

Abstract
Neurotrophic factors have well established roles in neuronal development, although their precise involvement in synapse formation and plasticity is yet to be fully determined. Using soma–soma synapses between identified Lymnaea neurons, we have shown recently that trophic factors are required for excitatory but not inhibitory synapse formation. However, neither the precise site (presynaptic versus postsynaptic cell) nor the underlying mechanisms have yet been defined. In the present study, synapse formation between the presynaptic cell visceral dorsal 4 (VD4) and its postsynaptic partner right pedal dorsal 1 (RPeD1) was examined to define the cellular mechanisms mediating trophic factor-induced excitatory synaptogenesis in cell culture. When paired in a soma–soma configuration in the presence of defined media (DM, nonproteinacious), mutually inhibitory synapses were appropriately reconstructed between VD4 and RPeD1. However, when cells were paired in the presence of increasing concentrations of Lymnaea brain-conditioned medium (CM), a biphasic synapse (initial excitatory synaptic component followed by inhibition) developed. The CM-induced excitatory synapse formation required trophic factor-mediated activation of receptor tyrosine kinases in the postsynaptic cell, RPeD1, and a concomitant modulation of existing postsynaptic nicotinic acetylcholine receptors (nAChRs). Specifically, when RPeD1 was isolated in DM, exogenously applied ACh induced a hyperpolarizing response that was sensitive to the AChR antagonist methyllycaconitine (MLA). In contrast, a single RPeD1 isolated in CM exhibited a biphasic response to exogenously applied ACh. The initial depolarizing phase of the biphasic response was sensitive to both mecamylamine and hexamethonium chloride, whereas the hyperpolarizing phase was blocked by MLA. In soma–soma-paired neurons, the VD4-induced synaptic responses in RPeD1 were sensitive to the cholinergic antagonists in a concentration range similar to that used to block cholinergic responses in single RPeD1 cells. Therefore, the modulation of postsynaptic nAChRs was sufficient to account for the trophic factor-induced excitatory synaptogenesis. This study thus provides the first direct evidence that trophic factors act postsynaptically to promote excitatory synapse formation.
Keywords: trophic factors, cell culture, synapse formation, synaptic plasticity, acetylcholine receptors, Lymnaea
Recent studies on both vertebrate and invertebrate models have extended the conventional roles of trophic factors during neurodevelopment to neurite outgrowth and synapse formation (Hamakawa et al., 1999; Woodin et al., 1999) and plasticity (McAllister et al., 1999; Schinder and Poo, 2000). Regarding synapse formation and synaptic plasticity, however, neither the precise sites of neurotrophic actions (presynaptic verses postsynaptic) nor the underlying mechanisms have yet been fully elucidated.
In the present study, we took advantage of a simple in vitromodel system consisting of soma–soma synapses. This experimental preparation enabled us to readily examine the mechanisms underlying synapse formation between defined sets of presynaptic and postsynaptic neurons from the mollusk Lymnaea. The soma–soma synapse model has been used previously by our laboratory to demonstrate the requirement of exogenous trophic factors for excitatory but not inhibitory synapse formation between identified neurons (Feng et al., 1997; Hamakawa et al., 1999). Moreover, we have demonstrated that in the absence of appropriate trophic factors, neurons, which normally form excitatory synapses in vivo, establish inappropriate inhibitory synapses in vitro. These inappropriate inhibitory synapses were subsequently corrected by the addition of trophic molecules to the culture medium (Woodin et al., 1999). Although these findings indicate that trophic factors play a significant role in synapse formation and plasticity of excitatory synapses, the underlying mechanisms remain unexplored.
To define the cellular mechanisms underlying trophic factor-induced excitatory synapse formation, we examined the effects of trophic factors on synapses that developed in a soma–soma configuration between neurons visceral dorsal 4 (VD4) and right pedal dorsal 1 (RPeD1). Feng et al. (1997) demonstrated previously that when juxtaposed in cell culture, inhibitory synapses develop in the absence of trophic factors. In the present study, however, increasing concentrations of trophic factors promoted the formation of an additional excitatory synaptic component, which preceded the inhibitory response (i.e., a biphasic synapse), whereas in vivo an inhibitory response was generally observed. The trophic factor-induced formation of this excitatory synaptic component presented us with an opportunity to define the mechanisms underlying both excitatory and inhibitory synapse formation. Using the soma–soma synapse model, we provide direct evidence that trophic factor-induced specificity and plasticity of excitatory synapse formation involves postsynaptic (RPeD1) modulation of nicotinic acetylcholine receptors (nAChRs). Furthermore, this postsynaptic neurotransmitter receptor modulation is both necessary and sufficient to account for the trophic factor-induced excitatory synapse formation.
MATERIALS AND METHODS
Animals. Lymnaea stagnalis were maintained at room temperature in a well-aerated aquarium containing filtered pond water. For experiments involving cell isolation, ∼1- to 2-month-old snails (shell length, 18–20 mm) were used, whereas conditioned medium (CM) was prepared from 2- to 3-month-old animals (shell length, 25–30 mm).
Cell culture. Neurons were isolated from the central ring ganglia and maintained in cell culture as described previously (Syed et al., 1990, 1999; Ridgway et al., 1991). In brief, snails were anesthetized with 10% Listerine solution (ethanol, 21.9%; and methanol, 0.042%) in normal Lymnaea saline (in mm: 51.3 NaCl, 1.7 KCl, 4.0 CaCl2, and 1.5 MgCl2) buffered to pH 7.9 with HEPES. The central ring ganglia were then washed several times (three washes, 15 min each) with normal saline containing antibiotic (gentamycin, 50 μg/ml). The central ring ganglia were then treated with enzyme (trypsin) followed by enzyme inhibitor (trypsin inhibitor) and pinned down in the bottom of a dissection dish. All procedures were performed under sterile culture conditions.
The identified neurons (somata and initial axon segment) were isolated by applying gentle suction through a fire-polished Sigmacote (Sigma, St. Louis, MO)-treated pipette. The isolated neurons were then plated on poly-l-lysine-pretreated glass coverslips (Ridgway et al., 1991) in the presence of either defined medium (DM; L-15; Life Technologies, Gaithersburg, MD; special order) or CM. Soma–soma synapses were prepared by juxtaposing the isolated somata of the identified neurons (Feng et al., 1997).
In some experiments, isolated cells were initially plated on hemolymph-pretreated glass coverslips (to prevent neuronal adhesion) in the presence of CM and receptor tyrosine kinase (RTK) antagonists. After 12–18 hr of incubation in the antagonist, the cells were transferred to and soma–soma paired on poly-l-lysine-pretreated glass coverslips in the presence of CM.
CM was prepared by incubating gentamycin (20 μg/ml)-treated ganglia in Sigmacote-treated glass Petri dishes containing DM. DM consisted of serum-free 50% L-15 medium with added inorganic salts (in mm: 40 NaCl, 1.7 KCl, 4.1 CaCl2, 1.5 MgCl2, and 10 HEPES, pH 7.9) and 20 μm gentamycin. The ganglia were incubated in a humidifier for 3–4 d (Wong et al., 1981; Syed et al., 1999), and the resulting CM was frozen (−20°C) until used. Heat-inactivated CM was prepared by boiling CM for 20 min.
