

Abstract
The purines, ATP and adenosine, control the rundown and termination of swimming in the Xenopus embryo. This intrinsic purinergic modulation, unavoidably present during every swimming episode, could lead to stereotyped inflexible behavior and consequently could jeopardize the survival of the embryo. To explore whether this control system can exhibit adaptability, I have used a minimal simulation in which a model neuron released ATP that (1) inhibited K+ currents and (2) was converted by ectonucleotidases to adenosine, which then inhibited Ca2+ currents. The model neuron exhibited an accommodating spike train controlled by the actions of ATP and adenosine. Feedforward inhibition by the upstream metabolite ADP of the ecto-5′-nucleotidase that converts AMP to adenosine introduced adaptability and allowed the resetting of spike accommodation. The strength of feedforward inhibition determined the extent to which resetting could occur. I have tested these predictions by examining swimming in the real embryo. The rundown of swimming was reset in a manner similar to that predicted by the single-neuron model. By blocking the purinoceptors, I have demonstrated that resetting in the embryo is attributable to the actions of the purines and results from feedforward inhibition of adenosine production. The resetting of rundown in the motor systems can be reformulated as an associative mechanism in which the temporal coincidence of two stimuli can prolong network activity if they fall within a particular time window. The length of the time window and the magnitude of the prolongation of neural activity both depend on the strength of the feedforward ADP-mediated inhibition of the ecto-5′-nucleotidase.
Keywords: ATP, adenosine, central pattern generator, ectonucleotidase, associative, purine
The purines ATP and adenosine are important neurotransmitters/modulators throughout the nervous system (Ralevic and Burnstock, 1998). They are implicated in the control of sleep (Porkka-Heiskanen et al., 1997), the transmission of pain (Poon and Sawynok, 1998; Ding et al., 2000), spinal motor circuits (Dale and Gilday, 1996), cardiorespiratory reflexes (Thomas and Spyer, 1999), and neuroprotection during transient ischemia (Rudolphi et al., 1992; Dale et al., 2000).
ATP released by neurons either as a cotransmitter or a principal transmitter (Edwards et al., 1992; Robertson et al., 2001) can act at two classes of postsynaptic receptor selective for ATP: the p2x ligand-gated channels and the p2y G-protein coupled receptors (Ralevic and Burnstock, 1998). The actions of ATP are terminated by ectonucleotidases, a heterogeneous collection of enzymes (Zimmermann, 1996; Zimmermann and Braun, 1999) that hydrolyze ATP to ADP or AMP. In most cases, adenosine is probably not released directly in the CNS (Zimmermann, 1996) but arises instead from the extracellular breakdown of ATP by ectonucleotidases. Clearly, the ectonucleotidases occupy a central role in purinergic signaling: they simultaneously terminate the actions of ATP and initiate the actions of adenosine. The kinetics of the ectonucleotidases must profoundly influence the interactions between ATP- and adenosine-mediated signaling. These kinetics can be complex, because the upstream metabolites ATP and ADP inhibit the ecto-5′-nucleotidase that converts AMP to adenosine (Gordon et al., 1986; Zimmermann, 1996). This feedforward inhibition delays the appearance of adenosine with respect to the previous release of ATP (James and Richardson, 1993; Dale, 1998).
In Xenopus embryos, ATP and adenosine control the rundown and termination of motor-pattern generation (Dale and Gilday, 1996;Dale, 1998; Brown and Dale, 2000, 2002a,b). ATP is released from spinal pattern-generating neurons and increases their excitability by inhibiting voltage-gated K+ currents. However, ATP is also converted by ectonucleotidases to adenosine, which diminishes neuronal excitability by inhibiting Ca2+ currents. Because the appearance of adenosine is delayed, the balance between the actions of ATP and adenosine changes with time and controls the slowing and eventual stopping of swimming (Dale, 1998). Direct measurement of adenosine production in the spinal cord during motor activity shows that it accumulates on a time course consistent with its production by ectonucleotidases (Dale, 1998). Indeed, the delay in the appearance of adenosine may result from the inhibition of the ecto-5′-nucleotidase by ATP and ADP. Simulations that incorporate the kinetics of ectonucleotidases in a detailed model of the spinal circuitry suggest that this mechanism can account for the delay in the production of adenosine (Dale, 1998). Indeed, ADP has been shown recently to inhibit the ecto-5′-nucleotidase in Xenopus spinal cord (Brown and Dale, 2002b).
Thus, the purines mediate intrinsic modulation of the central spinal network that generates swimming in Xenopus. Every time a tadpole swims, this modulatory system will act to ensure the termination of the activity. However, behavior must be adaptable to enhance survival of the organism. Therefore, how can an intrinsic modulatory system have sufficient flexibility to allow for the unexpected? This paper explores, through modeling and experimental tests, whether feedforward inhibition of the ecto-5′-nucleotidase might allow the necessary adaptability.
MATERIALS AND METHODS
Enzyme kinetics. The kinetic scheme for the conversion of ATP to adenosine was adapted from the study by Slakey et al. (1986) and is identical to that used in a previous study (Dale, 1998). In brief, the equations and kinetic parameters of Slakey et al. (1986) were used with appropriate scaling, which maintained their relative values. The release and processing of the purines was considered to occur in a unitary volume. Mixing was assumed to be instantaneous, and the diffusion of products and substrates was ignored. ATP was converted to adenosine by sequential steps, each described by Michaelis–Menten kinetics. The maximum velocity (Vmax) and Michaelis constant (Km) were, respectively: 2.2 μm/sec and 33.3 μm for the conversion of ATP to ADP; 0.32 μm/sec and 9.5 μm for the conversion of ADP to AMP; and 0.3 μm/sec and 0.94 μm for the conversion of AMP to adenosine. Adenosine was taken up with aVmax of 0.1 μm/sec and aKm of 1 μm. Because all of these reactions were considered to take place in a unitary volume, Vmax is given in units of concentration rather than mass per unit time (cf. Slakey et al., 1986). Feedforward inhibition of the conversion of AMP to adenosine (Ado) by ADP has been described previously (Slakey et al., 1986) as a first-order competitive interaction:
| Equation 1 |
where Ki is a constant of inhibition that was set to either 2 or 3 μm.
Model neurons. The model neuron was endowed with models of voltage-gated currents based on experimental descriptions and was identical to those specified previously (Dale, 1995a,b, 1998). The neuron released ATP in a voltage-dependent manner described by the following equation:
| Equation 2 |
where Kr is the rate of release, Kmax is the maximum possible rate, and V is the membrane potential of the neuron.Kmax was set to 20 μm/msec.
