

Abstract
Genetic deletion of a single allele of theBDNF gene affects hippocampal LTP and causes several behavioral phenotypes, including deficits in spatial learning. In the developing visual cortex, overexpression of BDNF accelerates the time course of the critical period for monocular deprivation (MD), and exogenous administration of BDNF alters the outcome of MD. We asked whether reduced levels of BDNF could affect visual cortex plasticity by studying long-term potentiation (LTP) induction and the effects of MD in heterozygous BDNF knock-out mice. We found that theta burst stimulation that induced LTP in the layer IV–III pathway of wild-type (wt) mice caused only a transient potentiation in BDNF+/− mice, and that this potentiation vanished in 25 min. In contrast, LTP elicited by stimulation of the white matter (WM), a form of LTP that can be induced only during the critical period, occurred normally in wt and BDNF+/− mice. The effects of MD during the critical period were similar in wt and BDNF+/− mice, indicating that layer IV-evoked, layer III LTP is not required for ocular dominance plasticity. We then asked whether reduction of cortical BDNF levels could prolong the critical period for MD and for the WM-evoked, layer III LTP induction. We found that in adult BDNF+/− mice, WM-evoked, layer III LTP was not inducible, and that the critical period for MD terminated normally. We conclude that deletion of one copy of the BDNF gene selectively impairs LTP of the layer IV–III pathway but does not alter ocular dominance plasticity.
Keywords: ocular dominance, neurotrophins, LTP, visual cortex, TrkB, visual deprivation
Monocular deprivation (MD) during the critical period causes a loss of responsiveness to the deprived eye in visual cortical neurons. Although the effects of MD have been widely studied, knowledge of the molecular mechanisms underlying the plastic changes induced by MD remains elusive. Evidence has emerged that neurotrophins regulate ocular dominance (OD) plasticity (McAllister et al., 1999). For instance, intracortical infusion of excess BDNF disrupts OD columns and counteracts the effects of MD (Cabelli et al., 1995; Galuske et al., 1996; Lodovichi et al., 2000). These actions are accompanied by a significant alteration of functional properties of cortical neurons. Recently, the analysis of a transgenic mouse exhibiting a precocious expression of BDNF in the forebrain has shown that these mice are susceptible to the effects of MD, but that their critical period terminates precociously (Hanover et al., 1999; Huang et al., 1999). A precocious cessation of plasticity is also present for the induction of long-term potentiation (LTP) in the white matter (WM)–layer III pathway, a form of LTP that can be elicited only during the critical period for MD (Kirkwood et al., 1995).
Although these experiments strongly suggest a key role for BDNF in visual cortical plasticity, they do not answer the question of whether endogenous BDNF is required under physiological circumstances for cortical plasticity. Experiments using tyrosine kinase receptor B (TrkB)–IgG scavenging molecules have indicated a role for endogenous TrkB ligands in the development of OD columns (Cabelli et al., 1997) and in cortical synaptic plasticity (Akaneya et al., 1997;Sermasi et al., 2000). However, these observations do not specifically address the role of BDNF, because TrkB–IgG binds BDNF and NT4 with equal affinity; NT4 is an important player in visual cortical plasticity because its administration prevents the anatomical and functional effects of MD (Riddle et al., 1995; Gillespie et al., 2000;Lodovichi et al., 2000).
To directly test the role of endogenous BDNF in visual cortical plasticity, we used mice with a genetic deletion of the BDNFgene (Lyons et al., 1999). Because BDNF null mutant mice die during the first few weeks after birth, we used the heterozygous animals that do not show abnormal mortality. In situ hybridization and RNase protection analysis reveals that BDNF mRNA expression is decreased in BDNF heterozygous mice (Linnarsson et al., 1997) (L. Tessarollo, unpublished observations). These mice present with deficits in feeding behavior, aggressiveness, and spatial learning (Linnarsson et al., 1997; but see Montkowski and Holsboer, 1997; Lyons et al., 1999; Kernie et al., 2000) and show an impaired hippocampal LTP (Korte et al., 1995;Patterson et al., 1996). Therefore, we have investigated whether the deletion of a single allele of BDNF could also affect plasticity in the visual cortex of BDNF+/− mice. We found that BDNF+/− mice have an impairment in LTP of the layer IV–III pathway but normal WM-evoked, layer III LTP. OD plasticity either during or after the critical period was unaffected.
