

Abstract
Chronic exposure to nicotine, as occurs during tobacco smoking, is one of several factors that have been reported to cause an upregulation of neuronal nicotinic acetylcholine receptors (nAChRs). Here, the influence of both chronic exposure to nicotine (10 μm, 24 hr) and the coexpression of subunit chimeras has been examined in cultured cell lines expressing recombinant α4β2 nAChRs, a major nicotinic receptor subtype expressed in the mammalian brain. Evidence is presented which demonstrates that both chronic exposure to nicotine and the coexpression of subunit chimeras upregulates levels of receptor expressed on the cell surface. Immunoblotting data indicate that neither chronic nicotine treatment nor coexpressed subunit partners greatly affect the level of total subunit protein. This finding, together with radioligand and antibody binding studies conducted on both intact and permeabilized cells, reveals that receptor upregulation corresponds to an increase in the proportion of total receptor expressed on the cell surface. It is also apparent that nicotine-induced nAChR upregulation is very strongly dependent on subunit composition and subunit domains. An important aspect of this study is that direct evidence has been obtained indicating that both chronic exposure to nicotine and coexpressed subunit partners can influence subunit conformation. The influence of chronic nicotine treatment on subunit folding may help to explain the phenomenon of nicotine-induced receptor upregulation. The finding that subunit conformation can be influenced by coassembled subunit partners is in agreement with models of receptor assembly which propose that subunit folding continues after initial subunit–subunit interactions.
Keywords: nicotinic, acetylcholine receptor, folding, conformation, assembly, chimera
Chronic exposure to nicotine, as occurs during tobacco smoking, has been widely reported to cause an upregulation of nicotinic acetylcholine receptors (nAChRs) in the brain. This has been demonstrated as an increase in the density of nicotinic radioligand binding sites both in postmortem human brain tissue of smokers (Benwell et al., 1988) and in the brains of animals after chronic exposure to nicotine (Marks et al., 1985, 1992; Schwartz and Kellar, 1985).
A major subtype of nAChRs expressed in mammalian brain contains the α4 and β2 subunits, and brain nAChRs coassembled from these subunits have been shown to be upregulated by chronic nicotine exposure (Flores et al., 1992). Several studies have reported a similar upregulation of nicotinic radioligand binding sites and cell-surface receptor levels in heterologous expression systems expressing recombinant α4β2 nAChRs after chronic exposure to nicotine (Peng et al., 1994; Zhang et al., 1994; Bencherif et al., 1995; Gopalakrishnan et al., 1996, 1997; Rothhut et al., 1996; Warpman et al., 1998;Whiteaker et al., 1998). It appears, however, that not all nAChR subtypes are upregulated to the same extent by chronic exposure to nicotine (Peng et al., 1997; Ke et al., 1998; Wang et al., 1998). The mechanisms underlying nicotine-induced upregulation of nAChRs are still unclear, but it is now generally accepted that this phenomenon, both in the brain and in heterologous expression systems, can be explained by post-transcriptional events (Marks et al., 1992; Peng et al., 1994;Zhang et al., 1994; Bencherif et al., 1995).
Heterologous expression of α4β2 nAChRs (and other nAChR subunits) frequently results in expression of only low levels of both nicotinic agonist binding sites and functional cell-surface receptors (Cooper and Millar, 1997, 1998; Kassner and Berg, 1997; Chen et al., 1998; Cooper et al., 1999; Sweileh et al., 2000). This has led to the conclusion that folding and assembly of neuronal nAChRs, in at least some host cell types, is a relatively inefficient process. Interestingly, very much higher levels of assembled cell-surface receptor can be generated by expressing chimeric subunits containing the extracellular domain of neuronal nAChR subunits fused to the transmembrane and intracellular region of the 5-hydroxytryptamine receptor 5-HT3Asubunit (Eiselé et al., 1993; Rangwala et al., 1997; Cooper et al., 1999). For example, coexpression of the nAChR β2 subunit with an α4–5HT3A subunit chimera (α4χ) results in an increase (of ∼20-fold) in the amount of β2 expressed on the cell surface and in the level of nicotinic radioligand binding sites (Cooper et al., 1999).
In this study, the effect of chronic nicotine treatment both on α4β2 nAChRs and on receptors assembled from chimeric subunits has been examined. The proportion of total subunit protein expressed on the cell surface and the proportion of radioligand binding sites on the cell surface have been examined. This has revealed changes in subunit conformation and distribution attributable to chronic nicotine treatment and as a consequence of coexpression of subunit chimeras.
MATERIALS AND METHODS
Materials. Rat neuronal nAChR α4 and β2 subunit cDNAs (Goldman et al., 1987; Deneris et al., 1988) were provided by Dr. Jim Patrick (Baylor College of Medicine). The mouse 5HT3AcDNA (Maricq et al., 1991) was provided by Dr. David Julius (University of California San Francisco). Chimeric nicotinic/serotonergic subunit cDNA constructs pRK5-α4/5HT3A and pRK5-β2/5HT3A (also referred to here as pRK5-α4χ and pRK5-β2χ, respectively) have been described previously (Cooper et al., 1999). All subunit cDNAs were subcloned into plasmid expression vector pRK5, as described previously (Cooper et al., 1999). An eight amino acid “FLAG” epitope-tag (Hopp et al., 1988) was introduced into the β2 cDNA at a position immediately after the predicted signal sequence cleavage site of the β2 subunit to create pRK5-β2FLAG. Monoclonal antibody (mAb) 270, which recognizes an extracellular epitope on the nAChR β2 subunit (Whiting and Lindstrom, 1987), was purified from the hybridoma cell line HB189 (obtained from the American Type Culture Collection, Rockville, MD). mAb299, which recognizes an extracellular epitope on the nAChR α4 subunit (Whiting and Lindstrom, 1988), was obtained from Sigma (Poole, UK). A polyclonal antiserum (pAb5HT3), raised against a fusion protein containing the intracellular loop region of the mouse 5HT3A receptor subunit (Turton et al., 1993), was provided by Dr. Ruth McKernan (Merck Sharp and Dohme Research Laboratories, Harlow, UK). A polyclonal antiserum (pAb120) raised against the extracellular region of the mouse 5HT3A receptor subunit (Spier et al., 1999) was obtained from Dr. Sarah Lummis (University of Cambridge). TSA201 cells, a derivative of the human embryonic kidney 293 cell line that expresses the simian virus 40 large T-antigen, were obtained from Dr. William Green (University of Chicago).
Cell culture and transfection. Cells were cultured in DMEM (Invitrogen) containing 2 mml-glutamine plus 10% heat-inactivated fetal calf serum (Sigma), penicillin (100 U/ml), and streptomycin (100 μg/ml) and maintained in a humidified incubator containing 5% CO2 at 37°C. Plasmid DNA was introduced into human TSA201 cells using the Effectene reagent (Qiagen) according to the manufacturer's instructions. Cells were transfected overnight and assayed for expression ∼42–44 hr after transfection. In all cases in which chronic exposure to nicotine was examined, nicotine (10 μm) was added to the cell culture medium for 24 hr (18–20 hr after transfection).
Radioligand binding. Binding studies with [3H]epibatidine (NEN Life Science; specific activity 67 Ci/mmol) or [3H]GR65630 (NEN Life Science; specific activity 76 Ci/mmol) were performed on cell membrane preparations as described previously (Lansdell et al., 1997). Binding studies with [3H]methylcarbamylcholine ([3H]MCC) (Tocris Cookson; specific activity 80 Ci/mmol) were used to determine the subcellular distribution of binding sites. Cells were harvested in isotonic saline (HBSS; Invitrogen), and binding was performed with intact cells in the presence of protease inhibitors (0.25 mmphenylmethylsulfonyl fluoride and 10 μg/ml each of leupeptin, aprotinin, and pepstatin; Sigma). Aliquots of the cell suspension were centrifuged and frozen rapidly, thawed, and then disrupted by passage three times through a 21 gauge needle in the presence of protease inhibitors. Samples were incubated with radioligand ([3H]epibatidine or [3H]MCC) for 2 hr at 4°C, and nicotine (2 mm) was used to define nonspecific binding. Samples were assayed by filtration onto Whatman GF/B filters presoaked in 0.5% polyethylenimine followed by rapid washing (three washes completed within <5 sec) using a Brandel cell harvester. Amounts of total cellular protein were determined by a Bio-Rad DC protein assay using BSA standards.
Enzyme-linked assay of cell-surface expression levels. Cell-surface antibody binding was assayed on cells grown on poly-l-lysine-coated glass coverslips, transfected, incubated in primary antibody, and fixed as described previously (Cooper et al., 1999). To measure total (cell surface and internal) antibody binding, fixed cells were exposed for 15 min to 0.1% Triton X-100. Coverslips were processed as above, but 0.1% Triton X-100 was included in all incubation buffers and in buffer used for the first two washing steps. Antibody solutions contained, additionally, 5% fetal calf serum. When surface and total antibody binding levels were compared, coverslips (with permeablized or intact cells) were fixed before addition of primary antibody and assayed in parallel. Additionally, lysine (25 mm) was added to buffers to reduce nonspecific binding. In all cases, coverslips were incubated with horseradish peroxidase-conjugated goat anti-rat IgG (Amersham Biosciences), goat anti-rabbit IgG (Pierce), or goat anti-mouse IgG (Pierce) and washed six times before incubation with 600 μl 3,3′,5,5′-tetramethylbenzidine (Sigma) for 1 hr. The supernatant was transferred to a cuvette, and absorbance was determined at 655 nm.
