

Abstract
Brain-derived neurotrophic factor (BDNF) plays a critical role in activity-dependent modifications of neuronal connectivity and synaptic strength, including establishment of hippocampal long-term potentiation (LTP). To shed light on mechanisms underlying BDNF-dependent synaptic plasticity, the present study was undertaken to characterize release of native BDNF from newborn rat hippocampal neurons in response to physiologically relevant patterns of electrical field stimulation in culture, including tonic stimulation at 5 Hz, bursting stimulation at 25 and 100 Hz, and theta-burst stimulation (TBS). Release was measured using the ELISA in situ technique, developed in our laboratory to quantify secretion of native BDNF without the need to first overexpress the protein to nonphysiological levels. Each stimulation protocol resulted in a significant increase in BDNF release that was tetrodotoxin sensitive and occurred in the absence of glutamate receptor activation. However, 100 Hz tetanus and TBS, stimulus patterns that are most effective in inducing hippocampal LTP, were significantly more effective in releasing native BDNF than lower-frequency stimulation. For all stimulation protocols tested, removal of extracellular calcium, or blockade of N-type calcium channels, prevented BDNF release. Similarly, depletion of intracellular calcium stores with thapsigargin and treatment with dantrolene, an inhibitor of calcium release from caffeine–ryanodine-sensitive stores, markedly inhibited activity-dependent BDNF release. Our results indicate that BDNF release can encode temporal features of hippocampal neuronal activity. The dual requirement for calcium influx through N-type calcium channels and calcium mobilization from intracellular stores strongly implicates a role for calcium-induced calcium release in activity-dependent BDNF secretion.
Keywords: activity, BDNF, caffeine, calcium channels, calcium-induced calcium release, electrical field stimulation, ELISAin situ, hippocampus, intracellular calcium stores, LTP, neurotrophins, patterned stimulation, ryanodine-sensitive stores, synaptic plasticity
Brain-derived neurotrophic factor (BDNF) plays a critical role in activity-dependent modulation of synaptic strength and neuronal connectivity at cortical and hippocampal synapses (Katz and Shatz, 1996; Black, 1999; McAllister et al., 1999;Lu and Gottschalk, 2000; Poo, 2001). For example, BDNF is required for activity-dependent synaptic competition and formation of ocular dominance columns in developing visual cortex (Cabelli et al., 1995) and can also regulate short- and long-term changes in synaptic strength. Specifically, exogenous BDNF can directly potentiate synaptic transmission in the hippocampus and cortex (Kang and Schuman, 1995;Akaneya et al., 1997), and endogenous BDNF is necessary for establishment of long-term potentiation (LTP) (Korte et al., 1995;Figurov et al., 1996; Patterson et al., 1996; Kang et al., 1997). However, despite these findings, relatively little is known about the cellular mechanisms regulating activity-dependent BDNF release from cortical and hippocampal neurons.
Most information about BDNF release from hippocampal neurons has been derived from studies using adenoviral gene-transfer techniques to overexpress BDNF to high concentrations (Goodman et al., 1996;Griesbeck et al., 1999; Gärtner and Staiger, 2002), or cells transfected with BDNF-green fluorescent protein (GFP) expression plasmids (Hartmann et al., 2001; Kohara et al., 2001). It is unclear, however, whether mechanisms governing sorting and release of BDNF in these artificial systems are the same or different from those governing sorting and release of native BDNF (Fawcett et al., 1997). In addition, most previous studies of BDNF release used continuous membrane depolarization to model neuronal activity (Goodman et al., 1996;Griesbeck et al., 1999). However, we showed previously, using primary sensory neurons, that patterned electrical stimulation, which more closely mimics neuronal activity in vivo, is much more effective at releasing native BDNF than continuous KCl depolarization (Balkowiec and Katz, 2000). Moreover, Hartmann et al. (2001) found that different mechanisms regulate release of BDNF-GFP in response to high-frequency presynaptic stimulation and continuous KCl depolarization, respectively. Finally, studies from this and other laboratories demonstrated that release of native BDNF from sensory neurons (Balkowiec and Katz, 2000; Lever et al., 2001) or of adenovirally overexpressed BDNF (AdVBDNF) from hippocampal neurons (Gärtner and Staiger, 2002) can be regulated by the frequency and pattern of stimulation. However, mechanisms underlying activity-dependent secretion of native BDNF from hippocampal neurons have not been defined.