The fast and slow components of the delayed rectifier K+ current were inhibited by ATP according to the following equation:
| Equation 3 |
where frac is the amount of inhibition and the IC50 was set to 5 μm. The maximum allowable inhibition was set to 20% (Dale and Gilday, 1996). Adenosine inhibited the Ca2+ current in a manner similar to that seen in Equation 3, but using the concentration of adenosine instead of ATP, with the IC50 set to 5 μm and the maximum inhibition set to 50% (cf. Brown and Dale, 2000).
The equations were integrated with a Cash–Karp embedded Runge–Kutta–Fehlberg algorithm (Press et al., 1992) and ran on a Sun Ultra170E (Sun Microsystems, Santa Clara, CA).
Analysis of model output. The output of the model was analyzed by measuring the duration of the spike trains before and after the resetting stimulus. A threshold-crossing method was used to measure the time of each spike in the train from which the relevant measures (train length, timing of the resetting stimulus, and duration of activity after the stimulus) could be calculated.
Experimental recordings from Xenopus embryos.Stage 37/38–40 Xenopus embryos (Nieuwkoop and Faber, 1956) were prepared for extracellular ventral-root recordings by well established techniques (Dale, 1995b). In accordance with the UK Animals (Scientific Procedures) Act (1986), embryos were anesthetized in tricaine methane sulfonate (1 mg/ml) until they no longer responded to stimuli. The dorsal fin was then slit, and they were treated with 0.77 mg/ml α-bungarotoxin until immobilized. The trunk skin was removed, and the muscles overlying the spinal cord were partially removed to facilitate drug access. The embryo was then spinalized around the level of the first postotic myotome. After this preparation, the embryo was transferred to a recording chamber that had a volume of ∼500 μl. Ventral-root recordings were made from the intermyotome cleft with small glass suction electrodes. The embryos were continually superfused with a saline solution consisting of (in mm): 115 NaCl, 2.4 NaHCO3, 3 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.4. Drugs were applied in the superfusate.
Swimming was evoked by a single stimulus to the skin (0.2–0.5 msec) delivered through a constant-current stimulus isolator via a bipolar suction electrode. Resetting stimuli (of the same intensity) were delivered in the same manner. However, the resetting stimulus consisted of four repetitions, separated by 200 msec, of a train of three stimuli each 10 msec apart. The strength of stimulation was left unaltered throughout the experiment. A Data Translation DT3010 (Data Translation, Marlboro, MA) was used to save the data to a hard disk at a rate of 6 kHz/sec. The computer also controlled the delivery of stimuli to the embryo. A 3 min rest period was given between the end of one swimming episode and the beginning of the next.
Analysis of swimming activity. Offline analysis was performed with specially written software that allowed cursors to measure the length of swimming episodes, the timing of the resetting stimulus (tpre), and the duration of activity occurring after the resetting stimulus (tpost). Resetting has to be judged according to the length of the episode that would have occurred had the second resetting stimulus not been given. This introduces an element of variability and uncertainty into the analysis, because we cannot know what the embryo would have done if the second stimulus had not been given, and the duration of swimming episodes often exhibits variability with time. I countered this in two ways. First, the experimental (resetting) and control episodes were interleaved so that any trends in episode length were possible to discern. Second, the length of control episodes was plotted against the episode number; because episodes were evoked at regular intervals, this is an approximately linear time axis. A regression line was then fitted to the data, and the slope and intercept of this line were used to interpolate the predicted length of episode for each reset episode (T), which would have occurred had no resetting stimulus been given. This had the advantage that it took into account trends in the duration of episodes (which a simple mean or median would not). Because each resetting experiment took at least 2–3 hr to complete, almost all embryos gradually swam for longer as the experiment progressed.
RESULTS
Model of enzyme kinetics demonstrates resetting of adenosine production
To test whether feedforward inhibition might allow resetting of purine levels, a simple model that used enzyme kinetics worked out for endothelium was used (Slakey et al., 1986). ATP was released at a constant rate, and the levels of the other metabolites were allowed to reach steady-state levels. At 2 sec, the rate of ATP released was greatly increased to a new level. This resulted in higher levels of accumulation of ADP and AMP (Fig. 1). Despite the dramatically raised levels of AMP, the concentration of adenosine fell (Fig. 1B) because the increased inhibition of the conversion of AMP to adenosine outweighed the greater availability of substrate. The levels of adenosine only exceeded those immediately before the step change in ATP release after an additional 1.8 sec (Fig. 1B). By considering just the enzyme kinetics divorced from any physiological context, we can see that feedforward inhibition will in principle allow flexibility of control over the levels of adenosine.
Fig. 1.

Analysis of enzyme kinetics shows that the resetting of adenosine (ADO) production is possible in principle. A, A simulation was run in which ATP was released at a constant rate of 100 μm/sec. After 2 sec, the rate of ATP release was elevated to 300 μm/sec.B, The same data as in A, but plotted on an expanded vertical axis to allow the changes in ATP and adenosine to be seen more clearly. The arrow highlights the fall in adenosine levels after the increase in ATP release. Note that the accumulation of adenosine over the first 2 sec is slowed by the presence of feedforward inhibition mediated by ADP.
Control of firing in a realistic self-modulating neuron
To examine whether feedforward inhibition might play this role in a more realistic but still highly simplified model, I simulated a single neuron based on models developed for Xenopus embryo spinal neurons (Dale, 1995a,b). This single neuron could release ATP with every action potential; the ATP was converted to adenosine via a scheme described by endothelial enzyme kinetics (Fig.2A). ATP inhibited the voltage-gated K+ currents and adenosine inhibited the voltage-gated Ca2+ currents within the model neuron (Dale and Gilday, 1996; Brown and Dale, 2000,2002b). The actions of adenosine were terminated by uptake according to a Michaelis–Menten scheme (see Materials and Methods). A highly simplified model of this type was chosen because it reduced the problem of purinergic modulation to its most basic formulation while still retaining some features of a real physiological system. In this highly simplified model, the single neuron releases sufficient ATP with each spike to self-modulate. In contrast, in the real spinal network, the purine levels will result from the parallel activity of many neurons.
Fig. 2.