MATERIALS AND METHODS
Animals. BDNF knock-out mice generated in the C57BL/6 genetic background were genotyped by PCR. BDNF+/− and control wild-type (wt) littermates were electrophysiologically analyzed.
In vitro electrophysiology. Brains were rapidly removed and immersed in ice-cold cutting solution containing (in mm): 130 NaCl, 3.1 KCl, 1.0 K2HPO4, 4.0 NaHCO3, 5.0 dextrose, 2.0 MgCl2, 1.0 CaCl2, 10 HEPES, 1.0 ascorbic acid, 0.5 myoinositol, 2 pyruvic acid, and 1 kynurenate, pH 7.3. Slices of visual cortex, 0.35 mm thick, were obtained using a vibratome (Leica, Nussloch, Germany) and transferred to a chamber containing cutting solution. The recording solution was identical to the cutting solution, with the following differences (in mm): 1.0 MgCl2, 2.0 CaCl2, 0.01 glycine, and no kynurenate. Slices were perfused at a rate of 2 ml/min with 35°C oxygenated recording solution. Electrical stimulation (100 μsec duration) was delivered with a bipolar concentric stimulating electrode (Frederick Haer Co., Bowdoinham, ME) placed either at the border between the WM and layer VI or in layer IV. Field potentials in layer III were recorded by means of a glass micropipette (1–3 MΩ) filled with NaCl (3 m). Baseline responses were obtained every 30 sec, with a stimulation intensity that yielded half-maximal responses. The amplitude of the maximum negative field potential in layer III was used as a measure of the evoked population excitatory synaptic current. After achievement of at least a 10 min stable baseline, theta burst stimulation (TBS) was delivered.
In vivo electrophysiology. Electrophysiological procedures were performed as described previously (Lodovichi et al., 2000). Briefly, animals were anesthetized with urethane (0.7 ml/kg , i.p., 20% solution in saline), and temperature was maintained at 37°C. A hole was drilled in the skull in correspondence with the binocular portion of the primary visual cortex (area Oc1B) contralateral to the deprived eye, the dura was removed, and a micropipette (1–3 MΩ) filled with NaCl (3 m) was inserted into the cortex (stereotaxic coordinates, 2.9–3.1 mm from the central fissure). For each animal, 8–10 cells were recorded in each of at least three tracks spaced evenly (>100 μm) across the mediolateral and anteroposterior extent of Oc1B, to avoid sampling bias. Only cells with a receptive field (RF) within 20° from the vertical meridian were included in our sample. Cell properties were determined from peristimulus time histograms (PSTHs) recorded in response to a computer-generated bar, averaged over at least 20 stimulus presentations. Cells were distributed in Wiesel and Hubel (1965) OD classes according to the following criteria: if no response was obtained from the ipsilateral eye, the unit was classified as 1; if the contralateral peak response (highest spiking frequency evoked by visual stimulation) was more than 1.3 times the peak response of the ipsilateral eye, the cell was classified as 2–3. The same criteria were applied for cells in which the ipsilateral eye was exclusively or predominantly driving the cell response; these cells were classified as 7 or 5–6, respectively. When the peak response of the dominant eye was less than 1.3 times the response of the other eye, the cell was classified as 4.
The OD distribution of each animal was summarized using the contralateral bias index (CBI) (Gordon and Stryker, 1996), as follows: CBI = ([(N(1) − N(7)] + 1/2[N(2/3) − N(5/6)] +N(tot))/2N(tot), where N(tot) is the total number of recorded cells and N(i) is the number of cells in class i. RF size was assumed to be the major axis of the area of a visual field eliciting a visual response higher than baseline discharge +2 SD. The baseline spiking frequency was measured in the absence of the visual stimulus.
Data from in vitro and in vivo electrophysiology are reported as averages ± SEM.
RESULTS
Selective impairment of layer IV-evoked, layer III LTP in BDNF+/− mice
To explore the role of BDNF in synaptic plasticity of the visual cortex during the critical period, we studied LTP induction in cortical slices of heterozygous BDNF+/− mice at postnatal day 21 (P21) to P30. A stimulating electrode was placed either in layer IV or in the WM; field potentials were recorded by an electrode positioned in layer III. Stimulus intensity was adjusted to values that elicited responses of amplitude approximately half (53 ± 6.3%) of the maximal response amplitude. Similar half-maximal amplitudes were evoked using equivalent stimulus intensities in wt and BDNF+/− mice (layer IV, 447 ± 20 μV with a stimulus intensity of 222 ± 31 μA for wt and 427 ± 35 μV with a stimulus intensity of 183 ± 21 μA for BDNF+/−; WM, 469 ± 35 μV with a stimulus intensity of 656 ± 273 μA for wt and 495 ± 43 μV with a stimulus intensity of 692 ± 139 μA for BDNF+/−).
