

Abstract
GABA transporter subtype 1 (GAT1) molecules were counted near GABAergic synapses, to a resolution of ∼0.5 μm. Fusions between GAT1 and green fluorescent protein (GFP) were tested in heterologous expression systems, and a construct was selected that shows function, expression level, and trafficking similar to that of wild-type (WT) GAT1. A strain of knock-in mice was constructed that expresses this mGAT1–GFP fusion in place of the WT GAT1 gene. The pattern of fluorescence in brain slices agreed with previous immunocytochemical observations. [3H]GABA uptake, synaptic electrophysiology, and subcellular localization of the mGAT1–GFP construct were also compared with WT mice. Quantitative fluorescence microscopy was used to measure the density of mGAT1–GFP at presynaptic structures in CNS preparations from the knock-in mice. Fluorescence measurements were calibrated with transparent beads and gels that have known GFP densities. Surface biotinylation defined the fraction of transporters on the surface versus those in the nearby cytoplasm. The data show that the presynaptic boutons of GABAergic interneurons in cerebellum and hippocampus have a membrane density of 800–1300 GAT1 molecules per square micrometer, and the axons that connect boutons have a linear density of 640 GAT1 molecules per micrometer. A cerebellar basket cell bouton, a pinceau surrounding a Purkinje cell axon, and a cortical chandelier cell cartridge carry 9000, 7.8 million, and 430,000 GAT1 molecules, respectively; 61–63% of these molecules are on the surface membrane. In cultures from hippocampus, the set of fluorescent cells equals the set of GABAergic interneurons. Knock-in mice carrying GFP fusions of membrane proteins provide quantitative data required for understanding the details of synaptic transmission in living neurons.
Keywords: GABA, synapse, transporter, green fluorescent protein, mouse, knock-in
When uptake by the GABA transporter GAT1 is inhibited, evoked IPSCs are prolonged at some synapses, presumably because transmitter molecules remain near receptors and reactivate them (Dingledine and Korn, 1985; Roepstorff and Lambert, 1992, 1994; Thompson and Gahwiler, 1992; Draguhn and Heinemann, 1996;Engel et al., 1998). However, miniature GABAergic IPSCs (mIPSCs) are not prolonged by GAT1 blockers. GAT1 also limits “spillover” to receptors outside the immediate synaptic cleft during repetitive presynaptic stimulation (Isaacson et al., 1993). In another spillover phenomenon, GAT1 inhibition produces tonic GABAAcurrents at high-affinity extrasynaptic receptors in hippocampus (Frahm et al., 2001; Overstreet and Westbrook, 2001; Tang et al., 2001) and cerebellum (Brickley et al., 1996).
Understanding the role of GABA uptake in these and other aspects of GABAergic transmission requires knowing key parameters and mechanisms. Quantitative studies of local GABAA receptor number (Nusser et al., 1997, 1998a,b) and of GABA receptor kinetics (Jones and Westbrook, 1995) are available. Although substantial mechanistic knowledge is available about the molecular physiology of the most widespread GABA transporter in the CNS, GAT1 (Guastella et al., 1990; Mager et al., 1993; Hilgemann and Lu, 1999), little is known about the density of this molecule at actual synapses. Detailed simulations of GABAergic transmission have assumed densities on the order of 500/μm2 (Jones and Westbrook, 1995; Jones et al., 1998, 1999).
This study was conducted to quantify the number and density of GAT1 near synapses. We constructed a strain of knock-in mice that express a GAT1–green fluorescent protein (GFP) fusion in place of the wild-type (WT) GAT1 gene. We calibrated the fluorescence measurements by exploiting previously described transparent beads that themselves have known GFP densities (Chiu et al., 2001) and by extending the calibrations with gels traceable to the beads. The resulting measurements show that GAT1 density is rather high at GABAergic presynaptic terminals. A complication is introduced by the fact that GAT1, like many neurotransmitter transporters, is dynamically regulated by surface/cytoplasmic partitioning (Corey et al., 1994; Beckman and Quick, 1998, 1999; Whitworth and Quick, 2001a,b); therefore, we have used surface biotinylation to define the fraction of transporters on the surface versus those in the nearby cytoplasm.
In addition to timely removal of GABA released spontaneously or by presynaptic impulses, other possible functions of GABA transporters include replenishing the supply of GABA in an inhibitory neuron and releasing GABA in a voltage-dependent but Ca2+-independent mechanism (Lester et al., 1996). To assess these roles, it is helpful to know which cells express GABA transporters. GABAergic interneurons are a heterogeneous population (Freund and Buzsaki, 1996), and there are reports that only some inhibitory interneurons express GAT1 (Engel et al., 1998) and that some excitatory neurons also express GAT1 (Yan et al., 1997). Furthermore, the ability to identify GABAergic neurons in culture would be a methodological contribution. We therefore performed synaptic electrophysiology on neurons in hippocampal cultures from knock-in mice, and we found that all GABAergic cells, and only GABAergic cells, fluoresce.
MATERIALS AND METHODS
mGAT1 and GFP fusion constructs. GFP37 was fused to the N terminus of mGAT1 (Liu et al., 1992) (GenBank M92378) by PCR cloning using SalI and EcoRI sites on the pGFP37 plasmid (Grabner et al., 1998; Chiu et al., 2001). This construct carries an eight-residue spacer between GFP37 and mGAT1. To fuse GFP to the C terminus of mGAT1, the mGAT1 open reading frame was subcloned into the HindIII and EcoRI sites of pcDNA3.1(+), and the GFP coding sequence was subcloned into the NotI andXbaI sites. A 12-residue spacer between mGAT1 and GFP was introduced by the multiple cloning site of the vector:
CGA ATT CTG CAG ATA TCC AGC ACA GTG GCG GCC GCC
R I L Q I S S T V A A A
The WT mGAT1 control and GFP control were subcloned between the HindIII and EcoRI and between theNotI and XbaI sites in pcDNA3.1(+), respectively.
Human embryonic kidney (HEK) 293T cells were plated on 35 mm plates to 50–70% confluence, transfected with the fusion constructs (Effectene, Qiagen), and incubated for an additional 60 hr. For GABA uptake assays, cells were washed twice with Krebs'–Ringer's (KRH) buffer (Ramamoorthy et al., 1998), incubated in 2.5 μm[3H]GABA (89 Ci/mmol; Amersham), in 2.2 [3H]GABA μCi/ml in KRH at room temperature for variable periods, or at variable [GABA] (2.2–8.8 μCi/ml) for 10 min, washed rapidly four times with KRH buffer containing 2.5 μm GABA, and solubilized in 1% SDS. [3H]GABA was assayed using liquid scintillation.
A lentivirus expression construct was constructed using the three-plasmid expression system as described (Naldini et al., 1996;Dull et al., 1998; Nadeau et al., 2000). The pHR′ vector was linearized with XhoI and MluI. The mGAT1–GFP fusion construct in pcDNA 3.1(+) was ligated into pHR′ by PCR using primers containing the appropriate restriction sites (XhoI andMluI). Aliquots of the lentivirus suspension were stored at −80°C, thawed once, and applied (15–20 μl) to Madin-Darby canine kidney (MDCK) cells.
Knock-in mouse targeting construct. The mGAT1 cDNA fragment digested with AccI and StuI (position 715–1785 in the coding sequence) was used to synthesize a random primed [α32P]dCTP probe using the protocol from the NEBlot kit (BioLabs). A 129 SVEV Tac FBR mouse spleen genomic library (Lambda FIX II genomic library; Stratagene) was screened. We isolated 26 genomic clones and identified one clone (#6) that carried intron 14 and exon 15 and had >4.0 kb of flanking genomic sequence on both arms.
Genomic clone #6 was amplified by subcloning into the NotI site in pBluescript. The amplified mGAT1 genomic clone #6 was subcloned into the HindIII site of pBluescriptSK(+) and subsequently subcloned into pKO vector (Lexicon Genetics), which carries the diphtheria toxin gene as a positive selection marker, using theSalI and EcoRI sites from pBluescript. The genomic DNA in the pKO vector was modified by inserting the 12-residue spacer plus GFP coding sequence just 5′ to the stop codon of mGAT1 (Exon #15; position 17617) by PCR using three pairs of primers. This construct was confirmed by sequencing. To eliminate restriction sites in the 12-residue spacer between mGAT1 and GFP, the spacer sequence was modified with silent mutations via the PCR primers:
CGC ATT CTC CAA ATC TCA AGC ACC GTA GCC GCA GCC
R I L Q I S S T V A A A
The floxed Neo cassette was inserted between position 17303 and 17311 (intron #14), where AscI sites were created by PCR. The floxed Neo cassette was modified by creating AscI sites on both ends by linkers. An additional 1.6 kb flanking arm at the 3′ end of this construct was created by replacing theApaI/EcoRI sites in the pKO vector with the corresponding mGAT1 genomic DNA sites. The final construct has ∼3.8 kb on the 5′ arm, –Neo-320 bp-GFP-, and ∼4.2 kb on the 3′ arm for homologous recombination (Fig.1A). The 320 bp sequence includes the 34 residues of the coding portion of exon 15.
Fig. 1.
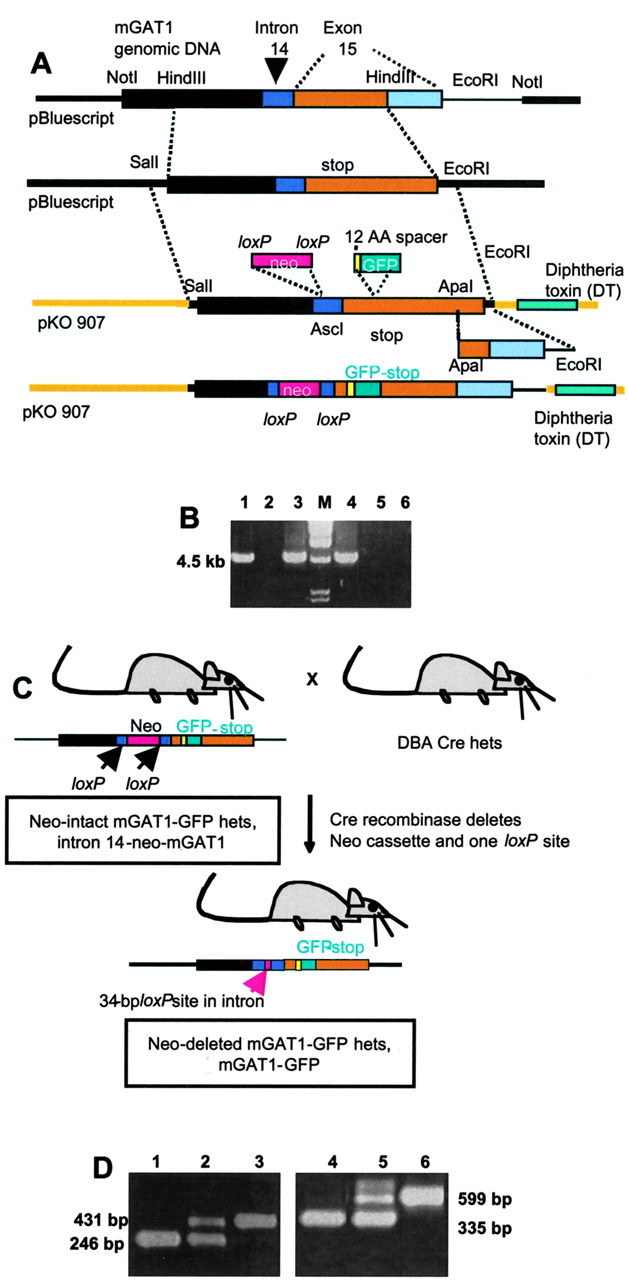
Generation and screening of knock-in strains.A, Modification of mGAT1 genomic DNA to generate a targeting plasmid that contains an mGAT1–GFP fusion sequence in an exon and a floxed neomycin selection cassette in an intron. See Materials and Methods for details. B, PCR screening to identify ES cells carrying the mutant gene. A 4.5 kb PCR product is expected. Lanes 1, 3, and4 represent positive ES cell clones; lane 2 is a negative clone. Lanes 5 and6 show negative controls with no PCR products from genomic DNA extracted from WT ES cells and from the final pKO plasmid construct shown in A. Lane M shows molecular length standards. C, Generation of the Neo-deleted mGAT1–GFP knock-in mouse. The intron 14-Neo-mGAT1 heterozygotes were mated with DBA mice carrying cre recombinase to eliminate the neomycin selection cassette. D, Exemplar PCR genotyping results. Lanes 1–3 show PCR products for mice that are homozygous for the presence of the GFP fusion, heterozygous, and WT, respectively. Lanes 4–6 represent the screening for mice that are homozygous for the presence of the Neo cassette, heterozygous, and WT, respectively.
