

Abstract
Traumatic brain injury (TBI) increases susceptibility to Alzheimer's disease (AD), but it is not known how TBI contributes to the onset or progression of this common late life dementia. To address this question, we studied neuropathological and behavioral consequences of single versus repetitive mild TBI (mTBI) in transgenic (Tg) mice (Tg2576) that express mutant human Aβ precursor protein, and we demonstrate elevated brain Aβ levels and increased Aβ deposition. Nine-month-old Tg2576 and wild-type mice were subjected to single (n = 15) or repetitive (n = 39) mTBI or sham treatment (n = 37). At 2 d and 9 and 16 weeks after treatment, we assessed brain Aβ deposits and levels in addition to brain and urine isoprostanes generated by lipid peroxidation in these mice. A subset of mice also was studied behaviorally at 16 weeks after injury. Repetitive but not single mTBI increased Aβ deposition as well as levels of Aβ and isoprostanes only in Tg mice, and repetitive mTBI alone induced cognitive impairments but no motor deficits in these mice. This is the first experimental evidence linking TBI to mechanisms of AD by showing that repetitive TBI accelerates brain Aβ accumulation and oxidative stress, which we suggest could work synergistically to promote the onset or drive the progression of AD. Additional insights into the role of TBI in mechanisms of AD pathobiology could lead to strategies for reducing the risk of AD associated with previous episodes of brain trauma and for preventing progressive brain amyloidosis in AD patients.
Keywords: Alzheimer's disease, amyloid plaques, brain injury, head trauma, APP mice, oxidative stress, cognitive function
Senile plaques (SPs) and neurofibrillary tangles are the neuropathological hallmarks of Alzheimer's disease (AD). SPs are formed by fibrillar Aβ peptides derived from amyloid precursor proteins (APPs) through sequential proteolytic cleavage of APP by β- and γ-secretases generating diverse species of Aβ with differential propensities to fibrillize and deposit in SPs (Saido, 1998; Mills and Reiner, 1999; Selkoe, 1999;Nunan and Small, 2000; Bayer et al., 2001). The 42–43 amino acid long Aβ variants known as Aβ(x-42/43) are thought to be the most amyloidogenic and critical in the onset and progression of AD amyloidosis, although the exact mechanisms underlying AD pathogenesis are incompletely understood. However, multiple mutations in theAPP and presinilin-1 and -2 genes have been shown to be pathogenic for familial AD (FAD; Price and Sisodia, 1998; Selkoe, 1999; Bayer et al., 2001). These mutations are thought to cause FAD by increasing production of Aβ(x-42/43) followed by accelerated AD amyloidosis (Jarrett and Lansbury, 1993; Felsenstein et al., 1994; Lannfelt et al., 1994; Hardy, 1997; Bayer et al., 2001).
Despite remarkable progress in understanding the genetics of FAD, less is known about genetic and epigenetic risk factors for sporadic AD (SAD) although ∼90% of AD cases are sporadic, and the incidence of SAD will continue to increase as life expectancy expands in coming decades (Martin, 1999). The apolipoprotein E ε4 allele is the most well documented genetic risk factor for SAD (Wisniewski et al., 1994), and traumatic brain injury (TBI) is the most robust environmental AD risk factor (Heyman et al., 1984; Mortimer et al., 1985; Guo et al., 2000; Plassman et al., 2000). Although recurrent TBI is thought to cause dementia pugilistica in career boxers, a mechanistic link between TBI and the induction or acceleration of AD has not yet been elucidated (Murai et al., 1998). This may reflect limitations in the available animal models used in previous studies (Nakagawa et al., 1999, 2000) of TBI.
In this article, we have used a recently developed mild TBI (mTBI) mouse model that does not require craniotomy and produces minimal structural brain damage (Laurer et al., 2001), and we use this model to determine whether single versus repetitive mTBI augments disease in a transgenic (Tg) mouse model (Tg2576) of AD-like amyloidosis (Hsiao et al., 1996). Accordingly, we conducted studies to examine the effects of single or repetitive mTBI on cognition and motor behavior as well as on the onset and progression of amyloidosis in Tg and wild-type (WT) mice at 2 d and 9 and 16 weeks after mTBI. We also monitored levels of isoprostanes, markers of lipid peroxidation (LPO), because of evidence linking oxidative damage and AD pathobiology (Praticòet al., 2000a, 2001). Significantly, these studies provide the first experimental evidence implicating TBI in mechanisms of AD by augmenting brain Aβ accumulation and LPO.
MATERIALS AND METHODS
Animals and surgical procedure
Tg APP695swe (Tg2576) mice and WT littermates were used in this study. At 9 months of age, both Tg and WT mice were subjected to mTBI as described previously (Laurer et al., 2001). Briefly, mice were anesthetized with sodium pentobarbital (65 mg/kg of body weight, i.p.); ointment was applied to their eyes to protect vision during surgery; and the mice were placed on a heating pad to maintain body temperature throughout the surgical procedures. All animals were mounted in a stereotactic frame; a skin incision was performed to expose the skull; and the mice remained in the stereotactic apparatus while subjected to mTBI using a pressure-driven instrument that is mechanically identical to a previously described controlled cortical impact device (Dixon et al., 1991; Smith et al., 1995) with minor modifications that have also been described previously (Laurer et al., 2001). The impounder was rigidly mounted at an angle of 20° from vertical, and because the depth and duration of the impact were kept constant, head movements were minimal while delivering the load. The procedure was completed with the closure of the incision using 4-0 silk sutures. The animals were removed from the stereotactic frame and placed in a heated cage, and after recovery from anesthesia (as evidenced by ambulation), they were returned to their home cages.
At 24 hr after the first mTBI, selected animals were reanesthetized as described above, and these mice were then subjected to a second mTBI in the same location over the left parietotemporal region. Sham-treated animals also were anesthetized and placed in the stereotactic frame; the skull was exposed, and the skin incision was sutured closed without brain injury on 2 consecutive days, thereby following exactly the surgical procedures of repetitive mTBI. All of these procedures were performed in strict accordance with the National Institutes of HealthGuide for the Care and Use of Laboratory Animals, and they were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Neurobehavioral analysis
Assessment of cognitive function. The testing paradigm for evaluation of cognitive function using the Morris water maze (MWM) has been described in detail previously (Fox et al., 1998;Pierce et al., 1998; Bareyre et al., 2000). Briefly, the MWM is a circular pool 1 m in diameter, painted white inside (Morris et al., 1982). The water (16–18°C) is made opaque by adding nontoxic, water-soluble white coloring. To test for mTBI-induced learning impairments, animals received no training in the MWM before injury and were trained to locate a stationary, submerged platform (0.5 cm below the surface) using external cues starting at 16 weeks after injury. The essential feature of the MWM is that mice can escape from the water onto the platform after being placed randomly at one of four sites in the pool. Latencies of four trials/d were recorded and averaged to obtain a measurement for the performance of each animal on a given day. Animals were tested for their ability to learn the visuospatial task in the MWM over an 8-d period that began 16 weeks after mTBI. Notably, previous studies to adapt the MWM to mice (see below) indicate that TBI does not cause changes in swim speed or visual acuity that influence latencies in the mouse version of the MWM, and this also was documented in the MWM studies here.