In wt animals, TBS of either layer IV or the WM induced LTP of layer III responses. The field potential amplitude 25 min after TBS was 115.3 ± 3.1% of the pre-TBS baseline for the layer IV–III pathway and 126.1 ± 6.8% for the WM–layer III pathway (Fig.1). In contrast, visual cortical slices taken from BDNF+/− mice showed only a transient potentiation in the layer IV–III pathway. Indeed, TBS did induce potentiation, but this potentiation decayed rapidly over time, with synaptic responses completely returning to the baseline level (101.5 ± 1.9%) within 25 min of induction (Fig. 1C). Typical waveforms recorded at different times during the experiment in wt and BDNF+/− mice are shown in Figure 1B,E.
Fig. 1.

BDNF+/− mice show impaired layer IV-evoked but normal WM-evoked layer III LTP. A–C (layers IV–III):A, Average time course of layer III field potential amplitude before and after TBS of layer IV in wt mice (n = 16 slices; 6 mice) and BDNF+/− mice (n = 10 slices; 6 mice). In contrast to wt animals, in which LTP persists for at least 30 min, BDNF+/− animals show a fast decay of response amplitude starting 5 min after TBS and returning to baseline values within 25 min. B, Average of 10 traces recorded from a wt and a BDNF+/− slice before, 3 min after TBS (BDNF+/−), and 25 min after TBS. C, Average and single cases of LTP in wt and BDNF+/− slices 25 min after TBS. BDNF+/− slices significantly differ from wt, showing no LTP at this time (t test). D–F (WM–layer III):D, Average time course of layer III field potential amplitudes before and after TBS of WM in wt mice (n= 6 slices; 2 mice) and BDNF+/− mice (n = 9 slices; 4 mice). Both experimental groups exhibit an LTP of synaptic responses after TBS. E, Average of 10 traces recorded from a wt and a BDNF+/− slice before and 25 min after TBS.F, Average and single cases of LTP in wt and BDNF+/− slices 25 min after TBS. There is no significant difference between the two experimental groups (t test). Solid linesrepresent prebaseline amplitude. Dashed lines show pre-TBS peak level.
In contrast to the decaying potentiation that we found in the layer IV–III pathway of BDNF+/− mice, TBS delivered to the WM of these mice was able to induce a long-lasting potentiation (Fig.1D–F). Figure 1E shows representative traces taken before and 25 min after TBS, when the response amplitude was 124.1 ± 4.5% for BDNF+/− mice. These data indicate that the decrease in BDNF levels causes a significant deficit in layer IV-evoked, layer III LTP while sparing normal LTP in the WM–layer III pathway.
OD plasticity during critical period is normal in BDNF+/− mice
To test the visual cortex plasticity of BDNF+/− mice in vivo, we evaluated the effect of MD during the critical period (Fig. 2). We monocularly deprived wt and BDNF+/− mice for 4–5 d at the peak of sensitivity to MD (P26) (Gordon and Stryker, 1996). At the end of the deprivation period, we assessed OD by comparing PSTHs recorded in response to stimulation of either eye. We found that both groups similarly shifted their OD distributions toward the nondeprived eye (Fig. 1B,C). To summarize the OD of each animal, we adopted the CBI, an index that is 0 for complete ipsilateral dominance and 1 for complete contralateral dominance (see Materials and Methods). CBIs of the two experimental groups were equally reduced by MD (Fig. 1D), demonstrating that there is no difference between wt and BDNF+/− mice in terms of sensitivity to MD during the critical period. We also analyzed several functional properties of cortical neurons such as peak response (wt, 54.9 ± 13.7 spikes/sec; BDNF+/−, 65.3 ± 39.3 spikes/sec), peak to baseline (wt, 11 ± 8.3; BDNF+/−, 8.6 ± 3.6), and receptive field size (wt, 27.9 ± 4.9°; BDNF+/−, 27.7 ± 3°) and found no differences between the two groups (t test). We conclude that the deletion of a singleBDNF allele does not impair the OD plasticity and response properties of cortical neurons during the critical period.