The single-neuron model for purinergic modulation.A, A single neuron possessing Na+(data not shown) and K+ and Ca2+channels modulated by ATP and adenosine (ADO), respectively. ADP could inhibit the conversion of AMP to adenosine.B, The duration of the spike train depended on the strength of the feedforward inhibition. As feedforward inhibition by ADP increased (Ki gets smaller), the duration of the spike train increased. Example records of spiking activity in the model neuron are shown next to the appropriate points on the graph.
This simple neuronal model gave an accommodating train of spikes in response to a pulse of current injection 50 pA in amplitude (Fig.2B). The length of this train of spikes depended on the strength of feedforward inhibition (Ki). IfKi was large, the train terminated quickly. As Ki became smaller, the length of the train increased steeply (Fig. 2B). The relationship between the train length andKi is similar to that described previously between the duration of motor activity in computational models of the intact spinal network andKi (Dale, 1998). This suggests that even this highly reduced model captures essential aspects of purinergic modulation in more complex spinal networks.
Resetting and rundown in the model neuron
To mimic a resetting sensory input, a second current pulse of 20 pA and 300 msec in duration was given on top of the first and timed to occur partway through the train of spikes (Fig.3). This is a more physiologically realistic manipulation analogous to the step change in ATP release in the previous simpler model. The resetting pulse caused an increased rate of firing and thus a greater amount of ATP release with a consequent accumulation of ADP and AMP.
Fig. 3.
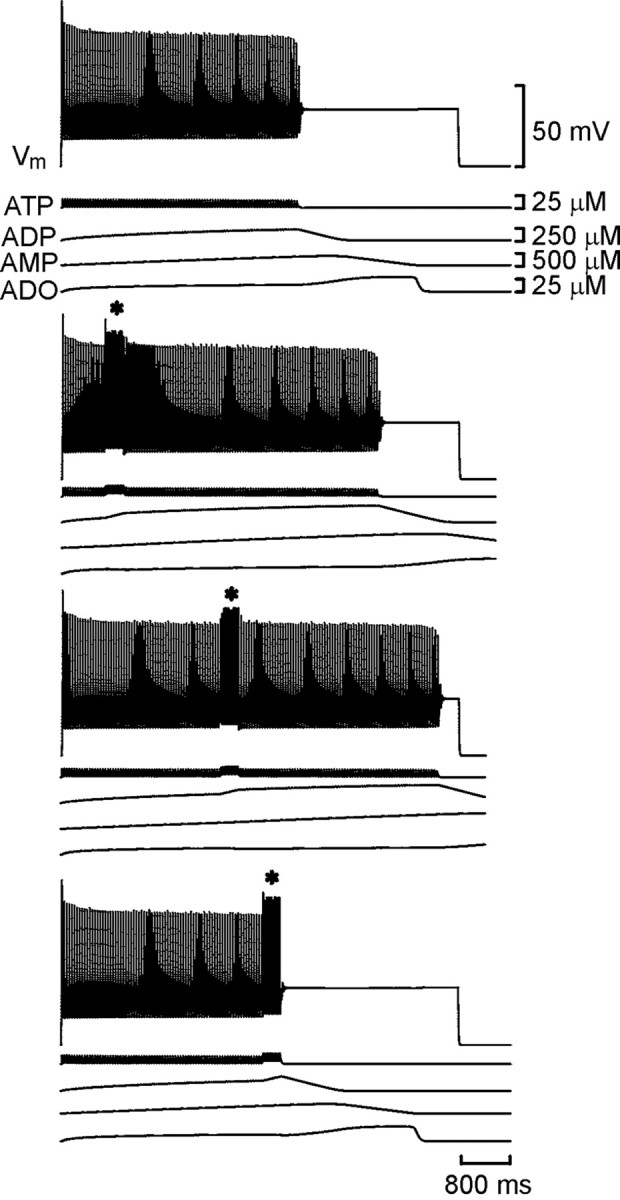
Resetting of the spike train in the model neuron. Each set of traces shows the membrane potential of the model neuron together with the concentrations of ATP, ADP, AMP, and adenosine (ADO). With Ki set to 2 μm, an accommodating train of spikes lasting ∼4 sec was evoked by current injection into the neuron (top trace). In the bottom sets oftraces, a second shorter current pulse was injected (*) during the first. Note that the total duration of spiking was prolonged compared with the control (middle two traces). If the second current pulse occurred past a certain time, the train was prematurely terminated (bottom trace).
Resetting was first examined with Ki = 2 μm. After the resetting pulse, the neuron continued to fire. Indeed, the total duration of firing now exceeded that of a neuron that did not receive the resetting pulse (Fig. 3). As the resetting pulse was moved later through the spike train, the total duration of firing increased. The additional activity reached a maximum of 51% of the control and had a mean value (calculated over all pulses that gave resetting) of 31% of the control. When the second resetting pulse was given toward the very end of the spike train, no prolongation of activity was seen, instead a premature shortening of activity occurred (Fig. 3). When shorter resetting pulses of 150 msec were given, lengthening of the spike train still occurred. However, the premature termination occurred when the resetting pulse was given at a much earlier stage, and the amount of additional activity was reduced to a mean of 17% of control.
To test whether the strength of feedforward inhibition affected the ability to reset the rundown of the spike train, additional simulations were performed with Ki = 3 μm. Under these conditions, spike trains were shorter, so the resetting pulse was shortened to either 100 or 200 msec. In both cases, the resetting pulse prolonged spiking activity if given early in the train but prematurely shortened it if given later (Fig. 4). The amount of lengthening of the spike train had a maximum value of 27% of control and a mean value of 18% of control.
Fig. 4.

With weaker feedforward inhibition (Ki = 3 μm), prolongation of the spike train and partial resetting of accommodation in response to a second stimulus (*) still occurred. However, the magnitude of the changes was less when compared with the stronger feedforward inhibition illustrated in Figure 3.
The relationship between the prolongation of activity and the timing and width of the resetting pulse was examined in more detail by normalizing the timing of tpre to the total duration of the spike train in the absence of T. The amount of activity that occurred tpostwas also normalized to T. The results for both strengths ofKi are summarized in Figure5. With stronger feedforward inhibition, and if the resetting pulse was given early in the train, complete resetting of activity occurred (tpost/T = ∼1) for both the 150 and 300 msec pulses. Astpre was lengthened, resetting became slightly less complete (tpost/T still remained >0.7). Although the magnitude of resetting appeared similar for both widths of the resetting pulse, the timing of premature termination appeared to depend strongly on the width of the resetting pulse. For the 300 msec pulse, premature termination occurred iftpre/T exceeded 0.75. When the resetting pulse was shorter, premature termination occurred aroundtpre/T = 0.4.