Homologous recombination, blastocyst injection, and mouse breeding. J7 embryonic stem (ES) cells from 129S3SvImJ mice were electroporated with the linearized construct and screened with Geneticin (G418; 180 μg/ml). The ES cell clones that survived this screening were further screened by PCR (Long template PCR kit; Roche) using a primer set that anneals to position 13017–43 and the neomycin resistance cassette (Neo): mGAT1 13017–43: 5′-GAC TGG TGG GAG AGG CAG ACT TTG AAC-3′; Neo-cassette 3′: 5′-CCA AGT TCT AAT TCC ATC AGA AGC TCC-3′.
The recombined mGAT1 gene showed a 4.5-kb PCR product (Fig.1B). Positive clones were confirmed by PCR using additional sets of primers. Two clones were injected into the C57 blastocyst. Chimeras that displayed >60% agouti coloring were mated with C57 females to generate heterozygous mice. These mice carry the Neo-cassette in intron 14 and were named intron 14-Neo-mGAT1. The loxP-flanked Neo-cassette was deleted by mating with DBA mice carrying cre recombinase. This deletion leaves the 34 bp of one loxP site plus restriction sites (SwaI-AscI) in the intron, and this Neo-deleted strain was named mGAT1-GFP (Fig. 1C).
To genotype the mice (Fig. 1D), three PCR primers were used for screening the GFP insertion: mGAT1 17399–430: 5′-GAC ATT TGG CTT ACT AGT GAG GAA ACA AGA GC-3′; mGAT1 17830–799: 5′-GCT AAG GGG CCT CTA CGG AAG CCT CCA GAG GC-3′; and GFP37 995–64: 5′-CCA TCT AAT TCA ACA AGA ATT GGG ACA ACT CC-3′.
To confirm deletion of the Neo-cassette, the primers were as follows: mGAT1 16922–54: 5′-CCA TGA GGT TGG CTG GAG GGA GAA TAA TGT AGC-3′; mGAT1 17521–54: 5′-GCA CAA TAT CTT CAC TGG GCT GAA TCA TGA CCT G-3′; and Neo-cassette 3′: 5′-CCA AGT TCT AAT TCC ATC AGA AGC TCC-3′.
GFP calibrations. Brain slices, MDCK cells, GFP-beads, and GFP in polyacrylamide gels were imaged using a Leica SP1 confocal microscope system. The confocal system was warmed up for >2 hr, so that the laser photopower fluctuated less than ±5%, monitored by a photopower meter (ThorLabs Inc.; model S20MM) and by the transmitted light detector in the microscope. During the ∼4 months of the quantitative imaging experiments, we monitored the stability of the photomultiplier tube (PMT) periodically by imaging the standardized GFP-beads at a standard photopower. Signals varied by <5%, indicating that the PMT is stable.
Images were taken with a 100× plan apochromatic objective, numerical aperture (NA) = 1.4 (Leica, #506038) using 2× zoom. The pinhole was set at 152 μm, as recommended by the manufacturer. The scan speed was 200 lines per second (slow mode), and the image size was 1024 × 1024 pixels. Each image was scanned with four repeats. The laser power was adjusted using the microscope's acousto-optical tunable filter, so that the fluorescence of a sample fell within the linear range of the detection system.
Bleaching is described by the relation, f(t) =f0e−kIt, where f0 andf(t) are the fluorescence at times zero andt(sec), respectively, k is the cross section for bleaching, measured previously as 4.7 × 10−3 (centimeters squared per Joule) (Chiu et al., 2001), and I is the laser intensity (Watts per centimeters squared). For instance, 0.57 μW laser power (a typical value) applied to a 50-μm-square region implies that the bleaching amounts to 8.7% at the conclusion of a 38-image z-stack series. In our experiments, bleaching actually occurred somewhat more slowly than this relation, probably because of scattering. Nonetheless, we used conditions characterized by kIt ≤ 0.12, so that the average bleaching for a structure within the imaged region was <6%. No formal bleaching corrections were applied.
GFP fluorescence was calibrated using two, well understood His6–GFP preparations. Our primary tool was a series of transparent Ni-NTA beads with known surface densities of His6–GFP (Chiu et al., 2001). GFP-beads prepared for this study from a stock >18 months old showed unchanged fluorescent intensity on the previously used imaging systems, indicating the stability of the His6–GFP solutions. For the present study, the beads were used to calibrate the Leica confocal microscope system. Using GFP-beads at densities at 300, 600, and 1500 GFP per square micrometer, we measured sensitivities of 0.10379 and 0.06944 (counts per pixel per GFP per square micrometer) at 1.15 and 0.77 μW, respectively (Fig.2A).
Fig. 2.

GFP characterization in beads, gels, and tissue samples. A, B, Calibration lines generated for His6–GFP beads (A) and His6–GFP in polyacrylamide gels (B) using the Leica confocal system. The measured slopes for His6–GFP beads are 0.10379 and 0.06944 (counts per pixel per GFP per square micrometer) at 1.15 and 0.77 μW, respectively. The measured slopes for His6–GFP in polyacrylamide gels were 0.02343, 0.01683, and 0.00718 (intensity per pixel per GFP per cubic micrometer) illuminated with 1.11, 0.75, and 0.31 μW, respectively.C, x–z projection of a slice from the cerebellar ML region. Scale bar, 10 μm.D, The profile of the decreased fluorescent intensity along the z-axis, averaged along thex-axis from C.
For GFP in polyacrylamide gels (Fig. 2B), the His6–GFP stock solution was diluted with 50 mm Tris-Cl, pH 8.0, 300 NaCl, 1.5 mg/ml BSA to various concentrations (a final volume of 50 μl) and then mixed with an equal volume of acrylamide solution (38%, 19:1) to generate a homogeneous polyacrylamide gel. The final concentrations of GFP in the gel were 6900, 5500, 4120, 2750, and 1375 GFP per cubic micrometer. The gels were sectioned at 60 μm slices using a vibratome and mounted for imaging. The sectioning and imaging were done within 30 min to prevent GFP diffusion. The sensitivities were proportional to photopower and equaled 0.02343, 0.01683, and 0.00718 (counts per pixel per GFP per cubic micrometer) at photopowers of 1.11, 0.75 and 0.31 μW, respectively (Fig. 2B). When these sensitivities are compared with those obtained with GFP-beads, it may be calculated that the microscope images fluoresce from an effectivez-axis thickness of 250 nm, probably because the microscope is gathering fluorescence from a z-axis range somewhat >250 nm, but with an efficiency somewhat less than that for the bead surface.
His6–GFP is stable in polyacrylamide (without SDS). We monitored His6–GFP fluorescent intensity before and after the gel was polymerized using a fluorometer and found no changes (data not shown). This agrees with a previously published paper that characterized GFP in polyacrylamide gels (Dickson et al., 1997). Exposing the GFP-beads to 4% paraformaldehyde, pH 8.0, did not interfere with GFP fluorescent intensity (data not shown) (Chalfie et al., 1994; Brock et al., 1999), indicating that no corrections are necessary for fixation with 4% paraformaldehyde in mouse brain.
Sample preparation. Mice were anesthetized with halothane (2-bromo-2-chloro-1,1,1-trifluorothane) and perfused with 4% paraformaldehyde in PBS, pH adjusted to 7.6 with Na2HPO4. Brains were dissected and kept in 4% paraformaldehyde for 1 hr in 4°C and then incubated in 30% sucrose in PBS for ∼20 hr. The brains were embedded in O.C.T. medium (Tissue-Tek) for either horizontal or sagittal sections and sliced by cryostat at 35 μm. Brain slices were stored in 11 mmNaH2PO4, 20 mmNa2HPO4, 30% ethylene glycol, and 30% glycerol, pH 7.5, at −20°C. Before imaging, slices were rinsed with PBS and mounted with Vectashield (refractive index 1.4577).
mGAT1–GFP intensity measurement using CCD camera. Surveys of mGAT1–GFP expression measurement in sagittal brain slices (see Fig.4B) were performed with an epifluorescence microscope (Nikon Eclipse 300) equipped with a CCD camera using a 2× objective. We chose 14 regions that represent the range of GFP intensity in the brain and gathered five images of each region using a 100× objective (NA 1.4). The averaged fluorescent intensity from each region was normalized to that of the Purkinje cell pinceaux.
Fig. 4.
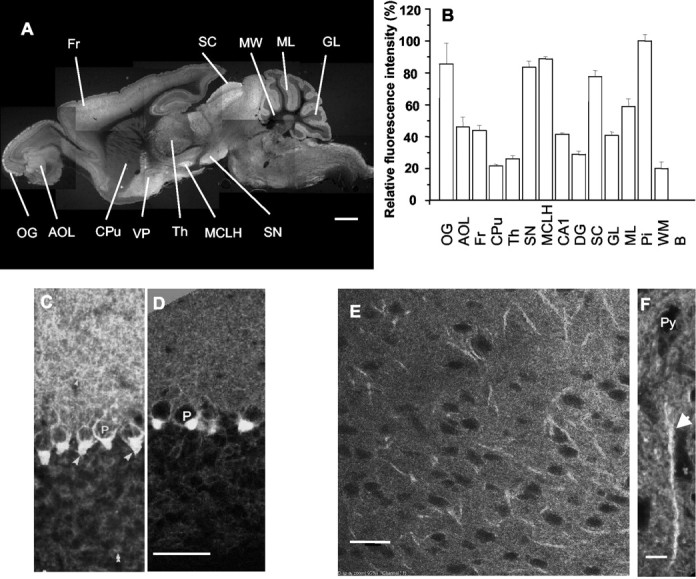
Overview of fluorescence in mGAT1–GFP mice.A, Montage forming a sagittal section ∼1.2 mm from the midline; homozygote. B, Relative mGAT1–GFP fluorescence intensity in various regions. C, D, Cerebellar cortex; comparison of previously published immunohistochemistry (C) (Radian et al., 1990), transformed to produce a negative image, with mGAT1–GFP expression (D); heterozygote, single confocal image from a 35 μm slice. E, F, Frontal cortex, layer 5/6. The linear objects are chandelier cell cartridges (F, arrow) surrounding the initial segments of pyramidal cells. E, Single confocal section from a heterozygote; F, projected stack (5 μm thick) from confocal sections of a homozygote. OG, Glomeruli of olfactory bulb; AOL, anterior olfactory nucleus;Fr, frontal cortex; Cpu, caudate putamen;Th, thalamus; SN, substantia nigra;MCLH, magnocellular nucleus of lateral hypothalamus;CA1, field of Ammon's horn in hippocampus;DG, dentate gyrus; SC, superior colliculus; GL, cerebellar granule layer;ML, cerebellar molecule layer; Pi, pinceaux; Py, pyramidal cell; WM, cerebellar white matter; EPI, external plexiform layer in olfactory bulb; VP, ventral pallidum. Scale bars:A, 1 mm; C–E, 50 μm;F, 10 μm.
Confocal imaging and quantification. For imaging Neo-deleted mice, the laser power was set between 0.75 and 1.1 μW. We estimated that the autofluorescence from a WT brain slice is equivalent to 10–20 GFP per square micrometer (Chiu et al., 2001), which is ∼1–2% of the density of mGAT1–GFP expression. No correction was made for this background fluorescence.
Using ImageJ, we selected boutons, axons, pinceaux, and cartridges for quantification from stacks of images (81 nm per step). The GFP volume density of a structure was determined by dividing (calculated average fluorescent intensity)/(slope of the standard curve, intensity per pixel per GFP per cubic micrometer) at similar photopower (Fig.2B). The total GFP molecules in a structure were calculated by multiplying the volume (total number of voxels) times the measured average density.
The GAT1–GFP surface density was calculated by dividing the surface area (sum of pixels) by the total number of GFP molecules. Heterozygotes and homozygotes gave equally precise data, but counts from the heterozygotes were half that from homozygotes and were multiplied by 2 for density measurements.
Fluorescence intensity decreases beneath the tissue surface along thez-axis with a space constant of ∼35 μm (Fig.2C,D), primarily because of refractive index mismatches and other causes of light-scattering effect that have been described and characterized previously (Hell et al., 1993; Pawley, 1995). We confined our measurements to the superficial 3–4 μm of brain slices, so that fluorescence decremented by <10%.
Biotinylation. Biotinylation experiments were performed essentially as described (Qian et al., 1997; Davis et al., 1998). Cerebellar slices (200 μm) from postnatal day (P) 30 mice were rinsed twice with 37°C PBS/Ca/Mg containing (in mm): 138 NaCl, 2.7 KCl, 1.5 KH2PO4, 9.6 Na2HPO4, 1 MgCl2, 0.1 CaCl2, pH 7.4. The tissue was next incubated with 2 ml of a solution containing 1 mg/ml sulfo-NHS biotin (Pierce) in PBS/Ca/Mg for 1 hr at 4°C with gentle shaking. The biotinylation solution was removed by three washes in PBS/Ca/Mg plus 100 mm glycine and quenched in this solution by incubating the cells at 4°C for 45 min with gentle shaking. The tissue was then homogenized in 1 ml RIPA buffer containing 100 mm Tris-Cl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 250 μm PMSF. The lysates were centrifuged at 20,000 × g at 4°C for 60 min. The supernatant fractions (300 μl) were incubated with an equal volume of Immunopure immobilized monomeric avidin beads (Pierce) for 60 min. The beads were washed three times with RIPA buffer, and adsorbed proteins were eluted with SDS sample buffer (62.5 mm Tris-Cl, pH 6.8, 2% SDS, 100 mm μ-mercaptoethanol) at room temperature for 30 min. Analysis was performed on aliquots (1) taken before incubation with beads (total cell lysate), (2) of the supernatant fraction after adsorption and centrifugation (the nonbiotinylated, “intracellular” fraction), and (3) of the bead eluate (the biotinylated, “surface” fraction). Samples were then run on a 10% acrylamide gel and transferred to nitrocellulose membrane. Western blotting was performed using an anti-GAT1 antibody (Corey et al., 1994) at 1:200 and an anti-GFP antibody (Chemicon) at 1:500.
