google Scholar]
- 34.Vannucci RC, Connor JR, Mauger DT, Palmer C, Smith MB, Towfighi J, Vannucci SJ. Rat model of perinatal hypoxic-ischemic brain damage. J Neurosci Res. 1999;55:158–163. doi: 10.1002/(SICI)1097-4547(19990115)55:2<158::AID-JNR3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 35.Villa PG, Henzel WJ, Sensenbrenner M, Henderson CE, Pettmann B. Calpain inhibitors, but not caspase inhibitors, prevent actin proteolysis and DNA fragmentation during apoptosis. J Cell Sci. 1998;111:713–722. doi: 10.1242/jcs.111.6.713. [DOI] [PubMed] [Google Scholar]
- 36.Volpe JJ. Brain injury in the premature infant: overview of clinical aspects, neuropathology, and pathogenesis. Semin Pediatr Neurol. 1998;5:135–151. doi: 10.1016/s1071-9091(98)80030-2. [DOI] [PubMed] [Google Scholar]
- 37.Volpe JJ. Neurology of the newborn, Ed 4. W.B. Saunders; Philadelphia: 2000. [Google Scholar]
- 38.Yang F, Sun X, Beech W, Teter B, Wu S, Sigel J, Vinter HV, Frautschy SA, Cole GM. Antibody to caspase-cleaved actin detects apoptosis in differentiated neuroblastoma and plaque-associated neurons and microglia in Alzheimer's disease. Am J Pathol. 1998;152:379–389. [PMC free article] [PubMed] [Google Scholar]