Fig. 5.

Summary graph demonstrating how the spike train can be reset by the second stimulus. Results for strong feedforward inhibition (Ki = 2 μm) are shown by circles (solid, 300 msec resetting pulse; open, 150 msec resetting pulse). Results for weaker feedforward inhibition (Ki = 3 μm) are shown bysquares (solid, 200 msec resetting pulse;open, 100 msec resetting pulse). Note how in both cases premature termination occurs earlier when the duration of the resetting pulse is shorter. The dashed line showstpre + tpost= T, and is the relationship expected when there is no resetting. Upward divergence from this line indicates resetting; downward divergence indicates premature termination.
With weaker feedforward inhibition, partial resetting occurred up to approximately tpre/T = 0.7–0.8. The magnitude of the partial resetting was similar for both the 100 and 200 msec resetting pulses. The effect of resetting, although by no means complete, was most obvious later in the train. Compare the points for Ki = 3 μm with the dotted line that specifies the condition in which no resetting occurs (tpre +tpost = T). When resetting pulses were given early, the points fell close to this line. However, when tpre/Texceeded 0.4, they lay appreciably above the line, demonstrating that partial resetting (0.5 <tpost/T < 0.7) occurred when the second input was given later (Fig. 5). As in the case with stronger feedforward inhibition, the width of the resetting pulse influenced the timing of the premature termination; the shorter pulse gives rise to earlier termination.
Resetting changes adenosine levels and the rate of adenosine production in the model
Close inspection of the trajectories of the concentrations of ATP, ADP, AMP, and adenosine reveals the reasons underlying the resetting of spiking activity. Figure 6 shows an example of how the resetting pulse changed the purine levels within the simulation. The amount of ATP released increased transiently because the frequency of firing increased. This resulted in a rapid change in the rate of production of ADP and a sustained increase in the level of ADP over the level that would have occurred in the absence of a resetting pulse. Not surprisingly, the greater availability of substrate resulted in an increase in AMP levels. In contrast, adenosine levels fell in a manner reminiscent of the much simpler simulation presented in Figure 1. This fall occurred because the higher levels of ADP resulted in greater inhibition of adenosine production, allowing the uptake of adenosine to lower the ambient adenosine. It took approximately 1.2 sec for the adenosine levels to return to the level before the resetting pulse (Fig. 6, 1). Because ADP levels remained higher after the resetting pulse, the greater inhibition of adenosine production meant that it took even longer (∼2 sec) for the adenosine levels to increase sufficiently to terminate the spike train (Fig. 6, 2).
Fig. 6.

Resetting of the spike train in the model neuron occurred because the elevated levels of ADP resulting from the second stimulus inhibit adenosine (ADO) production despite the greater levels of AMP present. A control trace(dashed lines) and reset trace(solid line) are shown superimposed. The vertical dotted lines show when the resetting stimulus was given.1 indicates the time taken for adenosine levels to recover to their pre-reset levels; 2 indicates the extra time taken for adenosine levels to rise high enough to terminate the spike train.
Inactivation of the Na+ current contributes to premature termination
Curiously, if the resetting pulse was given late in the train, no resetting occurred. Premature termination was seen instead. The threshold for spiking occurs at the membrane potential at which the net sum of all inward and outward currents equals zero. Premature termination of the spike train by late resetting pulses must arise because the resetting pulse causes a net loss of inward current and raises the threshold. This could occur if the enhanced firing during the resetting pulse increased the inactivation of the Na+ current (INa). Therefore, I examined the values of h, the Hodgkin–Huxley inactivation variable forINa (which has a value of 1 for no inactivation and 0 for total inactivation), when the spike train terminated normally and when a late resetting pulse caused premature termination (Fig. 7). During firing,h varied cyclically, with the membrane potential reaching almost zero during the spike and recovering to ∼0.75 during the afterhyperpolarization (AHP). When the spike train terminated normally (Fig. 7, dashed line), it was almost entirely attributable to the increase in adenosine levels and the consequent increased inhibition of the Ca2+ current: the dynamics of h remained almost unchanged throughout the train.
Fig. 7.

Increased inactivation ofINa causes premature termination of firing in the model neuron. Plots of membrane potential (Vmax), the Hodgkin–Huxley inactivation variable for INa(h), adenosine levels (Ado), percentage of block of the Ca2+ current (ICa) calculated from Equation 3, and injected current (I) versus time for a spike train that terminated normally (dashed lines) and one that terminated prematurely after a resetting pulse (solid line) are shown. The horizontal dotted linerepresents the maximal value of h achieved before the delivery of the resetting pulse. When premature termination after the resetting pulse did not occur (data not shown), hrecovered to the level of the dotted line.
During a resetting pulse, the higher firing rate (and consequently shorter AHPs) reduced the maximal recovery of h to ∼0.63. The available Na+ conductance was thus reduced by ∼16%. This reduction is initially offset by the greater amount of injected current during the resetting pulse and the drop in the levels of adenosine (Fig. 7), which leads to less inhibition of the Ca2+ current. Early in the spike train, when adenosine levels are lower, the net current after the removal of the resetting pulse is still inward, and firing continues. However, when the adenosine levels have risen sufficiently, even the decreased inhibition of the Ca2+ current resulting from the fall in adenosine levels (Fig. 7) cannot sufficiently offset the reduction in INa, and premature termination of firing occurs after the end of the resetting pulse. Thus, h contributes to the premature termination of firing after the resetting pulse but not to the normal termination process.
Interestingly, the timing of premature termination in the model was sensitive to the width of the resetting pulse (Fig. 5). The longer the resetting pulse, the more ADP will accumulate and hence give rise to greater feedforward inhibition of adenosine production. The resulting greater fall in adenosine levels will lead to a greater reduction of the inhibition of the Ca2+ currents and help to overcome the inactivation of the Na+ currents to a greater extent. Thus, after the longer resetting pulses, the total inward current will be higher for a longer period than after the shorter resetting pulses and will result in delayed premature termination.