Electrophysiology. Neuronal activity was monitored using conventional intracellular recording techniques, as described previously (Syed and Winlow, 1991b). Glass microelectrodes (1.5 μm internal diameter; World Precision Instruments, Sarasota, FL) were filled with a saturated solution of K2SO4 (resistance, 20–40 MΩ). An inverted microscope (Axiovert 135; Zeiss, Thornwood, NY) was used to view the neurons, which were impaled by Narashige (Tokyo, Japan) micromanipulators (MM202 and MM 204). Amplified electrical signals (NeuroData Instrument Corp.) were displayed on a digital storage oscilloscope (PM 3394; Philips, Eindhoven, The Netherlands) and recorded on a chart recorder (TA 240S; Gould, Cleveland, OH).
Receptor tyrosine kinase experiments. To test whether CM-derived trophic factors act via RTKs, the nonspecific RTK blocker lavendustin A (LavA; 10 μm) and its inactive analog lavendustin B (LavB; 10 μm) were used. Several other RTK inhibitors, including K252a, KT5296, and genistein, have been used previously to block CM-induced effects on synaptogenesis inLymnaea (Hamakawa et al., 1999). Although the above compounds effectively block CM-induced effects on neurite outgrowth and synapse formation, lavendustin A proved to be the most effective in blocking RTK activity, with the least effects on cell viability (Hamakawa et al., 1999). Thus, the lavendustin A and B concentrations used in the present study were based on previous studies in which these compounds were found to block trophic factor-induced effects onLymnaea neurons (Hamakawa et al., 1999; Woodin et al., 1999;Munno et al., 2000) as well as other invertebrate [leech (Catarsi and Drapeau, 1993; Catarsi et al., 1995) and Aplysia (Goldberg and Wu, 1995)] and vertebrate neurons [rat (Frerking et al., 1998)].
Acetylcholine chloride was obtained from Research Biochemicals (Natick, MA; product A-112). Phe-Met-Arg-Phe-amide (FMRFamide) (product P-6910), hexamethonium chloride (product H2138), methyllycaconitine (product M-168), and mecamylamine (mec; product M-9020) were obtained from Sigma.
RESULTS
We have demonstrated previously that synapse formation between excitatory but not inhibitory synaptic partners from the central ring ganglia of Lymnaea requires neurotrophic factors derived from CM (Hamakawa et al., 1999; Woodin et al., 1999). In this study, we sought to determine whether CM-derived trophic factors could modulate the efficacy of synapse formation between inhibitory partners VD4 and RPeD1.
To test the above possibility, we first examined synapse formation in the absence of trophic factors. Specifically, VD4 and RPeD1 were isolated and juxtaposed on poly-l-lysine-coated dishes containing DM only. Simultaneous intracellular recordings were made after 12–18 hr of soma–soma pairing. A burst of action potentials in VD4 either inhibited the spiking activity in RPeD1 (Fig.1A) or produced a compound IPSP (n = 26; Fig. 1B). These data demonstrate that inhibitory synapses, similar to those reported previously both in vitro (Feng et al., 1997) andin vivo (Syed et al., 1990; Syed and Winlow, 1991b), develop in DM between VD4 and RPeD1.
Fig. 1.

CM promotes the formation of an excitatory synaptic component from VD4 to RPeD1 at a normally inhibitory synapse. After 18 hr of cell pairing, simultaneous intracellular recordings from VD4 and RPeD1 somata revealed an inhibitory synapse in DM (A, B), whereas in 100% CM, an excitatory synaptic component (biphasic synapse) was detected (C, D).A, Specifically, in DM, a burst of action potentials in VD4 inhibited spontaneous action potentials in RPeD1. Similarly, action potentials in VD4 produced a compound IPSP in RPeD1 [B; membrane potential (VR), −55 mV]. In contrast, when this synapse was reconstructed in 100% CM, a burst of action potentials in VD4 initially enhanced the rate of spontaneous activity (C, solid line,e) in RPeD1, followed by an inhibition of the firing of action potentials (C, dotted line,i). Likewise, a burst of action potentials in VD4 produced an initial EPSP followed by an IPSP in RPeD1 (D; VR, −55 mV).Arrows indicate the injection of depolarizing current.
To determine whether CM could alter the efficacy of the VD4-induced inhibitory postsynaptic response in RPeD1, cells were paired in the presence of varying concentrations of CM. Interestingly, we found that although concentrations ranging from 25% (n = 8) to 50% (n = 8) CM did not significantly affect the inhibitory postsynaptic response in RPeD1, a novel excitatory component developed when the cells were paired in the presence of higher concentrations of CM (Fig. 2). Specifically, when paired overnight in the presence of 75% (n = 9) or 100% (n = 30) CM, induced action potentials in VD4 generated biphasic postsynaptic potentials (BPSPs, i.e., excitation followed by inhibition) in RPeD1. For instance, an induced train of action potentials in VD4 first increased the spike frequency of a spontaneously active RPeD1, followed by an inhibitory postsynaptic response (Fig. 1C). When maintained below its threshold for spiking, single action potentials in VD4 generated 1:1 BPSPs in RPeD1 (Fig. 1D). In addition, the excitatory component of the VD4-induced biphasic response in RPeD1 was often large enough to trigger spikes in its synaptic partner cell. It is important to note that increasing CM concentrations (0–100%) did not affect the inhibitory component of the postsynaptic response in RPeD1. These data thus demonstrate that CM induces a novel excitatory component, which is not observed in the absence of trophic factors.
Fig. 2.

The CM-induced excitatory synaptic component is concentration-dependent. VD4 and RPeD1 were soma–soma-paired in either DM (n = 26) or various concentrations of CM. Simultaneous electrophysiological recordings were made after 18 hr of cell pairing. The black bars represent the percentage of synapses with an excitatory synaptic component that developed in different concentrations of CM [25% (n = 8), 50% (n = 8), 75% (n = 9), and 100% (n = 30)]. The concentration of CM is indicated as a percentage. DM is represented as 0% CM.
Because RPeD1 makes a reciprocal inhibitory connection with VD4 bothin vivo (Syed and Winlow, 1991a) and in vitro(Feng et al., 1997), we next asked whether CM could also alter the nature of synaptic transmission between RPeD1 and VD4. In contrast with the VD4→RPeD1 synapse (Fig. 1), the nature of the synaptic transmission between RPeD1→VD4 remained unchanged (Fig.3). Specifically, the inhibitory synapse between RPeD1→VD4 remained inhibitory even in the presence of 100% CM (n = 6), and there were no differences between the amplitude or duration of the compound IPSPs from RPeD1→VD4 in DM (mean IPSP duration, 6.7 ± 2.9 sec; amplitude, 8.2 ± 2.3 mV) or CM (mean IPSP duration, 6.06 ± 1.3 sec; amplitude, 8.75 ± 2.9 mV).
Fig. 3.

CM does not modulate the RPeD1 VD4 inhibitory synapse. To determine whether CM could also modulate the synaptic transmission from RPeD1 to VD4, paired neurons were incubated in either DM or CM overnight. After 18 hr of cell pairing, simultaneous intracellular recordings revealed that inhibitory synapses developed from RPeD1 to VD4 in both DM (A) and CM (B). It is important to note that the CM-induced excitatory component observed at the VD4→RPeD1 synapse was not detected at the RPeD1→VD4 synapse. Furthermore, there was no statistical difference between the inhibitory RPeD1→VD4 synapse in either DM or CM (DM: mean IPSP duration, 6.7 ± 2.9 sec; amplitude, 8.2 ± 2.3 mV; CM: mean IPSP duration, 6.06 ± 1.3 sec; amplitude, 8.75 ± 2.9 mV). Arrows indicate the injection of depolarizing current.
