

Abstract
The ε4 allele of apolipoprotein E (APOE) and traumatic brain injury (TBI) are both risk factors for the development of Alzheimer's disease (AD). These factors may act synergistically, in that APOE4+ individuals are more likely to develop dementia after TBI. Because the mechanism underlying these effects is unclear, we questioned whetherAPOE4 and TBI interact either through effects on amyloid-β (Aβ) or by enhancing cell death/tissue injury. We assessed the effects of TBI in PDAPP mice (transgenic mice that develop AD-like pathology) expressing human APOE3(PDAPP:E3), human APOE4 (PDAPP:E4), or noAPOE (PDAPP:E−/−). Mice were subjected to a unilateral cortical impact injury at 9–10 months of age and allowed to survive for 3 months. Aβ load, hippocampal/cortical volumes, and hippocampal CA3 cell loss were quantified using stereological methods. All of the groups contained mice with Aβ-immunoreactive deposits (56% PDAPP:E4, 20% PDAPP:E3, 75% PDAPP:E−/−), but thioflavine-S-positive Aβ (amyloid) was present only in the molecular layer of the dentate gyrus in the PDAPP:E4 mice (44%). In contrast, our previous studies showed that in the absence of TBI, PDAPP:E3 and PDAPP:E4 mice have little to no Aβ deposition at this age. After TBI, all of the Aβ deposits present in PDAPP:E3 and PDAPP:E−/− mice were diffuse plaques. In contrast to the effect of APOE4 on amyloid, PDAPP:E3, PDAPP:E4, and PDAPP:E−/− mice did not differ in the amount of brain tissue or cell loss. These data support the hypothesis thatAPOE4 influences the neurodegenerative cascade after TBI via an effect on Aβ.
Keywords: Alzheimer's disease, amyloid, APP, traumatic brain injury, apoE, hippocampus
The ε4 allele of apolipoprotein E (APOE) and traumatic brain injury (TBI) are both risk factors for the development of Alzheimer's disease (AD). It was first found that APOE4 was a risk factor for AD in 1993 (Strittmatter et al., 1993), and numerous studies have confirmed this association. Several studies have found that individuals who sustained moderate to severe head injury are more likely to develop dementia and AD (Mayeux et al., 1993, 1995; Plassman et al., 2000). These risk factors appear to act synergistically, in that individuals who areAPOE4+ are even more likely to develop dementia if they sustain TBI at some time in their life. For example,APOE4+ individuals were 10 times more likely to develop AD after TBI than those who were APOE4−, whereasAPOE4 in the absence of injury was associated with only twice the risk (Mayeux et al., 1995; Tang et al., 1996).
Although the mechanisms underlying these effects are unclear, some evidence suggests that both APOE4 and TBI may influence the risk of AD via interactions with the amyloid-β (Aβ) peptide. For example, Aβ deposition can be found in ∼30% of people who die shortly after TBI (Roberts et al., 1991, 1994); a significant percentage of these patients are APOE4+ (Nicoll et al., 1995, 1996). In addition, analysis of CSF from TBI patients revealed elevated levels of Aβ1–42 for up to a week after TBI, in comparison with both controls and AD patients (Raby et al., 1998; Emmerling et al., 2000). In an evaluation of the effect of APOE3, APOE4, or APOE−/− in transgenic (TG) mice after TBI, greater mortality was observed inAPOE4 mice, whereas APOE3 mice exhibited better neurological function between 3 and 11 d after TBI (Sabo et al., 2000). Although APOE3 mice had less tissue loss after TBI than APOE−/− or wild-type mice, there was no significant difference in tissue loss comparing APOE3with APOE4 mice. The mice studied did not express human amyloid precursor protein, so Aβ deposition did not occur (Sabo et al., 2000).
Because the mechanism by which the APOE genotype and TBI interact to influence dementia remains unresolved, we questioned whether APOE4 and TBI interact through effects on Aβ, enhancement of cell death or tissue injury, or both. We used a well characterized model of TBI (Smith et al., 1995) and assessed the histological outcome in PDAPP mice (a TG mouse that develops AD-like pathology) that either lack apoE or express human APOE3 orAPOE4.