Resetting in the real embryo
The model suggests that feedback control by the purines can be reset by a second incoming stimulus of sufficient intensity to elevate firing and increase levels of ATP release. Therefore, I tested whether this might occur in a real physiological system by examining whether the rundown of swimming in Xenopus could be reset in an analogous manner. Swimming was initiated by a single stimulus to the skin; a second, stronger resetting stimulus was given to the skin partway through an episode. Because the duration of swimming episodes in Xenopus is variable, control and experimental episodes (in which the resetting stimulus was given) were interleaved. The time of the resetting stimulus (tpre) and the duration of activity after the stimulus (tpost) were normalized to the predicted length of the episode (T) (see Materials and Methods). If the sum of tpre andtpost was consistently greater thanT (over several consecutive points on a graph like those in Fig. 8), resetting was deemed to have occurred.
Fig. 8.

Resetting of the rundown of swimming (as monitored by ventral-root activity) in the real embryo. 1, Ventral-root activity during a control episode. All other episodes illustrated have a resetting stimulus delivered where indicated by anasterisk. Note that the total duration of activity was extended when the resetting stimulus occurred early (2–4) but was shortened when the stimuli occurred later (5, 6). The vertical dashed line indicates end of control episode.
Of 10 embryos tested, resetting occurred in seven (Figs. 8,9). In the embryos that demonstrated resetting, the mean episode duration (normalized toT) increased to 1.16 ± 0.02 (mean ± SEM;n = 7; p < 0.001). The embryos that demonstrated resetting could be subdivided into three categories. The first category demonstrated resetting early in the episode (up to approximately tpre/T = 0.4) (Fig. 9B). This resetting was almost complete (tpost/T = ∼1) and fell close to the line predicted by the model when feedforward inhibition was strong (Fig. 9B, gray lines). When the resetting stimulus was given at approximatelytpre/T = 0.4, premature termination occurred (Fig. 9B). This was remarkably similar to the value predicted by the model for a shorter resetting stimulus. The second category also demonstrated early resetting (up to approximatelytpre/T = 0.4) (Fig.9C) but did not exhibit premature termination. Instead, when the resetting stimulus was delivered later thantpre/T = 0.4, the resulting activity fell on or just below the linetpre +tpost = T. Consequently, no resetting of rundown occurred at these later stimulation times. The final category exhibited late resetting (Fig. 9D). Resetting was most evident whentpre/T > 0.3 and extended up to approximatelytpre/T = 0.8 when termination of activity occurred. This was very similar to the resetting predicted by the model when the feedforward inhibition was weak (Fig. 9D, gray line).
Fig. 9.
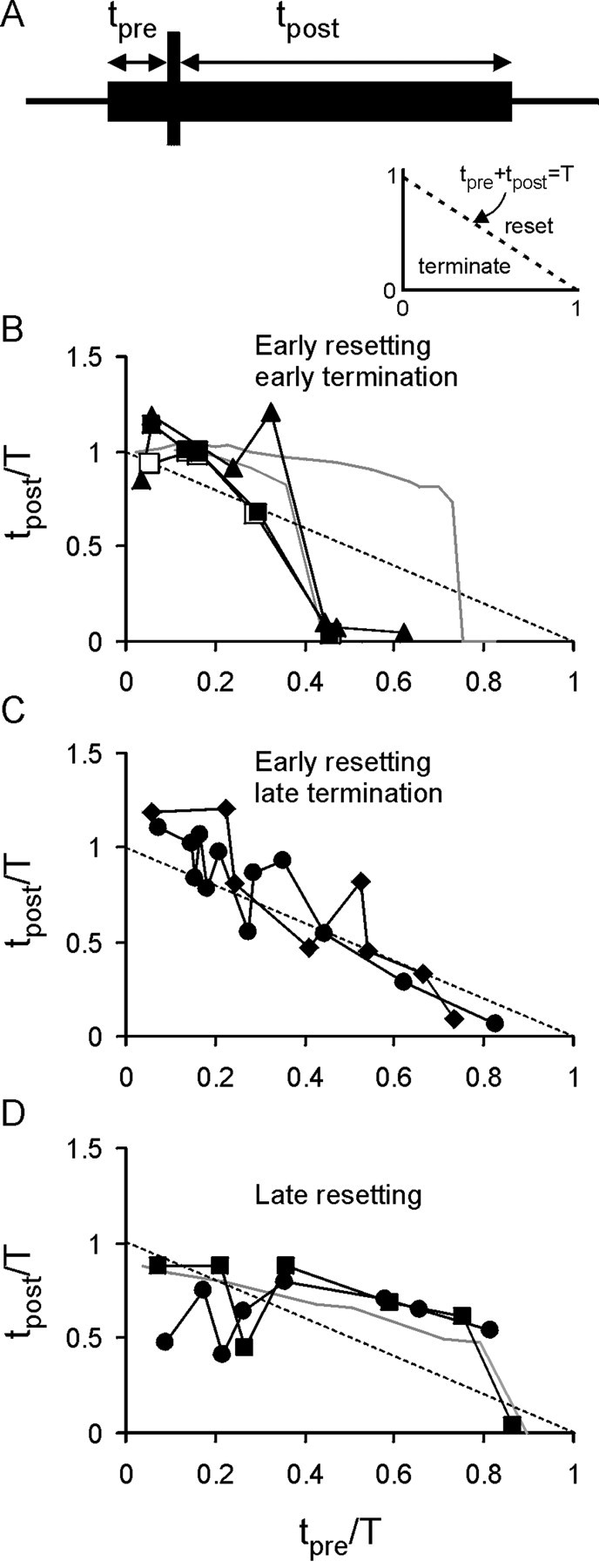
Analysis of resetting of swimming in embryos reveals that it is similar to that predicted by the model neuron.A, Schematic showing the measurement oftpre and tpostrelative to the resetting stimulus. The inset shows the normalized graph, with the dashed line indicating where no resetting takes place (tpre +tpost = T). Below this line, premature termination occurs; above the line, resetting occurs. B, Plots for three embryos, which showed a consistent pattern of early resetting followed by premature termination. This was very similar to the predictions from the model with strong feedforward inhibition (gray lines). C, Two additional embryos also exhibited early resetting but not premature termination.D, Two embryos in which late resetting took place. The resetting profile of these embryos was strikingly similar to that of the model with weaker feedforward inhibition (gray line).
Purines and resetting in the real embryo
Although I have demonstrated that resetting similar to that predicted by the model can occur in Xenopus, this is only a resemblance of a phenomenon and not a demonstration of the underlying mechanism. Therefore, I tested whether ATP and adenosine are involved in resetting by blocking their actions through the combined application of the antagonists pyridoxal-phosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS) and 8-phenyltheophylline (8PT), respectively (Dale and Gilday, 1996). Because these agents would completely block the activation of purinoceptors during motor activity, they should eliminate the resetting of rundown in the embryo if the mechanisms explored in the model neuron also contribute to resetting in the embryo.