Motor function. The composite neuroscore (NS), which includes a battery of motor tests, was obtained for mice before performing MWM evaluations as described previously for TBI in rats (McIntosh et al., 1987, 1989) after modifications for use in mice (Murai et al., 1998; Raghupathi et al., 1998; Nakamura et al., 1999;Scherbel et al., 1999). The NS measures the following tasks: (1) forelimb flexion response during suspension by the tail, (2) resistance to lateral pulsion, and (3) response of the hindlimb and toes (hindlimb flexion) when raised by the tail. Each animal was scored by an investigator blinded to the injury status of the animal using a scaling system ranging from 4 (preinjury control status) to 0 (afunctional).
Histological and immunohistochemical analysis
Tg2576 and WT littermate male and female mice used in this study received sham injury or single or repetitive mTBI, and the mice were killed 2 d or 9 or 16 weeks thereafter. Each experimental group consisted of five or six mice, except for the single mTBI 9 and 16 week post-mTBI groups (n = 0 and 3, respectively) and the sham and repetitive mTBI groups at 2 d after treatment (n = 3 for both of these cohorts). After the study of living mice was concluded, they were lethally anesthetized and perfused intracardially with PBS (0.1 m), pH 7.4, followed by phosphate-buffered 4% paraformaldehyde. Brains and spinal cords were removed, post-fixed overnight, sliced into 2-mm-thick coronal slabs, and embedded in paraffin in a frontal to occipital series of blocks, and the blocks were cut in a near serial array of 6-μm-thick coronal sections for analysis.
The histology and location of the mTBI site were examined by hematoxylin and eosin (H&E) as well as by Gomori's iron staining, and the distribution as well as the burden of Aβ deposits were demonstrated by immunostaining with the 4G8 anti-Aβ 17–24 monoclonal antibody. Before quantitative analysis, initial comparisons of several other well characterized antibodies, i.e., rabbit polyclonal antibodies 2332 and 2333 (both of which recognize multiple species of Aβ) and mouse monoclonal antibodies 6E10 (anti-Aβ 1–17), BA27 [anti-Aβ(x-40)], and BA05 [anti-Aβ(x-42)] were undertaken, and 4G8 was chosen for determining the Aβ burden because of its robust signal and optimal results for quantitative analysis. In addition, reactive astrocytes were visualized by an antibody to glial fibrillary acidic protein (GFAP; Dako, Carpinteria, CA). Finally, characteristic features of SPs also were identified in Tg mouse brains with and without mTBI by immunohistochemistry using anti-ubiquitin antibodies (Chemicon, Temecula, CA) and thioflavin-S staining.
Paraffin sections were subjected to immunohistochemistry as described previously (Murai et al., 1998; Nakagawa et al., 1999, 2000). Briefly, sections were deparaffinized in xylene, hydrated in a series of ethanol and deionized water, and subjected to an antigen retrieval step by immersing sections in 88% formic acid for 60 min before immunohistochemistry for Aβ. Sections were washed in water, and endogenous peroxidases were quenched using a freshly prepared mixture of methanol (150 ml) plus hydrogen peroxide (33%, 30 ml). The immunohistochemistry procedures have been described previously (Nakagawa et al., 1999), and the avidin–biotin complex method was used according to the instructions of the vendor (Vector Laboratories, Burlingame, CA). Negative controls included the application of the same immunohistochemistry protocol to sections, except preimmune serum was applied instead of primary antibody.
Image analysis
Brain sections immunostained with 4G8 from Tg2576 and WT littermate mice that survived for 2 d and 9 and 16 weeks after the last treatment (including sham and single and repetitive mTBI) were used for quantitative analysis, but the sections from the WT mice showed no amyloid deposits as reported previously (Hsiao et al., 1996).
Coronal brain sections from levels between the habenula nucleus and the posterior commissure of Tg mice subjected to sham or single or repetitive mTBI at longer postoperative survival intervals were subjected to quantitative analysis. For injured animals, the sections selected for analysis were subjacent to the mTBI site, as demonstrated by the presence of small iron deposits (resulting from minor blood vessel damage and release of red blood cells) revealed by Gomori's iron stain, and equivalent sections from sham-treated Tg mice were subjected to quantitative analysis. Eight sections from each animal, with 4G8 immunostaining but without a counterstain, were used for quantitative image analysis as described previously (Nakagawa et al., 1999, 2000).
Light microscopic images from the somatosensory cortex (SSC), perihippocampal cortex (PHC), and hippocampus (HP) from both ipsilateral (left) and contralateral (right) hemispheres to the mTBI site were captured from eight series of sections using a Nikon Microphot-FXA microscope with a 4× objective lens. Using a personal computer, each image was opened with image analysis software (Image Pro-plus; Media Cybernetics, Inc., Silver Spring, MD). Manual editing was then performed to eliminate nonspecific signals (e.g., blood vessels and staining artifacts). The areas occupied by Aβ-immunoreactive products in the regions of interest were measured, and the total area occupied by the outlined structures was measured to calculate (1) the total area with selected immunoreactive products and (2) the percentage of the area occupied by immunoreactive products over the outlined anatomical area in the image.
Sandwich Aβ ELISA
For quantitation of Aβ brain levels, Tg2576 and WT littermate male and female mice at 16 weeks postinjury survival times (n = 4–6) were perfused transcardially with PBS containing 0.01% of the antioxidant butylated hydroxytoluene (BHT). The cohort size for these studies was determined on the basis of previous studies of APP Tg mice using this ELISA (Murai et al., 1998;Nakagawa et al., 1999, 2000). The brain was removed, and each right and left cerebral cortex, hippocampus, and cerebellum were collected in individual test tubes, weighed, and frozen immediately with dry ice. Sequential extraction of samples was performed with high-salt buffer and formic acid to measure soluble and insoluble brain Aβ(x-40) and Aβ(x-42/43). The right and left cerebral cortices, hippocampus, and cerebellum were serially extracted in high-salt Re-assembly buffer (0.1 m Tris, 1 mmEGTA, 0.5 mm MgSO4, 0.75m NaCl, and 0.02 m NaF, pH 7.0) containing a protease inhibitor mixture (pepstatin A, leupeptin,N-tosyl-l-phenylalanine chloromethyl ketone,Nα-p-tosyl-l-lysine chloromethyl ketone, and soybean trypsin inhibitor, each at 1 μg/ml in 5 mm EDTA). Tissue was chopped into small pieces and then sonicated with 10 burst pulses (level 3) of a Fisher Scientific (Pittsburgh, PA) F60 Sonic Dismembrator. Homogenates were centrifuged at 100,000 × g for 1 hr at 4°C. Supernatants were removed, and pellets were resuspended in 70% formic acid and resonicated and centrifuged at 100,000 × gfor 1 hr at 4°C. Supernatants were removed and diluted 1:20 with 1m Tris base. The supernatant obtained in each extraction step was normalized to the original wet weight of the tissue sample and analyzed separately by ELISA. To do this, samples were diluted in buffer EC [0.02 m sodium phosphate, 0.2 m EDTA, 0.4m NaCl, 0.2% BSA, 0.05% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid, 0.4% Block-ace (Dainippon; Suita, Osaka, Japan), and 0.05% sodium azide, pH 7.0] and analyzed using Ban50 monoclonal antibody (mAb) to capture and BA-27 and BC-05 mAbs as reporters to detect Aβ1–40 and Aβ1–42/43, respectively, as described previously (Iwatsubo et al., 1994; Suzuki et al., 1994; Wang et al., 1999; Nakagawa et al., 2000).