Dissociated culture. Neuronal cultures were prepared as described (Li et al., 1998). Briefly, hippocampi from embryonic day (E) 16 embryo were digested with 0.25% trypsin and then triturated. Cells plated in polyethyleneimine-coated glass-bottom dishes (400 cells per square micrometer) were maintained in Neurobasal medium supplemented with B27, 500 μm Glutamax, and 5% horse serum (Invitrogen). Dual whole-cell recording was used after 8–14 d in culture. A presynaptic cell was current clamped at zero current except for 1 msec 0.5–1 nA outward stimulus pulses. Postsynaptic currents were recorded at membrane potentials ranging from −100 to 0 mV. The criterion for a monosynaptic response was a latency ≤3 msec. The presence or absence of fluorescence was established visually and confirmed by video images with an exposure time of 0.5 sec. Composition of solutions was as follows (in mm): bath: 140 NaCl, 5.4 KCl, 2 CaCl2, 1 MgCl2, 5 HEPES, 5 glucose, pH 7.4; pipette: 150 K gluconate, 5 NaCl, 0.1 CaCl2, 1 MgCl2, 5 EGTA, 5 HEPES, pH 7.2. Resting membrane potential ranged from −65 to −50 mV. Input resistances were 0.4–1 GΩ.
Synaptosomal preparation and GABA uptake assay. Mice were anesthetized with halothane, and brains were dissected and collected on ice. The cerebellum (∼50 mg) was homogenized in 20× (w/v) medium I (0.32 m sucrose, 0.1 mmEDTA, and 5 mm HEPES, pH 7.5) using a Teflon-glass homogenizer with 16 strokes. Synaptosomes were prepared (Nagy and Delgado-Escueta, 1984). The particulate fraction (P2) from the final centrifugation was suspended with 1 ml of medium I (∼1 mg/ml).
Twenty microliters of the suspension from the P2 particulate fraction were mixed with 280 μl of uptake buffer containing (in mm): 128 NaCl, 2.4 KCl, 3.2 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 10 glucose, 25 HEPES, pH 7.5 (Lu et al., 1998) and incubated at 37°C for 10 min. Subsequently, 100 μl of [3H]GABA at various concentrations was added and incubated for the indicated periods (final radioactive concentrations, 2.2–8.8 μCi/ml). Samples were placed on ice, washed with uptake buffer containing the same concentration of nonradioactive GABA at 10,000 × g, and counted. The GAT1 activity, defined by sensitivity to the specific GAT1 inhibitor NO-711 (30 μm final), accounted for 75–85% of WT activity.
Translocation treatments. For synaptosomal GABA uptake assays, synaptosomes were pretreated with 50 μmorthovanadate, 100 nm bisindolylmaleimide II, and 0.45 m sucrose at 37°C for 15 min and then transferred to synaptosomal GABA uptake buffer at 4°C. The GABA uptake assay lasted for 1 h at 4°C.
To prepare living brain slices for quantitative imaging and Western blot, mice were anesthetized, and brains were dissected free. Brains were then kept in ice-cold artificial CSF (ACSF) containing (in mm): 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 10 d-glucose, and 3 kynurenic acid, pH 7.3, bubbled with 95% O2 and 5% CO2. The brain was then sliced by vibratome at 120 μm thickness. The slices were incubated at 37°C in 50 μm orthovanadate, 100 nm bisindolylmaleimide II, and 0.45 m sucrose for 15 min. Slices were transferred to ice-cold ACSF. Some slices were subjected to biotinylation and quantitative Western blot as described in the previous section. Other slices were fixed in ice-cold ACSF containing 4% paraformaldehyde for 20 min. After a rinse with ACSF, slices were mounted and subjected to imaging.
Brain slice electrophysiology. Mice (P15–P25) were anesthetized with halothane before decapitation, according to University of California Los Angeles regulations. The brain was placed in ice-cold ACSF bubbled with 95% O2 and 5% CO2. Coronal slices (350 μm thick) were cut with a Leica VT1000S Vibratome and stored in bubbled ACSF for >1 hr.
Whole-cell recordings were made at 32–33°C from CA1 pyramidal cells (Zeiss Axioscope infrared-differential contrast videomicroscopy) with an Axopatch 200B amplifier. Electrodes were pulled (Narishige PP-83; Narishige, Tokyo, Japan) from borosilicate glass and filled with solution containing (in mm): 140 CsCl, 2 MgCl2, 10 HEPES, pH 7.2 with CsOH. Voltage-clamp recordings were made at a Vhold of −70 mV. The resistance of the electrodes was 3–6 MΩ, and series resistance was compensated by 70–85% (lag 7–8 μsec). Recordings were low-pass filtered at 3 kHz and digitized at 20 kHz. Synaptic events were detected with amplitude-based (threshold 6–7 pA) and kinetics-based criteria using custom-written software (LabView, National Instruments). Tonic GABAAreceptor-mediated currents were examined by applying the selective GABAA antagonist SR-95531 to the slice chamber in a final concentration of 100–150 μm (30–35 μl of a 6–8 mm SR-95531 solution; chamber volume 1.8–2.0 ml) (Brickley et al., 1996). The holding current was measured in 5 msec epochs at 100 msec intervals, and epochs were rejected if contaminating IPSCs were present. The mean holding current was calculated in three 5-sec-long periods (a, b, and c): a, 20 sec before SR-95531 application;b, just before SR-95531 application; c, 20 sec after the application. The tonic GABAA current was calculated as c − b.
RESULTS
cDNA constructs and functional assays
We fused the cDNA for a GFP mutant (GFP37, named for its stability at 37°C) (Grabner et al., 1998) to either the N or C terminal of the cDNA for mouse GAT1 (mGAT1) (Fig. 3A) and began by testing HEK 293T cells transfected with these constructs (Fig.3B–D). A control construct, expressing GFP alone, displayed uniform diffuse fluorescence throughout the cell including the nucleus (Fig. 3C), and the GABA uptake activity was indistinguishable from nontransfected cells (Fig.3E,F).
Fig. 3.

Testing and selecting the mGAT1–GFP fusion.A, The four constructs, mGAT1–GFP, mGAT1, and GFP in pcDNA3.1(+), and GFP–mGAT1 in pGFP37 were made to test the function of mGAT1–GFP fusions. The spacer sequences (8 residues for GFP–mGAT1, 12 residues for mGAT1–GFP) are shown in red. The C-terminal AYI sequence is shown in blue. B, The GFP–mGAT1 fusion is expressed mostly in the cytoplasm but not the nucleus of HEK cells and shows only slight increases in GABA uptake (data not shown). C, GFP expresses in cytoplasm and in the nucleus. D, The mGAT1–GFP fusion protein expresses on the membrane of HEK cells as well as in the cytoplasm, a typical situation for overexpressed proteins. E,F, Kinetics of GABA uptake by these fusion proteins expressed in HEK cells, tested at 2.5 μm GABA for time dependence (E) and in a 10 min assay for concentration dependence (F). mGAT1–GFP GABA uptake activity (○, green line) was indistinguishable from that of WT mGAT1 (●, black line). On the other hand, HEK cells expressing GFP (▴) have GABA uptake activity indistinguishable from noninfected cells. The GFP–mGAT1 fusion showed only slight increases in GABA uptake compared with untransfected cells (data not shown). G, H, Fluorescence on the apical membrane of MDCK cells transfected with a recombinant mGAT1–GFP lentivirus. The face-on view of an image stack (G) and the side view of the same stack (H) are illustrated. I, mGAT1 expressed in a cultured E18 hippocampal cell, infected after 12 d in culture with the recombinant lentivirus and imaged 5 d later. All labeled processes derive from one cell. Scale bars:B–D, 25 μm; G, H, 20 μm; I, 50 μm.
The C-terminal fusion mGAT1–GFP, which includes a 12-residue spacer between mGAT1 and the GFP, showed clear plasma membrane localization (Fig. 3D) as well as cytoplasmic localization, which is typically found with overexpressed membrane proteins. GABA uptake activity was similar to that of WT mGAT1 (Fig.3E,F). In Scatchard analysis, the EC50 values for mGAT1 and mGAT1–GFP were 10.9 and 9.3 μm, respectively, and the Vmax values were 3.6 and 3.9 nmol/mg protein per 10 min, respectively.
The N-terminal fusion protein, however, was localized primarily or exclusively in the cytoplasm rather than in the plasma membrane. The transfected cells displayed GABA uptake only slightly higher than untransfected cells and 10-fold less than WT mGAT1 or mGAT1–GFP (data not shown). The lack of nuclear expression suggests that GFP did indeed fuse with mGAT1, forming a protein that fails to pass through the nuclear pore (Fig. 3B).
Thus the C-terminal fusion, mGAT1–GFP, was the appropriate candidate for further study. We constructed a lentivirus expressing mGAT1–GFP and infected MDCK cells in culture. The mGAT1–GFP construct was directed to the apical membrane, as found previously for WT GAT1 expressed in MDCK cells (Fig. 3G,H) (Ahn et al., 1996). We also infected hippocampal neurons in primary culture and verified that the lentivirus-encoded mGAT1 construct was directed to the plasma membrane (Fig. 3I). These data showed that the mGAT1–GFP fusion protein fulfilled all of our qualitative and quantitative requirements, at least in heterologous expression systems. mGAT1–GFP functions like mGAT1 and is sorted like mGAT1.
mGAT1-GFP knock-in mouse: general pattern of fluorescence
A knock-in mouse strain carrying the mGAT1–GFP fusion was created by homologous recombination to replace the final coding exon of mGAT1 (Fig. 1). There are only two differences between genome of the mGAT1–GFP strain and the WT genome: the coding region of mGAT1 has been extended at the C terminus by the GFP37 sequence, and the adjacent exon retains a single 37 bp loxP site (a vestige of the selection procedure for the embryonic stem cells). Thus the encoded mGAT1–GFP gene matches the cDNA construct that we chose in the experiments of Figure 3. Homozygotes and heterozygotes from the mGAT1–GFP line display normal weight, development, and life span, and, on the basis of anecdotal observations, normal behavior.
Figure 4A presents a fluorescence image giving an overview of the brain of an adult mGAT1–GFP knock-in mouse. The pattern of fluorescence strongly resembles previous immunohistochemical findings: GAT1 is expressed in axons, in synapses, and in astrocytes in many brain regions (Radian et al., 1990; Ikegaki et al., 1994; Minelli et al., 1995; De Biasi et al., 1998; Yan and Ribak, 1998a,b; Wang and Ong, 1999; Ng and Ong, 2001). Wild-type littermates showed no detectable background fluorescence (Figs. 5C,6C). Figure4B presents a quantitation of the fluorescence at the cellular level in individual nuclei, normalized to the pinceaux in cerebellum, which give the highest signal (Fig. 4D). The absolute fluorescent intensities differed among animals by ± 7% (SEM, n = 3). In the glomeruli of the olfactory bulb, the superior colliculus, the substantia nigra, and the magnocellular nucleus of lateral hypothalamus, fluorescence was nearly as high (80–100%). The molecular layer of the cerebellum displayed ∼60% expression, whereas the hippocampus, cortex, granule layer of the cerebellum, and pons displayed 40–50% expression. The lowest level of expression, 20–30%, is observed in cerebellar white matter, thalamus, and striatum (Fig. 4B).
Fig. 5.