Fig. 2.
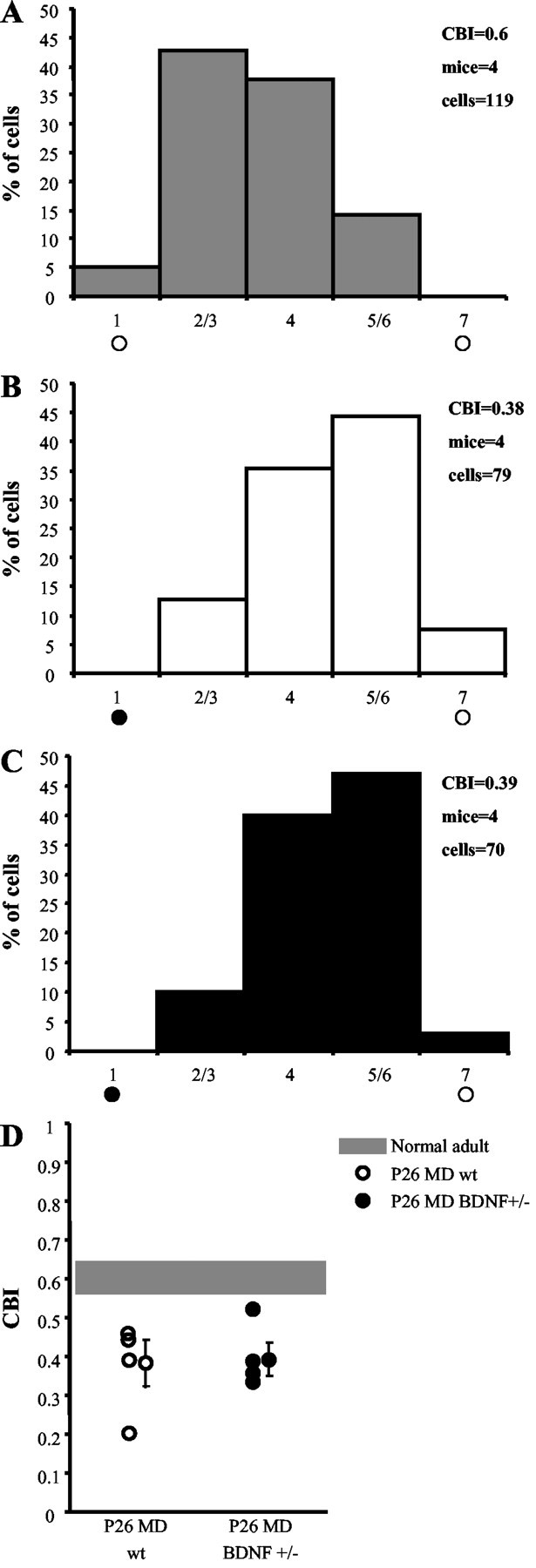
OD plasticity during the critical period is normal in BDNF+/− mice. A, OD distribution of normal wt mice. The CBI indicates a distribution bias in favor of the contralateral eye. Brief MD (4–5 d) beginning at P26 induces a strong OD shift toward the nondeprived eye in P26 MD wt mice (B) as well as in P26 MD BDNF+/− mice (C). D, Scatterplot of single animal CBIs. Left, P26 MD wt mice; right, P26 MD BDNF+/− mice; shaded region, range of normal adult wt mice. One-way ANOVA shows a significant difference (p = 0.012) between the three groups; thepost hoc Tukey test reveals a significant difference between normal adult and P26 MD wt (p = 0.029) or BDNF+/− (p = 0.006) mice but not between P26 MD wt and P26 MD BDNF+/− mice.
Critical period is normal in BDNF+/− mice
Because overexpression of BDNF causes a precocious closure of the critical period for MD and for induction of WM-evoked, layer III LTP (Huang et al., 1999), we investigated whether reduction of cortical BDNF levels was sufficient to prolong the period during which WM-evoked, layer III LTP can be induced and MD is effective.