Isoprostane analysis
Urine was collected from Tg and WT mice into plastic tubes containing BHT after various periods of survival (9, 12, and 16 weeks after injury; n = 4–6) from each injury group. Samples were spiked with a fixed amount of internal standard (d4-8,12-iso-iPF2α-VI) extracted on a C18 cartridge column. The eluted fraction was purified by thin-layer chromatography and finally assayed by negative ion chemical ionization gas chromatography and mass spectrometry, as described previously (Praticò et al., 1998, 2001; Praticò, 1999). Urine aliquots (0.1 ml) were used for measurement of creatinine levels by a commercially available standardized automated colorimetric assay (Sigma, St. Louis, MO). Levels were expressed as nanograms per milligram of creatinine. Finally, aliquots of brain tissue samples obtained for the Aβ ELISA were used for isoprostane analysis, including samples of cerebral cortex, hippocampus, and cerebellum collected 9 and 16 weeks after injury by the procedures described here. All the assays were performed without knowledge of the age or mTBI status of the mice.
Statistical analysis
Data for Aβ deposits, levels of isoprostanes (i.e., 8,12-iso-iPF2α-VI), and Aβ-40 and Aβ-42 concentrations from the studies described above were expressed as mean ± SEM. Areas of amyloid deposition and isoprostane and Aβ levels were assessed by ANOVA and subsequently by Student's unpaired two-tailed t test, taking into consideration survival time and type of injury for each animal. Results in the tests for neurological motor function are nonparametric data and were compared using a Kruskal–Wallis ANOVA by ranks. Data obtained in the MWM are parametric data and are given as mean ± SEM. These data were analyzed using a four-way ANOVA for overall effects (genotype, injury status, gender, and time) followed by multiple two-way ANOVAs for additional comparisons between particular groups within a given genotype. Significance was set at p < 0.05.
RESULTS
Neurobehavioral analysis
All mice underwent the MWM test, and all were able to swim without any sign of functional motor impairment. Sham WT animals (n = 14) demonstrated the ability to learn the visuospatial task with decreasing latencies to find the platform 16 weeks after injury (Fig.1A). WT animals subjected to either single (n = 20) or repetitive mTBI (n = 19) demonstrated a similar ability to learn the new visuospatial task with latencies that were not significantly different from those of WT sham animals (Fig. 1A), indicating that neither single nor repetitive mTBI influenced learning ability assessed in the chronic postinjury period in WT animals.
Fig. 1.
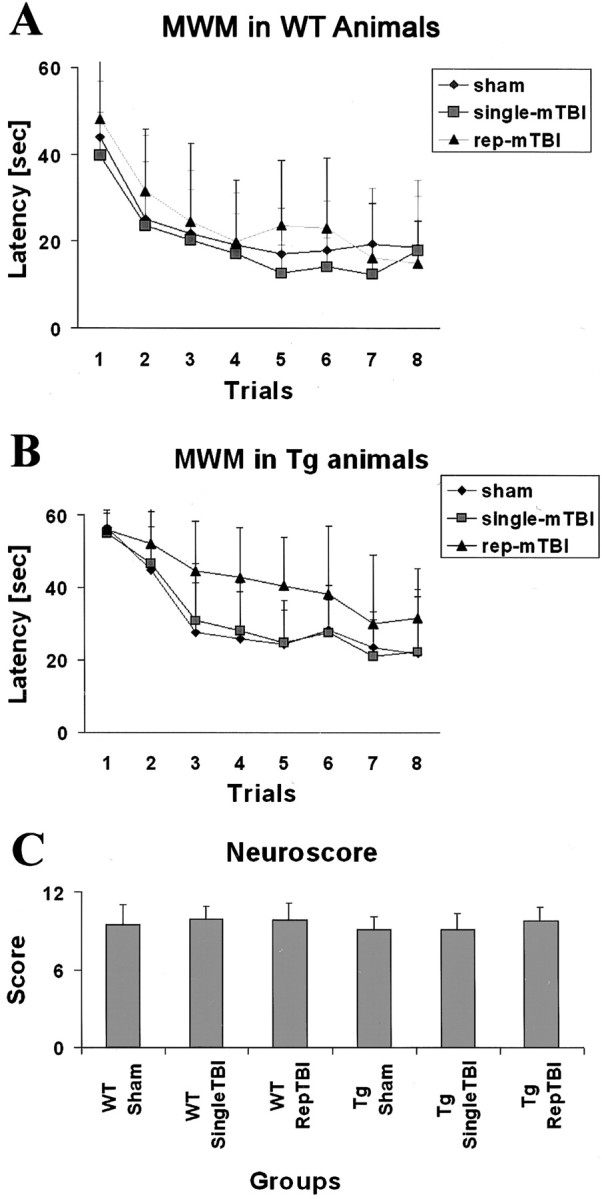
Data from behavioral and motor tests in WT and Tg2576 mice. A, B, Data from the MWM test in WT and Tg mice. Trials were made in consecutive 8 d sessions at 16 weeks after injury. C, Motor function tests performed at 16 weeks after injury. Rep, Repetitive.
Sham Tg animals (n = 9) demonstrated the ability to learn a visuospatial task with decreasing latencies to find the platform 16 weeks after surgery (Fig. 1B). Tg animals subjected to single mTBI (n = 15) demonstrated a similar ability to learn the new visuospatial task with latencies that were not significantly different from those of Tg sham animals, indicating that single mTBI did not result in cognitive dysfunction at 16 weeks after injury (ANOVA, p = 0.225; Fig.1B). In contrast, Tg animals subjected to repetitive mTBI (n = 13) demonstrated an altered ability to learn a visuospatial task with latencies that were significantly increased relative to sham-injured Tg mice (ANOVA, p = 0.0158) as well as to single mTBI Tg mice (ANOVA, p = 0.0116), indicating that repetitive mTBI resulted in a significant cognitive dysfunction at 16 weeks after injury (Fig. 1B). This interpretation is supported by evidence that neither WT nor Tg sham animals showed any motor impairments 16 weeks after surgery, and that neither single nor repetitive mTBI led to deficits in motor function in WT or Tg mice 16 weeks after trauma (Fig. 1C). Thus, impaired motor performance cannot account for the MWM performance deficits in the repetitive mTBI Tg mice described above.
Histology
No histopathological changes or evidence of cell loss after single mTBI injury were reported previously in the ipsilateral cortex, hippocampal CA3, or dentate hilus up to 8 weeks after injury of WT mice (Laurer et al., 2001), and we extended these observations to the effects of mTBI and sham treatment on the Tg mice described here (Fig.2). Notably, after single mTBI at 16 weeks after injury, there was no significant cortical neuropathology at the impact site (Fig. 2E), and the same was true of the sham-treated mice (Fig. 2C). However, single mTBI resulted in minimal iron deposits subjacent to the meninges at the impact site that were not evident at low-power magnification (Fig.2E), but repetitive mTBI caused mild edema of the cortical surface at the injury site 2 d after mTBI (Fig.2A) and more evident iron deposits at 16 weeks after mTBI (Fig. 2G), but there was no other evidence of pathology by H&E staining, and there was no indication that CA3 and other hippocampal areas were altered by single or repetitive mTBI.