To address this issue, the present study characterized mechanisms of native BDNF release from hippocampal neurons in response to physiologically relevant patterns of stimulation. To measure release of native BDNF, we developed an in vitro model using dissociated neuron cultures and a modified ELISA, termed ELISA in situ (Balkowiec and Katz, 2000). Our findings demonstrate that release of native BDNF from hippocampal neurons can be evoked directly by patterned electrical stimulation via activation of tetrodotoxin-sensitive sodium channels and calcium influx through N-type channels, in the absence of synaptic activity. Moreover, this release requires intact intracellular calcium stores, suggesting that calcium-induced calcium release (CICR) plays a role in activity-dependent secretion of native BDNF from hippocampal neurons.
Parts of this paper have been published previously in abstract form (Balkowiec and Katz; 2001; Katz and Balkowiec, 2001).
MATERIALS AND METHODS
Preparation of hippocampal cultures. Newborn rats (Sprague Dawley strain; Zivic-Miller, Zelienople, PA) were deeply anesthetized by hypothermia and decapitated. Hippocampi were rapidly and aseptically dissected from each brain in ice-cold Ca2+/Mg2+-free Dulbecco's phosphate-buffered salt solution (Mediatech, Herndon, VA), followed by removal of meninges and mincing to small pieces. The hippocampal tissue was next digested in 0.1% crystallized trypsin-3X (Worthington Biochemical, Lakewood, NJ) with 0.01% Deoxyribonuclease I (Sigma, St. Louis, MO) dissolved in Ca2+/Mg2+-free HBSS (Mediatech) for 15 min at 37°C in a humidified atmosphere of 5%CO2 and 95% air. After the enzymatic treatment, hippocampal tissue was rinsed twice: first in 0.1% soybean trypsin inhibitor (Worthington Biochemical) dissolved in Ca2+/Mg2+-containing Dulbecco's phosphate-buffered salt solution (Mediatech) and next in culture medium containing 10% fetal bovine serum (HyClone, Logan, UT). The tissue was next transferred to chilled Ca2+/Mg2+-free HBSS and triturated through siliconized fire-polished Pasteur pipettes. After trituration, undissociated tissue fragments were allowed to settle for 5 min, and the supernatant was transferred to a new tube and centrifuged at 200 × g for 1 min to form a cell pellet. The resulting supernatant was discarded, and the cells were gently resuspended in culture medium. Dissociated hippocampal cells were plated in UV-sterilized, 96-well flat-bottomed ELISA plates (MaxiSorp; Nalge Nunc, Naperville, IL) coated with poly-d-lysine (Sigma) and anti-BDNF capture antibody (BDNF Emax ImmunoAssay System; Promega, Madison, WI) (Balkowiec and Katz, 2000), after 30 min preplating in uncoated 35 mm Petri dishes. Hippocampal cultures were grown for 3 d in Neurobasal-A medium supplemented with B-27 serum-free supplement, 0.5 mml-glutamine, 1% penicillin–streptomycin–neomycin antibiotic mixture and 5 ng/ml human recombinant basic fibroblast growth factor (Invitrogen, Gaithersburg, MD).
BDNF immunoassay. BDNF protein was measured with a modified sandwich ELISA, termed ELISA in situ, as described previously (Balkowiec and Katz, 2000). Briefly, 96-well ELISA plates were (1) UV-sterilized for 15 min, (2) treated with poly-d-lysine (0.1 mg/ml; Sigma) for 30 min at room temperature, (3) rinsed with a sterile double-distilled water, and (4) coated with anti-BDNF monoclonal antibody (BDNF Emax ImmunoAssay System; Promega) at 4°C for 16.5 hr. Next, plates were washed and blocked, followed by two 1 hr incubations with culture medium to remove any residue of the ELISA washing solution. Then, the hippocampal neurons were prepared as described above, plated in anti-BDNF-coated wells, and grown for 3 d (see Results). The BDNF Emax ImmunoAssay System (Promega) was used according to the protocol of the manufacturer, except that the concentration of the anti-BDNF monoclonal and anti-human BDNF polyclonal antibody was 5 and 2 μg/ml, respectively, and the dilution of the anti-IgY-HRP antibody was 1:100. All reagents used before cell plating were sterilized with 0.2 μm Acrodisc syringe filters (Pall, Ann Arbor, MI). BDNF samples used to generate the standard curves were incubated in the same plate as the cells. At the end of the culture period, plates were extensively washed to remove all cells and cell debris, and the anti-human BDNF polyclonal antibody was applied, followed by subsequent steps according to the protocol of the manufacturer. Absorbance values were read at 450 nm in a plate reader (Vmax; Molecular Devices, Sunnyvale, CA). For control wells in which anti-BDNF monoclonal antibody was omitted, absorbance values were not significantly different from the absorbance of blank wells.