MATERIALS AND METHODS
PDAPP mice expressing amyloid precursor protein (APP) with a mutation that causes familial AD (APPV717F) (Games et al., 1995) but lacks the mouse APOE gene (PDAPP+/+, APOE−/−) (Bales et al., 1997) were bred with mice that express human APOEunder control of the glial fibrillary acidic protein (GFAP) promoter (GFAP-APOE3 and GFAP-APOE4) (Sun et al., 1998). The GFAP-APOE3 and GFAP-APOE4 mice were all on a C57BL/6 mouse APOE−/− background. The breeding produced three types of TG mice (Holtzman et al., 2000): PDAPP+/−,APOE−/− (PDAPP:E−/−), PDAPP+/−, APOE3+/− (PDAPP:E3), and PDAPP+/−, APOE4+/− (PDAPP:E4). HumanAPOE3 and APOE4 TG mouse lines used in this experiment were matched for APOE levels (Sun et al., 1998). The mice were subjected to a controlled cortical impact injury (left hemisphere) (Smith et al., 1995) at 9–10 months of age, while under pentobarbital (65 mg/kg) anesthesia. This injury results in underlying cortical damage with shrinkage of the hippocampus and CA3 cell loss. Mortality rates immediately after TBI were similar in all groups (∼25%), resulting in sample sizes of n = 4 PDAPP:E−/−,n = 10 PDAPP:E3, and n = 9 PDAPP:E4. At 12–13 months (3 months after injury), all mice were killed; their brains were removed, fixed, frozen, and sliced into 50 μm coronal sections from the genu of the corpus callosum through the caudal extent of the hippocampus (Holtzman et al., 2000). Three sets of sections, each containing every sixth slice, were collected from each brain. The sections were then mounted and stained with pan anti-Aβ antibody (Biosource, Camarillo, CA), cresyl violet, or 4′,6′-diamidino-2-phenylindole (DAPI). Animals with Aβ-immunoreactive (IR) deposits were further analyzed using the Cavalieri point-counting method with stereological software (Stereo Investigator;MicroBrightField Inc., Colchester, VT) to quantify the area covered by Aβ-IR deposition (Aβ load) as described previously (Holtzman et al., 2000).
To obtain volume estimates of the hippocampus and cortex, cresyl violet-stained sections were analyzed. Hippocampal measurements were taken from every sixth section through the entire structure; cortical measurements were taken from the first three anterior sections containing hippocampal tissue. The cortical region of interest was defined as cortical tissue dorsal to the superior extent of the thalamus. The volumes of the hippocampus and defined cortical region were determined using the Stereo Investigator software. The percentage of hippocampal and cortical tissue lost (ipsilateral vs contralateral to injury) was calculated for each group.
In most animals, the inferior blade of the CA3 hippocampal field exhibited a focal lesion ipsilateral to the injury, also noted in rodent models of TBI (Nakagawa et al., 1999). CA3 neuronal counts were obtained using DAPI-stained sections through the entire extent of the hippocampus. A randomly selected subset of brains was analyzed (n = 4 PDAPP:E−/−; n = 5 PDAPP:E3;n = 5 PDAPP:E4), and measurements were taken from all sections that contained CA3 neurons. In each section, the inferior blade of CA3 was traced using the computer. Estimates of neuronal numbers were obtained with the optical fractionator technique using Stereo Investigator software. The area was traced with a 4× lens, and neurons were counted throughout the traced area using systematic random sampling with a 100× lens.
The data were analyzed using Statistica 6.0 (Statsoft Inc., Tulsa, OK), and α levels of p < 0.05 were set for significance. The frequency of Aβ deposition was analyzed using χ2 analysis. The percentage of the hippocampus covered by Aβ-IR was analyzed with a two-way ANOVA that included one between-subjects factor (genotype: PDAPP:E3 vs PDAPP:E4 vs PDAPP:E−/−) and one within-subjects factor (hemisphere: ipsilateral vs contralateral to impact). Cortical and hippocampal volume estimate data were analyzed with two-way ANOVAs that included one between-subjects factor (genotype: PDAPP:E3 vs PDAPP:E4 vs PDAPP:E−/−) and one within-subjects factor (hemisphere: ipsilateral vs contralateral to impact). CA3 neuronal counts were analyzed with a two-way ANOVA that included one between-subjects factor (genotype: PDAPP:E3 vs PDAPP:E4 vs PDAPP:E−/−) and one within-subjects factor (hemisphere: ipsilateral vs contralateral to impact).