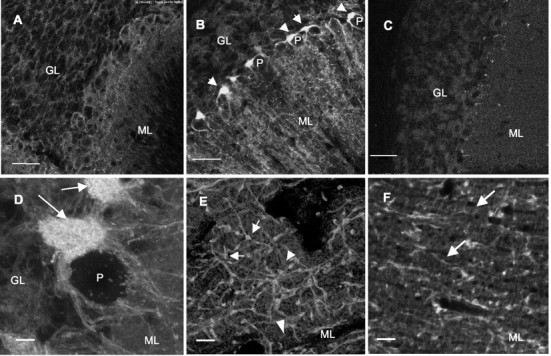
Details of mGAT1–GFP fluorescence in cerebellum, including individual boutons. A, In a P9 mGAT1–GFP mouse, mGAT1–GFP expresses in somata of several cell types; however, no obvious axonal expression was observed. B, In a P29 mGAT1–GFP mouse, mGAT1–GFP is clearly expressed in axons and boutons in the molecular layer (ML) and in pinceaux (arrows) surrounding the axonal hillock of Purkinje cells (P). The background stripe-like structures in ML are probably Bergmann glia.C, The P29 WT shows low background fluorescence and no obvious structures. This image was taken under >10-fold higher illumination photopower and has been further brightened digitally more than the other images. D, mGAT1–GFP expresses in pinceaux (arrows) in the hillock of a Purkinje cell (P). E, mGAT1–GFP fluorescence in axons and boutons in ML. Boutons with higher (arrows) and lower (arrowheads) levels of mGAT1–GFP are indicated. F, The diffuse stripe-like structures (arrows) represent the putative Bergmann glia expressing mGAT1–GFP. This structure is most evident in horizontal sections, probably revealing the glial palisades of Altman (Altman and Bayer, 1997). ML, Molecule layer;GL, granule cell layer; P, Purkinje. Scale bars: A–C, 50 μm;D–F, 5 μm. A,B, Single confocal slices from heterozygotes;D–F, projected stacks (6, 4, and 4 μm thick, respectively) from homozygotes.
Fig. 6.
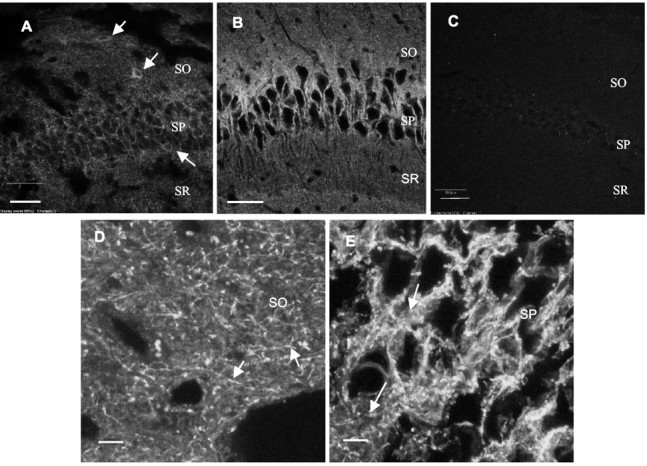
Details of mGAT1–GFP fluorescence in CA1 region of hippocampus. A, In a heterozygous P9 mGAT1–GFP mouse, fluorescence is observed both in somata of inhibitory interneurons (arrows) and at synapses near and in the pyramidal cell layer. B, In a 3 month postnatal homozygous mouse, fluorescence is observed only in axons and synapses but not in somata. C, A P29 WT mouse shows low background fluorescence and no obvious structures. This image was taken at >10-fold higher illumination photopower and has been further brightened digitally more than the other images. D, High-magnification view of stratum oriens, showing fluorescent axons (arrows) and boutons. The dimmer background fluorescence could be caused by expression in astrocytes. Brain tissues were prepared from homozygote, 60 d postnatal mouse. E, Neuropil (arrows) in pyramidal cell layers. The lack of clear axon and bouton images could be attributable to mGAT1–GFP expression in both astrocytes and axons (Yan et al., 1997). Brain tissues were prepared from homozygote, 60 d postnatal mouse.SO, Stratum oriens; SP, stratum pyramidale; SR, stratum radiatum. A,D, E, Projected stacks (5, 4, and 4 μm thick, respectively). Scale bars: B, 50 μm;C, 40 μm; A, D,E, 5 μm.
More detailed images show that mGAT1–GFP is expressed primarily on axons and presynaptic boutons of GABAergic inhibitory neurons throughout the brain, again as revealed in previous studies. Figure 4,C and D, compares a previously published immunocytochemical image of cerebellar cortex (Radian et al., 1990) (shown as the negative, for better comparison with the fluorescence) with a fluorescent image from an mGAT1–GFP knock-in mouse. mGAT1 fluorescence occurs in a reticular pattern in the molecular layer and in the pinceaux (named for their resemblance to the paintbrushes of the French Impressionists), which are processes of basket and stellate cells that surround the initial segment of the Purkinje cell axon (Fig.5D) (Palay and Chan-Palay, 1974; Morara et al., 1996; Ribak et al., 1996). The pinceaux were a focus of our quantitative studies (see below).
In other cerebellar cell types, mGAT1–GFP shows more diffuse fluorescence. For instance, in some sections of the molecular layer, we observed diffuse stripes of fluorescence (Fig.5B,F); these structures are probably the Bergmann glia (Rattray and Priestley, 1993; Barakat and Bordey, 2002). In the glomeruli of the granule cell layer, where GAT1 is expressed in the axons and boutons of Golgi cells (Radian et al., 1990; Yan and Ribak, 1998a,b), fluorescence is also slightly diffuse, and no clearly fluorescent axons or boutons were observed (Figs. 4D, 5B). Also, in younger mice (for instance, at P9) (Fig. 5A), low levels of diffuse, presumably intracellular expression were observed in many cell types (Rosina et al., 1999).
In forebrain (including hippocampus), GAT1 is thought to be expressed in GABAergic interneurons, including chandelier, basket, and dendritic inhibitory cells (Freund and Buzsaki, 1996) and is particularly prominent on the cartridges formed by chandelier cells on pyramidal neurons (Woo et al., 1998). Figure 4, E and F, shows mGAT1 expression in the cartridges.
Figure 6 shows details of fluorescence in hippocampus. Figure 6,A and B, compares GAT1 localization during development. The P9 knock-in mouse shows both synaptic and somatic expression, which agrees with other studies on young mice (Fig.6A) (Yan et al., 1997; Yan and Ribak, 1998a). mGAT1–GFP in P29 and older mice is no longer expressed in interneuron somata but is expressed in axons and synapses throughout the hippocampus and is especially high in the pyramidal cell layer (Fig.6B); this developmental pattern is similar to that in cerebellum (Fig. 5A,B). At higher magnification, images of stratum oriens (Fig. 6D) and stratum pyramidale (Fig. 6E) reveal axons and boutons originating from chandelier and basket cells (Freund and Buzsaki, 1996). In the excitatory cell layers in the CA regions and in dentate gyrus, high levels of fluorescence but no clear synaptic structures were observed. This is probably caused by the high level of astroglial expression, which reduced our resolution at individual synapses (Ribak et al., 1996) (Fig. 6E). Observations on hippocampal cultures provide additional evidence for glial GAT1 expression (see Fig. 10E). Slices from WT mice display little or no fluorescence, even at higher incident light intensity (Fig.6C).
Fig. 10.
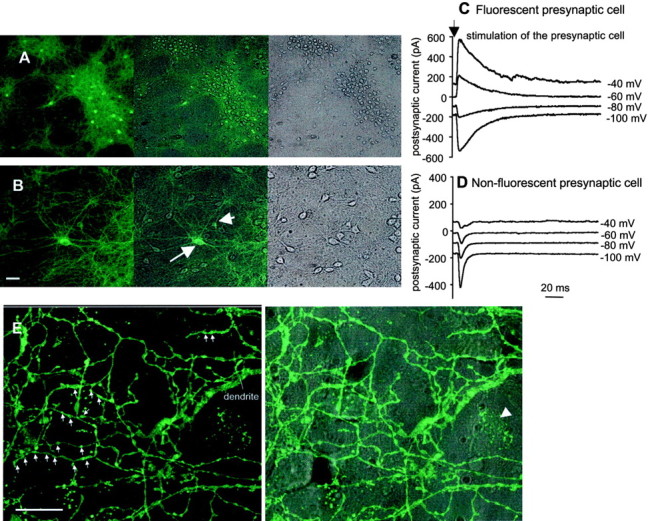
Electrophysiological measurements show that inhibitory interneurons fluoresce in hippocampal dissociated culture.A, B, Cultured hippocampal neurons were imaged with 10× (A) and 20× (B) objectives. The images show the fluorescent neurons under epifluorescence (left) and all neurons under transmitted light (right). The merged images are shown in the center panels. There are 16–18 fluorescent neurons in A and 5 (of which the 2 most obvious are marked with arrows) in B. Scale bars:A, 100 μm; B, 50 μm.C, D, Exemplar waveforms of voltage–clamp currents recorded from nonfluorescent postsynaptic cells studied at various holding potentials. C, Records during stimulation of a fluorescent presynaptic cell. D, Another cell; records during stimulation of a nonfluorescent presynaptic cell. E, The axon of a dissociated hippocampal interneuron forms synaptic boutons on nearby excitatory neurons. Scale bar, 20 μm. Left panel, Fluorescence only; arrows point to boutons that make contact on a nonfluorescent soma. Right panel, Fluorescence overlaid on Nomarski image of the culture. In the right-hand panel, the pointer identifies a glial cell expressing GAT1 (arrowhead).
Counting mGAT1 molecules in cerebellum
The data presented thus far indicate that mGAT1–GFP fluorescence occurs as expected from previous studies, with regard to the overall pattern of expression, the cellular localization, and the time course of expression. We therefore proceeded to count mGAT1–GFP molecules, with localization at the resolution of confocal microscopy. These counts were based on two sets of standards. The first standard was a set of transparent beads with calibrated surface densities of GFP. The beads themselves were calibrated on the basis of both single-molecule fluorescence measurements and mass analyses of the GFP preparations used to generate the beads (Chiu et al., 2001). The second standard was a set of polyacrylamide gels with calibrated volume densities of GFP, prepared from the same GFP preparations (see Materials and Methods). For the present measurements, we developed stereological procedures to quantify GFP fluorescence in micrometer-scale structures imaged in a series of confocal stacked sections from brain tissue. These methods accounted for the resolution of the microscope in thex-y plane and the z-axis (see Fig. 8), for the scattering and loss of signal in tissue sections, and for possible bleaching with prolonged exposure (see Materials and Methods).
Fig. 8.
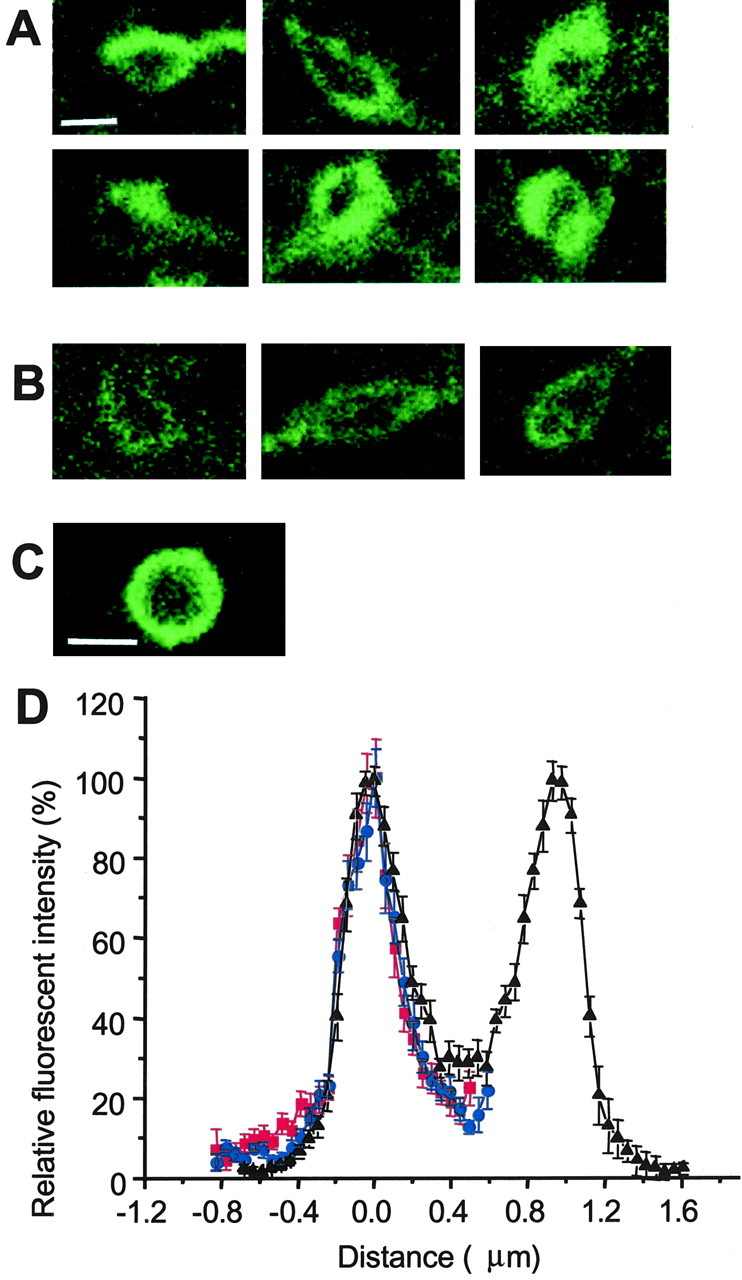
Single-bouton images and point-spread function (A, B). Projections are of confocal stacks in cerebellar ML, 1–1.5 μm thick. Scale bar, 1 μm.A, Boutons with higher total mGAT1–GFP expression show subregions of higher fluorescence. B, Boutons with lower fluorescence levels show more evenly distributed fluorescence.C, Single confocal image of an individual latex bead coated with His6–GFP. Scale bar, 1 μm. D, Profiles of GFP fluorescence in single confocal images of the beads (black), boutons from slices subjected to translocation treatment (red), and boutons from untreated slices (blue). Mean ± SEM; n = 16 each.