Immunoblotting. At 18–20 hr after transfection, cells were maintained in the absence or presence of nicotine (10 μm) for 24 hr. Cells were rinsed twice and harvested in isotonic saline, pelleted, and snap-frozen. Cell pellets were thawed on ice in the presence of protease inhibitors (0.25 mm phenylmethylsulfonyl fluoride and 10 μg/ml each of leupeptin, aprotinin, and pepstatin; Sigma), and 150 μg of total cellular protein was separated by 7.5% SDS-PAGE. Gels were equilibrated for 20 min in transfer buffer (25 mm Tris, 192 mm glycine, 20% methanol, pH 8.3) and electroblotted onto Hybond-C nitrocellulose membranes (Amersham Biosciences). Membranes were blocked by incubation in PBS containing 0.1% Tween 20 and 5% nonfat milk powder and then incubated with primary antibody in blocking solution for 2 hr at room temperature. The membrane was washed thoroughly, incubated with an HRP-conjugated secondary antibody (goat anti-rat IgG, goat anti-rabbit IgG, or goat anti-mouse IgG), and processed using the ECL detection system (Amersham Biosciences). Protein concentrations of cell pellets were determined by a Bio-Rad DC protein assay using BSA standards according to the manufacturer's instructions.
RESULTS
Influence of nicotine and subunit composition on radioligand binding
Specific high-affinity binding of the nicotinic radioligand [3H]epibatidine (Bmax = 0.15 ± 0.04 pmol/mg protein; n = 8) was detected when mammalian kidney (TSA201) cells were cotransfected with rat nAChR α4 and β2 subunit cDNAs. This is consistent with previous reports showing that α4 and β2 coassemble to generate a high-affinity agonist binding site and functional nAChRs when expressed in various mammalian cell lines (Whiting et al., 1991; Buisson et al., 1996; Ragozzino et al., 1997;Cooper et al., 1999). In agreement with our previously published findings (Cooper et al., 1999), significantly higher levels of radioligand binding sites were detected when either α4 or β2 was coexpressed with nAChR/5HT3R subunit chimeras (α4χ or β2χ). Coexpression of α4 with β2χ, rather than β2, generated a significant increase (4.5 ± 0.5-fold;n = 4; p < 0.05) in radioligand binding sites (Fig.1A). Similarly, coexpression of β2 with α4χ, rather than α4, generated a significant increase (17.6 ± 1.2-fold; n = 4;p < 0.001) in total specific radioligand binding (Fig. 1A). As reported previously, none of these subunits forms an agonist binding site when expressed alone, but all of the pair-wise subunit combinations generate a high-affinity binding site for [3H]epibatidine that is not significantly different from that of α4β2 (Kd = 41 ± 22 pm) (Cooper et al., 1999); thus, as discussed previously (Cooper et al., 1999), we conclude that this represents an increase in the proportion of α4 or β2 subunits coassembling to generate correctly folded agonist binding sites when coexpressed with chimeric subunits.
Fig. 1.
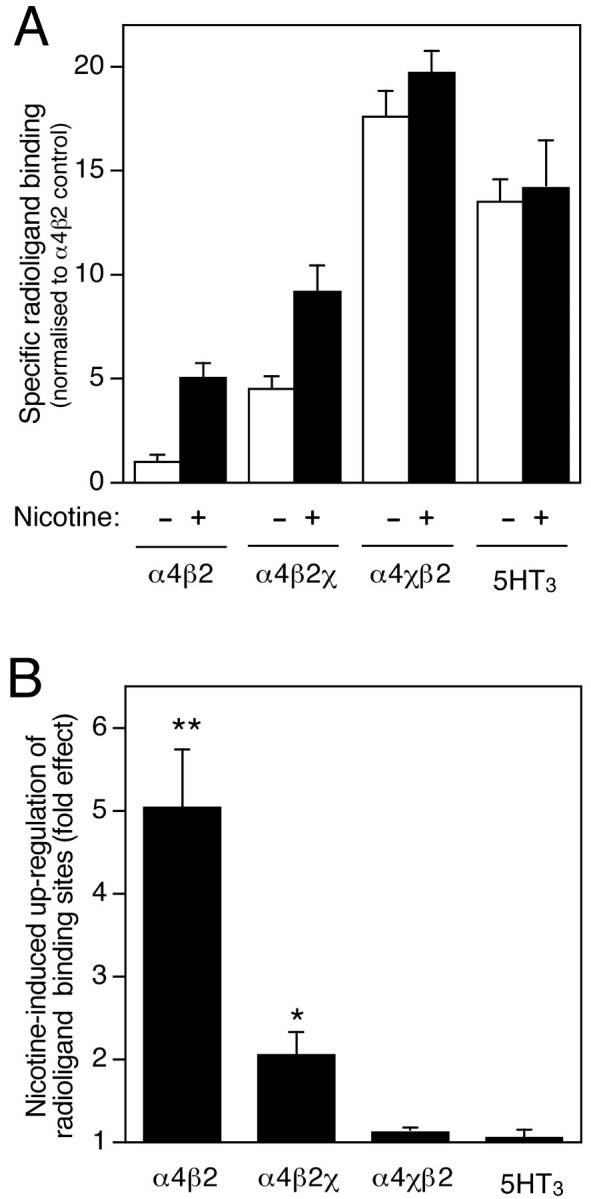
Influence of chronic nicotine treatment and subunit composition on radioligand binding to transfected TSA201 cells.A, Specific radioligand binding to cell homogenates from transfected cells was determined using a saturating concentration of [3H]epibatidine (2 nm, for α4β2, α4β2χ, and α4χβ2) or [3H]GR-65630 (10 nm, for 5HT3A). For all subunit combinations (α4β2, α4β2χ, α4χβ2, and 5HT3A), radioligand binding was determined in cells that had been grown in the absence (white bars) or presence (black bars) of nicotine (10 μm, 24 hr). Data represent means ± SEM of four independent experiments that have been normalized to the level of binding obtained with α4β2 in the absence of nicotine (0.15 ± 0.04 pmol/mg protein; n = 8). B, Data have been replotted to show the extent of nicotine-induced upregulation of radioligand binding for each subunit combination. Significant differences from control, determined by two-tailed Student'st test, are indicated (*p < 0.05; **p < 0.001).
Treatment of cells expressing α4β2 with nicotine (10 μm, 24 hr) resulted in an upregulation (5.1 ± 0.7-fold; n = 4; p < 0.001) in the level of specific [3H]epibatidine binding to cell homogenates (Fig. 1A). This is consistent with earlier studies which have demonstrated that treatment of cells expressing α4β2 with low concentrations of nicotine over relatively long periods (typically 1–100 μm,for 24–48 hr) produces an upregulation of nicotinic radioligand binding sites by a post-transcriptional mechanism (Peng et al., 1994;Bencherif et al., 1995; Gopalakrishnan et al., 1996; Whiteaker et al., 1998). In parallel experiments, cells expressing subunit combinations α4χβ2 or α4β2χ were treated with nicotine (10 μm, 24 hr). The extent of nicotine-induced upregulation of radioligand binding in transfected cells was found to be influenced strongly by subunit composition (Fig.1A). This has been emphasized by replotting the data to illustrate the level of radioligand binding in nicotine-treated cells relative to untreated cells (Fig. 1B). Chronic nicotine treatment produced a significant upregulation of radioligand binding with α4β2χ (2.1 ± 0.3-fold; n = 4;p < 0.05) but did not cause a significant upregulation of binding to α4χβ2. The concentration of nicotine used produces a maximal effect for all three subunit combinations (data not shown). As a control, the effect of chronic exposure to nicotine on the level of binding of the 5HT3R antagonist GR-65630 to cells transfected with the 5HT3A subunit was examined (Fig. 1). Incubation in 10 μm nicotine for 24 hr had no significant effect on the number of [3H]GR-65630 binding sites in cells expressing the 5HT3A subunit.
Influence of nicotine and subunit composition on cell-surface expression
An enzyme-linked antibody binding assay (Cooper et al., 1999) was used to determine the level of nAChR expressed on the surface of cells transfected with the α4β2, α4β2χ, and α4χβ2 subunit combinations. In agreement with previous findings (Cooper et al., 1999), coexpression of α4 with β2χ, rather than β2, did not significantly increase cell-surface expression of the α4 subunit, but coexpression of β2 with α4χ, rather than α4, caused a significant upregulation in the level of β2 expressed on the cell surface (8.5 ± 1.9-fold; n = 4; p< 0.001) (Fig.2A).
Fig. 2.