RESULTS
Aβ analysis
Frequency and pattern of Aβ deposition
We found previously that PDAPP mice expressing humanAPOE3 or APOE4 do not develop Aβ deposition until ∼15 months of age, when PDAPP:E4 mice in particular begin depositing Aβ and amyloid (Holtzman et al., 2000; Fagan et al., 2002). In contrast, after TBI, we found that a high percentage of brain-injured PDAPP:E4 mice had Aβ deposition by 12–13 months of age. In the PDAPP:E4 mice, 55.6% had Aβ-IR deposits within the hippocampus and 44% had thioflavine-S-positive Aβ (fibrillar amyloid) in the molecular layer (ML) of the dentate gyrus (Table1). Among the PDAPP:E3 mice, only 20% had hippocampal Aβ-IR deposits, all of which were diffuse plaques. No PDAPP:E3 mice had fibrillar amyloid deposition. Significantly more PDAPP:E4 mice had ML Aβ-IR deposits compared with PDAPP:E3 mice (χ2, p < 0.02) (Fig.1). This is notable because Aβ deposition in the ML of PDAPP mice coincides with the onset of fibrillar Aβ deposition and neuritic plaque formation (Holtzman et al., 2000; Fagan et al., 2002). Thus, only the PDAPP:E4 mice developed neuritic plaque formation after TBI at this age. Assessment of Aβ40 and Aβ42immunostaining of PDAPP:E3 and PDAPP:E4 mice revealed the same pattern of staining as that seen with the pan-Aβ antibody (data not shown). Qualitatively, the same differences between PDAPP:E3 and PDAPP:E4 mice were noted with these antibodies. As in previous studies with PDAPP and other human APP TG mice, neurofibrillary tangles were not seen. Because PDAPP:E3 and PDAPP:E4 mice have little to no Aβ deposits at 12–13 months of age in the absence of TBI (Holtzman et al., 2000; Fagan et al., 2002), TBI appears to accelerate Aβ deposition in the form of amyloid in the presence of human APOE4 to a greater extent than APOE3. Consistent with previous reports (Holtzman et al., 2000; Fagan et al., 2002), three of the four PDAPP:E−/− mice had hippocampal diffuse Aβ-IR deposits at 12 months of age; however, none were fibrillar.
Table 1.
Histological analysis after TBI
| PDAPP:E3 | PDAPP:E4 | PDAPP:E−/− | p < 0.05 | |
|---|---|---|---|---|
| Sample size | 10 | 9 | 4 | |
| Percentage of cortical loss | 24.9 | 21.4 | 21.9 | No |
| Percentage of hippocampal loss | 9.2 | 9.3 | 8.5 | No |
| Percentage of CA3 inferior blade neuron loss | 35.7 | 30.0 | 26.6 | No |
| Percentage of group with Aβ-IR deposits | 20 | 55.6 | 75 | No |
| Percentage of group with molecular layer Aβ-IR deposits | 0 | 44.4 | 0 | Yes |
| Contralateral to TBI: hippocampal Aβ load (%) | 0.1 | 0.9 | 13.8 | Yes |
| Ipsilateral to TBI: hippocampal Aβ load (%) | 0.1 | 2.3 | 10.3 | Yes |
| Contralateral to TBI: % of total Aβ in molecular layer | 0 | 28.1 | 0 | Yes |
| Ipsilateral to TBI: % of total Aβ in molecular layer | 0 | 29.8 | 0 | Yes |
Fig. 1.

A, Almost one-third of the total hippocampal Aβ load was contained in the ML of the dentate gyrus in PDAPP:E4 mice. Localization of Aβ deposition in the ML is associated with the formation of fibrillar amyloid. In contrast, no ML Aβ-IR deposits were found in PDAPP:E3 or PDAPP:E−/− mice 3 months after TBI. B, Photomicrographs show Aβ staining in the hippocampus (arrowheads delineate the borders of the ML).
Hippocampal Aβ load
Analysis of all three groups revealed no significant difference between the two hemispheres in the amount of Aβ immunoreactivity. There was a significant main effect of genotype in that, consistent with previous reports (Holtzman et al., 2000; Fagan et al., 2002), PDAPP:E−/− mice had a significantly greater Aβ load than PDAPP:E3 or PDAPP:E4 mice (p < 0.0001). However, the Aβ deposits present in PDAPP:E−/− mice consisted of only thioflavine-S-negative, diffuse Aβ (i.e., nonfibrillar, nonamyloid deposits). The amount of diffuse Aβ in PDAPP:E−/− mice after TBI was not clearly increased compared with PDAPP:E−/− mice in the absence of TBI (Holtzman et al., 2000; Fagan et al., 2002). A separate PDAPP:E3 versus PDAPP:E4 analysis revealed a significant main effect of genotype (PDAPP:E4 > PDAPP:E3; p < 0.05) but no significant hemisphere effect. Approximately 35% of the hippocampal Aβ load in PDAPP:E4 mice was contained within the ML, whereas no PDAPP:E3 or PDAPP:E−/− mice had ML deposition. Analysis of the percentage of Aβ deposition within the ML of the dentate gyrus revealed a significant main effect of genotype (PDAPP:E4 > PDAPP:E3 and PDAPP:E−/−; p < 0.006) (Fig. 1).