Our most detailed data are from cerebellar molecular layer (ML), where the relatively large size of basket cell and stellate cell boutons is well suited to our methods (Palay and Chan-Palay, 1974). The mGAT1–GFP fluorescent boutons have a diameter × length of 1–1.5 × 2–3 μm (Fig. 5E; also see Fig.8A,B). We noted two classes of structures with regard to fluorescent intensity and have quantified these classes separately. Some 50–70% of boutons and their connecting axons display the highest total mGAT1–GFP fluorescence (Fig.5E, arrows; also see Fig. 8A) and will be described first. The average bouton volume is 1.84 ± 0.14 μm3 and surface area is 6.73 ± 0.43 μm2. On the basis of calibrations from polyacrylamide gels, the average total mGAT1–GFP molecules in a bouton is 9000 ± 640, representing a concentration of 4960 ± 124 mGAT1–GFP per cubic micrometer. If all mGAT1–GFP molecules are on the membrane, the surface density is 1335 ± 37 mGAT1–GFP per square micrometer (n = 23). Quantifications based on the calibrated GFP-beads yield surface densities ∼16% lower, at 1120 ± 32 mGAT1–GFP per square micrometer.
In boutons with higher mGAT1–GFP expression, we observed higher mGAT1–GFP in subregions, ∼1.5–2-fold higher (see Fig.8A). In sections simultaneously immunostained for GABAA receptors and imaged for GFP fluorescence, we have noted that ∼90% of the higher-expression boutons are associated with GABAA receptors, suggesting that these are bona fide areas of synaptic contact.
We now describe the 30–50% of boutons with lower fluorescence (Fig.5E, arrowheads) (also typified by Fig.8B). The bouton volume is 1.35 ± 0.15 μm3, and surface area is 5.12 ± 0.48 μm2. On the basis of calibrations from polyacrylamide gels, the average total mGAT1–GFP molecules in a bouton is 5070 ± 527, representing a concentration of 3850 ± 126 GFP per cubic micrometer. If all mGAT1–GFP molecules are on the membrane, the surface density is 980 ± 18 mGAT1–GFP per square micrometer (n = 11). Quantifications based on the calibrated GFP-beads yield surface densities ∼16% lower, at 820 ± 40 mGAT1–GFP per square micrometer. Thus the lower-intensity class of boutons appear to express mGAT1–GFP at levels 64–75% of that of the higher-intensity class.
The axons connecting the boutons have an apparent diameter of ∼0.3 μm (Fig. 5E,F), but this figure is near the limit of resolution and must be considered approximate. We analyzed the linear fluorescence density of axons that ran along the z-axis for a distance >500 nm, or twice the theoretical z-axis resolution, so that adjacent sections were averaged with little distortion from edge effects. (We would prefer axons that run for >1 μm, but the tortuous course may vitiate such a search.) Axons in the higher-intensity class showed fluorescence corresponding to 636 ± 19 mGAT1–GFP molecules per micrometer (n = 4). If one assumes an actual diameter of 0.3 μm, the membrane density of mGAT1–GFP molecules is 677 per square micrometer, close to half that of the boutons. Axons that connect lower-intensity boutons also showed fluorescence at least 30% lower than the high-intensity class.
Each cerebellar pinceau consists of dozens of intertwined basket cell boutons and axons. Pinceaux vary in size and may sometimes associate as linked pairs. We have treated pinceaux as solid objects. The average volume and surface area of a pinceau are 1020 ± 83 μm3 and 470 ± 12 μm2 (n = 8). The total of mGAT1–GFP molecules in a pinceau is ∼7.8 × 106, and the average volume density is 7700 ± 443 GFP per cubic micrometer, or 55% higher than the volume density in the high-intensity class of bouton.
We estimate that the density of mGAT1–GFP in Bergmann glia is ∼1200–2000 GFP per cubic micrometer. Our observations on GAT1 expression in these cells will be described in a separate publication.
Counting mGAT1 molecules in cortex and hippocampus
GAT1 is expressed on axons and boutons in cortex; boutons have a diameter × length of ∼0.8 × ∼1.8 μm. The average bouton volume is 1.3 ± 0.1 μm3, and the average surface area is 3.3 ± 0.2 μm2. The volume and surface density of mGAT1–GFP expression are 3120 ± 140 GFP per cubic micrometer and 1180 ± 46 GFP per square micrometer (mean ± SEM;n = 8), respectively. The volume and surface of the measured cortex cartridge are 188 μm3and 233 μm2, respectively (n = 1). The total GFP is ∼4.3 × 105 per cartridge, and the average concentration is ∼2400 GFP per cubic micrometer.
In hippocampus, the neuropil in the pyramidal cell layer showed the highest levels of fluorescence, followed by stratum oriens (Fig. 6B). Presynaptic boutons of inhibitory neurons have only slightly greater diameter than do axons and similar fluorescence levels; therefore, in many cases the identification of a bouton is tentative. We estimate that a bouton in stratum oriens has dimensions of ∼0.4–0.5 × 1.2 μm, slightly larger than previous measurements (i.e., 0.3 × 0.9 μm) (Somogyi et al., 1983; Gulyas et al., 1993; Halasy and Somogyi, 1993; Freund and Buzsaki, 1996). On the basis of the polyacrylamide gel calibration, the average total number of mGAT1–GFP molecules in a bouton is 1200 ± 40, and the volume concentration is 6071 ± 200 GFP per micrometer cubed. If all mGAT1–GFP molecules are expressed on the surface, we calculate a density of 800 ± 24 GFP per micrometer squared. Our measurements could be distorted by fluorescence point spread. Direct measurements of surface fluorescence in boutons, based on the GFP-bead calibrations, yield a surface mGAT1–GFP density of 1218 ± 59 GFP per square micrometer; we consider this the more reliable measurement of surface density. Under the conditions used for quantification of fluorescence in cortex and hippocampus of mGAT1–GFP mice, there was little or no detectable fluorescence in WT mice (data not shown).
Surface/cytoplasmic partitioning of mGAT1–GFP in WT and mGAT1–GFP mice
The mGAT1–GFP construct was selected because it expresses, functions, and sorts like WT mGAT1 in heterologous expression systems (Fig. 3). The comparisons between mGAT1–GFP fluorescence and previous immunocytochemistry also show a good agreement with regard to localization in the animals (Fig. 4). Nonetheless, we found differences between the functional expression level of functional GAT1 in mGAT1–GFP knock-in animals versus WT animals. We assayed crude synaptosome preparations from knock-in and WT mice for [3H]GABA uptake (Fig.7A). In Scatchard analyses, the EC50 values for wild type, heterozygotes, and homozygotes were 9.41, 11.95, and 11.47 μm, respectively, reaffirming that the mGAT1–GFP fusion construct functions like the WT. However, theVmax values of the three genotypes were 6.2, 5.2, and 2.2 (nanomoles per milligram of protein per 5 min), respectively, consistent with the presence of approximately one-third as many functional mGAT1–GFP molecules on the plasma membrane as compared with WT (n = 3 for each genotype) (Fig.7A). In a total of six sets of synaptosome preparations each of the three genotypes (three experiments with complete concentrations series and three with a single [3H]GABA concentration), the homozygote and heterozygote knock-in mice displayed 32 ± 5% and 76 ± 7% (mean ± SEM) of the WT activity, respectively.
Fig. 7.

GABA uptake and biotinylation assays for mGAT1–GFP partitioning. A, Scatchard plot of [3H]GABA uptake for all three mGAT1–GFP genotypes (mean ± SEM; n = 3). Insetshows image of green fluorescent synaptosomes. B, Manipulation of membrane/cytoplasmic partitioning, assayed by [3H]GABA uptake. Data show NO-711-sensitive [3H]GABA uptake, measured over a time course of 1 hr at 4°C. Synaptosomes were preincubated for 10 min with control solution or subjected to “translocation treatment” with orthovanadate (50 μm), bisindolylmaleimide II (100 nm) to inactivate PKC, and 0.45 m sucrose, all at 4°C. C, Manipulation of membrane/cytoplasmic partitioning, assayed by surface biotinylation of cerebellar slices.Lanes 1–4, Tissue from mGAT1–GFP mice, probed with anti-GFP antibody. Lanes 5–8, Tissue from WT mice, probed with anti-GAT1 antibody. Lanes 1,2, 5, and 6 were also probed with anti-actin antibody. D, Quantitation of immunostaining for mGAT1–GFP and mGAT1 in lanes 1–8.y-axis shows percentage of total staining (intracellular + extracellular) in the pair of lanes denoted by similar patterns.Lane pairs 1 + 3, 2 +4, 5 + 7, and6 + 8 add up to 100%. Data are mean ± SEM from three experiments like that of C.E, Fluorescence intensity in single boutons, untreated or subjected to translocation treatment.
Because of this unexpected threefold decrease in surface transporter expression in mGAT1–GFP mice, we performed additional manipulations to ensure that fluorescence measurements were made on mice with mGAT1–GFP molecules that were all translocated to the cell surface (Corey et al., 1994; Beckman and Quick, 1998; Bernstein and Quick, 1999; Whitworth and Quick, 2001a,b). Synaptosomes were incubated for 10 min with orthovanadate to inactivate tyrosine phosphatases, with bisindolylmaleimide II to inactivate PKC, and with 0.45 msucrose to suppress endocytosis, all at 4°C to further suppress endocytosis (“translocation treatment”), followed by a 1 hr assay for [3H]GABA uptake. Under these conditions, the untreated mGAT1–GFP synaptosomes showed approximately one-third the WT [3H]GABA uptake levels (Fig. 7B), in agreement with the results for the assay at 37°C in Figure 7A. The translocation treatment increased the [3H]GABA uptake by ∼60% in WT synaptosomes (Fig. 7B), in agreement with previous data (Corey et al., 1994; Beckman and Quick, 1998; Bernstein and Quick, 1999; Whitworth and Quick, 2001a,b). However, the translocation treatment produced a much larger, ∼3.5-fold increase in the mGAT1–GFP synaptosomes, to a level not significantly different from the uptake activity of the orthovanadate-treated WT synaptosomes (Fig.7B). This suggested that the translocation treatment placed all the transporters on the surface in a functional state.
We performed surface biotinylation on cerebellar slices to confirm and explain these manipulations as well as to estimate the proportion of surface versus cytoplasmic transporters in WT mice (Fig. 7C) (Beckman et al., 1999; Bernstein and Quick, 1999; Horton and Quick, 2001). We found that the ratio of surface mGAT1 to subsurface mGAT1 differed in WT and knock-in mice. In WT mice, 63 ± 8% of the mGAT1 was on the surface membrane, but in mGAT1–GFP knock-in mice, only 24 ± 11% of the mGAT1 was on the surface. The translocation treatment placed >90% of the GAT1 on the surface in both mGAT1–GFP mice and WT mice (Fig. 7D) (this nearly complete pulldown also provides assurance that the data from other manipulations can be analyzed quantitatively to provide a surface/cytoplasmic ratio). These data are consistent with the lower level of [3H]GABA uptake in the mGAT1–GFP mice and with the increase produced by the translocation treatment (Fig.7A,B). Similar data were obtained in surface biotinylation experiments on hippocampal slices (data not shown).
We then performed additional fluorescence imaging experiments on boutons in cerebellar slices, the structures that we consider the most amenable to quantitative data. There was no significant difference (<3%) between the average mGAT1 fluorescence of individual untreated boutons (n = 35 boutons) and those that were subjected to the translocation treatment (n = 33 boutons) (Fig.7E). These data show that the parameters of Table1 may be interpreted as the maximal GAT1 density that can be translocated to the surface membrane.
Table 1.
Counts of mGAT1–GFP molecules in presynaptic structures of inhibitory interneurons
| Volume (μm3) | Surface area (μm2) | Total mGAT1–GFP | mGAT1–GFP density | |||
|---|---|---|---|---|---|---|
| Volume (/μm3) | Surface (/μm2) | Linear (/μm) | ||||
| Boutons, cerebellum | 1.8 ± 0.1 | 6.7 ± 0.4 | 9000 ± 640 | 4960 ± 124 | 1340 ± 95 | |
| Pinceaux, cerebellum | 1020 | 470 | (7.8 ± 0.45) × 106 | 7700 ± 443 | ||
| Axons, cerebellum | High: 677 ± 20 | High: 636 ± 19 | ||||
| Boutons, cortex | 1.3 ± 0.1 | 3.3 ± 0.2 | 3300 ± 326 | 2600 ± 140 | 1000 ± 100 | |
| Cartridge, cortex | 188 | 233 | 3.6 × 105 | 2000 | ||
| Boutons, hippocampus | ∼0.2 | ∼1.5 | 1200 ± 40 | 6070 ± 200 | 800 ± 24 | |
The cells in bold present the parameters that we consider most reliable.