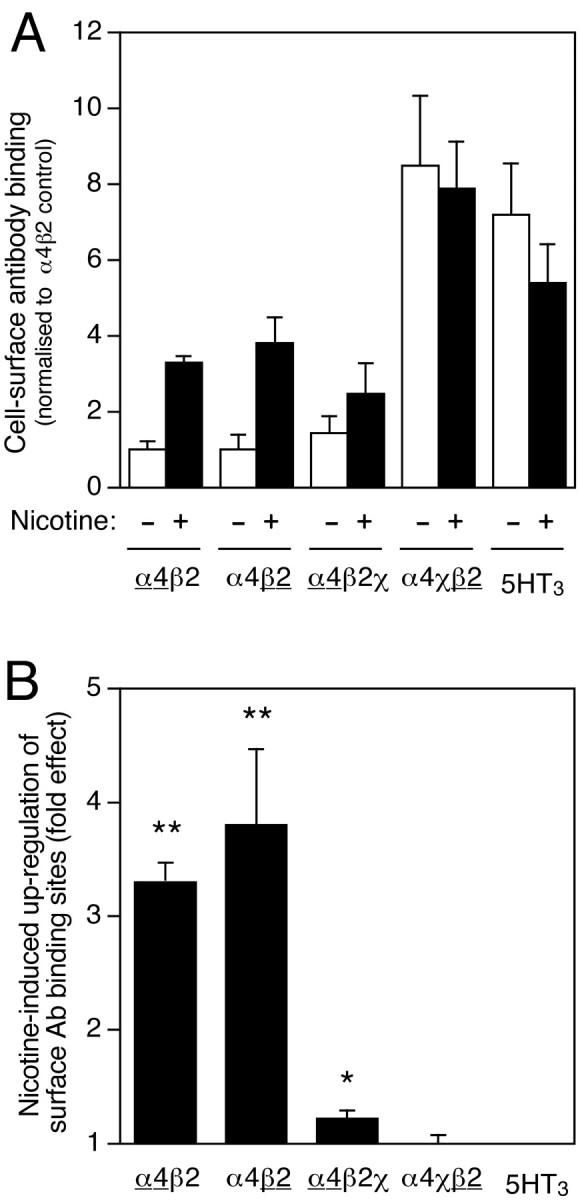
Influence of chronic nicotine treatment and subunit composition on cell-surface antibody binding. A, Surface antibody binding to intact cell monolayers was determined with mAb270 (anti-β2), mAb299 (anti-α4), or pAb120 (anti-5HT3A) using an enzyme-linked antibody binding assay (see Materials and Methods). Surface levels of α4χβ2 were assayed by mAb270 binding, and levels of α4β2χ were assayed by mAb299 binding, thus ensuring that only assembled cell-surface complexes were detected (neither α4 nor β2 is expressed on the cell surface when expressed individually). Surface levels of α4β2 were assayed separately with both mAb270 and mAb299. For all subunit combinations (α4β2, α4β2χ, α4χβ2, and 5HT3A), antibody binding was determined in cells that had been grown in the absence (white bars) or presence (black bars) of nicotine (10 μm, 24 hr). The background signal from mock-transfected coverslips has been subtracted. Data are the means ± SEM of four independent experiments and have been normalized to the level of surface antibody binding determined with α4β2 in the absence of nicotine.B, Data have been replotted to show the extent of nicotine-induced upregulation of antibody binding for each subunit combination. Where two subunits are coexpressed, the subunit assayed by mAb binding is underlined. Statistical significance of the results, determined by two-tailed Student's t test, is indicated (*p = 0.05; **p < 0.01).
Experiments were performed to examine the effect of chronic nicotine treatment (10 μm, 24 hr) on levels of cell-surface receptor in cells expressing α4β2, α4β2χ, and α4χβ2 subunit combinations (Fig. 2A). Because α4χ and β2χ are expressed on the cell surface as homomeric complexes (Cooper et al., 1999), surface levels of α4χβ2 were assayed by mAb270 binding, and levels of α4β2χ were assayed by mAb299 binding, thus ensuring that only assembled cell-surface complexes were detected. Surface levels of α4β2 were assayed separately with both mAb270 and mAb299. Replotting of these data (Fig. 2B) emphasizes that, as was found for radioligand binding (Fig. 1), the extent of nicotine-induced upregulation of cell-surface receptor is influenced strongly by subunit composition. Chronic nicotine treatment caused a significant upregulation of cell-surface α4β2 (3.3 ± 0.2-fold, n = 4, p < 0.01, when assayed by mAb299; 3.8 ± 0.7-fold, n = 4,p < 0.01, when assayed by mAb270) and of α4β2χ (1.2 ± 0.1-fold; n = 4; p = 0.05), but in contrast, nicotine did not significantly increase the level of cell-surface expression of either α4χβ2 or 5HT3A (Fig. 2B).
Determination of total subunit protein level by immunoblotting
Previous immunoblotting studies that examined the level of total subunit protein in transfected cells have shown that subunit chimeras (such as α4χ) and nonchimeric subunits (such as α4) are expressed at similar levels (Cooper et al., 1999). The same approach was used here to examine the effect of chronic nicotine treatment on levels of total subunit protein. Immunoblotting with mAb299, an antibody raised against an extracellular epitope on α4 (Whiting and Lindstrom, 1988), indicates that chronic nicotine treatment has no significant effect on the level of total α4 or α4χ subunit protein (Fig.3A). This experiment also confirms our previous finding (Cooper et al., 1999) that similar levels of total α4 and α4χ subunit protein are expressed in transfected TSA201 cells.
Fig. 3.

Influence of chronic nicotine treatment and subunit composition on total subunit protein levels determined by immunoblotting. Total cellular protein (150 μg) from TSA201 cells transfected with various subunit combinations and grown in the presence or absence of nicotine (10 μm, 24 hr) was separated by SDS-PAGE and immunoblotted with mAb299, which recognizes α4 and α4χ, mAbFLAG-M2, which recognizes epitope-tagged β2FLAG, or pAb5HT3, which recognizes β2χ. Specific immunoreactive bands (absent from mock-transfected control cells) were detected for α4 (∼70 kDa), α4χ (∼55 kDa), β2FLAG (∼55 kDa), and β2χ (∼55 kDa). The positions of protein molecular weight markers are shown.
Although mAb270, an antibody raised against an extracellular epitope of β2 (Whiting and Lindstrom, 1987), was used successfully to detect cell surface-expressed subunit (Fig. 2), it was not possible to use this antibody to detect β2 subunit protein on immunoblots, presumably because it recognizes a conformation-sensitive epitope. Therefore, to compare the levels of total β2 subunit expressed in the presence and absence of chronic nicotine treatment, mAbFLAG-M2 (Hopp et al., 1988) was used to detect an epitope-tagged β2 construct (β2FLAG) expressed in TSA201 cells (Fig.3B). The level of β2χ subunit protein expressed in the presence and absence of chronic nicotine treatment was examined by immunoblotting with pAb5HT3, a polyclonal antibody raised against the intracellular loop region of 5HT3A (Turton et al., 1993) (Fig. 3C). Immunoblotting with mAbFLAG-M2 and pAb5HT3indicates that chronic nicotine treatment does not upregulate the level of total β2 or β2χ subunit protein. It appears, therefore, that chronic nicotine treatment does not upregulate levels of total protein for any of the subunits examined (α4, α4χ, β2, and β2χ).
The results described above suggest that although chronic nicotine treatment and the expression of subunit chimeras can upregulate the level of cell-surface receptor and the number of radioligand binding sites, they do not upregulate the level of total subunit protein. The following series of experiments was aimed at investigating the effect of chronic nicotine treatment and chimeric subunits on the subcellular distribution of radioligand binding sites and of subunit protein.
Subcellular distribution of nicotinic binding sites
The proportion of nicotinic agonist binding sites expressed at the cell surface of transfected cells was determined by examining the level of radioligand binding with [3H]MCC, a membrane-impermeant nicotinic ligand that binds to α4β2 nAChRs with high affinity (Kd = 2.9 ± 0.4 nm) (Lansdell and Millar, 2000). This is a more direct approach than the method used previously to compare surface and total specific binding sites that used unlabeled impermeant ligands to selectively block surface binding sites for the membrane-permeant radioligand [3H]epibatidine (Whiteaker et al., 1998). The Bmax value determined for [3H]MCC binding to α4β2 cell homogenates (0.15 ± 0.05 pmol/mg protein; n = 7) is not significantly different from the value determined with [3H]epibatidine. In Figure4A, data determined for both cell-surface [3H]MCC binding and total specific [3H]MCC binding for three subunit combinations (α4β2, α4β2χ, and α4χβ2) have been used to calculate the proportion of binding sites on the cell surface (as a percentage of total specific binding). Although only 24.3 ± 2.2% (n = 6) of α4β2 binding sites were detected on the cell surface, a significantly higher proportion of the total specific binding sites was expressed on the cell surface for α4β2χ and α4χβ2 (44.3 ± 8.7 and 91.1 ± 6.2%, respectively; n = 4).
Fig. 4.