Taken together, the above data demonstrate that the CM-induced effects on VD4→RPeD1 synapses are exclusive to this synapse and are cell type-specific.
CM-induced excitatory synapse formation between VD4 and RPeD1 is mediated by postsynaptic receptor tyrosine kinases
Brain-conditioned medium contains a variety of secreted molecules, including those required for the formation of the excitatory synaptic component of the biphasic response observed between VD4 and RPeD1. To demonstrate the proteinacious nature of the molecules present in CM, VD4 and RPeD1 were soma–soma-paired in the presence of heat-inactivated CM. After 12–18 hr of cell pairing, intracellular recordings revealed that the paired cells had developed only inhibitory synapses (n = 5; Fig. 4), in a manner similar to that observed in DM (Fig. 1). These data demonstrate that the CM-derived molecule(s) responsible for the excitatory component of the BPSP is heat-labile and most likely proteinacious in nature.
Fig. 4.
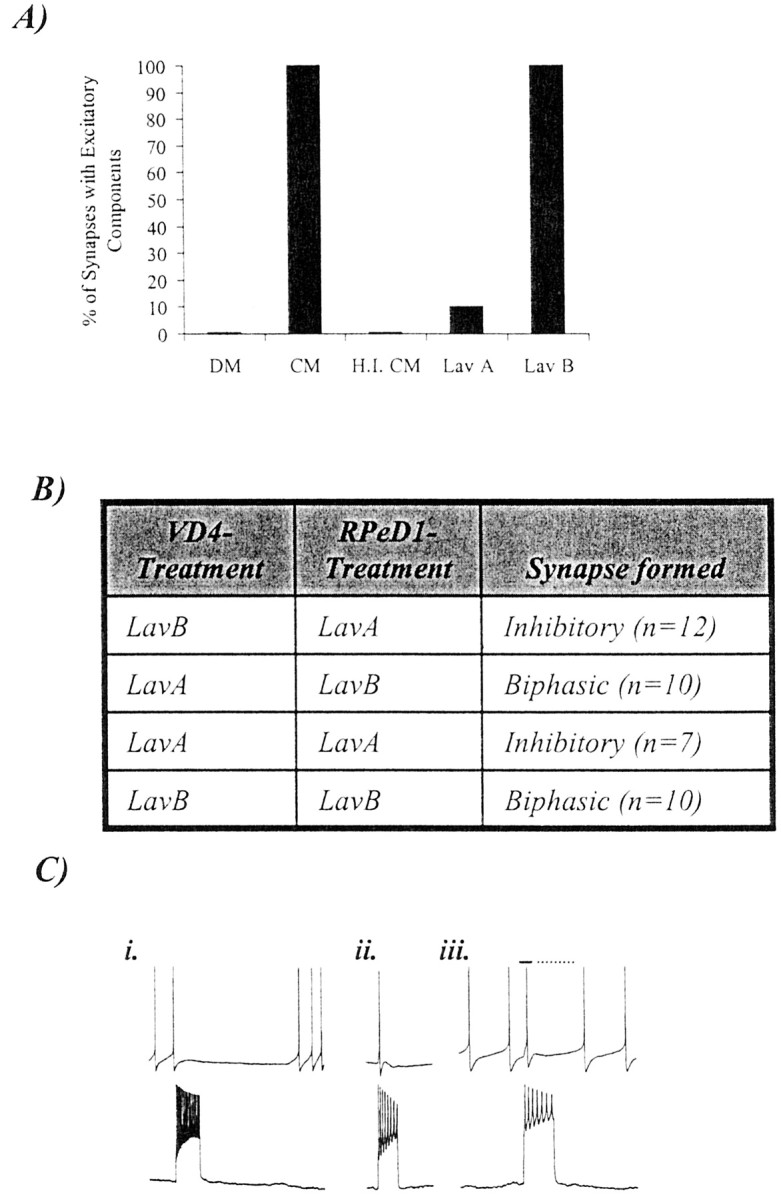
The CM-induced excitatory synaptic component between VD4 and RPeD1 requires postsynaptic activation of receptor tyrosine kinases. A, When paired in DM, the excitatory synaptic component of the biphasic synapse did not develop between VD4 and RPeD1 (n = 26). In 100% CM, however, a biphasic synapse developed reliably (n = 30). The excitatory synaptic component of this biphasic synapse failed to develop in heat-inactivated 100% CM (H.I. CM;n = 5). In addition, the CM-induced excitatory synaptic component was also blocked when pairs were incubated in CM containing a receptor tyrosine kinase inhibitor (Lav A;n = 12) but not by its inactive form (Lav B; n = 5). B, To determine whether the trophic factor-induced development of BPSPs required presynaptic or postsynaptic RTK activity, cells were selectively incubated in either LavA or LavB. Specifically, VD4 and RPeD1 were selectively incubated in either LavA or LavB and then soma–soma-paired in the following four configurations: (1) RTK activity blocked in postsynaptic neurons, (2) RTK activity blocked in presynaptic neurons, (3) RTK activity blocked in both neurons, and (4) RTK activity not blocked in either neuron. C, Selective block of RTK activity in the postsynaptic cell prevented the development of a biphasic synapse, and only an inhibitory synapse formed. A burst train of action potentials in VD4 under such conditions inhibited spontaneously firing of action potentials in RPeD1 (i). In contrast, when RTK activity was selectively blocked in the presynaptic neuron, there was no affect on the development of the biphasic synapse. Specifically, a burst of action potentials in VD4 induced an action potential in RPeD1 followed by an IPSP (ii; VR, −56 mV). Likewise, a burst of action potentials in VD4 initially enhanced the rate of spontaneous activity (iii,solid line) in RPeD1, followed by an inhibition of the firing of action potentials (iii, dotted line).
We have demonstrated previously that CM and trophic factor-induced excitatory synapse formation between Lymnaea neurons is mediated via RTKs (Hamakawa et al., 1999; Woodin et al., 1999). To demonstrate further that the CM-induced excitatory component of the BPSPs did indeed involve trophic factors and RTK activity, VD4 and RPeD1 were soma–soma-paired in CM containing the nonspecific RTK inhibitor LavA. After 12–18 hr of pairing, synapses were tested electrophysiologically. Specifically, when cultured in CM containing LavA (10 μm), the CM-induced excitatory component of the BPSPs failed to develop in soma–soma-paired cells (n = 12; Fig. 4A). LavB (10 μm), an inactive analog of this compound, however, did not block the formation of CM-induced excitatory synaptic component (n = 5; Fig. 4A). Neither LavA nor LavB affected the inhibitory component of the BPSPs.