Many images of synaptic boutons (Figs. 5E,8) show slightly elevated fluorescence intensity in the intracellular compartment. We asked whether this apparently intracellular fluorescence corresponds to the nonbiotinylated mGAT1. We compared the spatial profile of fluorescence of the mGAT1–GFP on boutons from the cerebellar molecular layer (Fig.8A,B) with His6–GFP on the surface of latex beads 1.5 μm in diameter (Fig. 8C). The beads also appeared to have a “tail” of internal fluorescence similar to that of the boutons, and this tail did not change after translocation treatment (Fig.8D). We have not systematically explored the cause of this fluorescence spread, but both the boutons and the beads have elevated refractive indices relative to the external solution, and this refractive index difference may distort the optical signals slightly. Regardless of the source of this optical effect, we conclude that our optical measurements do not resolve a component of mGAT1–GFP fluorescence away from the surface membrane. The data suggest that the cytoplasmic fraction of cellular mGAT1 remains within a few hundred nanometers of the surface membrane and cannot be distinguished from the fluorescence of surface-membrane mGAT1.
mGAT1–GFP mice display normal GABAergic transmission
We measured spontaneous IPSCs in whole-cell patch-clamp experiments on CA1 hippocampal pyramidal cells in slices from the brains of WT and mGAT1–GFP mice (Fig.9A). There were only minor changes in average frequency, amplitude, and waveform of these events between WT and mGAT1–GFP mice. We also tested the effects of blocking GAT1 with NO-711 and found few differences between the effects on WT and mGAT1 mice.
Fig. 9.

Electrophysiology of GABAergic transmission to CA1 pyramidal neurons in hippocampal slices from WT and mGAT1–GFP mice. A, Spontaneous GABAA-mediated IPSCs in Cl−-loaded CA1 pyramidal cells (holding potential, −70 mV). Averages of 50 IPSCs from WT and mGAT–GFP slices are shown. Right-hand panelshows mean ± SEM of several parameters. B, Tonic GABAA receptor-mediated currents recorded in Cl−-loaded CA1 pyramidal cells (holding potential, −70 mV). In the presence of NO-711 (10 μm), application of the GABAA receptor antagonist SR-95531 (>100 μm) blocked the spontaneous IPSCs and caused an outward shift in the baseline by 30–40 pA. This change represents the tonic GABAA receptor-mediated current and was observed in cells from both WT and mGAT1 mice. The right-hand panelshows mean ± SEM of the tonic current in WT and mGFP slices, in the presence and absence of NO-711.
In previous experiments on CA1 hippocampal pyramidal cells, pharmacological inhibition of GAT1 also produced tonic GABAA currents, corresponding to a steady GABA concentration estimated at ∼1 μm (Frahm et al., 2001;Overstreet and Westbrook, 2001) (Tang et al., 2001). We therefore compared measurements of tonic current in cells from WT and mGAT1–GFP mice (Fig. 9B). Perfusion of the GABAA antagonist SR-95531 (>100 μm) produced a complete silencing of spontaneous IPSCs as well as a positive shift in the holding current, corresponding to the tonic GABAA current (Fig.9B). mGAT1–GFP and WT cells displayed equal tonic currents, showing that GABAA receptors in mGAT1–GFP mice, like receptors in WT mice, can detect elevated extracellular GABA concentrations (Fig. 9B); these receptors are presumably located extrasynaptically. In the absence of NO-711, SR-95531 perfusion again silenced spontaneous IPSCs but produced much smaller (and equal) tonic current in either WT or mGAT1–GFP cells (Fig. 9B). This shows that mGAT1–GFP is able to prevent spillover of GABA onto receptors responsible for the tonic current.
Cultured neurons make inhibitory synapses if and only if they are fluorescent
Figure 10, A andB, shows micrographs of dissociated cultures from mGAT1–GFP knock-in embryo hippocampus, cultured at E16 and studied after 16 d in culture. Of the cultured neurons, 10–20% are fluorescent. mGAT1–GFP is expressed throughout these cells, similar to our observations of early postnatal mice (Fig.10A,B). Although the fluorescence in mice became restricted to axons and synapses after the third postnatal week (Figs. 5A,B,6A,B), the pattern of whole-cell fluorescence persisted after >29 d in culture, suggesting that the details of localization differ between cells in dissociated culture andin vivo. A previous report shows that GAT1 immunocytochemistry persists in the soma and dendrites of cultured rat hippocampal neurons, ruling out major effects of the GFP moiety in this case (Pietrini et al., 1994).
The major goal of the culture experiments was to determine whether the set of fluorescent neurons is identical to the set of GABAergic interneurons. Neurons of homozygous or heterozygous mGAT1–GFP mice were cultured for 8–14 d and then screened, in paired whole-cell patch-clamp recordings, for the presence of synaptic connections (Fig.10C,D). Thirty-nine fluorescent cells were stimulated in this series of experiments. Thirty-four of the fluorescent neurons induced postsynaptic currents in the second, nonfluorescent neuron of a cell pair (Fig. 10C). In all cases the reversal potential of this current, when corrected for the liquid junction potential, was approximately −80 mV, close to the equilibrium potential for Cl− under our experimental conditions. These putative IPSCs were blocked both by picrotoxin and by bicuculline (40 μm; each of four and six tested synapses, respectively). Furthermore, in one tested pair of fluorescent neurons, there were reciprocal inhibitory synaptic currents. These data indicate that all tested fluorescent cells were GABAergic inhibitory neurons.
In 30 tested pairs of nonfluorescent cells, there were 36 synapses. In all cases the reversal potential of the postsynaptic current induced by stimulation of nonfluorescent cells was more positive than −20 mV. This current was also characterized by faster decay when compared with the postsynaptic current induced by stimulation of fluorescent cells (Fig. 10D shows an example). Both reversal potential and more rapid decay of these postsynaptic currents indicate the excitatory nature of nonfluorescent cells. In all four tested cases, these putative excitatory currents were blocked by CNQX (10 μm), confirming their glutamatergic nature. In 39 cases, we stimulated a nonfluorescent neuron and recorded from a fluorescent neuron; there were 26 and 0 cases of EPSCs and IPSCs, respectively.
Thus the set of fluorescent neurons in hippocampal cultures equals the set of GABAergic interneurons. Fluorescent neurons formed boutons on nearby nonfluorescent neurons (Fig. 10E) as well as on themselves, and these boutons are probably some of the inhibitory synapses.
We make an additional, more technical point. The fluorescent interneurons were detected readily in a standard inverted microscope equipped with an Hg lamp, a GFP fluorescence cube, and a 20× (NA 0.4) objective. Even the heterozygotes produced unambiguous fluorescent neurons, leading us to expect that inhibitory neurons would be detectable if the homozygous mGAT1–GFP mice are crossed with other mouse strains. In data not shown, we have confirmed that inhibitory neurons were detected in cultured hippocampus when the mGAT1–GFP line was crossed to lines carrying point-mutant nicotinic receptors (Labarca et al., 2001) or point-mutant serotonin 5-HT3 receptors.
DISCUSSION
This paper describes, to our knowledge, the first knock-in mouse strain that carries a GFP-tagged protein for the purpose of counting and localizing molecules of that protein. We conclude that GAT1 molecules are present at surface densities of 800–1300 per square micrometer in presynaptic boutons, that a single basket cell bouton in cerebellum contains 9000 mGAT1 molecules, and that a pinceau contains 7.8 × 106 molecules. Additional biotinylation experiments show that 61–63% of these molecules are on the surface membrane; the remainder are within a few hundred nanometers in the cytoplasm. These impressively high numbers agree with previous guesses (see introductory remarks).
The study uses the absolute calibrations provided by previously developed transparent agarose beads with known surface densities of GFP (Chiu et al., 2001). In the present study, the bead method was extended to gels containing known densities of GFP (Hack et al., 2000).
The knock-in GFP method, accompanied by biochemical analysis of surface/cytoplasmic partitioning, complements several other techniques appropriate for quantifying synaptic proteins. Quantitative immunogold electron microscopy has provided quite useful estimates for GABAA and AMPA receptor density (Nusser et al., 1997, 1998a,b), at the resolution of the electron microscope, but involves uncertainties arising from the efficiency of antibody binding and section thickness. A volume measurement method yields the average density of a specific protein on the plasma membrane (Lehre and Danbolt, 1998) but cannot give local densities of a variably distributed protein (for instance, at synapses). Autoradiography with a specific ligand (Anglister et al., 1994) requires a specific, irreversibly bound ligand; these are unavailable for most channels and receptors, including GAT1, and in any case cannot be used on living tissues.
The mGAT1–GFP mouse displays apparently normal life span, reproductive behavior, and weight, and mostly normal tonic and phasic inhibitory synaptic transmission (Fig. 9), despite a threefold decrease in surface GAT1 function. These observations agree generally with clinical observations that the anti-epileptic GAT1 inhibitors, such as tiagabine, are well tolerated at doses that produce only partial mGAT1 blockade. The present project has also unexpectedly generated a genuine mGAT1 knock-out, the homozygous intron 14-Neo-mGAT1 mice (Jensen et al., 2001) (Fig. 1). We will report elsewhere on extensive electrophysiological and behavioral differences between homozygous intron 14-Neo-mGAT1 and WT mice. The intron 14-Neo-mGAT1 heterozygote, however, which has half-normal GAT1 function, displays few if any deficits. This is consistent with the normal phenotype of the mGAT1–GFP mice despite their threefold lower GABA uptake.
Sorting and membrane/cytoplasmic partitioning of mGAT1–GFP
We performed preliminary experiments with heterologous expression to define the proper mGAT1–GFP fusion (Fig. 3). On the basis of GABA uptake assays in HEK cells, the 12-residue spacer followed by GFP preserved the EC50 for GABA and the expression level (Fig. 3). On the basis of localization in MDCK cells, the additional sequences also preserved cellular localization (Fig. 3). However, synaptosomes from mGAT1–GFP mice display only approximately one-third as much surface functional GAT1 as WT mice, and surface biotinylation shows that most of the mGAT1–GFP remains intracellular (Fig. 7). GAT1 is dynamically regulated in neurons, moving from the cytoplasmic vesicles to the surface membrane on a time scale of minutes in response to regulation by several pathways, including phosphorylation and interactions with syntaxin (Corey et al., 1994;Beckman and Quick, 1998; Bernstein and Quick, 1999; Whitworth and Quick, 2001a,b). We used pharmacological manipulations of this trafficking to localize all of the GAT1 on the surface membrane (Fig.7), and then repeated the measurements under these same conditions, to remove cytoplasmic/membrane partitioning as a possible source of ambiguity in the total (cytoplasmic plus membrane) measured densities.
The source of the abnormal cytoplasmic/membrane partitioning in mGAT1–GFP mice is suggested by a recent study showing that the final three residues of GAT1, AYI, interact with pals1, an endogenous PDZ domain protein (McHugh et al., 2001). In HEK cells, which do not express pals1, WT GAT1 shows the same functional expression level as GAT1 minus the terminal AYI sequence or as GAT1 minus the terminal AYI sequence with coexpression of pals1. However, HEK cells coexpressing pals1 and WT GAT1 display three times greater GABA transport. We do not yet know whether the lack of a PDZ interaction decreases the rate of exocytosis or increases that of endocytosis. Nonetheless, these data explain how the intact mouse displays threefold lower GAT1 activity, despite the functional data of Figure 3. Thus, the present knock-in mouse strain has one important difference from WT mice, but importantly, the present data also suggest that a PDZ interaction enhances surface localization of GAT1, extending previous conclusions based on a heterologous expression system.
Any GAT1–GFP knock-in mouse would also be subject to dynamic regulation of membrane/cytoplasmic partitioning, although the equilibrium is shifted toward membrane expression in the presence of a proper PDZ interaction (Corey et al., 1994; Beckman and Quick, 1998;Bernstein and Quick, 1999; Whitworth and Quick, 2001a,b). The data of Figure 7 show that an average of 39% (based on the [3H]GABA uptake data) or 37% (based on the biotinylation data) of GAT1 is intracellular in WT mice. Because untreated synapses and translocation treatment synapses showed indistinguishable densities and fluorescence profiles (Fig. 8), we suggest that the intracellular mGAT1–GFP remains near the surface membrane (within the resolution of the confocal microscope). The entire pool of 1000–1300 GAT1 molecules per square micrometer (Table 1) is potentially available for surface function. This value may vary locally, leading to uncertainty that exceeds the systematic factors discussed in the next section.