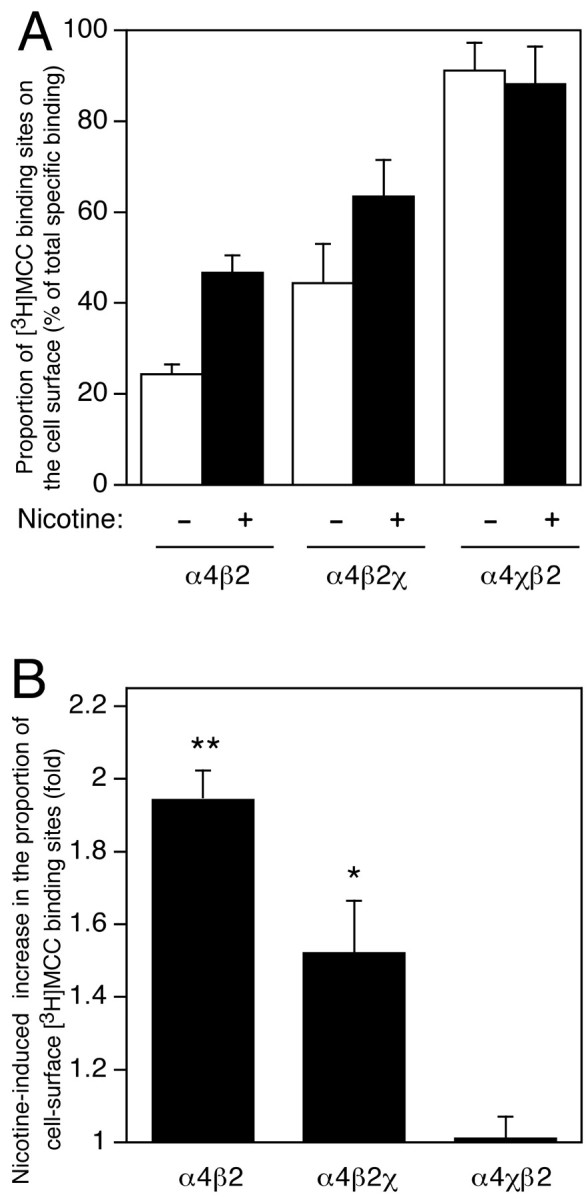
Influence of chronic nicotine treatment and subunit composition on the proportion of radioligand binding sites expressed on the cell surface. A, Specific binding of the membrane-impermeant nicotinic radioligand [3H]MCC was determined for intact and homogenized cells in the absence (white bars) and presence (black bars) of nicotine (10 μm, 24 hr). Data are presented as the proportion of binding sites on the cell surface (as a percentage of total specific binding). Data are means ± SEM of four to six independent experiments using a saturating concentration of ligand (25 nm).B, Data have been replotted to show the extent of nicotine-induced upregulation of cell-surface radioligand binding for each subunit combination. Statistical significance of the results, determined by two-tailed Student's t test, is indicated (*p = 0.067; **p < 0.01).
The influence of chronic nicotine treatment on the subcellular distribution of nicotinic radioligand binding sites was examined for α4β2, α4β2χ, and α4χβ2 subunit combinations (Fig. 4). In cells transfected with α4β2, the proportion of cell-surface [3H]MCC binding sites increased (from 24.3 ± 2.2%) to 46.6 ± 3.9% after chronic nicotine treatment. Levels of total specific binding to α4β2 were upregulated to a similar extent by nicotine treatment, whether assayed by [3H]MCC (4.0 ± 1.0-fold;n = 6) or by [3H]epibatidine (5.1 ± 0.7-fold;n = 4) (Fig. 1). The proportion of α4β2χ cell-surface binding sites increased (from 44.3 ± 8.7%) to 63.4 ± 8.1%. In contrast, no significant difference was seen in the proportion of cell-surface α4χβ2 binding sites (91.1 ± 6.2% in untreated cells and 88.1 ± 8.4% in nicotine-treated cells). The extent of nicotine-induced upregulation of cell-surface binding sites is emphasized by replotting data as fold increase in surface binding (Fig. 4B). For α4β2 and α4β2χ, the fold increase in the percentage of cell-surface binding sites after nicotine treatment was 1.9 ± 0.2-fold (n = 6; p < 0.01) and 1.5 ± 0.3-fold (n = 4; p = 0.067), respectively. The influence of nicotine treatment and coexpressed subunits on the proportion of radioligand binding sites on the cell surface is summarized in Table 1.
Table 1.
Influence of chronic nicotine and subunit chimeras on cell-surface expression
| Cotransfected subunits | Cell-surface radioligand binding (% of total specific binding)1-a | Cell-surface antibody binding (% of total mAb binding)1-a | ||
|---|---|---|---|---|
| − Nicotine | + Nicotine | − Nicotine | + Nicotine | |
| α4 + β2 | 24.3 ± 2.2 | 46.6 ± 3.9 | 2.1 ± 0.7 (α4) | 8.5 ± 2.0 (α4) |
| 12.2 ± 2.1 (β2) | 17.7 ± 4.5 (β2) | |||
| α4 + β2χ | 44.3 ± 8.7 | 63.4 ± 8.1 | 3.2 ± 0.8 (α4) | 6.5 ± 1.5 (α4) |
| 60.1 ± 5.9 (β2χ) | 25.4 ± 2.6 (β2χ) | |||
| α4χ + β2 | 91.1 ± 6.2 | 88.1 ± 8.4 | 55.7 ± 3.9 (α4χ) | 63.0 ± 10.9 (α4χ) |
| 90.5 ± 4.6 (β2) | 93.0 ± 4.4 (β2) | |||
Cell-surface radioligand binding (as a percentage of total specific binding) was determined by comparing levels of specific radioligand binding with a saturating concentration of [3H]MCC (25 nm) to intact cell preparations and to cell homogenates (see Materials and Methods). Data are means ± SEM of four to six independent experiments performed in triplicate on cells grown in the presence and absence of nicotine (10 μm, 24 hr). Cell-surface antibody binding (as a percentage of binding to permeabilized cells) was determined by comparing levels of binding to intact and permeabilized cells with an enzyme-linked assay (see Materials and Methods). Data are means of 5–11 independent experiments performed in duplicate on cells grown in the presence and absence of nicotine (10 μm, 24 hr). The data summarize results presented in Figure 4A([3H]MCC binding), Figure 5B (mAb299 binding), and Figure 6B (mAb270 binding). Subunits were assayed with antibodies recognizing extracellular epitopes (mAb299 for α4 and α4χ, mAb270 for β2 and β2χ). The subunit being assayed is indicated in brackets. Because α4χ and β2χ (but not α4 and β2) can form homomeric as well as heteromeric cell-surface complexes (Cooper et al. 1999), cell-surface antibody binding with mAbs targeting α4χ and β2χ will detect a heterogeneous receptor population.
The data presented refer to the proportion of binding sites (either radioligand binding sites or antibody binding sites) on the cell surface. Because the amount of total (surface + internal) subunit protein that is in a conformation recognized by mAb270 or mAb299 will not necessarily be the same as the proportion of subunit folded and assembled into a conformation capable of forming a high-affinity radioligand binding site, it is not unexpected that the values for the proportion of radioligand and antibody binding sites on the cell surface may differ. Additionally, mAb270 and mAb299, but not [3H]MCC, are able to detect unassembled intracellular subunits. Surface antibody and radioligand binding are a percentage of total subunit protein that is in a conformation recognized by mAb270, mAb299, or [3H]MCC. This is not necessarily equivalent to the number of binding sites (radioligand or antibody) located on the cell surface as a percentage of total subunit protein in all conformations.
Because we assume that subunits on the cell surface are folded correctly into their “mature” conformation, it would be reasonable to expect that the absolute (fold) increase in cell-surface receptor caused by nicotine treatment would be similar, whether assayed by antibody or by radioligand binding. As was illustrated in Figure2B, chronic nicotine treatment produced a three- to fourfold increase in the amount of α4β2 on the cell surface and a 1.2-fold increase in coassembled α4β2χ complexes on the cell surface. The absolute increase in cell-surface [3H]MCC binding caused by chronic nicotine treatment was 7.3 ± 2.4-fold (n = 4) for α4β2 and 1.6 ± 0.5-fold (n = 4) for α4β2χ. There was considerable variability between experiments in the fold increase in surface [3H]MCC binding to α4β2 that is probably attributable to the relatively large experiment-to-experiment variability in the (low) basal levels of [3H]MCC binding in cells expressing α4β2 nAChRs. As a consequence, the nicotine-induced increase in cell-surface [3H]MCC binding in cells expressing α4β2 nAChRs is not significantly different from the increase in surface receptor assayed by mAb binding. Despite the experiment-to-experiment variability in the absolute increase in [3H]MCC binding, a very consistent and reproducible increase was observed in the proportion of total α4β2 nAChR on the cell surface (assayed by [3H]MCC binding) (see data presented in Table 1).