Applied separately, PPADS shortens and 8PT lengthens swimming episodes (Dale and Gilday, 1996). The effect of these two antagonists applied simultaneously at 10 and 2 μm, respectively, was to lengthen swimming episodes (214 ± 45 sec in control; 375 ± 94 sec in the presence of 8PT and PPADS; n = 6;p = 0.05) as the opposing effects of purinergic modulation were removed. Little resetting was evident in the embryos in the presence of 8PT and PPADS (Fig.10A). Almost all of the points plotted on a normalized time graph fell on or below the linetpre +tpost = T (Fig.10B). Indeed, the mean sum oftpre/T +tpost/T was 0.97 ± 0.04 (n = 6), indicating that overall no resetting took place. Blockade of the effects of the purines thus removed the ability of a second stimulus to reset rundown. This suggests very strongly that the mechanisms underlying resetting in the embryo are the same as those explored in the simple single-neuron model.
Fig. 10.
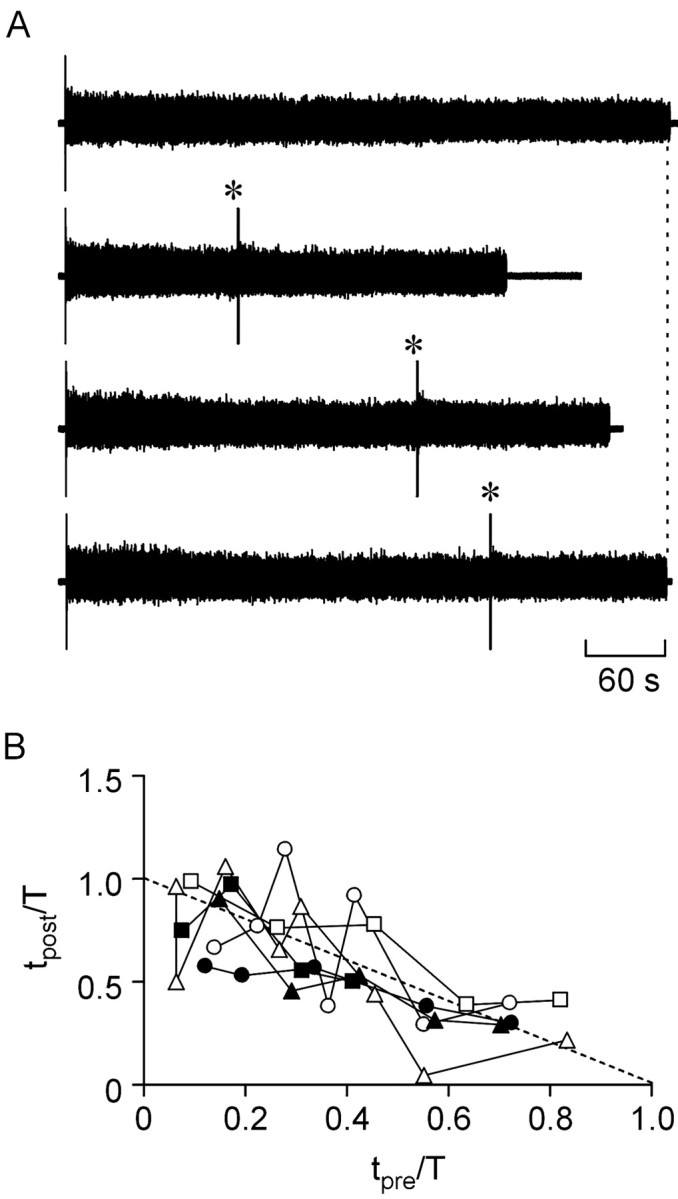
Blockade of purine receptors prevented resetting of the rundown of swimming. Resetting was examined when the embryo was superfused with 10 μm PPADS and 2 μm 8PT to block the p2y and A1 receptors. A, Exampletraces showing a control episode (top trace) and three episodes in which a resetting stimulus was delivered (*). Note how resetting does not occur. Thevertical dashed line indicates end of control episode.B, Summary graphs analogous to those of Figure 9, showing the results for all six embryos. Note how the majority of points fall on or below the dashed line, which indicatestpre + tpost= T.
DISCUSSION
This paper addresses how flexibility can be introduced into an intrinsic control system so that stereotyped behavior can be adapted to the unpredictable. In Xenopus, ATP release is indissolubly wedded to the operation of the locomotor network. ATP is released from spinal pattern-generating neurons by two mechanisms: first, as a cotransmitter in conventional spike-mediated transmission (Brown and Dale, 2002a) and, second, by a spike-independent mechanism as a result of the activation of glutamate receptors (Brown and Dale, 2002a). Because glutamate plays a central role in the operation of the locomotor network (Dale and Roberts, 1984, 1985), the release of ATP is an unavoidable consequence of circuit operation. Once ATP has been released, the presence of ectonucleotidases will ensure that its conversion to adenosine is only a matter of time. Because the embryo cannot swim without the purinergic control system being activated, how can the rundown program controlled by ATP and adenosine be reset by new sensory inputs during motor activity?
Minimal models of purinergic control
I have used a highly simplified single-neuron model, which constitutes the ultimate reduction of the spinal network inXenopus. Previous modeling studies using 200–400 neurons have demonstrated the sufficiency of the known purinergic mechanisms to control rundown (Dale, 1998). The much simpler model used in this paper reduces the problem to its essence: activity-dependent release of ATP, conversion of ATP to adenosine with feedforward inhibition by ADP, and ATP- and adenosine-dependent modulation of outward and inward currents, respectively. This reduction makes interpretation of the model output and underlying mechanisms much simpler. Furthermore, by using a highly reduced model, the conclusions and predictions are likely to be of more general use and applicability than a model that incorporates biological complexity that ties it to a particular physiological system.
The single-cell model demonstrates that feedforward inhibition controls the length of the spike train evoked by current injection in a manner analogous to the rundown of motor episodes in more complex network models (Dale, 1998). This suggests that the reduced model does indeed capture the essentials of the purinergic control system. One reason that such a simple model may exhibit behavior similar to the real system may be that the component classes of neurons in theXenopus spinal network have similar properties and receive common inputs (Dale and Kuenzi, 1997). Therefore, the firing behavior of just one neuron is likely to approximate that of a larger population.