Other sources of variation and systematic error
Several additional factors introduce variation into our range of mGAT1 counts. In cortex and hippocampus, differential expression of several genes defines subsets of inhibitory interneurons (Seress et al., 1993; Acsady et al., 1996; Gulyas et al., 1990, 1996; Hajos et al., 1996, 2000; Kiss et al., 1997; Blasco-Ibanez et al., 1998; Katona et al., 1999; Emri et al., 2001). In hippocampus, we usually found that stratum radiatum had lower fluorescence than stratum oriens or stratum lacunosum moleculare. Although the culture experiments show that all GABAergic neurons express mGAT1 robustly, this difference warrants systematic study in view of observations in rats that, in stratum radiatum, evoked IPSCs are prolonged to a greater extent by tiagabine (Engel et al., 1998).
The accuracy of volume and surface measurements on boutons depends on their size and the point-spread function. For the larger boutons, such as those in ML of cerebellum, we have relatively accurate measurements that define the relations among integrated fluorescence, volume density, and surface density. For smaller boutons, such as those in hippocampus, the resolution in the x-y plane andz-axis are limiting factors. Although integrated numbers of mGAT1–GFP molecules have good accuracy, the uncertainties about membrane density in hippocampus may approach a factor of 2.
Several other factors are primarily related to instrumentation and might introduce systematic errors. Previous measurements on the GFP beads indicate that we understand their surface densities to within 20% (Chiu et al., 2001). The bead-based and polyacrylamide gel-based measurements involve substantially different analyses and independent calibration series, except that the polyacrylamide gel analysis assumes that 100% of the His6–GFP molecules are active, a point proven only for the His6–GFP molecules tethered to the beads (Chiu et al., 2001). We are encouraged that these two measurement series agree within <20% when both are applied to determine the membrane density of mGAT1–GFP molecules at cerebellar boutons, which have good geometric properties for both analyses. Nevertheless, possible sources of systematic errors include mismatch of refractive index and other sources of light scattering. We measured the decrement in fluorescence intensity within a tissue slice and concluded that, within the first 4 μm, the fluorescent intensity dropped <10% (see Materials and Methods). Stability of GFP during sample preparation has been monitored and appears to add little error (see Materials and Methods). Background subtraction is straightforward. Bleaching corrections are straightforward based on laser power, which we monitored; fluctuations were on the order of ±5%. We consider it likely that systematic errors of the type discussed in this paragraph amount to <30%. The measurements are now limited principally by instrumentation (∼20%) and perhaps by a series of systematic errors that might total 30%. The usual methods for propagating errors suggest that these fractions should be added in quadrature, for a total error of ∼36%.
GABAergic interneurons in culture
The experiments on hippocampal cultures emphasize the precision of the knock-in construct by showing that virtually all GABAergic neurons, and only GABAergic neurons, show robust mGAT1–GFP fluorescence. GAT1 thus takes its place as one of the genes common to all inhibitory interneurons.
Some interneurons are readily identifiable in hippocampal culture because they are round and unusually large. However, physiological researchers routinely find that many cultured inhibitory neurons resemble pyramidal cells. The mGAT1–GFP mouse provides an unambiguous method to choose living GABAergic neurons for various studies.
Implications of the measurements
The present number for the average density of GAT1 in a presynaptic bouton is 800–1300 per square micrometer. Because of dynamic partitioning between cytoplasm and surface membrane, 61–63% of this number (average value) is on the surface membrane of WT mice in the absence of special treatments. We also observe localized zones of 50–100% higher density, perhaps corresponding to synapses. These densities roughly equal our expectation when we began the project (see introductory remarks). This density is not limited by membrane packing, because GAT1 can be expressed in Xenopus oocytes at densities exceeding 10,000 per square micrometer (Mager et al., 1993). Glutamate transporters occur at densities three- to fivefold higher than GAT1 in cerebellar molecular layer (Lehre and Danbolt, 1998), yet these GAT1 densities are sufficient to shape the time course of the evoked IPSC and to suppress tonic conductance at extrasynaptic GABAA receptors.
Acetylcholinesterase is present in several-fold excess at nerve–muscle synapses (Anglister et al., 1994). One cannot yet conclude, simply because the mGAT1–GFP mice serendipitously display normal synaptic transmission but approximately fourfold lower overall GAT1 surface expression, that a similar excess applies to GAT1, because the decrement could vary by cell type and by region on the cell. Both the present knock-in mice expressing fluorescent mGAT1 and the related mGAT1 knock-out strain provide an appropriate source of preparations to approach such questions systematically.
Footnotes
This research was supported by grants from National Institutes of Health (DA-09121, DA-010509, NS-11756, MH-49176, MH-61468, NS-030549, and National Research Service Award to M.L.) and the National Science Foundation (0119493), and by a Della Martin Fellowship (C.-S.C.). We are indebted to Cesar Labarca and other members of the Caltech and University of California Los Angeles groups for much advice, Nathan Nelson for the mGAT1 cDNA, Tau-Mu Yi and M. Simon for use and help with the confocal microscope, and Melinda Turner and Robert Farley for discussion.
Correspondence should be addressed to Henry A. Lester, Division of Biology, M/C 156-29, California Institute of Technology, Pasadena, CA 91125. E-mail: lester@caltech.edu.
REFERENCES
- 1.Acsady L, Gorcs TJ, Freund TF. Different populations of vasoactive intestinal polypeptide-immunoreactive interneurons are specialized to control pyramidal cells or interneurons in the hippocampus. Neuroscience. 1996;73:317–334. doi: 10.1016/0306-4522(95)00609-5. [DOI] [PubMed] [Google Scholar]
- 2.Ahn J, Mundigl O, Muth TR, Rudnick G, Caplan MJ. Polarized expression of GABA transporters in Madin-Darby canine kidney cells and cultured hippocampal neurons. J Biol Chem. 1996;271:6917–6924. doi: 10.1074/jbc.271.12.6917. [DOI] [PubMed] [Google Scholar]
- 3.Altman J, Bayer S . . Development of the cerebellar system in relation to its evolution, structure, and functions. CRC; Boca Raton, FL: 1997. [Google Scholar]
- 4.Anglister L, Stiles JR, Salpeter MM. Acetylcholinesterase density and turnover number at frog neuromuscular junctions, with modeling of their role in synaptic function. Neuron. 1994;12:783–794. doi: 10.1016/0896-6273(94)90331-x. [DOI] [PubMed] [Google Scholar]
- 5.Barakat L, Bordey A. GAT-1 and reversible GABA transporter in Bergmann glia in slices. J Neurophysiol. 2002;88:1407–1419. doi: 10.1152/jn.2002.88.3.1407. [DOI] [PubMed] [Google Scholar]
- 6.Beckman ML, Quick MW. Neurotransmitter transporters: regulators of function and functional regulation. J Membr Biol. 1998;164:1–10. doi: 10.1007/s002329900388. [DOI] [PubMed] [Google Scholar]
- 7. Beckman ML, Bernstein EM, Quick MW. Multiple G-protein-coupled receptors initiate protein kinase C redistribution of GABA transporters in hippocampal neurons. J Neurosci 19 1999. RC9(1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstein EM, Quick MW. Regulation of γ-aminobutyric acid (GABA) transporters by extracellular GABA. J Biol Chem. 1999;274:889–895. doi: 10.1074/jbc.274.2.889. [DOI] [PubMed] [Google Scholar]
- 9.Blasco-Ibanez JM, Martinez-Guijarro FJ, Freund TF. Enkephalin-containing interneurons are specialized to innervate other interneurons in the hippocampal CA1 region of the rat and guinea-pig. Eur J Neurosci. 1998;10:1784–1795. doi: 10.1046/j.1460-9568.1998.00190.x. [DOI] [PubMed] [Google Scholar]
- 10.Brickley SG, Cull-Candy SG, Farrant M. Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J Physiol (Lond) 1996;497:753–759. doi: 10.1113/jphysiol.1996.sp021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brock R, Hamelers IH, Jovin TM. Comparison of fixation protocols for adherent cultured cells applied to a GFP fusion protein of the epidermal growth factor receptor. Cytometry. 1999;35:353–362. doi: 10.1002/(sici)1097-0320(19990401)35:4<353::aid-cyto8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 12.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 13.Chiu CS, Kartalov E, Unger M, Quake S, Lester HA. Single-molecule measurements calibrate green fluorescent protein surface densities on transparent beads for use with “knock-in” animals and other expression systems. J Neurosci Methods. 2001;105:55–63. doi: 10.1016/s0165-0270(00)00354-x. [DOI] [PubMed] [Google Scholar]
- 14.Corey JL, Davidson N, Lester HA, Brecha N, Quick MW. Protein kinase C modulates the activity of a cloned gamma-aminobutyric acid transporter expressed in Xenopus oocytes via regulated subcellular redistribution of the transporter. J Biol Chem. 1994;269:14759–14767. [PubMed] [Google Scholar]
- 15.Davis KE, Straff DJ, Weinstein EA, Bannerman PG, Correale DM, Rothstein JD, Robinson MB. Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J Neurosci. 1998;18:2475–2485. doi: 10.1523/JNEUROSCI.18-07-02475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Biasi S, Vitellaro-Zuccarello L, Brecha NC. Immunoreactivity for the GABA transporter-1 and GABA transporter-3 is restricted to astrocytes in the rat thalamus. A light and electron-microscopic immunolocalization. Neuroscience. 1998;83:815–828. doi: 10.1016/s0306-4522(97)00414-4. [DOI] [PubMed] [Google Scholar]
- 17.Dickson RM, Cubitt AB, Tsien RY, Moerner WE. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 18.Dingledine R, Korn SJ. γ-Aminobutyric acid uptake and the termination of inhibitory synaptic potentials in the rat hippocampal slice. J Physiol (Lond) 1985;366:387–409. doi: 10.1113/jphysiol.1985.sp015804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Draguhn A, Heinemann U. Different mechanisms regulate IPSC kinetics in early postnatal and juvenile hippocampal granule cells. J Neurophysiol. 1996;76:3983–3993. doi: 10.1152/jn.1996.76.6.3983. [DOI] [PubMed] [Google Scholar]
- 20.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emri Z, Antal K, Gulyas AI, Megias M, Freund TF. Electrotonic profile and passive propagation of synaptic potentials in three subpopulations of hippocampal CA1 interneurons. Neuroscience. 2001;104:1013–1026. doi: 10.1016/s0306-4522(01)00136-1. [DOI] [PubMed] [Google Scholar]
- 22.Engel D, Schmitz D, Gloveli T, Frahm C, Heinemann U, Draguhn A. Laminar difference in GABA uptake and GAT-1 expression in rat CA1. J Physiol (Lond) 1998;512:643–649. doi: 10.1111/j.1469-7793.1998.643bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frahm C, Engel D, Draguhn A. Efficacy of background GABA uptake in rat hippocampal slices. NeuroReport. 2001;12:1593–1596. doi: 10.1097/00001756-200106130-00016. [DOI] [PubMed] [Google Scholar]
- 24.Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 25.Grabner M, Dirksen RT, Beam KG. Tagging with green fluorescent protein reveals a distinct subcellular distribution of L-type and non-L-type Ca2+ channels expressed in dysgenic myotubes. Proc Natl Acad Sci USA. 1998;95:1903–1908. doi: 10.1073/pnas.95.4.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guastella JG, Nelson N, Nelson H, Czyzyk L, Keynan S, Midel MC, Davidson N, Lester HA, Kanner B. Cloning and expression of a rat brain GABA transporter. Science. 1990;249:1303–1306. doi: 10.1126/science.1975955. [DOI] [PubMed] [Google Scholar]
- 27.Gulyas AI, Gorcs TJ, Freund TF. Innervation of different peptide-containing neurons in the hippocampus by GABAergic septal afferents. Neuroscience. 1990;37:31–44. doi: 10.1016/0306-4522(90)90189-b. [DOI] [PubMed] [Google Scholar]