Influence of nicotine and subunit composition on total subunit levels
The cell-surface antibody binding data presented earlier (Fig. 2) indicates that chronic nicotine causes an upregulation in the level of cell-surface α4β2 and α4β2χ receptor in transfected cells. To determine the effect of chronic nicotine treatment on the proportion of total subunit protein on the cell surface, the level of antibody (mAb270 and mAb299) binding was examined in both intact and permeabilized cell monolayers. It should be borne in mind that what is being measured in these experiments is not necessarily the proportion of total subunit protein that is expressed on the cell surface but rather the proportion of subunit protein recognized by either mAb270 or mAb299 (i.e., the proportion of subunit protein folded into a conformation recognized by these antibodies). As mentioned earlier, mAb270 is strongly conformationally sensitive (it does not recognize β2 after SDS denaturation), and although mAb299 does recognize SDS-denatured α4 subunit, suggesting that it recognizes a linear epitope, the mAb299 epitope will be masked if the subunit adopts a conformation in which the epitope is buried or inaccessible.
As can be seen in Figure 5A, the level of α4χ protein detected in permeabilized cells was significantly higher (4.2 ± 0.5-fold; n = 7;p < 0.001) than that of α4. This is in contrast to immunoblotting data (Fig. 3A) and suggests that, compared with the α4 subunit, a greater proportion of total α4χ subunit protein is folded into a conformation recognized by mAb299. The β2 subunit appears to have a dominant negative effect when coexpressed with α4χ, illustrated by a small but consistent (and significant) reduction in the level of mAb299 binding to permeabilized cells when α4χ is coexpressed with the β2 subunit (26 ± 3.5% decrease; n = 4; p < 0.02). There is no change in mAb299 binding to α4, whether α4 is expressed alone or with β2 or with β2χ (Fig. 5A). Examination of mAb270 binding to transfected cells (Fig. 6) reveals several interesting phenomena. In the absence of nicotine, the level of total mAb270 binding to cells transfected with β2χ alone is significantly higher (4.3 ± 0.2-fold; n = 3;p < 0.001) than to those transfected with β2 alone. Significantly less β2χ is detected when β2χ is coexpressed with α4 (mAb270 binding is reduced by 50.9 ± 4.5%;n = 3; p < 0.01). Thus, α4 exerts a strongly dominant negative effect on mAb270 binding (when coexpressed with β2χ). There is no change in mAb270 binding to β2, whether β2 is expressed alone or with β2; however, mAb270 detects significantly higher levels of β2 subunit (5.4 ± 0.7 fold;n = 5; p < 0.001) in permeabilized cells when β2 is coexpressed with α4χ (Fig.6A). Hence, although both α4χ and β2χ yield high levels of mAb299/mAb270 binding sites when expressed alone, antibody binding to both chimeric subunits is reduced by coexpression of wild-type (α4 or β2) subunits.
Fig. 5.
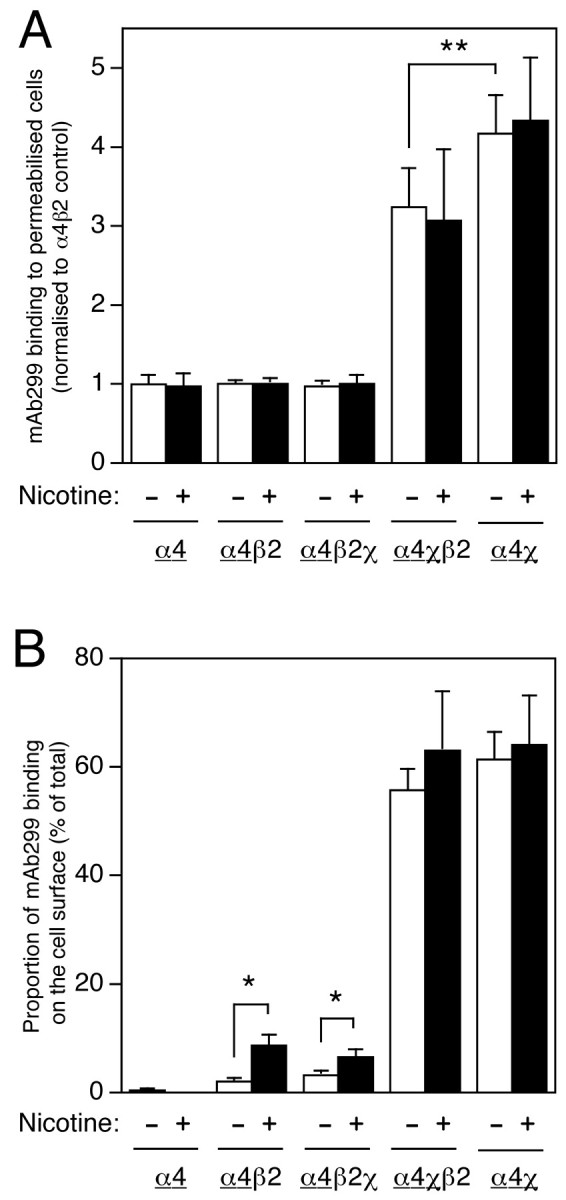
Influence of chronic nicotine treatment and subunit composition on subcellular distribution of α4 and α4χ subunits. Transfected TSA201 cells, grown on coverslips, were labeled with mAb299 (specific for α4 and α4χ subunits) either after membrane permeabilization or as intact cell monolayers. Data are presented as total antibody binding to permeabilized cells (A) and as the proportion of antibody binding sites on the cell surface (B). The level of mAb299 binding was determined in an enzyme-linked assay (see Materials and Methods). For all subunit combinations, mAb299 binding was determined in cells that had been grown in the absence (white bars) or presence (black bars) of nicotine (10 μm, 24 hr). Data are the means of five to nine independent experiments, each performed in duplicate, and have been normalized to the level of mAb299 binding determined for permeabilized cells transfected with α4β2 in the absence of nicotine. The background signal from mock-transfected coverslips has been subtracted. B, The proportion of mAb299 binding detected on the cell surface for each subunit combination has been determined from parallel experiments performed on permeabilized and intact cell monolayers. Because α4χ can form homomeric (as well as heteromeric) cell-surface complexes (Cooper et al., 1999), mAb299 would be expected to detect a heterogeneous population of cell-surface complexes in cells cotransfected with α4χ + β2. To emphasize which subunit is being assayed by mAb299 binding (α4 or α4χ, rather than β2 or β2x), the appropriate subunit is underlined. Significant differences, determined by two-tailed Student's t test, are indicated (*p < 0.05; **p < 0.02).
Fig. 6.
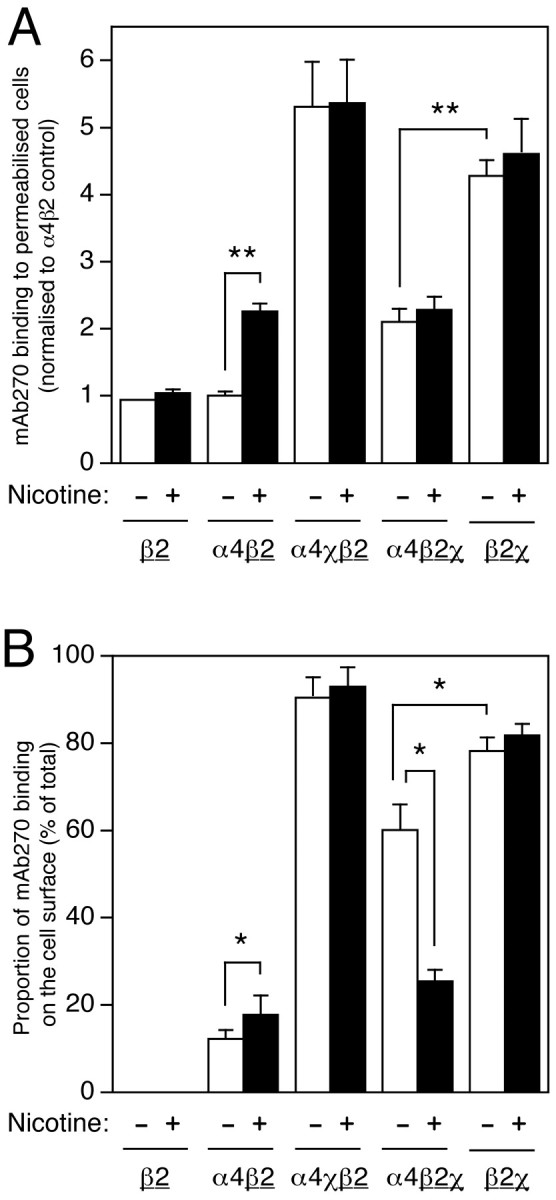
Influence of chronic nicotine treatment and subunit composition on subcellular distribution of β2 and β2χ subunits. Transfected TSA201 cells, grown on coverslips, were labeled with mAb270 (specific for β2 and β2χ subunits) either after membrane permeabilization or as intact cell monolayers. Data are presented as total antibody binding to permeabilized cells (A) and as the proportion of antibody binding sites on the cell surface (B). The level of mAb270 binding was determined in an enzyme-linked assay (see Materials and Methods). For all subunit combinations, mAb270 binding was determined in cells that had been grown in the absence (white bars) or presence (black bars) of nicotine (10 μm, 24 hr). Data are the means of 5–11 independent experiments, each performed in duplicate, and have been normalized to the level of mAb270 binding determined with permeabilized cells transfected with α4β2 in the absence of nicotine. The background signal from mock-transfected coverslips has been subtracted. B, The proportion of mAb270 binding detected on the cell surface for each subunit combination has been determined from parallel experiments performed on permeabilized and intact cell monolayers. Because β2χ can form homomeric (as well as heteromeric) cell-surface complexes (Cooper et al., 1999), mAb270 would be expected to detect a heterogeneous population of cell-surface complexes in cells cotransfected with α4 + β2χ. To emphasize which subunit is being assayed by mAb270 binding (β2 or β2χ, rather than α4 or α4χ), the appropriate subunit is underlined. Significant differences, determined by two-tailed Student'st test, are indicated (*p < 0.05; **p < 0.01).