To decipher the precise site of trophic factor action and RTK activity (i.e., presynaptic versus postsynaptic) for the development of the excitatory component of the BPSP, RTK activity was selectively blocked in either the presynaptic or postsynaptic cell. Specifically, VD4 and RPeD1 neurons were isolated and selectively incubated in the hemolymph-pretreated dishes (to prevent neural adhesion) containing CM and either LavA (10 μm) or LavB (10 μm). The cells were maintained under different experimental conditions, and the RTK activity was blocked in (1) postsynaptic, (2) presynaptic, or (3) both cells, or (4) the cells were maintained in the presence of LavB (no RTK block; Fig. 4B). The neurons were incubated in the RTK antagonist LavA or its inactive analog LavB for 12–18 hr, after which the cells were transferred and juxtaposed in the soma–soma configuration on poly-l-lysine-coated dishes containing CM. Approximately 4–6 hr after soma–soma pairing, the neurons were tested electrophysiologically for synapses. When RTK activity was selectively blocked in the postsynaptic RPeD1 neuron, a biphasic synapse failed to develop (n = 12; Fig.4C, i). In contrast, when RTK activity was selectively blocked in the presynaptic VD4 neuron, the biphasic synapse developed normally in 90% of the pairs tested (n = 10; (Fig. 4C, ii, iii). The data obtained under control conditions demonstrated that when RTK activity was blocked in both VD4 and RPeD1 before pairing, no biphasic synapses were detected (n = 7), whereas neurons incubated in the inactive analog of the RTK inhibitor LavB before pairing formed a biphasic synapse in 100% of the cases (n = 10).
Taken together, these data demonstrate that the inhibitory and excitatory components of the BPSPs are differentially regulated, such that only the excitatory component is contingent on trophic factor-induced activation of RTKs. Furthermore, RTK activity in the postsynaptic but not presynaptic neuron is required for trophic factor-induced excitatory synapse formation.
Trophic factors are required for the maintenance of the excitatory synapse
To determine whether CM-derived trophic factors were necessary for the maintenance of the excitatory component of the BPSPs, cells were first paired overnight (12–18 hr) in CM. Before intracellular recordings, the medium providing the trophic factor(s) was replaced with DM (CM→DM). Neuron pairs were tested at three different time intervals after CM→DM replacement as follows: in condition 1, cells were tested between 30 min and 2 hr; in condition 2, neurons were tested between 2 and 4 hr; and in condition 3, neurons were tested between 4 and 6 hr. In condition 1 (30 min to 2 hr), 54% of the VD4–RPeD1 pairs retained a biphasic synapse (n = 11; Fig. 5). In condition 2 (2–4 hr), only 33% of the pairs retained a biphasic synapse (n = 12), whereas none of the pairs displayed a biphasic synapse in condition 3 (4–6 hr; n = 11; Fig. 5). Although the excitatory component was completely lost 6 hr after the withdrawal of trophic factor, the inhibitory synapses persisted under all experimental conditions.
Fig. 5.

Trophic factors are required for both the formation and maintenance of the CM-induced excitatory synaptic component of the biphasic synapse between VD4 and RPeD1.A, CM was effective in inducing the excitatory component of the BPSP only if it was added to the culture dish within 4–8 hr of initial pairing in DM [condition A; DM (4–8 hr)→CM;n = 6]. CM addition after 12–18 hr of pairing in DM failed to induce an excitatory component [condition B; DM (12–18 hr)→CM; n = 7]. B, Removal of the trophic support after the biphasic synapse has already formed results in the gradual loss of the excitatory synaptic component. In condition 1, 30 min to 2 hr after removal of trophic support [CM→DM (30 min to 2 hr)], 54% of the pairs retained their biphasic synapse (n = 11). In condition 2, after the removal of trophic support [CM→DM (2–4 hr)], only 33% of the pairs still displayed a biphasic synapse (n = 12). In condition 3, when the trophic support had been removed for ≥4–6 hr [CM→DM (4–6 hr)], none of the VD4→RPeD1 synapses exhibited a biphasic response (n = 11). The loss of the excitatory component of the biphasic synapse was independent of new protein synthesis. The removal of trophic support coupled with the addition of the protein synthesis inhibitor anisomycin (12.5 μg/ml; CM→DM + anisomycin) did not affect the loss of the excitatory component, such that none of the pairs retained their biphasic synapse (n = 12). White bars represent inhibitory synapses; black bars represent biphasic synapses.
To test whether longer incubation in CM would stabilize the excitatory component of the BPSP such that it would no longer require trophic factors for its maintenance, cells were initially paired in CM for 36–40 hr. Electrophysiological recordings at 36–40 hr revealed a biphasic synapse between the cells, as was observed at 18–24 hr. After recording at 36–40 hr, the CM was subsequently replaced with DM. After 4–6 hr of medium exchange, the synapses were retested. We discovered that the excitatory component of the BPSP completely disappeared under these conditions (data not shown). These data demonstrate that the trophic factors are required at all times for the maintenance of the excitatory but not the inhibitory component of the BPSP.
To determine whether the loss of the excitatory component of the BPSP occurred through a protein synthesis-dependent step, VD4–RPeD1 pairs were incubated in CM overnight (12–18 hr), which was subsequently replaced with DM containing the membrane-permeable protein synthesis inhibitor anisomycin (12.5 μg/ml; Hamakawa et al., 1999). We reasoned that if the loss of the excitatory component of the synapse required a protein synthesis-dependent step (such as the synthesis and subsequent insertion or new receptors or receptor subunits), then we would expect VD4–RPeD1 pairs to retain biphasic synapses for a much longer period after the removal of trophic factors. Contrary to this prediction, however, at 6 hr after CM→DM plus anisomycin replacement, none of the pairs exhibited a biphasic synapse (n = 12). These data suggest that the switch from biphasic to inhibitory synapses did not require a protein synthesis-dependent step.
The excitatory component of the BPSPs requires trophic factor(s) at the time of soma–soma pairing
To determine the time dependency of the effectiveness of CM in inducing the excitatory component of the biphasic synapse, the juxtaposed cells were exposed to trophic factors at two different time intervals after their initial pairing in DM. Specifically, in condition A, the neurons were initially cultured in DM for 4–8 hr. The soma–soma-paired cells were then incubated in the CM for an additional 6–12 hr (total time in culture, 12–18 hr), and their synapses were tested. We found that the exposure to CM-derived trophic factors in condition A induced the excitatory component of the biphasic synapse in 83% of the cell pairs tested (n = 6; Fig. 5). In condition B, the cell pairs were initially cultured in DM for 12–18 hr (100% of the neuron pairs establish inhibitory synapses under these conditions; Fig. 1). The DM was then replaced with CM, and the pairs were incubated for an additional 6–12 hr before electrophysiological recording (total time in culture, 18–24 hr). The pairs in condition B did not reliably develop a biphasic response (14%; n = 7; Fig. 5), even after as long as 18 hr of incubation in CM. These data demonstrate that there is a critical period during which the cells must be exposed to trophic factors to develop the excitatory component of the BPSPs. After this critical period, the cells undergo a refractory period during which they lose their ability to respond to trophic factors.
CM-induced excitatory synapse formation does not involve a trophic factor-mediated change in the neurotransmitter phenotype
On the basis of previously published studies in vertebrates (Landis, 1990; Zhou and Bradford, 1997), we reasoned that the CM-induced excitatory component may be attributable to a trophic factor-induced alteration in the transmitter phenotype of the presynaptic neuron VD4. To explore this possibility, VD4 and RPeD1 were soma–soma paired in either DM or CM. Because excitatory synaptic transmission between VD4 and RPeD1 was shown previously to be cholinergic (Grigoriev et al., 1999), various AChR antagonists were used to block the VD4→RPeD1 synaptic response. The AChR antagonists were selected on the basis of previous studies that characterized the sensitivity of different ACh-induced anionic (hyperpolarizing) and cationic (depolarizing) responses in the related molluskAplysia (Kehoe and McIntosh, 1998).