Cortical and hippocampal volume estimates
Cortex
After TBI, the cortical volume ipsilateral to impact was significantly less than the contralateral, nonimpacted hemisphere for all groups (p < 0.0001) (Fig.2). A main effect of genotype revealed that PDAPP:E4 mice had slightly but significantly more cortical tissue bilaterally than PDAPP:E3 or PDAPP:E−/− mice, which did not differ (p < 0.001). The hemisphere–genotype interaction was not significant. The percentage of tissue loss in the cortex ipsilateral versus contralateral to injury revealed no significant genotype differences.
Fig. 2.

A, No group differences were found for percentage of area loss in either the cortex or the hippocampus.B, Photomicrograph shows a cresyl violet-stained brain section with atrophy of the cortex and hippocampus in the injured hemisphere 3 months after TBI.
Hippocampus
After TBI, the hippocampus ipsilateral to impact was significantly smaller than the contralateral hippocampus for all groups (p < 0.008) (Fig. 2). A main effect of the genotype revealed that PDAPP:E3 mice had slightly but significantly less overall hippocampal tissue bilaterally than PDAPP:E4 or PDAPP:E−/− mice, which did not differ (p < 0.001). The hemisphere–genotype interaction was not significant. The percentage of tissue loss in the hippocampus ipsilateral versus contralateral to injury revealed no significant genotype differences.
Neuronal counts (CA3 inferior blade)
After TBI, the inferior blade of CA3 ipsilateral to injury had significantly fewer neurons (∼35% less) than the uninjured hemisphere for all groups (p < 0.0005) (Fig.3). There were no significant genotype main effects or interactions. The percentage of CA3 cell loss revealed no significant genotype differences.
Fig. 3.
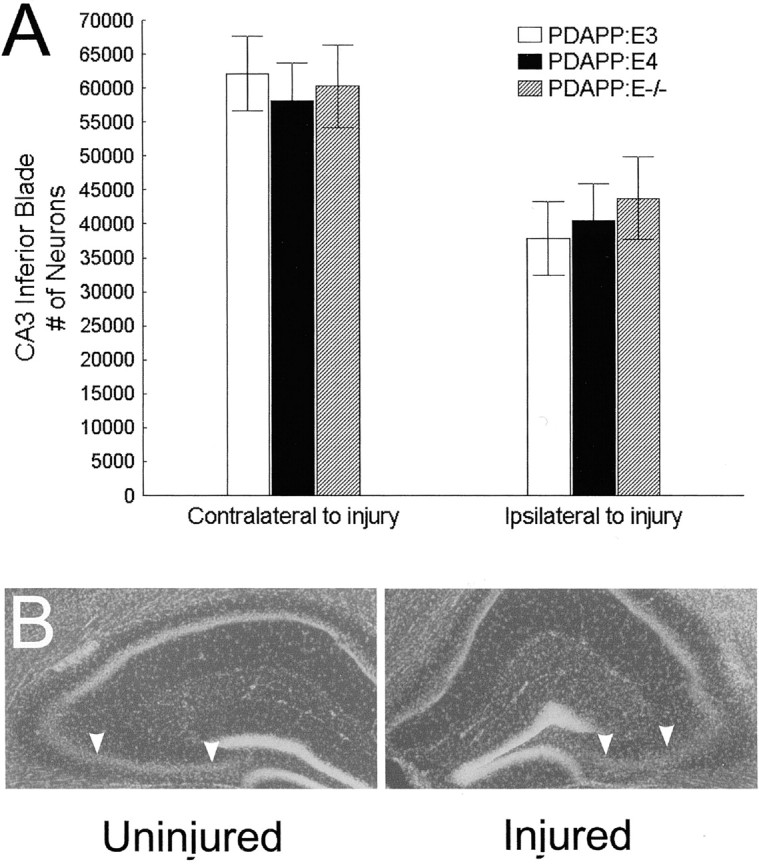
A, No group differences were found for neuronal loss within the inferior blade of CA3. B, Photomicrograph shows a DAPI-stained brain section revealing atrophy of the CA3 region (arrowheads delineate the borders of the CA3 inferior blade) 3 months after TBI.