Fig. 2.
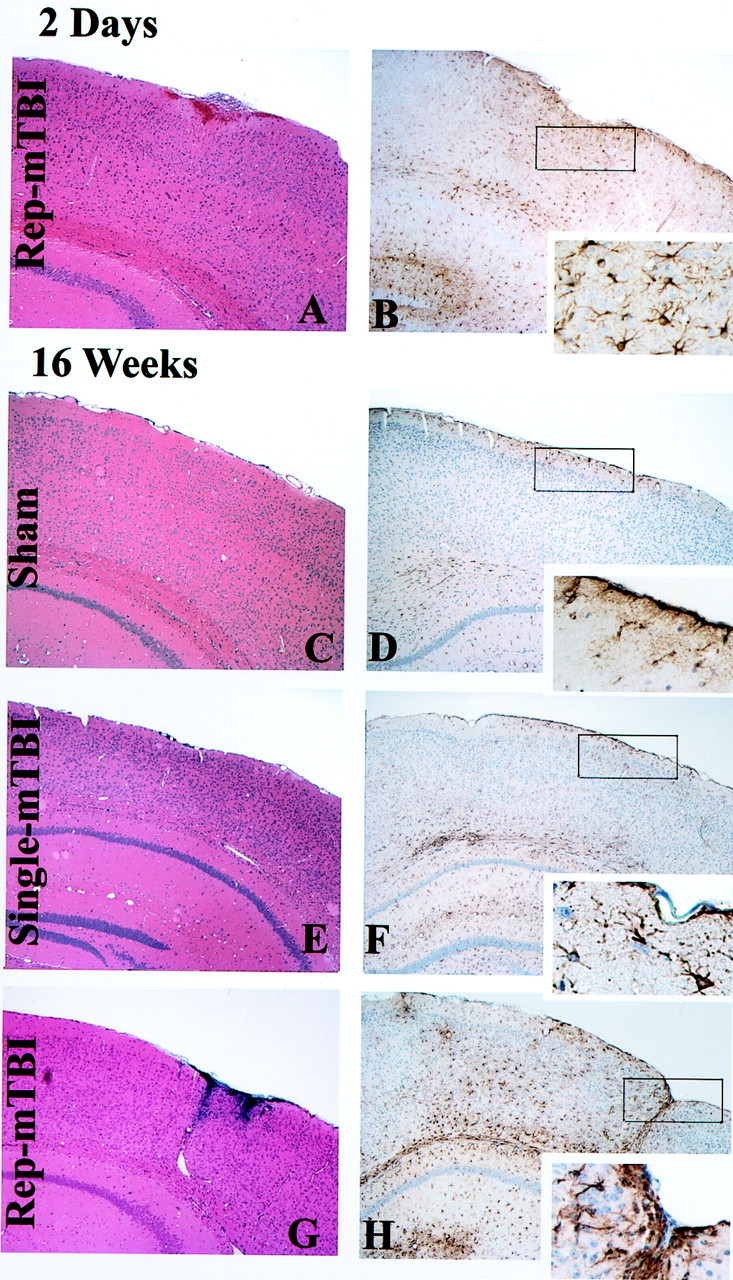
Histological sequelae of single or repetitive (Rep) mTBI and sham treatment in Tg2576 mice. H&E staining with Gomori's iron stain (A, C, E, G) and GFAP staining (B, D, F, H, insets) are shown. Single and repetitive mTBI resulted in no or very mild damage at the impact site in the brain at 2 d (A, B), and 16 weeks (C–H) after the injury. Eachinset indicates a high-power view of the rectangular area shown in the images in B, D, F, andH. Sham treatment resulted in no overt damage (C, D). A, B, Repetitive mTBI (2 d after TBI).C, D, Sham (16 weeks after treatment). E, F, Single mTBI (16 weeks after TBI). G, H, Repetitive mTBI (16 weeks after TBI).
GFAP staining
For Tg and WT mice subjected to repetitive mTBI, there was evidence of early and mild reactive gliosis detected by GFAP staining 2 d after the injury in cortex subjacent to the impact site, and a few GFAP-positive reactive astrocytes also were seen in the subjacent hippocampus (Fig. 2B). At 9 weeks after surgery, these reactive astrocytes were limited to the surface of the cortex below the impact site after repetitive mTBI (data not shown), but 16 weeks after sham treatment, there was no evidence of cortical gliosis (Fig. 2C,D), whereas single mTBI mice showed very mild astrocytosis and GFAP staining predominantly in the white matter after 16 weeks (Fig. 2F). Repetitive mTBI induced GFAP-positive reactive astrocytes confined to the impact site of both WT and Tg mice, and Figure 2, G and H, shows representative images of this in the Tg2576 mice. Apart from the impact site, infrequent reactivate astrocytes were found primarily in the white matter in the WT mice, and this was the case with the Tg mice, which also showed reactive astrocytes around SPs in addition to variable amounts of gliosis in the cortex, subcortical white matter, corpus callosum, and hippocampus.
Amyloid deposition
Aβ deposition was detectable in the cerebral cortex and hippocampus in the Tg2576 mice at 9 months of age and thereafter as reported previously (Hsiao et al., 1996; Takeuchi et al., 2000). Between 2 d and 9 weeks after injury, the burden and distribution pattern of Aβ deposits were scattered and infrequent in all groups of Tg mice, whereas there were no Aβ deposits in any of the WT mice. There was a mild increase in the Aβ burden in repetitive mTBI mice at 9 weeks after injury, but only at 16 weeks (i.e., in 12-month-old Tg mice) was the Aβ burden increased in both single and repetitive mTBI mice relative to sham-treated Tg mice (Fig.3). These amyloid deposits were detectable in selected brain regions, i.e., the olfactory bulb, all cortical regions, including the frontal, cingulate, and perihippocampal cortex, as well as the hippocampus, but not in areas such as the striatum, thalamus, cerebellum, brainstem, and spinal cord. Amyloid deposition visualized by 4G8 immunohistochemistry in sham-treated and single or repetitive mTBI mice also was confirmed using both thioflavin-S staining and ubiquitin immunohistochemistry (data not shown).
Fig. 3.

Amyloid deposition in Tg2576 mice with sham or repetitive (Rep) mTBI (B, D) with 4G8 immunohistochemistry at 9 (A, C) and 16 (B, D) weeks after mTBI. SPs increased in an age-dependent manner in both sham and injured mice, but the largest number of Aβ-positive SPs are seen in the 16 week postrepetitive mTBI mice (D).
To determine the effects of mTBI on progressive amyloid deposition at various periods after injury, the area occupied by 4G8-immunopositive deposition in the SSC, PHC, and HP both ipsilateral and contralateral to the impact site was analyzed (Fig. 4). The study groups consisted of sham-treated mice, repetitive mTBI mice (2 d and 9 and 16 weeks after TBI), and single mTBI mice (16 weeks after surgery). Comparison of the burden of Aβ positive deposits between the side of the impact and the contralateral side revealed that all three regions analyzed from both hemispheres showed a comparable accumulation of Aβ of the Tg2576 mice, and this was true across all groups of Tg mice throughout the time points analyzed.
Fig. 4.