Chronic nicotine treatment does not have a significant effect on the level of total mAb299 binding to α4 or α4χ in pemeabilized cells (Fig. 5A). Similarly, there was no effect of nicotine treatment on the level of β2χ, when expressed alone or when coexpressed with either α4 or α4χ (Fig. 6A). However, nicotine treatment of α4β2-transfected cells caused a significant increase (2.3 ± 0.1-fold, n = 9;p < 0.001) in the level of β2 subunit detected in permeabilized cells (Fig. 6A). These findings indicate that the amount of total β2 subunit protein folded into a conformation recognized by mAb270 is influenced both by coassembled partner subunits (e.g., α4 or α4χ) and by chronic nicotine treatment (e.g., when coexpressed with α4).
To confirm that the discrepancy between antibody-binding data derived from studies with permeabilized cells (Figs. 5A,6A) and from immunoblotting data (Fig. 3) is not an artifact caused by the permeabilized cell binding assay, the experiment was repeated with β2FLAG and assayed with mAbFLAG-M2 (which recognizes a linear epitope tag). In contrast to earlier results with mAb270 binding to β2 (Fig. 6), no significant difference was seen in the level of mAbFLAG-M2 binding to β2FLAG, either after chronic nicotine treatment or as a consequence of coexpression of β2FLAG with α4χ rather than α4 (Fig. 7). In agreement with the earlier data (Fig. 6), the influence of subunit composition and nicotine treatment on α4β2FLAG was observed with mAb270 binding to β2FLAG (Fig. 7). Together with the immunoblotting data (Fig. 3), this provides strong support for our conclusion that although factors such as nicotine treatment and coassembled subunit partners do not influence levels of total β2 subunit protein, they do influence the proportion of subunit protein folded into a conformation recognized by conformation-sensitive antibodies (such as mAb270) and by nicotinic ligands (such as [3H]epibatidine and [3H]MCC). Experiments were also performed to confirm that nicotine-induced upregulation of cell-surface receptor could be detected by mAbFLAG-M2 binding to cells transfected with β2FLAG. Chronic nicotine treatment of cells expressing α4β2FLAG resulted in the upregulation of cell-surface mAbFLAG-M2 binding (2.7 ± 0.3-fold; n = 3). This is similar to the level of receptor upregulation detected in experiments in which cells transfected with α4β2 were assayed by mAb270 binding (Fig. 2), and argues that, when expressed on the cell surface, most if not all of the assembled β2 subunit is in a conformation recognized by the conformationally sensitive antibody mAb270.
Fig. 7.

Influence of chronic nicotine treatment and subunit composition on levels of β2FLAG detected in permeabilized cells. Transfected TSA201 cells, grown on coverslips, were labeled with either mAb270 or mAbFLAG-M2 (both of which recognize β2FLAG). The level of antibody binding was determined after membrane permeabilization in an enzyme-linked assay using an HRP-linked secondary antibody (see Materials and Methods). For all subunit combinations, antibody binding was determined in cells that had been grown in the absence (white bars) or presence (black bars) of nicotine (10 μm, 24 hr). Data are the means of two to three independent experiments, each performed in duplicate, and have been normalized to the level of binding determined with cells transfected with α4χβ2FLAG in the absence of nicotine. The background signal from mock-transfected coverslips has been subtracted. Significant differences, determined by two-tailed Student'st test, are indicated (*p < 0.05).
Influence of nicotine and subunit composition on nAChR subunit distribution
Data from antibody binding experiments conducted with intact and permeabilized cells were used to determine the proportion of total folded subunit protein (i.e., total subunit protein recognized by mAb270 or mAb299) that is expressed on the cell surface (Figs.5B, 6B). It is apparent that chronic nicotine treatment upregulates the proportion of α4 subunit on the cell surface when it is coexpressed with either β2 (from 2.1 ± 0.7 to 8.5 ± 2.0%; n = 3; p < 0.05) or β2χ (from 3.2 ± 0.8 to 6.5 ± 1.5%;n = 3; p < 0.05) (Fig. 5B). A much higher proportion of α4χ is expressed on the cell surface in all combinations examined (∼60%) and is unaffected by chronic nicotine treatment (Fig. 5B).
Although a relatively small proportion of β2 is expressed on the cell surface when it is coexpressed with α4, ∼90% of β2 is detected on the cell surface when coexpressed with α4χ (Fig.6B). Particularly striking is the finding that coexpression of α4 substantially reduces the proportion of β2χ expressed on the cell surface (from 78.2 ± 3.1 to 60.1 ± 5.9%; n = 3; p < 0.02) and that this is reduced yet further by chronic nicotine treatment (to 25.4 ± 2.6%; n = 3; p < 0.05) (Fig.6B). The influence of both nicotine treatment and coexpressed subunits on the proportion of total subunit detected on the cell surface by antibody binding is summarized in Table 1.
DISCUSSION
It has been reported previously that relatively low levels of α4β2 nAChR are expressed on the cell surface of transfected cell lines, but coexpression of either α4 or β2 with subunit chimeras (α4χ and β2χ) containing the C-terminal domain of the 5HT3A subunit can upregulate levels of α4 and β2 on the cell surface (Cooper et al., 1999). Data presented in the current study allow us to interpret such observations in terms of subunit conformation and subcellular distribution.
Neither α4 nor β2 is expressed at detectable levels on the cell surface when expressed alone (Cooper et al., 1999;Harkness and Millar, 2001). In cells cotransfected with α4 and β2, ∼10% or less of total α4 or β2 subunit protein (recognized by mAb299 or mAb270, respectively) is on the cell surface. However, coexpression of β2 with α4χ (rather than with α4) results in a dramatic upregulation of cell-surface levels of β2 (∼8.5-fold) (Fig. 2), corresponding to 90% of total β2 subunit protein recognized by mAb270 (Fig. 6). Of the relatively low number of radioligand binding sites detected for α4β2 in the absence of nicotine, ∼25% are on the cell surface (Fig. 4). In cells transfected with α4β2χ, there are approximately five times more radioligand binding sites, and of these, ∼50% are expressed on the cell surface (Fig. 4). In cells transfected with α4χβ2, there are ∼20 times more radioligand binding sites, of which ∼90% are at the cell surface (Fig. 4). A more dramatic effect on radioligand binding and cell-surface expression is caused by replacing α4 with α4χ than by replacing β2 with β2χ (Figs.1, 2), suggesting that the low level of binding and cell-surface expression observed for α4β2 is influenced primarily by the α4 subunit.
Immunoblotting experiments with mAb299 comparing total cellular levels of SDS-denatured α4 and α4χ subunit protein indicate that when transfected alone these two subunits are expressed at similar levels (Cooper et al., 1999). In contrast, when the same antibody is used to determine total levels of α4 and α4χ subunit protein in permeabilized cells (without SDS denaturation of subunits), α4 is detected at significantly lower levels (approximately fourfold lower) than α4χ (Fig. 5A). This would suggest that a significant proportion of the total α4 subunit protein is in a conformation not recognized by mAb299, perhaps because of inappropriate subunit folding masking the mAb299 epitope.
An important aspect of the present study is the evidence that subunit folding can be influenced by the coassembled partner subunit. This was implied by our previous studies examining coassembly of chimeric α4χ and β2χ subunits (Cooper et al., 1999; Harkness and Millar, 2001), but the current findings present more direct evidence for an influence of coassembled subunit partners on subunit conformation. Immunoblotting (Fig. 3) and antibody binding to cell monolayers (Fig.7) with an epitope-tagged β2 subunit (β2FLAG) suggest that the level of total β2 subunit protein is similar, whether coexpressed with α4 or α4χ. In contrast, in permeabilized cells, the level of total mAb270 binding to β2 is approximately fivefold lower when it is coexpressed with α4 than with α4χ (Fig. 6A). A plausible explanation for this would be that when coassembled with α4χ, a greater proportion of the total β2 subunit protein is in a conformation recognized by mAb270. The idea that subunit conformation can be influenced by coassembly with other subunits (rather than subunits adopting their fully folded native conformation before assembly) is a feature of current models of nAChR assembly (Green and Millar, 1995; Green, 1999; Keller and Taylor, 1999) and has been proposed as a mechanism by which nAChRs and other hetero-oligomeric complexes achieve their appropriate subunit composition and stoichiometry via sequential addition of subunits to partially folded “assembly intermediate” complexes (Green and Claudio, 1993; Green and Millar, 1995; Green, 1999).