Feedforward inhibition and slowed adenosine production
Resetting of accommodation in the single-neuron model occurred because the additional ATP released during the resetting stimulus was converted to ADP, which was able to inhibit the production of adenosine, leading to a net fall in adenosine levels. This fall occurred despite the greater accumulation of AMP, the substrate for the production of adenosine. Thus, feedforward inhibition by ADP is critical for resetting adenosine production. Because ADP levels remain elevated after the resetting stimulus, the subsequent rate of adenosine production is slower. These two factors (the fall in absolute levels and the fall in the rate of production) ensure that the spike train is prolonged and that the duration of activity after the resetting pulse can equal the duration of a control train in which no resetting pulse had occurred.
Resetting in the Xenopus embryo
Sensory stimuli were able to reset rundown and termination of swimming in seven of the 10 embryos tested. The duration of extra activity evoked by the resetting stimulus was comparable with that predicted by the single-neuron model. There was some variability in the amount of resetting observed between embryos. In some embryos, almost complete resetting was seen. In others, the resetting profile was very similar to that seen in the model for weaker inhibition. Presumably, the strength of feedforward inhibition or the amount of ATP release varies between different embryos; this may account for why the extent of resetting also varied and why it was absent in a few cases. Premature termination also occurred in six of the 10 embryos examined. This could have the same underlying cause as premature termination in the model (which is based on kinetic models of Xenopusembryo neurons). The additional excitation provided by the sensory input would plausibly increase the levels of Na+ channel inactivation in the motor pattern-generating neurons, leading in turn to premature termination.
Purinergic transmission underlies resetting of rundown in the embryo
Although resetting in the embryo was strikingly similar to that predicted by the model, this is not sufficient to conclude that the resetting of swimming in the embryo is attributable to feedforward inhibition and the dynamics of adenosine accumulation. However, the evidence suggests that the purines and feedforward inhibition by ADP of the ecto-5′-nucleotidase, known to occur in the Xenopusspinal cord (Brown and Dale, 2002b), do play a key role. If the resetting of rundown was attributable to feedforward inhibition of adenosine production in the manner explored by the model, the pharmacological removal of the effects of ATP and adenosine should diminish the ability of the system to reset rundown. Blockade of the A1 and p2y receptors by 8PT and PPADS had two effects: it significantly lengthened swimming episodes, and it abolished the ability of a second stimulus occurring partway through the swimming episode to reset the rundown of motor activity.
The removal of the inhibitory action of adenosine must predominate over the removal of the excitatory action of ATP, as might be expected because adenosine accumulation is ultimately responsible for the termination of motor activity. Furthermore, because the combined action of the two antagonists completely removed the ability to reset rundown, the purinergic modulation must introduce the adaptability previously absent from the basic circuit. Significantly, the combined blockade of purinoceptors lengthened motor episodes so that they could last >10 min. Presumably, a variety of mechanisms led to their eventual termination (e.g., depletion of transmitter stores, accumulation of Ca2+, and activation of the slow Ca2+-dependent K+ current) (Wall and Dale, 1995), but episodes this long could represent an upper limit for the sustained activity possible within the embryonic network without a period of rest. Thus, by shortening the characteristic length of motor episodes below this putative upper limit, the purinergic control system may introduce adaptability. An analogy would be to compare a fully stretched spring with one only half stretched: whereas the former can only compress or break, the latter can compress and extend easily. The purines ensure that the “motor spring” is only partly stretched under normal circumstances.
The ectonucleotidases, a heterogeneous collection of ecto-enzymes, play a key role in this control mechanism. They are a rich, novel, and surprising locus for the control of activity in neural circuits. First, by regulating the breakdown of ATP into adenosine, the ectonucleotidases control the relative balance of these two opposing modulators. Second, feedforward inhibition by ADP of the ecto-5′-nucleotidase delays the appearance of adenosine and is an important determinant of the length of motor episodes (Dale, 1998). Third, feedforward inhibition is critical for the introduction of adaptability into the regulatory mechanism. More detailed knowledge of the particular types of ectonucleotidase present in the spinal cord is important, especially because some of these enzymes can be secreted and have selectivity for diphosphate nucleotides (Mulero et al., 1999;Braun et al., 2000).
An associative mechanism?
The experimental paradigm used here could be reformulated as a form of association between two stimuli. Instead of considering the activity of motor networks, imagine a cortical circuit capable of transient reverberatory activity triggered by at least two afferent inputs and that dies away under the control of the purines through the mechanisms explored in this paper. If one input started an episode of activity and a second input arrived within a time period T, activity within the circuit could be prolonged by the resetting mechanism. If the second input were to fall outside time T, activity either would not be reset or would be prematurely terminated. Therefore, T could be considered a time window of association between the two inputs. Crucially, the length ofT and the degree of resetting or prolongation of activity would depend on the strength of feedforward inhibition and the characteristics of the ectonucleotidases involved.
The associative mechanism proposed here has the distinction that it can operate over much longer time scales than those associated with mechanisms based on NMDA receptor-dependent long-term potentiation (typically, ∼100 msec). Furthermore, unlike NMDA receptor-based mechanisms, which are tied to the properties of the receptor, the length of the time window of association is adaptable on a time scale from seconds to minutes, depending on the strength of feedforward inhibition, the amount of ATP release, and the dynamics of ATP breakdown. Given that some ectonucleotidases are secretable, physiological regulation of this associative mechanism is feasible. Obviously, this is highly speculative, but it will be interesting to see whether transgenic mice lacking key components of purinergic signaling show deficiencies in behavioral plasticity.
Footnotes
This work was supported by the Wellcome Trust. I thank Drs. Bruno Frenguelli, Richard Baines, and David Spanswick for helpful comments on previous drafts of this paper.
Correspondence should be addressed to Nicholas Dale, Department of Biological Sciences, University of Warwick, Coventry, CV4 7AL, UK. E-mail: n.e.dale@warwick.ac.uk.