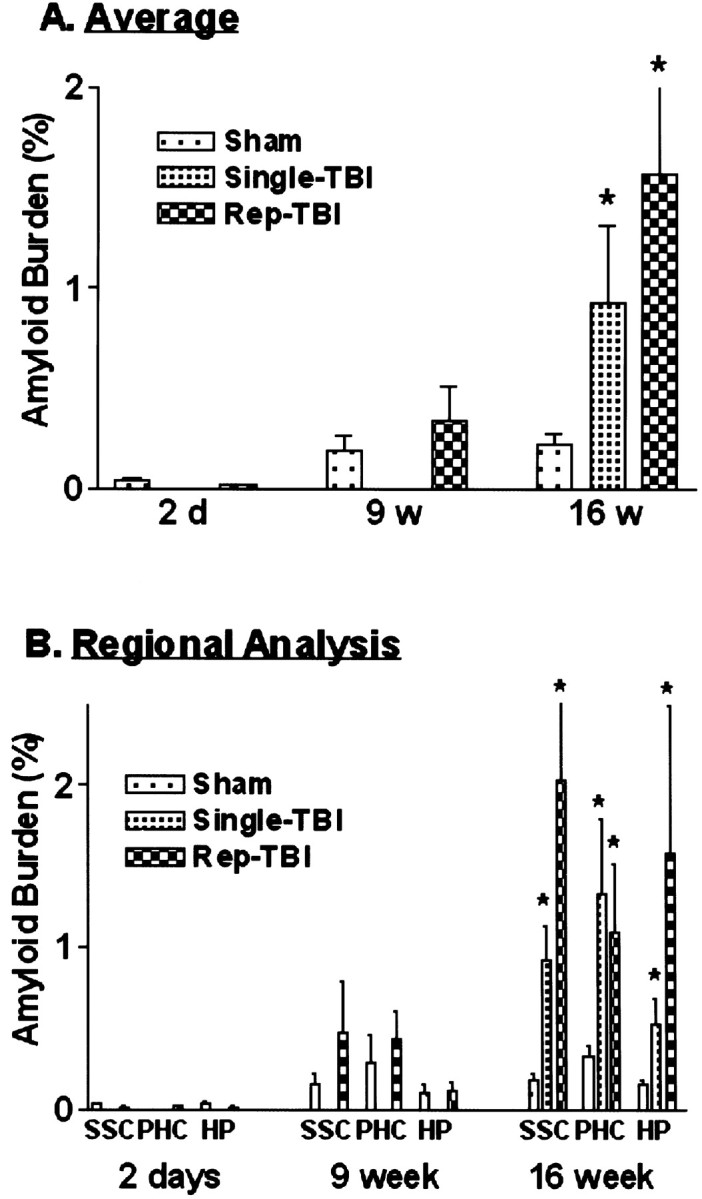
Average percentages of the area occupied by Aβ in three brain areas of interest, including the PHC, SSC, and HP.A, Data from the total number of mice in each group, including male and female for all regions of interest.B, Data in A plotted for each region (n = 4–6). *p < 0.05 in comparison with sham treatment. Rep, Repetitive.
Averaging the value of amyloid burden between right and left hemispheres of each anatomical region revealed that single mTBI (average = 0.93%; ANOVA, F = 12.04;p = 0.046) and repetitive mTBI (average = 1.57%; ANOVA, F = 6.25; p = 0.025) accelerated amyloid deposition by 16 weeks after trauma compared with the sham group (Fig. 4A), although the differences between sham treatment versus repetitive mTBI at 9 weeks were not significant. Because gender effects on amyloid burden were shown to be significant (Callahan et al., 2001), we performed analyses on the subgroup of male Tg2576 mice, and the outcome of these analyses showed that the burden was higher in Tg2576 female versus male mice. Most notably, however, there was a significant difference only at 16 weeks after injury (single mTBI, average = 0.93%; ANOVA, F = 12.04;p = 0.046; repetitive mTBI, average = 1.36%; ANOVA, F = 6.64; p = 0.032) in comparison with sham-treated Tg mice. Finally, further analysis of these data demonstrated that all regions (i.e., SSC, PHC, and HP) showed trends with respect to the effect of mTBI on amyloid burden (Fig. 4B), and by 16 weeks after injury, both single and repetitive mTBI mice accumulated a 4- to 10-fold higher Aβ burden than the sham-treated Tg2576 group.
Aβ ELISA
To independently assess the burden of brain Aβ40 and Aβ42, ELISA was used to analyze brain ipsilateral and contralateral to the hemisphere subjected to sham treatment or single versus repetitive mTBI at 16 weeks after surgery. As additional controls, right and left cerebellar hemispheres were collected from each Tg2576 mouse and analyzed individually. ELISA analysis showed that the cerebral cortex consistently had the most Aβ40 and Aβ42, the hippocampus had smaller amounts of both peptides than the cortex, whereas the cerebellum consistently had the least, and the concentrations of Aβ40 or Aβ42 did not differ between either hemisphere in all regions examined (Fig. 5). In single mTBI Tg2576 mice, concentrations of Aβ40 and Aβ42 were increased in the soluble fraction, but this group failed to show a significant difference in the Aβ40 levels compared with the sham-treated Tg2576 mice, although there was a significant increase in Aβ42 concentrations (Fig.5A). Indeed, neither insoluble Aβ40 nor Aβ42 in single mTBI Tg2576 mice showed a significant difference in comparison with cortices from sham-treated mice (Fig. 5B). However, in the repetitive mTBI group, Aβ40 and Aβ42 levels in both the soluble and insoluble fractions of neocortex from all Tg2576 mice were significantly higher than in the sham-treated Tg2576 mouse cortex (Fig.5).
Fig. 5.

Concentration of brain Aβ as determined by sandwich ELISA. Aβ1–40 and Aβ1–42 peptides were measured in soluble (A) and insoluble (B) fractions of cortex (CTX), hippocampus (HP), and cerebellum (CBL) from sham and single and repetitive (Rep) mTBI Tg2576 mice (n = 4–6). *p < 0.05; **p < 0.001 in comparison with sham injury.
Isoprostane analysis
8,12-iso-iPF2α-VI levels in urine and brain from selected WT and Tg mice were measured to assess the extent of oxidative stress after mTBI or sham treatment (Fig.6). Urinary 8,12-iso-iPF2α-VI levels in the sham group revealed a slight nonsignificant increase in this isoprostane isomer because of aging, but there was a significant difference in single mTBI Tg mice at 12 weeks after injury (single mTBI, ANOVA,p = 0.0292; F = 6.13 compared with the sham group), although this no longer was noticeable at 16 weeks after injury (Fig. 6A). In contrast, the repetitive mTBI group revealed high levels of 8,12-iso-iPF2α-VI as early as 9 weeks after injury (repetitive mTBI, ANOVA, p < 0.0001; F = 92.69 compared with the sham group), which were maintained until 16 weeks after injury (12 weeks after TBI, ANOVA,p < 0.0001; F = 85.62; 16 weeks after TBI, p < 0.0001, F = 165.6 compared with the sham group), and these levels were higher than in the single mTBI Tg2576 mice (Fig. 6A).
Fig. 6.

Concentration of 8,12-iso-iPF2α-VI in urine (A) and brain (B). Urine was collected at 9, 12, and 16 weeks after TBI or sham treatment. Brain tissue was collected from hippocampus (HP), cortex (CTX), and cerebellum (CBL) at 16 weeks after mTBI or sham treatment. All samples came from Tg2576 mice.Rep, Repetitive.