The dominant influence of the α4 subunit is illustrated further by comparing levels of mAb270 binding to β2χ when expressed alone or when coexpressed with α4 (Fig. 6A). Coexpression of the α4 subunit with β2χ reduces the amount of β2χ detected on the cell surface, suggesting that coassembly of α4 with β2χ causes substantial intracellular retention of the β2χ subunit. A similar dominant negative effect of α4 was observed for α4 coexpressed with the 5HT3A subunit (Harkness and Millar, 2001).
Immunoblotting of SDS-denatured cell extracts (Fig. 3) reveals that chronic nicotine treatment has little, if any, influence on total nAChR α4 or β2 subunit protein levels in transfected cells. This suggests that the observed upregulation in radioligand binding detected in cell homogenates as a consequence of chronic nicotine treatment (up to an approximately fivefold increase) (Fig. 1) is attributable to an increase in the proportion of total subunit protein that is folded and assembled into a conformation capable of generating an agonist binding site. Similarly, the observed upregulation of cell-surface nAChRs as a consequence of chronic nicotine treatment (up to ∼3.5-fold increase) (Figs. 2, 5) reflects a greater proportion of total subunit protein expressed on the cell surface. These findings are consistent with previous studies which concluded that chronic nicotine-induced upregulation occurs by a post-translational mechanism (Peng et al., 1994; Bencherif et al., 1995; Rothhut et al., 1996).
In comparison with its effect on α4β2, chronic nicotine treatment caused significantly less upregulation (of either radioligand or surface antibody binding) in cells transfected with chimeric subunits, presumably reflecting the higher levels of binding and cell-surface expression for such subunit combinations in the absence of nicotine. In cells transfected with α4β2χ, nicotine caused a twofold increase in total radioligand binding (Fig. 1) and increased the proportion of radioligand binding sites expressed on the cell surface from 44 to 63% (Fig. 4). Nicotine treatment also caused a 1.2-fold increase in the proportion of α4β2χ expressed on the cell surface. Chronic nicotine treatment had no significant effect on the proportion of cell-surface radioligand or antibody binding to α4χβ2.
For most subunit combinations examined, chronic nicotine treatment does not alter total subunit protein levels estimated by antibody binding to nondenatured subunit protein in permeabilized cells (Figs.5A, 6A), the only exception being a significant twofold increase in the amount of total mAb270 binding to β2 in cells transfected with α4β2 (Fig. 6). Because nicotine treatment does not substantially alter the total subunit protein level for any SDS-denatured subunits examined by immunoblotting (Fig. 3), this suggests that chronic nicotine treatment can alter the amount of β2 subunit folded into a conformation recognized by mAb270 when β2 is coexpressed with α4. In contrast, chronic nicotine treatment does not increase the amount of “correctly” folded β2 subunit (assayed by total mAb270 binding) when it is coexpressed with α4χ. As discussed earlier, a smaller proportion of the total β2 subunit protein is folded into a conformation recognized by mAb270 when coexpressed with α4 than with α4χ (Fig. 6). Chronic nicotine does not cause an upregulation of either radioligand binding or cell-surface expression when β2 is coexpressed with α4χ (Figs. 1, 2, 4, 5, 6). These findings suggest that changes in subunit conformation may underlie nicotine-induced upregulation of α4β2 nAChRs. It is also noteworthy that nicotine treatment causes a reduction in the level of β2χ subunit expressed on the cell surface when it is coexpressed with α4 but not when it is expressed alone (Fig. 6). This may reflect the fact that nicotine can enhance levels of α4 subunit folded into a conformation that can readily coassemble with β2χ. Because of the dominant negative effect of α4 (see reduced levels of surface β2χ when coexpressed with α4 discussed in Results) (Fig. 6), if more “correctly folded” α4 were available to bind to β2χ, after nicotine treatment, this would result in enhanced intracellular retention of β2χ.
The influence of coexpressed subunits on subunit conformation, coassembly, and upregulation is initiated, presumably, by events occurring in the endoplasmic reticulum (where initial subunit interactions occur) (Green and Millar, 1995). Because nicotine is a membrane-permeant ligand, it is quite plausible that the consequences of chronic nicotine treatment are caused, at least in part, by its interaction with intracellular receptors. Previous studies have demonstrated, however, that α4β2 upregulation can be induced by exposure to membrane-impermeant ligands. This has led to the suggestion that upregulation is mediated primarily by interaction with cell-surface receptors (Whiteaker et al., 1998), possibly by changes in subunit turnover (Peng et al., 1994).
Several conclusions can be drawn from this study. Neither chronic nicotine treatment nor coexpressed subunit partners greatly affect the level of total subunit protein detected by immunoblotting (Fig. 3), but both of these factors can influence subunit folding (assayed by antibody binding to permeabilized cells) (Figs. 5A,6A). Both chronic nicotine treatment and coexpressed subunits exert a dramatic effect on levels of radioligand binding (which reflects steady-state levels of coassembled subunits) (Fig. 1) and on the proportion of receptor expressed on the cell surface [assayed either by radioligand binding (Figs. 1, 4) or by antibody binding (Figs. 2, 5, 6)]. It is also evident that the effect of chronic nicotine on these parameters is strongly dependent on subunit composition.
Footnotes
This work was supported by grants from the Wellcome Trust. We thank Harriet Sale and Shehan Wevita for assistance with some experiments and Dr. Ian Parsons for construction of pRK5-β2FLAG.
Correspondence should be addressed to Dr. Neil S. Millar, Department of Pharmacology, University College London, Gower Street, London, WC1E 6BT, UK. E-mail: n.millar@ucl.ac.uk.
REFERENCES
- 1.Bencherif M, Fowler K, Lukas R, Lippiello PM. Mechanisms of up-regulation of neuronal nicotinic acetylcholine receptors in clonal cell lines and primary cultures of fetal rat brain. J Pharmacol Exp Ther. 1995;275:987–994. [PubMed] [Google Scholar]
- 2.Benwell MEM, Balfour DJK, Anderson JM. Evidence that tobacco smoking increases the density of (−)-[3H]nicotine binding sites in human brain. J Neurochem. 1988;50:1243–1247. doi: 10.1111/j.1471-4159.1988.tb10600.x. [DOI] [PubMed] [Google Scholar]
- 3.Buisson B, Gopalakrishnan M, Arneric SP, Sullivan JP, Bertrand D. Human α4β2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: a patch-clamp study. J Neurosci. 1996;16:7880–7891. doi: 10.1523/JNEUROSCI.16-24-07880.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen D, Dang H, Patrick JW. Contributions of N-linked glycosylation to the expression of a functional α7-nicotinic receptor in Xenopus oocytes. J Neurochem. 1998;70:349–357. doi: 10.1046/j.1471-4159.1998.70010349.x. [DOI] [PubMed] [Google Scholar]
- 5.Cooper ST, Millar NS. Host cell-specific folding and assembly of the neuronal nicotinic acetylcholine receptor α7 subunit. J Neurochem. 1997;68:2140–2151. doi: 10.1046/j.1471-4159.1997.68052140.x. [DOI] [PubMed] [Google Scholar]
- 6.Cooper ST, Millar NS. Host cell-specific folding of the neuronal nicotinic receptor α8 subunit. J Neurochem. 1998;70:2585–2593. doi: 10.1046/j.1471-4159.1998.70062585.x. [DOI] [PubMed] [Google Scholar]
- 7.Cooper ST, Harkness PC, Baker ER, Millar NS. Upregulation of cell-surface α4β2 neuronal nicotinic receptors by lower temperature and expression of chimeric subunits. J Biol Chem. 1999;274:27145–27152. doi: 10.1074/jbc.274.38.27145. [DOI] [PubMed] [Google Scholar]
- 8.Deneris ES, Connolly J, Boulter J, Wada E, Wada K, Swanson LW, Patrick J, Heinemann S. Primary structure and expression of β2: a novel subunit of neuronal nicotinic acetylcholine receptors. Neuron. 1988;1:45–54. doi: 10.1016/0896-6273(88)90208-5. [DOI] [PubMed] [Google Scholar]
- 9.Eiselé J-L, Bertrand S, Galzi J-L, Devillers-Thiéry A, Changeux J-P, Bertrand D. Chimaeric nicotinic-serotonergic receptor combines distinct ligand binding and channel specificities. Nature. 1993;366:479–483. doi: 10.1038/366479a0. [DOI] [PubMed] [Google Scholar]