Electrical field stimulation of hippocampal neurons. After an initial 3 d incubation, two adjacent culture wells were connected to each other through a thin strip of 1% agarose gel permeated with culture medium and to the stimulator (MultiStim System; Digitimer, Hertfordshire, UK) through Ag/AgCl stimulating electrodes (Balkowiec and Katz, 2000) (modified from Brevet et al., 1976;McDonough et al., 1994). Two additional wells were also connected to each other by agarose bridges but were not connected to the stimulator and served as controls. The plate was put back to the incubator, and the neurons were stimulated for 30 min with biphasic rectangular pulses delivered at various patterns (see Results). In experiments comparing the effects of patterned electrical stimulation with potassium-induced continuous depolarization, KCl was added to three additional wells, to a final concentration of 50 mm, as described previously (Ghosh et al., 1994; Androutsellis-Theotokis et al., 1996;Goodman et al., 1996; Heymach et al., 1996; Griesbeck et al., 1999;Balkowiec and Katz, 2000).
Drugs used. Tetrodotoxin (TTX) (Sigma),d,l-2-amino-5-phosphonovaleric acid (APV) (Sigma), 1-aminoindan-1,5-dicarboxylic acid (AIDA) (Sigma),trans-(1S,3R)-1-amino-1,3-cyclopentane-dicarboxylic acid (trans-[1S,3R]-ACPD) (Sigma), ω-Conotoxin GVIA (Sigma), and caffeine (Sigma) were dissolved in PBS and used at final concentrations of 1.5, 100, 500, 100, 1, and 30 μm, respectively. 6-Cyano-7-nitroquinoxaline-2,3-(1H,4H)-dione (CNQX) (Sigma) was dissolved in 0.1 m NaOH and used at a final concentration of 20 μm. LY341495 (Tocris Cookson, Ellisville, MO) was dissolved in 15 mmNaOH and used at a final concentration of 100 μm. Nimodipine (Sigma) and ionomycin (Calbiochem, La Jolla, CA) were dissolved in methyl alcohol and used at final concentrations of 2 and 1 μm, respectively. Bay K-8644 (Calbiochem), dantrolene (Sigma), and thapsigargin (Sigma), dissolved in DMSO, were used at final concentrations of 5, 50, and 10 μm, respectively, and the final concentration of DMSO was 0.02%.
Immunocytochemistry. Cultures for immunocytochemical staining were fixed with 4% paraformaldehyde in 0.1m sodium phosphate buffer, pH 7.4, for 30 min at room temperature. Neuron-specific enolase (NSE) immunostaining was performed as described previously for protein gene product 9.5 (Brady et al., 1999), using rabbit polyclonal anti-NSE (1:2000 dilution; Polysciences, Warrington, PA). The number of neurons in each culture was evaluated by counting all NSE-immunoreactive cells per well. Experiments were repeated twice with four cultures per experimental group. Values were compared using ANOVA, followed by Duncan's multiple comparison procedure, and p < 0.05 was considered significant. Phosphorylated cAMP response element-binding protein (P-CREB) immunostaining was performed as described previously (Balkowiec and Katz, 2000). Control cultures, in which primary antibody was omitted, were completely devoid of staining.
Calculations and statistical analysis. BDNF levels were calculated from the standard curve prepared for each plate, using SOFTmax PRO version 3.0 software (Molecular Devices, Sunnyvale, CA). The standard curves were linear within the range used (0–500 pg/ml), and the quantities of BDNF in experimental samples were always within the linear range of the standard curve. Data are expressed as mean ± SE. Samples were compared using ANOVA, followed by Duncan's multiple comparison procedure, and p < 0.05 was considered significant.