Analyses of the cortex, hippocampus, and cerebellum from the sham and injured Tg2576 mice (Fig. 6B) revealed that the cerebral cortex and hippocampus, but not the cerebellum, showed significantly higher 8,12-iso-iPF2α-VI levels in both single and repetitive mTBI groups at 16 weeks after surgery (cortex, single vs sham ANOVA, p = 0.027; F = 14.16; repetitive vs sham ANOVA, p = 0.0006;F = 21.30; hippocampus, single vs sham ANOVA,p = 0.0256; F = 6.48; repetitive vs sham ANOVA, p = 0.0002; F = 26.93). Interestingly, 8,12-iso-iPF2α-VI levels in the cortex were significantly higher than levels in the hippocampus in all treatment groups (Fig. 6B).
DISCUSSION
The present study shows for the first time that there is a clear positive correlation between episodes of TBI and increased amyloid deposition in a Tg mouse model of AD amyloidosis. Specifically, we demonstrated that repetitive mTBI in the Tg2576 mice resulted in a significant acceleration of amyloid deposition by image analysis of immunohistochemically stained brain sections and increased Aβ40 and Aβ42 production and accumulation in soluble and insoluble brain homogenates by a sensitive Aβ ELISA. Moreover, these findings also were associated with a significantly greater impairment in cognitive function in MWM tests and elevated brain and urinary levels of an isoprostane isomer (i.e., 8,12-iso-iPF2α-VI) that is a well characterized and reliable index of oxidative stress (Praticò et al., 1998, 2000a; Praticò and Delanty, 2000). Thus, these data provide the first compelling mechanistic linkage between previous episodes of TBI and subsequent Aβ amyloidosis as well as cognitive impairment and LPO similar to that observed in living AD patients. Furthermore, these data strongly support previous epidemiological studies implicating TBI as one of the most robust environmental risk factors for AD.
Both the image analysis and ELISA studies here establish that AD-like amyloidosis, including Aβ deposition and increasing levels of insoluble Aβ, was accelerated by both single and repetitive mTBI. This is consistent with previous studies in deceased acute human head trauma victims, which demonstrated a positive correlation between TBI and Aβ deposits (Roberts et al., 1994), but it contrasts with previous animal model studies that did not show similar correlations, although TBI causes acute and chronically progressive neurodegenerative changes in the brains of WT and Tg animals expressing WT or mutant human APP (Smith et al., 1997; Murai et al., 1998; Pierce et al., 1998;Nakagawa et al., 1999, 2000). The explanation for these discrepancies among the current and previous studies of animal models of TBI and AD is not clear, but these differences might be attributable to the fact that TBI induced a selective loss of neurons that secrete Aβ in Tg mice generated by using neuron-specific promoters to drive WT or mutant APP transgene expression or to one or more species differences between the brains of humans and other mammals. Alternatively, the effects reported here suggest that mild TBI may more closely model the types of injuries in humans that predispose individuals who survive episodes of head trauma to develop AD later in life.
For this reason, we developed a mild TBI mouse model to elucidate the role of head trauma in Tg mice that have been shown to develop an age-dependent AD-like amyloidosis attributable to expression of a double APP mutation found in Swedish FAD patients (Hsiao et al., 1996). This strategy also enabled us to dissect out differences in the effect of single versus repetitive mTBI in the Tg2576 mouse model of AD amyloidosis. Indeed, the present study demonstrated some similarities and differences in histological and pathological sequelae of mTBI between Tg mice with single versus repetitive mTBI. For example, both injuries resulted in a trend toward acceleration of amyloid production and deposition 9 weeks after mTBI and beyond, whereas there were differences in the extent of histological damage produced by these injuries, because single mTBI induced milder pathology in the neocortex compared with repetitive mTBI, consistent with initial studies of this new model of TBI (Laurer et al., 2001). Furthermore, single injury transiently increased LPO to a lesser extent than the more sustained and greater effect on oxidative stress that followed repetitive mTBI in the Tg mice. Moreover, there were different functional consequences of single versus repetitive mTBI such that impaired cognitive function occurred exclusively in Tg mice subjected to repetitive mTBI at 16 weeks after injury. Finally, it is highly noteworthy that repetitive mTBI augmented key pathological features found in the brains of AD patients, whereas single mTBI failed to do so, and this is consistent with previous epidemiological studies suggesting that the more severe the brain injury, the greater the possibility of developing AD (Plassman et al., 2000). Because there is no similar linkage of Aβ amyloidosis to other forms of brain damage, including stroke, it appears that TBI has unique effects on Aβ metabolism, clearance, or both. However, there is growing evidence to suggest that the risk of developing neurodegenerative disease is enhanced by repetitive brain injury, and although a single head injury might not be sufficient to result in functional impairments, the cumulative effects of multiple traumatic insults to the brain could lead to CNS dysfunction and degeneration (Gentleman et al., 1993; Geddes et al., 1999).
Consistent with previous reports (Nunomura et al., 2000; Praticòet al., 2000a, 2001), the present study supports the notion of a close linkage between elevated levels of brain oxidative stress and the development of a neurodegenerative disorder as well as amyloid pathology, but our study is the first to show that LPO is enhanced by TBI and intimately linked to increased amyloid deposition and accumulation in an experimental model. However, although there are no comparable data on isoprostanes beyond those presented here on the effects of TBI in WT rodents, the assessment of isoprostane isomers such as 8,12-iso-iPF2α-VI can be exploited to analyze the extent of LPO in AD and in animal models of this disorder (Praticò et al., 2001). Indeed, in our previous studies of Tg2576 mice, we showed that isoprostane levels started increasing a few months (i.e., at ∼4 months) before amyloid plaques appeared in brain parenchyma (i.e., at ∼9 months) of these mice, and here we showed that the isoprostane levels were significantly higher in the Tg2576 mice subjected to repetitive mTBI compared with sham-treated Tg mice at 9 weeks after surgery and well before there was significant augmentation of amyloid deposition at 16 weeks after mTBI. The reasons for differences in the levels of isoprostanes in the single versus repetitive mTBI results here in the Tg mice are unclear, but they probably reflect the greater extent of cellular stress or blood–brain barrier damage in the repetitive mTBI experiments. A number of other reports support the hypothesis that oxidative damage is mechanistically involved in AD brain degeneration (Markesbery and Carney, 1999;Praticò and Delanty, 2000). Moreover, our previous study of the same line of Tg mice (Praticò et al., 2001) and other studies of aging Down's syndrome patients (Praticò et al., 2000b) suggest that increasing oxidative stress precedes amyloid deposition in brain. Consistent with these findings, our data here demonstrated that mTBI produced a rapid increase in LPO in the Tg2576 mice, as reflected by measures of brain and urinary 8,12-iso-iPF2α-VI, and this was paralleled by an increase in the production and accumulation of Aβ in brain. Because the effects of TBI extend beyond the site of impact and evolve over many months after an episode of TBI, it is plausible that these diffuse and sustained effects of TBI may be mediated in part by ongoing LPO induced by TBI.