WM-evoked, layer III LTP is not present in slices from adult (older than P63) wt or BDNF+/− mice. Figure3A shows the average time course of the response amplitudes before and after TBS. Figure3C represents the response amplitude change 25 min after TBS for each experimental slice. The field potential amplitude remained at pre-TBS levels for both wt (103.6 ± 0.9%) and BDNF+/− (104.3 ± 2.0%) mice. Typical traces for wt and BDNF+/− mice before and 25 min after TBS are shown in Figure 3B.
Fig. 3.

WM-evoked, layer III LTP is normal in adult BDNF+/− mice. A, Average time course of layer III field potential amplitude before and after TBS in wt mivr (n = 10 slices; 3 mice) and BDNF+/− mice (n = 10 slices; 4 mice). The field potential amplitude remained at pre-TBS levels in both experimental groups.B, Average of 10 traces recorded from a wt and a BDNF+/− slice before and 25 min after TBS. C, Average and single cases of LTP in wt and BDNF+/− slices 25 min after TBS. There is no significant difference between the two experimental groups (t test). Solid lines represent prebaseline amplitude. Dashed lines show pre-TBS peak level.
To investigate the closure of the critical period for MD in BDNF+/− mice, we recorded adult mice (older than P51) of both genotypes after 4–5 d of MD (Fig. 4). We found that both wt and BDNF+/− mice were insensitive to MD, showing no shift of OD distribution, and we also found CBIs comparable with those of normal (nondeprived) adult mice. Peak response, peak to baseline, and receptive field size were also indistinguishable between monocularly deprived BDNF+/− and wt mice, either deprived or nondeprived (data not shown; t test). To exclude the possibility that the critical period could be slightly prolonged, we assessed the effect of 4–5 d of MD beginning at P35 and P42–P45, when the critical period is ending. At these ages, MD induced a similar slight OD shift in both genotypes (Fig. 4D). Two-way ANOVA on CBIs of all groups (P26, P35, P42–P45, and adults of both genotypes) showed a significant effect of age (p= 0.004) but no significant effect of genotype and no interaction of genotype with age. These data indicate that the time course of the offset of the critical period is normal in BDNF+/− mice.
Fig. 4.

The critical period is normal in BDNF+/− mice. A, In adult wt mice (older than P51), MD does not induce an OD shift. B, In adult BDNF+/− mice (older than P70), MD is also ineffective in inducing an OD shift.C, CBIs of single animals. Left, Adult MD wt mice; right, adult MD BDNF+/− mice; shaded region, nondeprived mice. No significant difference is present between the three groups (one-way ANOVA). D, Developmental regulation of MD effects in wt and BDNF+/− mice. CBIs of wt and BDNF+/− mice monocularly deprived at different ages (P26, P35, P42, adult) for 4–5 d are shown. Both genotypes exhibit the same sensitivity to MD during the critical period, become increasingly less sensitive to MD at the tail of the critical period, and are completely insensitive to MD as adults. Two-way ANOVA on CBIs of all groups (P26, P35, P42, adult; both genotypes) showed a significant effect of age (p = 0.004) but not of genotype, and no significant interaction between age and genotype.
DISCUSSION
The present study shows that reduction by half of BDNF levels in heterozygous BDNF mice did not affect the outcome of MD either during or after the critical period. In contrast, BDNF+/− mice showed different effects on two forms of cortical LTP. Namely, WM-evoked, layer III LTP, a form of LTP known to be coregulated with OD plasticity during the critical period (Kirkwood et al., 1995), was normally inducible in BDNF+/− mice, whereas layer IV-evoked, layer III LTP was impaired. WM-evoked, layer III LTP could not be induced in adult wt and BDNF+/− mice.
LTP deficiency in BDNF+/− mice
The presence of an impairment of layer IV-evoked, layer III LTP in BDNF+/− mice strengthens the notion that BDNF is a central player in the mechanisms of synaptic plasticity. Indeed, a number of experiments have shown an involvement of BDNF in LTP, primarily in the hippocampus (Kang and Schuman, 1995; Ying et al., 2002). Moreover, different lines of BDNF knock-out mice generated independently in different laboratories have shown a severe defect in hippocampal LTP in both homozygous and heterozygous mice (Korte et al., 1995; Patterson et al., 1996). It has been suggested that the LTP impairment present in the hippocampus of BDNF knock-outs is the result of a more pronounced synaptic fatigue at the level of CA1 synapses during high-frequency tetanic stimulation (Pozzo-Miller et al., 1999). In our experiments, we did not find deficits of the response to the TBS used for LTP induction (data not shown). It is possible that either the defect is present in a minority of synapses in the visual cortex or that our TBS is a less challenging stimulation with respect to tetanic stimulation.