REFERENCES
- 1.Braun N, Fengler S, Ebeling C, Servos J, Zimmermann H. Sequencing, functional expression and characterization of rat NTPDase6, a nucleoside diphosphatase and novel member of the ecto-nucleoside triphosphate diphosphohydrolase family. Biochem J. 2000;351:639–647. [PMC free article] [PubMed] [Google Scholar]
- 2.Brown P, Dale N. Adenosine A1 receptors modulate high voltage-activated Ca2+ currents and motor pattern generation in the Xenopus embryo. J Physiol (Lond) 2000;525:655–667. doi: 10.1111/j.1469-7793.2000.00655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown P, Dale N. Modulation of K+ currents in Xenopus spinal neurons by p2y receptors: a role for ATP and ADP in motor pattern generation. J Physiol (Lond) 2002a;540:843–850. doi: 10.1113/jphysiol.2001.013192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown P, Dale N. Spike-independent release of ATP from Xenopus spinal neurons evoked by activation of glutamate receptors. J Physiol (Lond) 2002b;540:851–860. doi: 10.1113/jphysiol.2001.013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dale N. Kinetic characterization of the voltage-gated currents possessed by Xenopus embryo spinal neurons. J Physiol (Lond) 1995a;489:473–488. doi: 10.1113/jphysiol.1995.sp021066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dale N. Experimentally derived model for the locomotor pattern generator in the Xenopus embryo. J Physiol (Lond) 1995b;489:489–510. doi: 10.1113/jphysiol.1995.sp021067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dale N. Delayed production of adenosine underlies temporal modulation of swimming in frog embryo. J Physiol (Lond) 1998;511:265–272. doi: 10.1111/j.1469-7793.1998.265bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dale N, Gilday D. Regulation of rhythmic movements by purinergic neurotransmitters in frog embryos. Nature. 1996;383:259–263. doi: 10.1038/383259a0. [DOI] [PubMed] [Google Scholar]
- 9.Dale N, Kuenzi FM. Ion channels and the control of swimming in the Xenopus embryo. Prog Neurobiol. 1997;53:729–756. doi: 10.1016/s0301-0082(97)00048-8. [DOI] [PubMed] [Google Scholar]
- 10.Dale N, Roberts A. Excitatory amino acid receptors in Xenopus embryo spinal cord and their role in the activation of swimming. J Physiol (Lond) 1984;348:527–543. doi: 10.1113/jphysiol.1984.sp015123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dale N, Roberts A. Dual-component amino-acid-mediated synaptic potentials: excitatory drive for swimming in Xenopus embryos. J Physiol (Lond) 1985;363:35–59. doi: 10.1113/jphysiol.1985.sp015694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dale N, Pearson T, Frenguelli BG. Direct measurement of adenosine release during hypoxia in the CA1 region of the rat hippocampal slice. J Physiol (Lond) 2000;526:143–155. doi: 10.1111/j.1469-7793.2000.00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding Y, Cesare P, Drew L, Nikitaki D, Wood JN. ATP, P2X receptors and pain pathways. J Autonom Nerv Syst. 2000;81:289–294. doi: 10.1016/s0165-1838(00)00131-4. [DOI] [PubMed] [Google Scholar]
- 14.Edwards FA, Gibb AJ, Colquhoun D. ATP receptor-mediated synaptic currents in the central nervous system. Nature. 1992;359:144–147. doi: 10.1038/359144a0. [DOI] [PubMed] [Google Scholar]
- 15.Gordon EL, Pearson JD, Slakey LL. The hydrolysis of extracellular adenine nucleotides by cultured endothelial cells from pig aorta: feed-forward inhibition of adenosine production at the cell surface. J Biol Chem. 1986;261:15496–15507. [PubMed] [Google Scholar]
- 16.James S, Richardson PJ. Production of adenosine from extracellular ATP at the striatal cholinergic synapse. J Neurochem. 1993;60:219–227. doi: 10.1111/j.1471-4159.1993.tb05841.x. [DOI] [PubMed] [Google Scholar]
- 17.Mulero JJ, Yeung G, Nelken ST, Ford JE. CD39–L4 is a secreted human apyrase, specific for the hydrolysis of nucleoside diphosphates. J Biol Chem. 1999;274:20064–20067. doi: 10.1074/jbc.274.29.20064. [DOI] [PubMed] [Google Scholar]
- 18.Nieuwkoop PD, Faber J. Normal tables of Xenopus laevis (Daudin). North-Holland; Amsterdam: 1956. [Google Scholar]
- 19.Poon A, Sawynok J. Antinociception by adenosine analogs and inhibitors of adenosine metabolism in an inflammatory thermal hyperalgesia model in the rat. Pain. 1998;74:235–245. doi: 10.1016/s0304-3959(97)00186-3. [DOI] [PubMed] [Google Scholar]
- 20.Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Press WH, Flannery BP, Teukolsky SA, Vetterling WT. Numerical recipes in C: the art of scientific computing, Ed 2. Cambridge UP; Cambridge, UK: 1992. [Google Scholar]
- 22.Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharm Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- 23.Robertson SJ, Ennion SJ, Evans RJ, Edwards FA. Synaptic P2X receptors. Curr Opin Neurobiol. 2001;11:378–386. doi: 10.1016/s0959-4388(00)00222-1. [DOI] [PubMed] [Google Scholar]
- 24.Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB. Neuroprotective role of adenosine in cerebral-ischemia. Trends Pharmacol Sci. 1992;13:439–445. doi: 10.1016/0165-6147(92)90141-r. [DOI] [PubMed] [Google Scholar]
- 25.Slakey LL, Cosimini K, Earls JP, Thomas C, Gordon EL. Simulation of extracellular nucleotide hydrolysis and determination of kinetic constants for the ectonucleotidases. J Biol Chem. 1986;261:15505–15507. [PubMed] [Google Scholar]
- 26.Thomas T, Spyer KM. A novel influence of adenosine on ongoing activity in rat rostral ventrolateral medulla. Neuroscience. 1999;88:1213–1223. doi: 10.1016/s0306-4522(98)00296-6. [DOI] [PubMed] [Google Scholar]
- 27.Wall MJ, Dale N. A slowly activating Ca2+-dependent K+ current that plays a role in termination of swimming in Xenopus embryos. J Physiol (Lond) 1995;487:557–572. doi: 10.1113/jphysiol.1995.sp020900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimmermann H. Biochemistry, localization and functional roles of ecto-nucleotidases in the nervous system. Prog Neurobiol. 1996;49:589–618. doi: 10.1016/0301-0082(96)00026-3. [DOI] [PubMed] [Google Scholar]
- 29.Zimmermann H, Braun N. Ecto-nucleotidases: molecular structures, catalytic properties, and functional roles in the nervous system. Prog Brain Res. 1999;120:371–385. [PubMed] [Google Scholar]