DISCUSSION
Our previous work (Holtzman et al., 2000; Fagan et al., 2002) demonstrated that Aβ deposition in PDAPP mice that express humanAPOE normally does not begin until ∼15 months of age, ≥6 months later than in animals expressing murine apoE. The appearance of Aβ-IR deposits by 12–13 months in the current study suggests that TBI accelerated the Aβ deposition process in the presence of human APOE4. Furthermore, only PDAPP:E4 mice had significant Aβ-IR deposits in the ML of the dentate gyrus within 3 months of TBI. These ML deposits are associated with thioflavine-S-positive staining, indicating the conversion of soluble Aβ to a β-sheet conformation and neuritic plaque formation. As in our previous studies, PDAPP:E−/− mice had higher levels of Aβ deposition than PDAPP:E3 or PDAPP:E4 mice, yet none of these deposits consisted of true amyloid. Thus, although the presence of APOE facilitates Aβ fibril formation, human APOE is likely also to play a role in Aβ clearance. Our results suggest that TBI andAPOE4 (compared with APOE3) interact to result in greater and earlier amyloid deposition. Overall, these data suggest that the association with APOE4 and higher risk for cognitive impairment and AD after TBI may in part be attributable to APOE–Aβ interactions.
Human studies have shown that both short- and long-term recovery from TBI seem to be influenced by APOE. APOE4+ individuals scored significantly worse on neuropsychological tests 3 weeks after mild to moderate TBI than APOE4− individuals (Liberman et al., 2002), and APOE4 was predictive of longer periods of unconsciousness and worse clinical outcome after TBI (Friedman et al., 1999). Furthermore, APOE4+ individuals were twice as likely as APOE4− individuals to be dead, comatose, or severely disabled 6 months after TBI (Teasdale et al., 1997). In addition to the poor general clinical outcome associated with APOE4, memory performance within 6 months of head injury was worse inAPOE4+ patients compared with APOE4− patients (Crawford et al., 2002), whereas APOE4 led to worse motor function after TBI (Lichtman et al., 2000). Mild, repetitive head injury also appears to interact with APOE. APOE4+ professional boxers had significantly worse neurological scores on a test of chronic brain injury that encompassed cognitive, motor, and behavioral domains than boxers who were APOE4− (Jordan et al., 1997). Similarly, older APOE4+ professional football players scored lower on cognitive tests than APOE4− players (Kutner et al., 2000).
Clinical and experimental TBI is also associated with accelerated Aβ deposition (Roberts et al., 1991), with an even greater effect observed in APOE4+ individuals on both parenchymal and vascular Aβ deposits (Nicoll et al., 1995, 1996; Macfarlane et al., 1999; Leclercq et al., 2002). Aβ deposition is also accelerated after seizure-induced neurodegeneration, even in young APOE4+ subjects (Gouras et al., 1997). In addition to human studies, the effects of TBI on Aβ and AD pathology have also been studied using TG mouse models of AD. Smith et al. (1998) reported that TBI in PDAPP mice resulted in an 84% loss of CA3 neurons compared with only a 36% loss in non-TG mice. Nakagawa et al. (1999, 2000) have reported that TBI in both young and old PDAPP mice induces atrophy and reduces Aβ deposition in the ipsilateral versus contralateral hippocampus. Aβ deposition after repetitive brain injury using different APP TG mice (Tg2576) and milder cortical impact has also been reported (Uryu et al., 2002). Our study extends these findings and demonstrates the amyloid-promoting effects of human APOE4.
How TBI results in an isoform-dependent increase in amyloid deposition is not clear. Both in vitro and in vivo studies demonstrate that APOE can interact with Aβ and influences the probability of whether Aβ will aggregate in a β-sheet conformation, resulting in neuritic toxicity (for review, see Wisniewski et al., 1997; Holtzman, 2001). The level of apoE plays a significant role in this effect, because mouse apoE regulates Aβ deposition in a gene dose-dependent manner in vivo (Bales et al., 1997). The effects of TBI on APOE–Aβ interactions may be secondary to an increase in APOE levels after TBI as well as alterations in APOE-dependent Aβ clearance. An increase in APOE levels has been noted after multiple types of brain injury coincident with glial activation (Teter, 2000). In addition to neuronal degeneration, there is cellular reorganization with increased gliosis and alterations in the vasculature. APOE can potentially interact with different apoE receptors as well as the extracellular matrix. Because both of these factors change in regions of injury, APOE-mediated Aβ clearance may be reduced after TBI, thereby favoring amyloid deposition. It is interesting that amyloid deposits were increased not unilaterally but bilaterally after TBI in the presence of APOE4. This suggests that mechanisms such as changes in APOE expression and alterations in APOE-dependent clearance are likely to occur bilaterally in this model of TBI.