Previous studies have shown that BDNF is a powerful regulator of LTP and long-term depression plasticity in the developing visual cortex either in vitro or in vivo (Akaneya et al., 1996, 1997; Huber et al., 1998; Sermasi et al., 2000; Jiang et al., 2001). However, none of these studies has addressed the issue of a selective role of BDNF in LTP of different intracortical pathways. Our study indicates that endogenous BDNF differentially regulates the synaptic plasticity of cortical circuits. Indeed, although LTP could be readily induced by WM stimulation, layer IV-evoked, layer III responses potentiated only transiently after TBS in BDNF+/− mice. This selective impairment of layer IV-evoked, layer III LTP suggests that TBS stimulation of layer IV, but not of WM, activates mechanisms that are strongly dependent on BDNF for the stabilization of LTP. Although the nature of these mechanisms is still unknown, the deficit in layer IV-evoked, layer III LTP present in BDNF+/− mice is not sufficient to alter OD plasticity in vivo. Indeed, BDNF+/− mice exhibited a normal OD shift after MD during the critical period. Thus, BDNF heterozygous mice represent an example in which an OD shift is caused by MD even if LTP of the layer IV–layer III pathway is impaired, suggesting that this form of synaptic plasticity is not a good model for mechanistic studies of OD plasticity.
Normal OD plasticity in BDNF+/− mice
OD plasticity was normal in BDNF heterozygous mice during, at the end of, and after cessation of the critical period, demonstrating a normal maturation of the molecular mechanisms responsible for OD plasticity in BDNF+/− mice. Functional properties of visual cortical neurons were also indistinguishable among the two genotypes. Overall, our in vivo data show that heterozygous BDNF mice exhibit a haplosufficient phenotype for visual cortical development. This differs from other aspects of BDNF function, such as its regulation of feeding behavior, aggressiveness (Lyons et al., 1999;Kernie et al., 2000), and spatial learning (Linnarsson et al., 1997; but see Montkowski and Holsboer, 1997). Indeed, all of these behaviors are affected in BDNF+/− mice. The lack of deficits in OD plasticity in the BDNF+/− mice is surprising considering the data showing that BDNF is an important factor in the cellular mechanisms underlying the developmental plasticity of OD. The exogenous supply of BDNF disrupts OD columns in kittens (Cabelli et al., 1995) and alters the outcome of MD in rats and kittens (Galuske et al., 1996; Lodovichi et al., 2000). The application of TrkB–IgG also affects the anatomical segregation of OD columns in kittens (Cabelli et al., 1997). However, at least some of the effects of TrkB–IgG could be attributable to their action on NT4, which is also bound by TrkB–IgG. NT4 regulates OD plasticity, because its application counteracts the effects of MD in various species (Riddle et al., 1995; Gillespie et al., 2000; Lodovichi et al., 2000). Therefore, it is possible that in BDNF+/− mice the endogenous levels of NT4 are able to compensate for the reduction in BDNF, resulting in a normal phenotype with respect to visual cortical plasticity. Alternatively, BDNF cortical levels, although reduced, are still sufficient for normal plasticity of the visual cortex.
Analyzing various forms of plasticity in heterozygous BDNF+/− mice, we have shown that the reduction of endogenous BDNF affects one form of cortical LTP but does not impair OD plasticity. The generation of conditional cortical knock-outs of BDNF is needed to understand whether the complete elimination of the endogenous BDNF can have a more general effect on the developmental plasticity of the visual cortex.
A.B. and L.C. contributed equally to this paper.
This work was supported by the Cofinanziamento Ministero Istruzione Università e Ricerca, Consiglio Nazionale delle Ricerche targeted project in Biotechnology SP-5, Progetto Strategico Neuroscienze.
Correspondence should be addressed to Dr. Tommaso Pizzorusso, Scuola Normale Superiore, Istituto Neuroscienze Consiglio Nazionale delle Ricerche, via Moruzzi, 1, 56100 Pisa, Italy. E-mail:tommaso@in.pi.cnr.it.