The synaptic transmission between cells paired in either DM or CM was first tested under normal conditions. In DM, an inhibitory synapse was initially detected (Fig.6A). Using a fast perfusion system (Feng et al., 2002), methyllycaconitine (MLA) was applied to the VD4–RPeD1 pair (concentration range, 100 nm to 5 μm). When 100 nm or 1 μm MLA was applied to the VD4–RPeD1 pairs, a partial block of the inhibitory synapse was observed (data not shown). Perfusion of 5 μm MLA, however, completely blocked the VD4-induced IPSPs in RPeD1 (n = 5; Fig.6B). Neither mec (n = 6; 1 μm; Fig. 6C) nor hexamethonium chloride (HMC; n = 6; 500 μm;Fig. 6D) was effective in blocking the inhibitory synapse in DM, although a slight reduction in the amplitude of VD4-induced IPSPs in RPeD1 was noted in the presence of these antagonists. The inhibitory synapse in these experiments recovered fully after washout with DM (Fig. 6F).
Fig. 6.

Both excitatory and inhibitory components of the BPSP are blocked by nAChR antagonists. Bath perfusion with the nAChR antagonists MLA (5 μm), mec (1 μm), and HMC (500 μm) blocks the synaptic transmission from VD4 to RPeD1 in both DM and CM. A, In DM, an inhibitory synapse develops from VD4 to RPeD1. B, Perfusion of the nAChR inhibitor MLA completely blocked the synaptic transmission between VD4 and RPeD1. Application of the nAChR inhibitors mec (C) and HMC (D) only partially reduced the VD4-induced IPSP in RPeD1. Furthermore, co-perfusion of MLA and mec completely blocked the synaptic response in RPeD1 (E). Normal synaptic response recovered on drug washout with normal DM (F). In CM, a biphasic synapse was recorded in RPeD1 in response to VD4 stimulation as expected (G). Perfusion of MLA completely eliminated the inhibitory response (H). In contrast, perfusion of either mec (I) or HMC (J) selectively blocked the excitatory component of the biphasic synaptic response from VD4 to RPeD1. Co-perfusion of MLA and mec completely blocked both components of the biphasic synapse (K). Washout with normal DM removed the nAChR block (L). As a control, the RPeD1 to VD4 synapse was monitored in the presence of each of the nAChR antagonists (M). None of the antagonists affected the RPeD1 to VD4 synapse (the example shown is from neurons cultured in DM in the presence of HMC).Arrows indicate the injection of depolarizing current. For all traces, the postsynaptic neuron [RPeD1 (A–L) or VD4 (M)] was held at −56 mV.
For pairs cultured in CM, a BPSP was initially detected (Fig.6G). MLA (5 μm) selectively blocked the inhibitory phase of the biphasic synapse (n = 6; Fig. 6H) but did not affect the excitatory component. The excitatory phase was, however, selectively blocked by both mec (1 μm;n = 6; Fig.6I) and HMC (500 μm;n = 15; Fig. 6J). Simultaneous perfusion of MLA and either HMC (data not shown) or mec (Fig.6K) blocked both components of the synapse (n = 6). After washout with DM, the control response returned (Fig. 6L).
To confirm the specificity of the AChR antagonists forLymnaea neurons, the reciprocal synapse, from RPeD1→VD4 (which is dopaminergic), and the response to other exogenously applied neurotransmitters were tested in the presence of HMC or MLA. Neither HMC (500 μm;n = 7; Fig.6J) nor MLA (5 μm;n = 6; data not shown) affected the RPeD1→VD4 synapse. Furthermore, the responsiveness of the RPeD1 neuron to serotonin (1 μm) was not affected in the presence of AChR antagonists. Specifically, application of serotonin induced an inhibitory and hyperpolarizing response when applied exogenously to either single or paired RPeD1 neurons (see Fig.8K) in the presence of HMC (500 μm).
Fig. 8.

ACh produces biphasic nonsynaptic response in RPeD1 isolated in CM. RPeD1 was cultured alone for 18 hr in either DM or CM before testing the electrophysiological response to neurotransmitter. For neurons cultured in DM, exogenous application of 1 μm ACh (solid line) induced an inhibitory and hyperpolarizing response in a spontaneously firing cell (A, i) or if the cell were held at constant membrane potential (−56 mV; A, ii). MLA (1 μm) blocked the inhibitory response of ACh in RPeD1 neurons cultured in DM (B, i, ii). Cholinergic responses in RPeD1 were partially reduced but not completely blocked by mec (C, i, ii). Co-perfusion of both MLA and mec also completely blocked the ACh-induced nonsynaptic response in RPeD1 (D, i, ii). The hyperpolarizing response to ACh was restored immediately (1–5 min) after drug washout with normal DM (E, i, ii). For neurons cultured in CM, exogenous application of 1 μm ACh (solid line) onto RPeD1 produced a biphasic response in CM (F). Perfusion with the nAChR antagonist blocked these responses (B, C) in the same manner as described for the synapse in Figure 6. That is, MLA (1 μm) blocked the hyperpolarizing component of the biphasic response in CM (G), whereas mec (1 μm) abolished the depolarizing phase of the biphasic response (H). Co-perfusion of these two nAChR antagonists completely blocked the ACh-induced nonsynaptic response in RPeD1 (I). The biphasic response of RPeD1 to ACh recovered immediately after washout with normal DM (J). The action of the nAChR blockers was specific to the ACh response. HMC failed to block the hyperpolarizing response of exogenously applied 1 μmserotonin (5-HT; solid line), seen in this trace as inhibiting spontaneous action potentials in RPeD1 (K). The effect of CM was specific to the postsynaptic RPeD1 neuron. The application of 1 μmACh onto VD4 had the same inhibitory effect regardless of whether VD4 was cultured in DM (L) or CM (M). Solid lines indicate the duration of application ACh, except where noted for 5-HT. RPeD1 neurons were held at a constant membrane potential of −56 mV (A, ii–E, ii, F–J) or allowed to fire spontaneously with no current injection (A, i–E, i, K–M).
Because previous reports have suggested that VD4 may also contain FMRFamide-like peptides (Saunders et al., 1992; Santama et al., 1995), the above data do not rule out the possibility that such peptides may also be responsible for either component of the biphasic response. To demonstrate that the CM-induced BPSPs did not involve FMRFamide-like peptides and to further ensure the specificity of the AChR antagonists for cholinergic receptors, FMRFamide was exogenously applied to single or paired RPeD1 maintained in either DM or CM. After 18 hr of isolation in either DM or CM, FMRFamide (1 μm) was applied exogenously to RPeD1. Under both experimental conditions, FMRFamide exerted inhibitory effects on RPeD1 (Fig.7A,D). HMC (500 μm) and MLA (1 μm) were perfused separately into the culture dish, and the FMRFamidergic responses were retested. In both CM (n = 5) and DM (n = 5), FMRFamide (1 μm)-induced inhibitory responses remained unperturbed by the nAChR antagonists (Fig.7B,C,E,F).
Fig. 7.

FMRFamide application to isolated RPeD1 produces an inhibitory response in both DM and CM. FMRFamide (10−6m) exogenously applied to RPeD1 (solid line), which had been isolated in cell culture for 18 hr, produced an inhibitory response in both DM (A) and CM (D). Bath perfusion with either HMC (B, E) or MLA (C, F) did not perturb the FMRFamide-induced (10−6m) effects in RPeD1 in either DM or CM. Solid lines indicate the duration of FMRFamide application.