The current study, in which the only known difference between the groups of PDAPP mice was the presence or absence of human APOE isoforms, provides evidence that isoform-specific APOE–Aβ interactions contribute to the premature development of AD pathology. Although the promotion of amyloid deposition per se is unlikely to lead to accelerated dementia, the neuritic dystrophy associated with amyloid as well as other events coincident with or downstream of amyloid formation in humans are likely to contribute to cognitive dysfunction. These processes include Aβ oligomer formation, tangle formation, cell loss, and synaptic loss. Some in vivo studies have found that apoE influences aspects of brain function and plasticity after different forms of injury (Fagan et al., 1998; Sheng et al., 1998;Stone et al., 1998; Buttini et al., 1999; Genis et al., 2000; Sabo et al., 2000), including TBI (Chen et al., 1997), and it is possible that APOE influences the outcome after different forms of brain injury via more than one mechanism. Our data suggest that understanding the mechanism(s) by which TBI promotes APOE isoform-dependent amyloid deposition will lead to important insights into how accelerated Aβ-related AD-like changes occur and potential ways to prevent it.
Footnotes
This work was supported by National Institutes of Health Grants AG13956, AG05681, AG11355 (D.M.H.), DA07261 (R.E.H.), and NS08803; a Veterans Administration Merit Review; and National Football League Charities (T.K.M.)
Correspondence should be addressed to Dr. David M. Holtzman, Washington University School of Medicine, Department of Neurology 660 South Euclid Avenue, Box 8111, St. Louis, MO 63110. E-mail:holtzman@neuro.wustl.edu.
REFERENCES
- 1.Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM. Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nat Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 2.Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW. Expression of human apolipoprotein E3 or E4 in the brains of Apoe −/− mice: isoform-specific effects on neurodegeneration. J Neurosci. 1999;19:4867–4880. doi: 10.1523/JNEUROSCI.19-12-04867.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y, Lomnitski L, Michaelson DM, Shohami E. Motor and cognitive deficits in apolipoprotein E-deficient mice after closed head injury. Neuroscience. 1997;80:1255–1262. doi: 10.1016/s0306-4522(97)00007-9. [DOI] [PubMed] [Google Scholar]
- 4.Crawford FC, Vanderploeg RD, Freeman MJ, Singh S, Waisman M, Michaels L, Abdullah L, Warden D, Lipsky R, Salazar A, Mullan MJ. APOE genotype influences acquisition and recall following traumatic brain injury. Neurology. 2002;58:1115–1118. doi: 10.1212/wnl.58.7.1115. [DOI] [PubMed] [Google Scholar]
- 5.Emmerling MR, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM, Mehta PD, Spiegel K, Kuo YM, Roher AE, Raby CA. Traumatic brain injury elevates the Alzheimer's amyloid peptide Aβ42 in human CSF: a possible role for nerve cell injury. Ann NY Acad Sci. 2000;903:118–122. doi: 10.1111/j.1749-6632.2000.tb06357.x. [DOI] [PubMed] [Google Scholar]
- 6.Fagan AM, Murphy BA, Patel SN, Kilbridge JF, Mobley WC, Bu G, Holtzman DM. Evidence for normal aging of the septo-hippocampal cholinergic system in apoE (−/−) mice but impaired clearance of axonal degeneration products following injury. Exp Neurol. 1998;151:314–325. doi: 10.1006/exnr.1998.6818. [DOI] [PubMed] [Google Scholar]
- 7.Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine apoE markedly influence Aβ metabolism both prior and subsequent to plaque formation in a mouse model of Alzheimer's disease. Neurobiol Dis. 2002;9:305–318. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- 8.Friedman G, Froom P, Sazbon L, Grinblatt I, Shochina M, Tsenter J, Babaey S, Yehuda B, Groswasser Z. Apolipoprotein E-ε4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology. 1999;52:244–248. doi: 10.1212/wnl.52.2.244. [DOI] [PubMed] [Google Scholar]