REFERENCES
- 1.Akaneya Y, Tsumoto T, Hatanaka H. Brain-derived neurotrophic factor blocks long-term depression in rat visual cortex. J Neurophysiol. 1996;76:4198–4201. doi: 10.1152/jn.1996.76.6.4198. [DOI] [PubMed] [Google Scholar]
- 2.Akaneya Y, Tsumoto T, Kinoshita S, Hatanaka H. Brain-derived neurotrophic factor enhances long-term potentiation in rat visual cortex. J Neurosci. 1997;17:6707–6716. doi: 10.1523/JNEUROSCI.17-17-06707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 4.Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of trkB inhibits formation of ocular dominance columns. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- 5.Galuske RA, Kim DS, Castren E, Thoenen H, Singer W. Brain-derived neurotrophic factor reversed experience-dependent synaptic modifications in kitten visual cortex. Eur J Neurosci. 1996;8:1554–1559. doi: 10.1111/j.1460-9568.1996.tb01618.x. [DOI] [PubMed] [Google Scholar]
- 6.Gillespie DC, Crair MC, Stryker MP. Neurotrophin-4/5 alters responses and blocks the effect of monocular deprivation in cat visual cortex during the critical period. J Neurosci. 2000;20:9174–9186. doi: 10.1523/JNEUROSCI.20-24-09174.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon JA, Stryker MP. Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci. 1996;16:3274–3286. doi: 10.1523/JNEUROSCI.16-10-03274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hanover JL, Huang ZJ, Tonegawa S, Stryker MP. Brain-derived neurotrophic factor overexpression induces precocious critical period in mouse visual cortex. J Neurosci 19 1999. RC40(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- 10.Huber KM, Sawtell NB, Bear MF. Brain-derived neurotrophic factor alters the synaptic modification threshold in visual cortex. Neuropharmacology. 1998;37:571–579. doi: 10.1016/s0028-3908(98)00050-1. [DOI] [PubMed] [Google Scholar]
- 11.Jiang B, Akaneya Y, Ohshima M, Ichisaka S, Hata Y, Tsumoto T. Brain-derived neurotrophic factor induces long-lasting potentiation of synaptic transmission in visual cortex in vivo in young rats, but not in the adult. Eur J Neurosci. 2001;14:1219–1228. doi: 10.1046/j.0953-816x.2001.01751.x. [DOI] [PubMed] [Google Scholar]
- 12.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 13.Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirkwood A, Lee HK, Bear MF. Co-regulation of long-term potentiation and experience-dependent synaptic plasticity in visual cortex by age and experience. Nature. 1995;375:328–331. doi: 10.1038/375328a0. [DOI] [PubMed] [Google Scholar]
- 15.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linnarsson S, Bjorklund A, Ernfors P. Learning deficit in BDNF mutant mice. Eur J Neurosci. 1997;9:2581–2587. doi: 10.1111/j.1460-9568.1997.tb01687.x. [DOI] [PubMed] [Google Scholar]
- 17.Lodovichi C, Berardi N, Pizzorusso T, Maffei L. Effects of neurotrophins on cortical plasticity: same or different? J Neurosci. 2000;20:2155–2165. doi: 10.1523/JNEUROSCI.20-06-02155.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci USA. 1999;96:15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 20.Montkowski A, Holsboer F. Intact spatial learning and memory in transgenic mice with reduced BDNF. NeuroReport. 1997;8:779–782. doi: 10.1097/00001756-199702100-00040. [DOI] [PubMed] [Google Scholar]
- 21.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 22.Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knock-out mice. J Neurosci. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riddle DR, Lo DC, Katz LC. NT-4-mediated rescue of lateral geniculate neurons from effects of monocular deprivation. Nature. 1995;378:189–191. doi: 10.1038/378189a0. [DOI] [PubMed] [Google Scholar]
- 24.Sermasi E, Margotti E, Cattaneo A, Domenici L. TrkB signalling controls LTP but not LTD expression in the developing rat visual cortex. Eur J Neurosci. 2000;12:1411–1419. doi: 10.1046/j.1460-9568.2000.00014.x. [DOI] [PubMed] [Google Scholar]
- 25.Wiesel TN, Hubel DH. Comparison of the effects of unilateral and bilateral eye closure on cortical unit responses in kittens. J Neurophysiol. 1965;28:1029–1040. doi: 10.1152/jn.1965.28.6.1029. [DOI] [PubMed] [Google Scholar]
- 26.Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci. 2002;22:1532–1540. doi: 10.1523/JNEUROSCI.22-05-01532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]