- 10.Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of α4 and β2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol. 1992;41:31–37. [PubMed] [Google Scholar]
- 11.Goldman D, Deneris E, Luyten W, Kochhar A, Patrick J, Heinemann S. Members of a nicotinic acetylcholine receptor gene family are expressed in different regions of the mammalian central nervous system. Cell. 1987;48:965–973. doi: 10.1016/0092-8674(87)90705-7. [DOI] [PubMed] [Google Scholar]
- 12.Gopalakrishnan M, Monteggia LM, Anderson DJ, Molinari EJ, Piattoni-Kaplan M, Donnelly-Roberts D, Arneric SP, Sullivan JP. Stable expression, pharmacologic properties and regulation of the human neuronal nicotinic acetylcholine α4β2 receptor. J Pharmacol Exp Ther. 1996;276:289–297. [PubMed] [Google Scholar]
- 13.Gopalakrishnan M, Molinari EJ, Sullivan JP. Regulation of human α4β2 neuronal nicotinic acetylcholine receptors by cholinergic channel ligands and second messenger pathways. Mol Pharmacol. 1997;52:524–534. [PubMed] [Google Scholar]
- 14.Green WN. Ion channel assembly: creating structures that function. J Gen Physiol. 1999;113:163–169. doi: 10.1085/jgp.113.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green WN, Claudio T. Acetylcholine receptor assembly: subunit folding and oligomerization occur sequentially. Cell. 1993;74:57–69. doi: 10.1016/0092-8674(93)90294-z. [DOI] [PubMed] [Google Scholar]
- 16.Green WN, Millar NS. Ion-channel assembly. Trends Neurosci. 1995;18:280–287. [PubMed] [Google Scholar]
- 17.Harkness PC, Millar NS. Inefficient cell-surface expression of hybrid complexes formed by the co-assembly of neuronal nicotinic acetylcholine receptor and serotonin receptor subunits. Neuropharmacology. 2001;41:79–87. doi: 10.1016/s0028-3908(01)00042-9. [DOI] [PubMed] [Google Scholar]
- 18.Hopp TP, Prickett KS, Price VL, Libby RT, March CJ, Cerretti DP, Urdal DL, Conlon PJ. A short polypeptide marker sequence useful for recombinant protein identification and purification. Biotechnology. 1988;6:1204–1210. [Google Scholar]
- 19.Kassner PD, Berg DK. Differences in the fate of neuronal acetylcholine receptor protein expressed in neurons and stably transfected cells. J Neurobiol. 1997;33:968–982. doi: 10.1002/(sici)1097-4695(199712)33:7<968::aid-neu8>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 20.Ke L, Eienhour CM, Bencherif M, Lukas RJ. Effects of chronic nicotine treatment on expression of diverse nicotinic acetylcholine receptor subtypes. I. Dose- and time-dependent effects of nicotine treatment. J Pharmacol Exp Ther. 1998;286:825–840. [PubMed] [Google Scholar]
- 21.Keller ST, Taylor P. Determinants responsible for assembly of the nicotinic acetylcholine receptor. J Gen Physiol. 1999;113:171–176. doi: 10.1085/jgp.113.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lansdell SJ, Millar NS. The influence of nicotinic receptor subunit composition on agonist, α-bungarotoxin and insecticide (imidacloprid) binding affinity. Neuropharmacology. 2000;39:671–679. doi: 10.1016/s0028-3908(99)00170-7. [DOI] [PubMed] [Google Scholar]
- 23.Lansdell SJ, Schmitt B, Betz H, Sattelle DB, Millar NS. Temperature-sensitive expression of Drosophila neuronal nicotinic acetylcholine receptors. J Neurochem. 1997;68:1812–1819. doi: 10.1046/j.1471-4159.1997.68051812.x. [DOI] [PubMed] [Google Scholar]
- 24.Maricq AV, Peterson AS, Brake AJ, Myers RM, Julius D. Primary structure and functional expression of the 5HT3 receptor, a serotonin-gated ion channel. Science. 1991;254:432–437. doi: 10.1126/science.1718042. [DOI] [PubMed] [Google Scholar]
- 25.Marks MJ, Stitzel JA, Collins AC. Time course study of the effects of chronic nicotine infusion on drug response and brain function. J Pharmacol Exp Ther. 1985;235:619–628. [PubMed] [Google Scholar]
- 26.Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC. Nicotine binding and nicotine receptor subunit RNA after chronic nicotine treatment. J Neurosci. 1992;12:2765–2784. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J. Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Mol Pharmacol. 1994;46:523–530. [PubMed] [Google Scholar]
- 28.Peng X, Gerzanich V, Anand R, Wang F, Lindstrom J. Chronic nicotine treatment up-regulates α3 and α7 acetylcholine receptor subtypes expressed by the human neuroblastoma cell line SH-SY5Y. Mol Pharmacol. 1997;51:776–784. doi: 10.1124/mol.51.5.776. [DOI] [PubMed] [Google Scholar]
- 29.Ragozzino D, Fucile S, Giovannelli A, Grassi F, Mileo AM, Ballivet M, Alema S, Eusebi F. Functional properties of neuronal nicotinic acetylcholine receptor channels expressed in transfected human cells. Eur J Neurosci. 1997;9:480–488. doi: 10.1111/j.1460-9568.1997.tb01625.x. [DOI] [PubMed] [Google Scholar]
- 30.Rangwala F, Drisdel RC, Rakhilin S, Ko E, Atluri P, Harkins AB, Fox AP, Salman SB, Green WN. Neuronal α-bungarotoxin receptors differ structurally from other nicotinic acetylcholine receptors. J Neurosci. 1997;17:8201–8212. doi: 10.1523/JNEUROSCI.17-21-08201.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rothhut B, Romano SJ, Vijayaraghavan S, Berg DK. Post-translational regulation of neuronal acetylcholine receptors stably expressed in a mouse fibroblast cell line. J Neurobiol. 1996;29:115–125. doi: 10.1002/(SICI)1097-4695(199601)29:1<115::AID-NEU9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz RD, Kellar KJ. In vivo regulation of [3H]acetylcholine recognition sites in brain by nicotinic cholinergic drugs. J Neurochem. 1985;45:427–433. doi: 10.1111/j.1471-4159.1985.tb04005.x. [DOI] [PubMed] [Google Scholar]
- 33.Spier AD, Wotherspoon G, Nayak SV, Nichols RA, Priestly JV, Lummis SCR. Antibodies against the extracellular domain of the 5-HT3 receptor label both native and recombinant receptors. Mol Brain Res. 1999;67:221–230. doi: 10.1016/s0169-328x(99)00055-8. [DOI] [PubMed] [Google Scholar]
- 34.Sweileh W, Wenberg K, Xu J, Forsayeth J, Hardy S, Loring RH. Multistep expression and assembly of neuronal nicotinic receptors is both host-cell- and receptor-subtype-dependent. Mol Brain Res. 2000;75:293–302. doi: 10.1016/s0169-328x(99)00302-2. [DOI] [PubMed] [Google Scholar]
- 35.Turton S, Gillard NP, Stephenson FA, McKernan RM. Antibodies against the 5-HT3-A receptor identify a 54 kDa protein affinity-purified from NCB20 cells. Mol Neuropharmacol. 1993;3:167–171. [Google Scholar]
- 36.Wang F, Nelson ME, Kuryatov A, Olale F, Cooper J, Keyser K, Lindstrom J. Chronic nicotine treatment up-regulates human α3β2 but not α3β4 acetylcholine receptors stably transfected in human embryonic kidney cells. J Biol Chem. 1998;273:28721–28732. doi: 10.1074/jbc.273.44.28721. [DOI] [PubMed] [Google Scholar]
- 37.Warpman U, Friberg L, Gillespie A, Hellström-Lindahl E, Zhang X, Nordberg A. Regulation of nicotinic receptor subtypes following chronic nicotinic agonist exposure in M10 and SH-SY5Y neuroblastoma cells. J Neurochem. 1998;70:2028–2037. doi: 10.1046/j.1471-4159.1998.70052028.x. [DOI] [PubMed] [Google Scholar]
- 38.Whiteaker P, Sharples CGV, Wonnacott S. Agonist-induced up-regulation of α4β2 nicotinic acetylcholine receptors in M10 cells: pharmacological and spatial definition. Mol Pharmacol. 1998;53:950–962. [PubMed] [Google Scholar]
- 39.Whiting P, Lindstrom J. Purification and characterization of a nicotinic acetylcholine receptor from rat brain. Proc Natl Acad Sci USA. 1987;84:595–599. doi: 10.1073/pnas.84.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whiting PJ, Lindstrom JM. Characterization of bovine and human neuronal nicotinic acetylcholine receptors using monoclonal antibodies. J Neurosci. 1988;8:3395–3404. doi: 10.1523/JNEUROSCI.08-09-03395.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whiting P, Schoepfer R, Lindstrom J, Priestley T. Structural and pharmacological characterization of the major brain nicotinic acetylcholine receptor subtype stably expressed in mouse fibroblasts. Mol Pharmacol. 1991;40:463–472. [PubMed] [Google Scholar]
- 42.Zhang X, Gong Z-H, Hellstrom-Lindahl E, Nordberg A. Regulation of α4β2 nicotinic acetylcholine receptors in M10 cells following treatment with nicotinic agents. NeuroReport. 1994;6:313–317. doi: 10.1097/00001756-199501000-00022. [DOI] [PubMed] [Google Scholar]