- 28.Gulyas AI, Miles R, Hajos N, Freund TF. Precision and variability in postsynaptic target selection of inhibitory cells in the hippocampal CA3 region. Eur J Neurosci. 1993;5:1729–1751. doi: 10.1111/j.1460-9568.1993.tb00240.x. [DOI] [PubMed] [Google Scholar]
- 29.Gulyas AI, Hajos N, Freund TF. Interneurons containing calretinin are specialized to control other interneurons in the rat hippocampus. J Neurosci. 1996;16:3397–3411. doi: 10.1523/JNEUROSCI.16-10-03397.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hack NJ, Billups B, Guthrie PB, Rogers JH, Muir EM, Parks TN, Kater SB. Green fluorescent protein as a quantitative tool. J Neurosci Methods. 2000;95:177–184. doi: 10.1016/s0165-0270(99)00178-8. [DOI] [PubMed] [Google Scholar]
- 31.Hajos N, Acsady L, Freund TF. Target selectivity and neurochemical characteristics of VIP-immunoreactive interneurons in the rat dentate gyrus. Eur J Neurosci. 1996;8:1415–1431. doi: 10.1111/j.1460-9568.1996.tb01604.x. [DOI] [PubMed] [Google Scholar]
- 32.Hajos N, Katona I, Naiem SS, MacKie K, Ledent C, Mody I, Freund TF. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci. 2000;12:3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- 33.Halasy K, Somogyi P. Distribution of GABAergic synapses and their targets in the dentate gyrus of rat: a quantitative immunoelectron microscopic analysis. J Hirnforsch. 1993;34:299–308. [PubMed] [Google Scholar]
- 34.Hell S, Reiner G, Cremer C, Stelzer EHK. Aberrations in confocal fluorescence microscopy induced by mismatches in refractive-index. J Microsc. 1993;169:391–405. [Google Scholar]
- 35.Hilgemann DW, Lu CC. GAT1 (GABA:Na+:Cl−) cotransport function. Database reconstruction with an alternating access model. J Gen Physiol. 1999;114:459–476. doi: 10.1085/jgp.114.3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horton N, Quick MW. Syntaxin 1A up-regulates GABA transporter expression by subcellular redistribution. Mol Membr Biol. 2001;18:39–44. [PubMed] [Google Scholar]
- 37.Ikegaki N, Saito N, Hashima M, Tanaka C. Production of specific antibodies against GABA transporter subtypes (GAT1, GAT2, GAT3) and their application to immunocytochemistry. Brain Res Mol Brain Res. 1994;26:47–54. doi: 10.1016/0169-328x(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 38.Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- 39.Jensen K, Chiu C-S, Labarca C, Deshpande P, Lester HA, Mody I. GABA-transporter (GAT-1) deficient mice show tremor and increased GABAergic tonic conductance. Soc Neurosci Abstr. 2001;27:1317. [Google Scholar]
- 40.Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 41.Jones MV, Jonas P, Westbrook GL. Free GABA declines rapidly at granule cell synapses in hippocampal slices. Soc Neurosci Abstr. 1998;24:1317. [Google Scholar]
- 42.Jones MV, Overstreet LS, Jonas P, Westbrook GL. Temporal and spatial focusing of inhibition through the interaction of GABA diffusion with GABAA receptor kinetics. Soc Neurosci Abstr. 1999;25:1246. [Google Scholar]
- 43.Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kiss J, Magloczky Z, Somogyi J, Freund TF. Distribution of calretinin-containing neurons relative to other neurochemically identified cell types in the medial septum of the rat. Neuroscience. 1997;78:399–410. doi: 10.1016/s0306-4522(96)00508-8. [DOI] [PubMed] [Google Scholar]
- 45.Labarca C, Schwarz J, Deshpande P, Schwarz S, Nowak MW, Fonck C, Nashmi R, Kofuji P, Dang H, Shi W, Fidan M, Khakh BS, Chen Z, Bowers BJ, Boulter J, Wehner JM, Lester HA. Point mutant mice with hypersensitive α4 nicotinic receptors show dopaminergic deficits and increased anxiety. Proc Natl Acad Sci USA. 2001;98:2786–2791. doi: 10.1073/pnas.041582598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lester HA, Cao Y, Mager S. Listening to neurotransmitter transporters. Neuron. 1996;17:807–810. doi: 10.1016/s0896-6273(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 48.Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci. 1998;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu QR, Mandiyan S, Nelson H, Nelson N. A family of genes encoding neurotransmitter transporters. Proc Natl Acad Sci USA. 1992;89:6639–6643. doi: 10.1073/pnas.89.14.6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu Y, Grady S, Marks MJ, Picciotto M, Changeux JP, Collins AC. Pharmacological characterization of nicotinic receptor-stimulated GABA release from mouse brain synaptosomes. J Pharmacol Exp Ther. 1998;287:648–657. [PubMed] [Google Scholar]
- 51.Mager S, Naeve J, Quick M, Labarca C, Davidson N, Lester HA. Steady states, charge movements, and rates for a cloned GABA transporter expressed in Xenopus oocytes. Neuron. 1993;10:177–188. doi: 10.1016/0896-6273(93)90309-f. [DOI] [PubMed] [Google Scholar]
- 52.McHugh EM, Zhu W, Mager S. Identification and characterization of the interaction of the GABA transporter GAT-1 with pals-1. Soc Neurosci Abstr. 2001;27:2429. [Google Scholar]
- 53.Minelli A, Brecha NC, Karschin C, DeBiasi S, Conti F. GAT-1, a high-affinity GABA plasma membrane transporter, is localized to neurons and astroglia in the cerebral cortex. J Neurosci. 1995;15:7734–7746. doi: 10.1523/JNEUROSCI.15-11-07734.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morara S, Brecha NC, Marcotti W, Provini L, Rosina A. Neuronal and glial localization of the GABA transporter GAT-1 in the cerebellar cortex. NeuroReport. 1996;7:2993–2996. doi: 10.1097/00001756-199611250-00039. [DOI] [PubMed] [Google Scholar]
- 55.Nadeau H, McKinney S, Anderson DJ, Lester HA. ROMK1 (Kir1.1) causes apoptosis and chronic silencing of hippocampal neurons. J Neurophysiol. 2000;84:1062–1075. doi: 10.1152/jn.2000.84.2.1062. [DOI] [PubMed] [Google Scholar]
- 56.Nagy A, Delgado-Escueta AV. Rapid preparation of synaptosomes from mammalian brain using nontoxic isoosmotic gradient material (Percoll). J Neurochem. 1984;43:1114–1123. doi: 10.1111/j.1471-4159.1984.tb12851.x. [DOI] [PubMed] [Google Scholar]
- 57.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 58.Ng CH, Ong WY. Increased expression of gamma-aminobutyric acid transporters GAT-1 and GAT-3 in the spinal trigeminal nucleus after facial carrageenan injections. Pain. 2001;92:29–40. doi: 10.1016/s0304-3959(00)00468-1. [DOI] [PubMed] [Google Scholar]
- 59.Nusser Z, Cull-Candy S, Farrant M. Differences in synaptic GABAA receptor number underlie variation in GABA mini amplitude. Neuron. 1997;19:697–709. doi: 10.1016/s0896-6273(00)80382-7. [DOI] [PubMed] [Google Scholar]
- 60.Nusser Z, Hajos N, Somogyi P, Mody I. Increased number of synaptic GABAA receptors underlies potentiation at hippocampal inhibitory synapses. Nature. 1998a;395:172–177. doi: 10.1038/25999. [DOI] [PubMed] [Google Scholar]
- 61.Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron. 1998b;21:545–559. doi: 10.1016/s0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- 62.Overstreet LS, Westbrook GL. Paradoxical reduction of synaptic inhibition by vigabatrin. J Neurophysiol. 2001;86:596–603. doi: 10.1152/jn.2001.86.2.596. [DOI] [PubMed] [Google Scholar]
- 63.Palay SL, Chan-Palay V. Cerebellar cortex: cytology and organization. Springer; New York: 1974. [Google Scholar]
- 64.Pawley JB. Image contrast in confocal light microscopy. In: Cheng PC, Kriete A, editors. Handbook of biological confocal microscopy. Plenum; New York: 1995. pp. 281–310. [Google Scholar]
- 65.Pietrini G, Suh YJ, Edelmann L, Rudnick G, Caplan MJ. The axonal γ-aminobutyric acid transporter GAT-1 is sorted to the apical membranes of polarized epithelial cells. J Biol Chem. 1994;269:4668–4674. [PubMed] [Google Scholar]
- 66.Qian Y, Galli A, Ramamoorthy S, Risso S, DeFelice LJ, Blakely RD. Protein kinase C activation regulates human serotonin transporters in HEK-293 cells via altered cell surface expression. J Neurosci. 1997;17:45–57. doi: 10.1523/JNEUROSCI.17-01-00045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Radian R, Ottersen OP, Storm-Mathisen J, Castel M, Kanner BI. Immunocytochemical localization of the GABA transporter in rat brain. J Neurosci. 1990;10:1319–1330. doi: 10.1523/JNEUROSCI.10-04-01319.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramamoorthy S, Giovanetti E, Qian Y, Blakely RD. Phosphorylation and regulation of antidepressant-sensitive serotonin transporters. J Biol Chem. 1998;273:2458–2466. doi: 10.1074/jbc.273.4.2458. [DOI] [PubMed] [Google Scholar]
- 69.Rattray M, Priestley JV. Differential expression of GABA transporter-1 messenger RNA in subpopulations of GABA neurones. Neurosci Lett. 1993;156:163–166. doi: 10.1016/0304-3940(93)90463-u. [DOI] [PubMed] [Google Scholar]
- 70.Ribak CE, Tong WM, Brecha NC. GABA plasma membrane transporters, GAT-1 and GAT-3, display different distributions in the rat hippocampus. J Comp Neurol. 1996;367:595–606. doi: 10.1002/(SICI)1096-9861(19960415)367:4<595::AID-CNE9>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 71.Roepstorff A, Lambert JD. Comparison of the effect of the GABA uptake blockers, tiagabine and nipecotic acid, on inhibitory synaptic efficacy in hippocampal CA1 neurones. Neurosci Lett. 1992;146:131–134. doi: 10.1016/0304-3940(92)90060-k. [DOI] [PubMed] [Google Scholar]
- 72.Roepstorff A, Lambert JDC. Factors contributing to the decay of the stimulus-evoked IPSC in rat hippocampal CA1 neurons. J Neurophysiol. 1994;72:2911–2926. doi: 10.1152/jn.1994.72.6.2911. [DOI] [PubMed] [Google Scholar]
- 73.Rosina A, Morara S, Provini L. GAT-1 developmental expression in the rat cerebellar cortex: basket and pinceau formation. NeuroReport. 1999;10:1613–1618. doi: 10.1097/00001756-199905140-00041. [DOI] [PubMed] [Google Scholar]
- 74.Seress L, Gulyas AI, Ferrer I, Tunon T, Soriano E, Freund TF. Distribution, morphological features, and synaptic connections of parvalbumin- and calbindin D28k-immunoreactive neurons in the human hippocampal formation. J Comp Neurol. 1993;337:208–230. doi: 10.1002/cne.903370204. [DOI] [PubMed] [Google Scholar]
- 75.Somogyi P, Nunzi MG, Gorio A, Smith AD. A new type of specific interneuron in the monkey hippocampus forming synapses exclusively with the axon initial segments of pyramidal cells. Brain Res. 1983;259:137–142. doi: 10.1016/0006-8993(83)91076-4. [DOI] [PubMed] [Google Scholar]
- 76.Tang CY, Nusser Z, Mody I. Selective regulation of tonic and phasic GABAA conductances in hippocampal neurons. Soc Neurosci Abstr. 2001;27:1317. [Google Scholar]
- 77.Thompson SM, Gahwiler BH. Effects of the GABA uptake inhibitor tiagabine on inhibitory synaptic potentials in rat hippocampal slice cultures. J Neurophysiol. 1992;67:1698–1701. doi: 10.1152/jn.1992.67.6.1698. [DOI] [PubMed] [Google Scholar]
- 78.Wang XS, Ong WY. A light and electron microscopic study of GAT-1 in the monkey basal ganglia. J Neurocytol. 1999;28:1053–1061. doi: 10.1023/a:1007056608820. [DOI] [PubMed] [Google Scholar]
- 79.Whitworth TL, Quick MW. Substrate-induced regulation of γ-aminobutyric acid transporter trafficking requires tyrosine phosphorylation. J Biol Chem. 2001a;276:42932–42937. doi: 10.1074/jbc.M107638200. [DOI] [PubMed] [Google Scholar]
- 80.Whitworth TL, Quick MW. Upregulation of γ-aminobutyric acid transporter expression: role of alkylated γ-aminobutyric acid derivatives. Biochem Soc Trans. 2001b;29:736–741. doi: 10.1042/0300-5127:0290736. [DOI] [PubMed] [Google Scholar]
- 81.Woo TU, Whitehead RE, Melchitzky DS, Lewis DA. A subclass of prefrontal γ-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci USA. 1998;95:5341–5346. doi: 10.1073/pnas.95.9.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yan XX, Ribak CE. Developmental expression of gamma-aminobutyric acid transporters (GAT-1 and GAT-3) in the rat cerebellum: evidence for a transient presence of GAT-1 in Purkinje cells. Brain Res Dev Brain Res. 1998a;111:253–269. doi: 10.1016/s0165-3806(98)00144-8. [DOI] [PubMed] [Google Scholar]
- 83.Yan XX, Ribak CE. Increased expression of GABA transporters, GAT-1 and GAT-3, in the deafferented superior colliculus of the rat. Brain Res. 1998b;783:63–76. doi: 10.1016/s0006-8993(97)01157-8. [DOI] [PubMed] [Google Scholar]
- 84.Yan XX, Cariaga WA, Ribak CE. Immunoreactivity for GABA plasma membrane transporter, GAT-1, in the developing rat cerebral cortex: transient presence in the somata of neocortical and hippocampal neurons. Brain Res Dev Brain Res. 1997;99:1–19. doi: 10.1016/s0165-3806(96)00192-7. [DOI] [PubMed] [Google Scholar]