These data demonstrate that under both CM and DM conditions, FMRFamide-induced inhibitory responses remain unchanged. Furthermore, these data suggest that ACh mediates the novel excitatory component in CM as well as the inhibitory postsynaptic response observed in both DM and CM. These data do not, however, rule out the possibility that VD4 may also release a second, unidentified transmitter (e.g., glutamate or FMRFamide) in CM.
Trophic factor-induced excitatory synaptic transmission involves a switch in postsynaptic receptor responsiveness
To determine whether trophic factor-induced expression of the excitatory component of the BPSPs involved postsynaptic receptor mechanisms, the responsiveness of RPeD1 to exogenously applied ACh was examined in either the presence or absence of trophic factors. RPeD1 was cultured alone in either DM or CM for 12–18 hr before its responsiveness to neurotransmitter was examined electrophysiologically. Exogenous application of ACh (1 μm) to RPeD1 neurons maintained in DM produced an inhibitory response (n = 19; Fig. 8A). As in the synaptic response, MLA (1 μm) reversibly blocked the ACh-induced inhibitory response in RPeD1 (n= 7; Fig. 8B). The ACh-induced inhibitory response was somewhat sensitive to both HMC (500 μm;data not shown) and mec (1 μm; Fig.8C). Application of these antagonists partially blocked the ACh-induced inhibitory response.
When RPeD1 was cultured alone in CM, ACh (1 μm) application via pressure pulses (0.5 sec) induced a biphasic response (n = 20; Fig. 8F) that was sensitive to the AChR antagonists (Fig. 8G–I) in a manner similar to that observed at the VD4→RPeD1 synapse. That is, 500 μm HMC (n = 11; data not shown) and 1 μm mec (n = 7; Fig.8H) specifically blocked the excitatory component of the response (Fig. 8F), whereas 1 μm MLA (n = 7) selectively blocked the inhibitory component (Fig. 8G). Co-perfusion of MLA and either mec or HMC (n = 7) blocked both components of the biphasic response (Fig. 8I). The effects of all the antagonists were reversible on washout with DM (Fig.8J). The differential responsiveness of RPeD1 (in CM and DM) to ACh could not have resulted from changes in the resting membrane potential, because cells under both experimental conditions were maintained at the same level (−56 mV; DM, Fig. 8A, ii–E, ii; CM, 8F–J).
These data indicate that isolated RPeD1 is responsive to exogenously applied ACh in both DM and CM. The difference is that in DM, ACh produces a hyperpolarizing response, whereas in CM a biphasic response is elicited. The different responses induced by exogenous application of ACh to isolated RPeD1 neurons indicate that postsynaptic cholinergic receptors are the likely targets of trophic factor-induced modulation of excitatory synaptic responses.
The above data demonstrated that, although isolated RPeD1 was responsive to ACh in both DM and CM, the excitatory response was observed only under the latter experimental conditions. The AChR antagonists used in this study have been shown previously to block specific nicotinic AChR currents. Because the excitatory and inhibitory components of the cholinergic responses in RPeD1 were sensitive to different AChR antagonists, we suggest that they involve different ACh receptors. The above data are, therefore, consistent with our hypothesis that the neurotrophic factor-induced excitatory response in RPeD1 involves postsynaptic changes in the ACh receptors.
To rule out the possibility that CM-induced excitatory synapse formation may involve modulation of presynaptic autoreceptors in VD4, we next sought to determine (1) whether VD4 itself possesses ACh receptors and (2) whether these receptors were differentially regulated by neurotrophic factors.
Trophic factors do not modulate the presynaptic response to ACh
To determine the responsiveness of VD4 to ACh and to test whether this cell exhibits differential responses to exogenously applied ACh in both DM and CM, VD4 was isolated and maintained under these distinct experimental conditions. After 12–18 hr, ACh was applied exogenously, and the nonsynaptic responses were determined electrophysiologically. Regardless of the experimental conditions (i.e., DM vs CM;n = 6 for both), ACh induced an identical inhibitory and hyperpolarizing response in VD4 (Fig.8L,M).
The above data are important not only because they rule out the possibility of trophic factor-induced modification of autoreceptors as a mechanism for excitatory synapse formation but also because they indicate that the actions of trophic factors are specific to the postsynaptic cell.
DISCUSSION
This study is the first to demonstrate that brain-derived trophic factors can induce the formation of a novel, excitatory synaptic component at a synapse that is normally inhibitory. In addition, we showed that the mechanism underlying this CM-induced excitatory synapse formation is a trophic factor-mediated modulation of the postsynaptic nAChRs. This trophic factor-induced effect is specific to the postsynaptic cell, because blockade of CM-induced RTK activity specifically in the postsynaptic but not the presynaptic cell prevents development of the excitatory synaptic component. Furthermore, the trophic factor-induced modulation of nAChRs appears to be specific to these receptors, because CM does not modulate the response of postsynaptic FMRFamide receptors. Finally, the CM-derived trophic factors do not modify the response of presynaptic nAChRs.
Initially, we hypothesized that the CM-derived trophic factors may induce a switch in the presynaptic transmitter phenotype; this hypothesis was partially based on reports that neurotrophic factors are capable of modulating the neurotransmitter phenotype of neurons during development (Landis, 1990). For example, sweat glands have been shown to induce a switch in the transmitter phenotype of their in vivo partner sympathetic neurons from noradrenergic to cholinergic (Landis and Keefe, 1983).
The presynaptic neuron VD4 is reported to contain and release an FMRFamide-like peptide (Skingsley et al., 1993; Santama et al., 1995;Lovell, 2000). Exogenous application of FMRFamide, however, does not mimic all components of the VD4→RPeD1 synapse in CM (Nesic et al., 1996), leading to speculation that VD4 may contain a second neurotransmitter. Because preliminary evidence from our laboratory has implicated ACh as a putative neurotransmitter used by VD4 (Grigoriev et al., 1999), we rationalized that trophic factor-induced excitatory synapse formation may have resulted from a switch in the neurotransmitter phenotype from an FMRFamide-like peptide to ACh (or vice versa). The results from the present study have clearly demonstrated that the synaptic transmission from VD4 to RPeD1 is cholinergic in both DM and CM. Therefore, a trophic factor-induced switch in the VD4 transmitter phenotype is unlikely to account for the appearance of the CM-induced excitatory component.
The above data do not, however, preclude the possibility that VD4 may also contain and release FMRFamide-like peptides, which can be co-released with classic transmitters (Whim et al., 1993) or other transmitters. However, for the purposes of this study, it is important to recognize that trophic factors failed to alter the FMRFamide-induced nonsynaptic responses.
The ability of trophic factors to modulate the responsiveness of an isolated RPeD1 neuron to exogenously applied ACh provides convincing evidence that trophic factors act on postsynaptic neurotransmitter receptors to promote excitatory synapse formation. In the presence of DM, exogenous application of ACh produced a hyperpolarizing response, whereas in CM, a biphasic response with initial depolarization resulted. Because these cells were in isolation, we can conclude that the differing responses to the exogenous application of ACh in both DM and CM are independent of either cell–cell contact or signaling from the presynaptic cell. Rather, the ACh-induced depolarizing component of the biphasic response in RPeD1 maintained in CM develops only after its exposure to trophic factors. Moreover, the postsynaptic nAChRs that mediate the excitatory response in CM are influenced directly by trophic factors. Strengthening this notion are our data that demonstrate that selective blockade of RTK activity in the postsynaptic cell prevents the development of the excitatory component of the biphasic synapse.