- 9.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 10.Genis L, Chen Y, Shohami E, Michaelson DM. Tau hyperphosphorylation in apolipoprotein E-deficient and control mice after closed head injury. J Neurosci Res. 2000;60:559–564. doi: 10.1002/(SICI)1097-4547(20000515)60:4<559::AID-JNR15>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 11.Gouras GK, Relkin NR, Sweeney D, Munoz DG, Mackenzie IR, Gandy S. Increased apolipoprotein Eε4 in epilepsy with senile plaques. Ann Neurol. 1997;41:402–404. doi: 10.1002/ana.410410317. [DOI] [PubMed] [Google Scholar]
- 12.Holtzman DM. Role of apoE/Aβ interactions in the pathogenesis of Alzheimer's disease and cerebral amyloid angiopathy. J Mol Neurosci. 2001;17:147–155. doi: 10.1385/JMN:17:2:147. [DOI] [PubMed] [Google Scholar]
- 13.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jordan BD, Relkin NR, Ravdin LD, Jacobs AR, Bennett A, Gandy S. Apolipoprotein Eε4 associated with chronic traumatic brain injury in boxing. JAMA. 1997;278:136–140. [PubMed] [Google Scholar]
- 15.Kutner KC, Erlanger DM, Tsai J, Jordan B, Relkin NR. Lower cognitive performance of older football players possessing apolipoprotein Eε4. Neurosurgery. 2000;47:651–657. doi: 10.1097/00006123-200009000-00026. [DOI] [PubMed] [Google Scholar]
- 16.Leclercq PD, Graham DI, Nicoll JA, Gentleman SM. Influence of ApoE genotype on cerebral amyloid angiopathy after closed head-injury. Neuropathol Appl Neurobiol. 2002;28:161–162. [Google Scholar]
- 17.Liberman JN, Stewart WF, Wesnes K, Troncoso J. Apolipoprotein Eε4 and short-term recovery from predominantly mild brain injury. Neurology. 2002;58:1038–1044. doi: 10.1212/wnl.58.7.1038. [DOI] [PubMed] [Google Scholar]
- 18.Lichtman SW, Seliger G, Tycko B, Marder K. Apolipoprotein E and functional recovery from brain injury following postacute rehabilitation. Neurology. 2000;55:1536–1539. doi: 10.1212/wnl.55.10.1536. [DOI] [PubMed] [Google Scholar]
- 19.Macfarlane DP, Nicoll JA, Smith C, Graham DI. APOEε4 allele and amyloid β-protein deposition in long term survivors of head injury. NeuroReport. 1999;10:3945–3948. doi: 10.1097/00001756-199912160-00040. [DOI] [PubMed] [Google Scholar]
- 20.Mayeux R, Ottman R, Tang MX, Noboa-Bauza L, Marder K, Gurland B, Stern Y. Genetic susceptibility and head injury as risk factors for Alzheimer's disease among community-dwelling elderly persons and their first-degree relatives. Ann Neurol. 1993;33:494–501. doi: 10.1002/ana.410330513. [DOI] [PubMed] [Google Scholar]
- 21.Mayeux R, Ottman R, Maestre G, Ngai C, Tang MX, Ginsberg H, Chun M, Tycko B, Shelanski M. Synergistic effects of traumatic head injury and apolipoprotein-ε4 in patients with Alzheimer's disease. Neurology. 1995;45:555–557. doi: 10.1212/wnl.45.3.555. [DOI] [PubMed] [Google Scholar]
- 22.Nakagawa Y, Nakamura M, McIntosh TK, Rodriguez A, Berlin JA, Smith DH, Saatman KE, Raghupathi R, Clemens J, Saido TC, Schmidt ML, Lee VM, Trojanowski JQ. Traumatic brain injury in young, amyloid-β peptide overexpressing transgenic mice induces marked ipsilateral hippocampal atrophy and diminished Aβ deposition during aging. J Comp Neurol. 1999;411:390–398. [PubMed] [Google Scholar]
- 23.Nakagawa Y, Reed L, Nakamura M, McIntosh TK, Smith DH, Saatman KE, Raghupathi R, Clemens J, Saido TC, Lee VM, Trojanowski JQ. Brain trauma in aged transgenic mice induces regression of established aβ deposits. Exp Neurol. 2000;163:244–252. doi: 10.1006/exnr.2000.7375. [DOI] [PubMed] [Google Scholar]
- 24.Nicoll JA, Roberts GW, Graham DI. Apolipoprotein Eε4 allele is associated with deposition of amyloid β-protein following head injury. Nat Med. 1995;1:135–137. doi: 10.1038/nm0295-135. [DOI] [PubMed] [Google Scholar]