To date, numerous studies have suggested that oxidative stress promotes amyloid aggregation and fibril formation in vivo (Yanagisawa et al., 1995; Koppaka and Axelsen, 2000). From this point of view, high isoprostane levels could indicate that the oxidative damage promotes or reflects Aβ fibrillization, in addition to its putative role in promoting APP processing to favor production of amyloidogenic Aβ peptides. It also has been suggested that fibrillar Aβ could increase oxidative stress (Lorenzo and Yankner, 1994; Hensley et al., 1995;Yatin et al., 1999). Thus, it is plausible that the higher levels of oxidative stress caused by mTBI in the Tg2576 mice could promote APP processing to generate more Aβ, thereby augmenting Aβ fibrillization and fibril aggregation into SPs. Subsequently, as Aβ fibril formation, aggregation, and deposition continue, this might serve to further increase LPO and the overall levels of oxidative stress in brain. However, additional studies are clearly needed to elucidate the precise cascade of events that link oxidative stress to Aβ amyloidosis and brain degeneration in AD. Moreover, although this and other models of AD amyloidosis do not fully recapitulate the complete AD phenotype, and there are numerous other differences in the motor and cognitive abilities in mice and humans, animal models of AD brain pathology provide important experimental systems for elucidating mechanisms of Aβ- and TBI-induced neurodegeneration.
In summary, we tested the hypothesis that brain injury is an environmental risk factor for AD, and we provided critical experimental evidence in support of this notion. Although there have been dramatic advances in understanding the etiology of FAD, ∼90% of AD is sporadic, and it is clear that distinctively different initiating events cause SAD in contrast to genetic mutations that cause FAD. Although multiple genetic and epigenetic factors might act synergistically to predispose individuals to develop SAD (Kurochkin and Goto, 1994; Qiu et al., 1998; Chesneau et al., 2000; Iwata et al., 2000; Vekrellis et al., 2000), the experimental data presented here add considerable credibility to previous indirect epidemiological evidence linking head trauma to mechanisms of AD.
Footnotes
This work was supported by grants from the National Institutes of Health (National Institute on Aging; Head Injury Center Grant P50-NS08803). We thank Takeda Chemical Industries, Ltd., for providing antibodies, Dr. K. Hsiao for Tg2576 mice, Sanjay Kasturi for technical assistance, and colleagues in the Departments of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research, and the Head Trauma Center for assistance and advice regarding the studies described here.
REFERENCES
- 1.Bareyre FM, Saatman KE, Raghupathi R, McIntosh TK. Postinjury treatment with magnesium chloride attenuates cortical damage after traumatic brain injury in rats. J Neurotrauma. 2000;17:1029–1039. doi: 10.1089/neu.2000.17.1029. [DOI] [PubMed] [Google Scholar]
- 2.Bayer TA, Wirths O, Majtenyi K, Hartmann T, Multhaup G, Beyreuther K, Czech C. Key factors in Alzheimer's disease: beta-amyloid precursor protein processing, metabolism and intraneuronal transport. Brain Pathol. 2001;11:1–11. doi: 10.1111/j.1750-3639.2001.tb00376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Callahan MJ, Lipinski WJ, Bian F, Durham RA, Pack A, Walker LC. Augmented senile plaque load in aged female beta-amyloid precursor protein-transgenic mice. Am J Pathol. 2001;158:1173–1177. doi: 10.1016/s0002-9440(10)64064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chesneau V, Vekrellis K, Rosner MR, Selkoe DJ. Purified recombinant insulin-degrading enzyme degrades amyloid beta-protein but does not promote its oligomerization. Biochem J. 2000;351:509–516. [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 6.Felsenstein KM, Hunihan LW, Roberts SB. Altered cleavage and secretion of a recombinant beta-APP bearing the Swedish familial Alzheimer's disease mutation. Nat Genet. 1994;6:251–255. doi: 10.1038/ng0394-251. [DOI] [PubMed] [Google Scholar]
- 7.Fox GB, Fan L, LeVasseur RA, Faden AI. Effect of traumatic brain injury on mouse spatial and nonspatial learning in the Barnes circular maze. J Neurotrauma. 1998;15:1037–1046. doi: 10.1089/neu.1998.15.1037. [DOI] [PubMed] [Google Scholar]
- 8.Geddes JF, Vowles GH, Nicoll JA, Revesz T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol (Berl) 1999;98:171–178. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- 9.Gentleman SM, Graham DI, Roberts GW. Molecular pathology of head trauma: altered beta APP metabolism and the aetiology of Alzheimer's disease. Prog Brain Res. 1993;96:237–246. doi: 10.1016/s0079-6123(08)63270-7. [DOI] [PubMed] [Google Scholar]
- 10.Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316–1323. doi: 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- 11.Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 12.Hensley K, Butterfield DA, Mattson M, Aksenova M, Harris M, Wu JF, Floyd R, Carney J. A model for beta-amyloid aggregation and neurotoxicity based on the free radical generating capacity of the peptide: implications of “molecular shrapnel” for Alzheimer's disease. Proc West Pharmacol Soc. 1995;38:113–120. [PubMed] [Google Scholar]
- 13.Heyman A, Wilkinson WE, Stafford JA, Helms MJ, Sigmon AH, Weinberg T. Alzheimer's disease: a study of epidemiological aspects. Ann Neurol. 1984;15:335–341. doi: 10.1002/ana.410150406. [DOI] [PubMed] [Google Scholar]
- 14.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 15.Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee H-J, Hama E, Sekine-Aizawa Y, Saido TC. Identification of the major A beta (1–42)-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 16.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 17.Jarrett JT, Lansbury PT. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 18.Koppaka V, Axelsen PH. Accelerated accumulation of amyloid beta proteins on oxidatively damaged lipid membranes. Biochemistry. 2000;39:10011–10016. doi: 10.1021/bi000619d. [DOI] [PubMed] [Google Scholar]
- 19.Kurochkin IV, Goto S. Alzheimer's beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994;345:33–37. doi: 10.1016/0014-5793(94)00387-4. [DOI] [PubMed] [Google Scholar]
- 20.Lannfelt L, Bogdanovic N, Appelgren H, Axelman K, Lilius L, Hansson G, Schenk D, Hardy J, Winblad B. Amyloid precursor protein mutation causes Alzheimer's disease in a Swedish family. Neurosci Lett. 1994;168:254–256. doi: 10.1016/0304-3940(94)90463-4. [DOI] [PubMed] [Google Scholar]
- 21.Laurer H, Bareyre FM, Lee VM-Y, Trojanowski JQ, Longhi L, Hoover R, Saatman KE, Raghupathi R, Hoshino S, Grady MS, McIntosh TK. Mild head injury increases the brain's vulnerability to a second traumatic impact. J Neurosurg. 2001;95:859–870. doi: 10.3171/jns.2001.95.5.0859. [DOI] [PubMed] [Google Scholar]
- 22.Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Markesbery WR, Carney JM. Oxidative alterations in Alzheimer's disease. Brain Pathol. 1999;9:133–146. doi: 10.1111/j.1750-3639.1999.tb00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin JB. Molecular basis of the neurodegenerative disorders. N Engl J Med. 1999;340:1970–1980. doi: 10.1056/NEJM199906243402507. [DOI] [PubMed] [Google Scholar]