Because the exogenous application of a single neurotransmitter, ACh, mimicked the synaptic response, our data also suggest that this transmitter alone is responsible for the synaptic transmission at the VD4→RPeD1 synapse. Our results from single cells and synaptic pairs support this conclusion and demonstrate that the inhibitory component of the biphasic response is indeed blocked by one AChR antagonist (MLA), whereas the excitatory component is completely blocked by two different AChR antagonists (HMC and mec). The concentrations and the kinetics of the antagonists used here are consistent with those in previous studies in other mollusks (Kehoe and McIntosh, 1998). For example, MLA selectively blocked cholinergic anionic currents in cultured Aplysia neurons at concentrations of <1 μm, whereas mec and HMC selectively blocked cationic currents at concentrations of ∼1 and 100–500 μm, respectively (Kehoe and McIntosh, 1998). Thus, the sensitivity of molluscan neurons to various pharmacological agents is conserved between the mollusk species Lymnaea andAplysia. Although the AChR antagonists have characteristics similar to those from other invertebrate species, the precise pharmacological profiles of Lymnaea nAChRs remain to be fully examined. For example, the differential sensitivity of nonsynaptic (single-cell) and synaptic (paired) cholinergic responses can likely be attributed to either the kinetics of the synaptic versus nonsynaptic AChRs or more restricted access of the antagonist to the receptors in the synaptic cleft.
It is important to point out that these experiments do not preclude the possibility that trophic factors may also modulate the presynaptic cell in other ways (e.g., potentiation of neurotransmitter release), in addition to their demonstrated effects on postsynaptic neurons. However, the data presented here clearly indicate that any presynaptic modification that may have resulted from CM is not required for the trophic factor-induced excitatory synapse formation.
The present results are supported by previous studies, which showed that the neurotrophins can indeed modify the postsynaptic cell during synapse development. For example, application of BDNF triggers an increase in the expression of AMPA-type glutamate receptor proteins (Narisawa-Saito et al., 1998). Similarly, interruptions in TrkB-mediated signaling result in the loss of postsynaptic receptor clusters at the neuromuscular junction, indicating the role of neurotrophin signaling on the maintenance of postsynaptic AChR clusters (Gonzalez et al., 1999).
The data presented in this study showed that the mechanisms via which trophic factors induced the excitatory component of the biphasic response involved RTKs. However, the loss of the excitatory component of the biphasic response, after removal of trophic factors, did not require de novo protein synthesis. These results thus rule out the possibility of new synapse formation that might be contingent on new protein synthesis. Our data thus favor the idea that trophic factors may either act to modulate the conductance of AChR ion channels by short-term changes such as phosphorylation or by altering the subunit composition of nAChRs. These possibilities, however, remain to be investigated.
It is interesting to point out that whatever the nature of these trophic factor-induced modulatory effects might be, these changes can only be brought about within a particular time window during synapse formation. When neurons are isolated from the intact brain and paired in culture, they form a mature synapse within 12–18 hr after pairing (D. W. Munno and N. I. Syed, unpublished observations). We have shown previously that if an inappropriate inhibitory synapse forms within this time window, it can be corrected to the appropriate excitatory synapse by adding trophic factors to the culture medium (Woodin et al., 1999). For the VD4–RPeD1 pairs, however, the trophic factors were no longer effective in inducing an excitatory component after an appropriate inhibitory synapse had formed. Therefore, these data suggest that a critical period exists before synapse maturation, during which the cells must be exposed to trophic factors to develop the excitatory component of the biphasic synapse.
One of the main advantages of using identified neurons fromLymnaea is that the functional significance of many of the cells is well defined in vivo. This is especially true for VD4 and RPeD1, which form part of a three-cell central pattern generator (CPG) that controls respiration (Syed et al., 1990). This respiratory CPG is based primarily on reciprocal inhibitory synapses that normally exist between VD4 and RPeD1. This synapse has also been reported at times to be biphasic (Benjamin, 1984; Magoski and Bulloch, 2000). However, neither the functional significance of this synaptic plasticity nor the underlying mechanisms have yet been defined. The present study has, thus, attempted to resolve a long-lasting controversy regarding the variable nature of the synaptic connections between RPeD1 and VD4 observed by different investigators. For instance, studies have reported a purely excitatory (Magoski and Bulloch, 2000), purely inhibitory (Syed and Winlow, 1991b), or biphasic synapse (Benjamin, 1984; Magoski and Bulloch, 2000) between VD4 and RPeD1 in vivo. Although these variations in the nature of synaptic connections were attributed to seasonal plasticity (Copping et al., 2000), the precise mechanisms involved in this phenomenon remain unknown. The results of the present study implicate trophic factor-induced modulation of postsynaptic receptors as a mechanism by which a neuron can switch from a purely inhibitory to a biphasic synapse. On the basis of our data, we propose that a biphasic synapse between VD4 and RPeD1 may arise either during development or as a result of seasonal adaptations (Lymnaea in its natural habitat, i.e., pond water, becomes dormant during winter months and active during spring and summer months), which may in turn cause either an upregulation or downregulation of trophic factors, thus switching the sign of synaptic transmission from pure inhibitory to biphasic. The results from the present study provide support for a mechanism that could account for changes in neuronal circuitry to accommodate long-lasting changes in the behavioral program.
In conclusion, this study provides the first direct evidence that trophic factors are required postsynaptically for excitatory synapse formation. The mechanism underlying trophic factor-induced excitatory synapse formation involves a chronic modification of postsynaptic nAChRs. Moreover, because VD4 and RPeD1 are identified neurons of the respiratory CPG, we are now in a position to examine how trophic factors may induce plasticity of synaptic connections within a rhythm-generating network.
Footnotes
This work was supported by the Canadian Institutes of Health Research (CIHR) and by the Canadian Neurotrauma Research Program. N.I.S. is an Alberta Heritage Foundation for Medical Research (AHFMR) senior scholar. M.A.W. was supported by studentship awards from the AHFMR, CIHR, and Neuroscience Canada Foundation. D.W.M. was supported by a studentship award from the AHFMR. Excellent technical support by Wali Zaidi is also acknowledged.