- 25.Nicoll JA, Roberts GW, Graham DI. Amyloid β-protein, APOE genotype and head injury. Ann NY Acad Sci. 1996;777:271–275. doi: 10.1111/j.1749-6632.1996.tb34431.x. [DOI] [PubMed] [Google Scholar]
- 26.Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 27.Raby CA, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM, Mehta PD, Spiegel K, Kuo YM, Roher AE, Emmerling MR. Traumatic brain injury increases β-amyloid peptide 1–42 in cerebrospinal fluid. J Neurochem. 1998;71:2505–2509. doi: 10.1046/j.1471-4159.1998.71062505.x. [DOI] [PubMed] [Google Scholar]
- 28.Roberts GW, Gentleman SM, Lynch A, Graham DI. βA4 amyloid protein deposition in brain after head trauma. Lancet. 1991;338:1422–1423. doi: 10.1016/0140-6736(91)92724-g. [DOI] [PubMed] [Google Scholar]
- 29.Sabo T, Lomnitski L, Nyska A, Beni S, Maronpot RR, Shohami E, Roses AD, Michaelson DM. Susceptibility of transgenic mice expressing human apolipoprotein E to closed head injury: the allele E3 is neuroprotective whereas E4 increases fatalities. Neuroscience. 2000;101:879–884. doi: 10.1016/s0306-4522(00)00438-3. [DOI] [PubMed] [Google Scholar]
- 30.Sheng H, Laskowitz DT, Bennett E, Schmechel DE, Bart RD, Saunders AM, Pearlstein RD, Roses AD, Warner DS. Apolipoprotein E isoform-specific differences in outcome from focal ischemia in transgenic mice. J Cereb Blood Flow Metab. 1998;18:361–366. doi: 10.1097/00004647-199804000-00003. [DOI] [PubMed] [Google Scholar]
- 31.Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma. 1995;12:169–178. doi: 10.1089/neu.1995.12.169. [DOI] [PubMed] [Google Scholar]
- 32.Smith DH, Nakamura M, McIntosh TK, Wang J, Rodriguez A, Chen XH, Raghupathi R, Saatman KE, Clemens J, Schmidt ML, Lee VM, Trojanowski JQ. Brain trauma induces massive hippocampal neuron death linked to a surge in β-amyloid levels in mice overexpressing mutant amyloid precursor protein. Am J Pathol. 1998;153:1005–1010. doi: 10.1016/s0002-9440(10)65643-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stone DJ, Rozovsky I, Morgan TE, Anderson CP, Finch CE. Increased synaptic sprouting in response to estrogen via an apolipoprotein E-dependent mechanism: implications for Alzheimer's disease. J Neurosci. 1998;18:3180–3185. doi: 10.1523/JNEUROSCI.18-09-03180.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Y, Wu S, Bu G, Onifade MK, Patel SN, LaDu MJ, Fagan AM, Holtzman DM. GFAP-apoE transgenic mice: astrocyte specific expression and differing biological effects of astrocyte-secreted apoE3 and apoE4 lipoproteins. J Neurosci. 1998;18:3261–3272. doi: 10.1523/JNEUROSCI.18-09-03261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang MX, Maestre G, Tsai WY, Liu XH, Feng L, Chung WY, Chun M, Schofield P, Stern Y, Tycko B, Mayeux R. Effect of age, ethnicity, and head injury on the association between APOE genotypes and Alzheimer's disease. Ann NY Acad Sci. 1996;802:6–15. doi: 10.1111/j.1749-6632.1996.tb32593.x. [DOI] [PubMed] [Google Scholar]
- 37.Teasdale GM, Nicoll JAR, Murray G, Fiddes M. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet. 1997;350:1069–1071. doi: 10.1016/S0140-6736(97)04318-3. [DOI] [PubMed] [Google Scholar]
- 38.Teter B. Apolipoprotein E isotype-specific effects in neurodegeneration. Alzheimer Rep. 2000;3:199–212. [Google Scholar]
- 39.Uryu K, Laurer H, McIntosh T, Pratico D, Martinez D, Leight S, Lee VM, Trojanowski JQ. Repetitive mild brain trauma accelerates Aβ deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci. 2002;22:446–454. doi: 10.1523/JNEUROSCI.22-02-00446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wisniewski T, Ghiso J, Frangione B. Biology of Aβ amyloid in Alzheimer's disease. Neurobiol Dis. 1997;4:313–328. doi: 10.1006/nbdi.1997.0147. [DOI] [PubMed] [Google Scholar]