- 25.McIntosh TK, Noble L, Andrews B, Faden AI. Traumatic brain injury in the rat: characterization of a midline fluid-percussion model. Cent Nerv Syst Trauma. 1987;4:119–134. doi: 10.1089/cns.1987.4.119. [DOI] [PubMed] [Google Scholar]
- 26.McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AL. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- 27.Mills J, Reiner PB. Regulation of amyloid precursor protein cleavage. J Neurochem. 1999;72:443–460. doi: 10.1046/j.1471-4159.1999.0720443.x. [DOI] [PubMed] [Google Scholar]
- 28.Morris RG, Garrud P, Rawlins JN, O'Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- 29.Mortimer JA, French LR, Hutton JT, Schuman LM. Head injury as a risk factor for Alzheimer's disease. Neurology. 1985;35:264–267. doi: 10.1212/wnl.35.2.264. [DOI] [PubMed] [Google Scholar]
- 30.Murai H, Pierce JE, Raghupathi R, Smith DH, Saatman KE, Trojanowski JQ, Lee VM-Y, Loring JF, Eckman C, Younkin S, McIntosh TK. Twofold overexpression of human beta-amyloid precursor proteins in transgenic mice does not affect the neuromotor, cognitive, or neurodegenerative sequelae following experimental brain injury. J Comp Neurol. 1998;392:428–438. doi: 10.1002/(sici)1096-9861(19980323)392:4<428::aid-cne2>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 31.Nakagawa Y, Nakamura M, McIntosh TK, Rodriguez A, Berlin JA, Smith DH, Saatman KE, Raghupathi R, Clemens J, Saido TC, Schmidt ML, Lee VM-Y, Trojanowski JQ. Traumatic brain injury in young, amyloid-beta peptide overexpressing transgenic mice induces marked ipsilateral hippocampal atrophy and diminished Abeta deposition during aging. J Comp Neurol. 1999;411:390–398. [PubMed] [Google Scholar]
- 32.Nakagawa Y, Reed L, Nakamura M, McIntosh TK, Smith DH, Saatman KE, Raghupathi R, Clemens J, Saido TC, Lee VM-Y, Trojanowski JQ. Brain trauma in aged transgenic mice induces regression of established abeta deposits. Exp Neurol. 2000;163:244–252. doi: 10.1006/exnr.2000.7375. [DOI] [PubMed] [Google Scholar]
- 33.Nakamura M, Saatman KE, Galvin JE, Scherbel U, Raghupathi R, Trojanowski JQ, McIntosh TK. Increased vulnerability of NFH-LacZ transgenic mouse to traumatic brain injury-induced behavioral deficits and cortical damage. J Cereb Blood Flow Metab. 1999;19:762–770. doi: 10.1097/00004647-199907000-00006. [DOI] [PubMed] [Google Scholar]
- 34.Nunan J, Small DH. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000;483:6–10. doi: 10.1016/s0014-5793(00)02076-7. [DOI] [PubMed] [Google Scholar]
- 35.Nunomura A, Perry G, Pappolla MA, Friedland RP, Hirai K, Chiba S, Smith MA. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J Neuropathol Exp Neurol. 2000;59:1011–1017. doi: 10.1093/jnen/59.11.1011. [DOI] [PubMed] [Google Scholar]
- 36.Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–369. doi: 10.1016/s0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- 37.Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 38.Praticò D. F(2)-isoprostanes: sensitive and specific non-invasive indices of lipid peroxidation in vivo. Atherosclerosis. 1999;147:1–10. doi: 10.1016/s0021-9150(99)00257-9. [DOI] [PubMed] [Google Scholar]
- 39.Praticò D, Delanty N. Oxidative injury in diseases of the central nervous system: focus on Alzheimer's disease. Am J Med. 2000;109:577–585. doi: 10.1016/s0002-9343(00)00547-7. [DOI] [PubMed] [Google Scholar]
- 40.Praticò D, Lee VM-Y, Trojanowski JQ, Rokach J, FitzGerald GA. Increased F2-isoprostanes in Alzheimer's disease: evidence for enhanced lipid peroxidation in vivo. FASEB J. 1998;12:1777–1783. doi: 10.1096/fasebj.12.15.1777. [DOI] [PubMed] [Google Scholar]
- 41.Praticò D, Clark CM, Lee VM-Y, Trojanowski JQ, Rokach J, FitzGerald GA. Increased 8,12-iso-iPF2α-VI in Alzheimer's disease: correlation of a noninvasive index of lipid peroxidation with disease severity. Ann Neurol. 2000a;48:809–812. [PubMed] [Google Scholar]
- 42.Praticò D, Iuliano L, Amerio G, Tang LX, Rokach J, Sabatino G, Violi F. Down's syndrome is associated with increased 8,12-iso-iPF2α-VI levels: evidence for enhanced lipid peroxidation in vivo. Ann Neurol. 2000b;48:795–798. [PubMed] [Google Scholar]
- 43.Praticò D, Uryu U, Leight S, Trojanowski JQ, Lee VM-Y. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Price DL, Sisodia SS. Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- 45.Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J Biol Chem. 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 46.Raghupathi R, Fernandez SC, Murai H, Trusko SP, Scott RW, Nishioka WK, McIntosh TK. BCL-2 overexpression attenuates cortical cell loss after traumatic brain injury in transgenic mice. J Cereb Blood Flow Metab. 1998;18:1259–1269. doi: 10.1097/00004647-199811000-00013. [DOI] [PubMed] [Google Scholar]
- 47.Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1994;57:419–425. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saido TC. Alzheimer's disease as proteolytic disorders: anabolism and catabolism of beta-amyloid. Neurobiol Aging. 1998;19:S69–S75. doi: 10.1016/s0197-4580(98)00033-5. [DOI] [PubMed] [Google Scholar]
- 49.Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW, McIntosh TK. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Natl Acad Sci USA. 1999;96:8721–8726. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399:A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 51.Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma. 1995;12:169–178. doi: 10.1089/neu.1995.12.169. [DOI] [PubMed] [Google Scholar]
- 52.Smith DH, Chen XH, Pierce JE, Wolf JA, Trojanowski JQ, Graham DI, McIntosh TK. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 1997;14:715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L, Jr, Eckman C, Golde TE, Younkin SG. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 54.Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao AK, Hasegawa M, Mann DM, Hyman BT, Iwatsubo T. Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol. 2000;157:331–339. doi: 10.1016/s0002-9440(10)64544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V, Rosner MR, Selkoe DJ. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J Neurosci. 2000;20:1657–1665. doi: 10.1523/JNEUROSCI.20-05-01657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Dickson DW, Trojanowski JQ, Lee VM-Y. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- 57.Wisniewski T, Lalowski M, Levy E, Marques MR, Frangione B. The amino acid sequence of neuritic plaque amyloid from a familial Alzheimer's disease patient. Ann Neurol. 1994;35:245–246. doi: 10.1002/ana.410350223. [DOI] [PubMed] [Google Scholar]
- 58.Yanagisawa K, Odaka A, Suzuki N, Ihara Y. GM1 ganglioside-bound amyloid beta-protein (A beta): a possible form of preamyloid in Alzheimer's disease. Nat Med. 1995;1:1062–1066. doi: 10.1038/nm1095-1062. [DOI] [PubMed] [Google Scholar]
- 59.Yatin SM, Varadarajan S, Link CD, Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer's amyloid beta-peptide (1–42). Neurobiol Aging. 1999;20:325–330. doi: 10.1016/s0197-4580(99)00056-1. [DOI] [PubMed] [Google Scholar]