REFERENCES
- 1.Benjamin PR. Interneuronal network acting as snail neurosecretory cells (yellow cells and yellow-green cells of Lymnaea). J Exp Biol. 1984;113:165–185. [Google Scholar]
- 2.Catarsi S, Drapeau P. Tyrosine kinase-dependent selection of transmitter responses induced by neuronal contact. Nature. 1993;363:353–355. doi: 10.1038/363353a0. [DOI] [PubMed] [Google Scholar]
- 3.Catarsi S, Ching S, Merz DC, Drapeau P. Tyrosine phosphorylation during synapse formation between identified leech neurons. J Physiol (Lond) 1995;485:775–786. doi: 10.1113/jphysiol.1995.sp020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Copping J, Syed NI, Winlow W. Seasonal plasticity of synaptic connections between identified neurons in Lymnaea. Acta Biol Hung. 2000;51:205–210. [PubMed] [Google Scholar]
- 5.Feng ZP, Klumperman J, Lukowiak K, Syed NI. In vitro synaptogenesis between the somata of identified Lymnaea neurons requires protein synthesis but not extrinsic growth factors or substrate adhesion molecules. J Neurosci. 1997;17:7839–7849. doi: 10.1523/JNEUROSCI.17-20-07839.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng ZP, Grigoriev N, Munno DW, Lukowiak K, MacVicar B, Goldberg JI, Syed NI (2002) Development of Ca2+ hotspots between Lymnaeaneurons during synaptogenesis. J Physiol (Lond), in press. [DOI] [PMC free article] [PubMed]
- 7.Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. J Neurophysiol. 1998;80:3383–3386. doi: 10.1152/jn.1998.80.6.3383. [DOI] [PubMed] [Google Scholar]
- 8.Goldberg DJ, Wu DY. Inhibition of formation of filopodia after axotomy by inhibitors of protein tyrosine kinases. J Neurobiol. 1995;27:553–560. doi: 10.1002/neu.480270409. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez M, Ruggiero FP, Chang Q, Shi YJ, Rich MM, Kraner S, Balice-Gordon RJ. Disruption of Trkb-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions [see comments]. Neuron. 1999;24:567–583. doi: 10.1016/s0896-6273(00)81113-7. [DOI] [PubMed] [Google Scholar]
- 10.Grigoriev NG, Woodin MA, Spafford JD, Lukowiak K, Syed NI. Dual-cell patch clamp analysis of synaptic interactions between soma-soma paired Lymnaea neurons. Soc Neurosci Abstr. 1999;25:78.13. [Google Scholar]
- 11.Hamakawa T, Woodin MA, Bjorgum MC, Painter SD, Takasaki M, Lukowiak K, Nagle GT, Syed NI. Excitatory synaptogenesis between identified Lymnaea neurons requires extrinsic trophic factors and is mediated by receptor tyrosine kinases. J Neurosci. 1999;19:9306–9312. doi: 10.1523/JNEUROSCI.19-21-09306.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kehoe J, McIntosh JM. Two distinct nicotinic receptors, one pharmacologically similar to the vertebrate α7-containing receptor, mediate Cl currents in Aplysia neurons. J Neurosci. 1998;18:8198–8213. doi: 10.1523/JNEUROSCI.18-20-08198.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Landis SC. Target regulation of neurotransmitter phenotype. Trends Neurosci. 1990;13:344–350. doi: 10.1016/0166-2236(90)90147-3. [DOI] [PubMed] [Google Scholar]
- 14.Landis SC, Keefe D. Evidence for neurotransmitter plasticity in vivo: developmental changes in properties of cholinergic sympathetic neurons. Dev Biol. 1983;98:349–372. doi: 10.1016/0012-1606(83)90365-2. [DOI] [PubMed] [Google Scholar]
- 15.Lovell P. PhD thesis. University of Calgary; 2000. Modulation of transmitter release during early stages of synapse formation between identified Lymnaea neurons. [Google Scholar]
- 16.Magoski NS, Bulloch AG. Stability and variability of synapses in the adult molluskan CNS. J Neurobiol. 2000;42:410–423. [PubMed] [Google Scholar]
- 17.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 18.Munno DW, Woodin MA, Lukowiak K, Syed NI, Dickinson PS. Different extrinsic trophic factors regulate neurite outgrowth and synapse formation between identified Lymnaea neurons. J Neurobiol. 2000;44:20–30. doi: 10.1002/1097-4695(200007)44:1<20::aid-neu3>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 19.Narisawa-Saito M, Carnahan J, Araki K, Yamaguchi T, Nawa H. Brain-derived neurotrophic factor regulates the expression of AMPA receptor proteins in neocortical neurons. Neuroscience. 1998;88:1009–1014. doi: 10.1016/s0306-4522(98)00496-5. [DOI] [PubMed] [Google Scholar]
- 20.Nesic OB, Magoski NS, McKenney KK, Syed NI, Lukowiak K, Bulloch AG. Glutamate as a putative neurotransmitter in the mollusc, Lymnaea stagnalis. Neuroscience. 1996;75:1255–1269. doi: 10.1016/0306-4522(96)00241-2. [DOI] [PubMed] [Google Scholar]
- 21.Ridgway RL, Syed NI, Lukowiak K, Bulloch AG. Nerve growth factor (NGF) induces sprouting of specific neurons of the snail, Lymnaea stagnalis. J Neurobiol. 1991;22:377–390. doi: 10.1002/neu.480220406. [DOI] [PubMed] [Google Scholar]
- 22.Santama N, Wheeler CH, Skingsley DR, Yeoman MS, Bright K, Kaye I, Burke JF, Benjamin PR. Identification, distribution and physiological activity of three novel neuropeptides of Lymnaea: EFLRlamide and pQFYRlamide encoded by the FMRFamide gene, and a related peptide. Eur J Neurosci. 1995;7:234–246. doi: 10.1111/j.1460-9568.1995.tb01059.x. [DOI] [PubMed] [Google Scholar]
- 23.Saunders SE, Kellett E, Bright K, Benjamin PR, Burke JF. Cell-specific alternative RNA splicing of an FMRFamide gene transcript in the brain. J Neurosci. 1992;12:1033–1039. doi: 10.1523/JNEUROSCI.12-03-01033.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23:639–645. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- 25.Skingsley DR, Bright K, Santama N, van Minnen J, Brierley MJ, Burke JF, Benjamin PR. A molecularly defined cardiorespiratory interneuron expressing SDPFLRFamide/GDPFLRFamide in the snail Lymnaea: monosynaptic connections and pharmacology. J Neurophysiol. 1993;69:915–927. doi: 10.1152/jn.1993.69.3.915. [DOI] [PubMed] [Google Scholar]
- 26.Syed NI, Winlow W. Coordination of locomotor and cardiorespiratory networks of Lymnaea stagnalis by a pair of identified interneurones. J Exp Biol. 1991a;158:37–62. doi: 10.1242/jeb.158.1.37. [DOI] [PubMed] [Google Scholar]
- 27.Syed NI, Winlow W. Respiratory behaviour in the pond snail Lymnaea stagnalis. II. Neural elements of the central pattern generator (CPG). J Comp Physiol [A] 1991b;169:557–568. [Google Scholar]
- 28.Syed NI, Bulloch AG, Lukowiak K. In vitro reconstruction of the respiratory central pattern generator of the mollusk Lymnaea. Science. 1990;250:282–285. doi: 10.1126/science.2218532. [DOI] [PubMed] [Google Scholar]
- 29.Syed NI, Zaidi H, Lovell P. In vitro reconstruction of neuronal networks: a simple model system approach. In: Windhurst U, Johansson H, editors. Modern techniques in neuroscience research. Springer; Berlin: 1999. pp. 361–377. [Google Scholar]
- 30.Whim MD, Church PJ, Lloyd PE. Functional roles of peptide cotransmitters at neuromuscular synapses in Aplysia. Mol Neurobiol. 1993;7:335–347. doi: 10.1007/BF02769181. [DOI] [PubMed] [Google Scholar]
- 31.Wong R, Hadley R, Kater S, Hauser G. Neurite outgrowth in molluscan organ and cell cultures: the role of conditioning factor(s). J Neurosci. 1981;1:1008–1021. doi: 10.1523/JNEUROSCI.01-09-01008.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woodin MA, Hamakawa T, Takasaki M, Lukowiak K, Syed NI. Trophic factor-induced plasticity of synaptic connections between identified Lymnaea neurons. Learn Mem. 1999;6:307–316. [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou J, Bradford HF. Nerve growth factors and the control of neurotransmitter phenotype selection in the mammalian central nervous system. Prog Neurobiol. 1997;53:27–43. doi: 10.1016/s0301-0082(97)00030-0. [DOI] [PubMed] [Google Scholar]