RESULTS
The goal of the present study was to define the role of action potentials and voltage-activated calcium channels in activity-dependent BDNF release from hippocampal neurons. In view of the fact that BDNF can be released from hippocampal neurons by activation of glutamate receptors (GluR) (Ghosh et al., 1994; Griesbeck et al., 1999; Hartmann et al., 2001), we sought to examine release in the absence of potentially confounding trans-synaptic interactions. We therefore used 3-d-old cultures of newborn hippocampal neurons, taking advantage of the fact that synaptic connections between hippocampal neurons in culture do not form until >4 d in vitro (Bito et al., 1996), and functional synapses do not appear until 6–8 d in vitro (Deisseroth et al., 1996). Consistent with these observations, blockade of glutamatergic transmission did not affect electrical stimulation-evoked BDNF release in this study (for details, see below).
Short-term patterned electrical stimulation, but not short-term continuous KCl depolarization, significantly increases native BDNF release from hippocampal neurons
Initial studies sought to determine whether release of native BDNF from hippocampal neurons is regulated by patterned neuronal activity, as in sensory neurons (Balkowiec and Katz, 2000). We compared the effects of theta-burst electrical field stimulation, which mimics the typical firing mode of hippocampal pyramidal cells during learning (O'Keefe and Recce, 1993), and continuous depolarization by 50 mm KCl (Ghosh et al., 1994; Goodman et al., 1996; Griesbeck et al., 1999; Hoener, 2000) on BDNF release from newborn hippocampal neurons. BDNF levels, measured by ELISA in situ, were compared after 30 min of control conditions, theta-burst stimulation (TBS) (trains of 25 bursts, each burst of four pulses at 100 Hz, delivered with an interburst interval of 200 msec and an intertrain interval of 20 sec) (see Fig. 2A, TBS), or KCl-induced continuous depolarization. After theta-burst stimulation, there was a highly significant increase in BDNF release from hippocampal neurons (44.32 ± 3.08 pg/ml, n = 30 vs 3.18 ± 1.29 pg/ml in control, n = 48;p < 0.0001) (Fig. 1). In contrast, 50 mm KCl-induced continuous depolarization over the same time period did not result in a detectable increase in BDNF release (5.44 ± 1.87 pg/ml; n = 39; p = 0.3154) (Fig. 1). To determine whether, under our experimental conditions, continuous membrane depolarization activated hippocampal neurons, we performed immunostaining with an antibody against P-CREB, a marker of neuronal depolarization (Ghosh et al., 1994; Moore et al., 1996). After 30 min treatment with 50 mm KCl, there was a marked increase in P-CREB staining in the vast majority of cells (data not shown), indicating that this treatment was effective at activating neurons in our cultures. Together, these data indicate that patterned neuronal activity is significantly more effective than continuous depolarization at releasing native BDNF from hippocampal neurons.
Fig. 2.

High-frequency patterns of electrical stimulation that are known to induce LTP are significantly more effective at releasing native BDNF from hippocampal neurons than low-frequency stimulation. A, Schematic representation of the stimulation pattern applied to each group of cultures.B, Mean levels of BDNF released in sister cultures of newborn hippocampal neurons during 30 min of electrical field stimulation with 100 biphasic rectangular pulses of 4 msec, delivered at 5 Hz (n = 16), 10 Hz (n = 14), 25 Hz (n = 40), 100 Hz (n= 42), or TBS (n = 30), with interburst intervals, respectively, of 0, 10, 16, 19, and 15 sec, as shown inA. *p < 0.05; **p < 0.01; ***p < 0.001;n.s., not significant.
Fig. 1.

Short-term patterned electrical stimulation, but not short-term continuous KCl depolarization, significantly increases release of native BDNF from hippocampal neurons. Mean levels of BDNF measured in sister cultures of newborn hippocampal neurons after 3 d in vitro plus 30 min of the following: (1) control conditions (no stimulation; n = 48), (2) TBS (25 bursts of 4 pulses at 100 Hz, delivered at 5 Hz, every 20 sec;n = 30), or (3) continuous depolarization with 50 mm KCl (n = 39). ***p < 0.001; n.s., not significant.
High-frequency patterns of electrical stimulation that are known to induce hippocampal LTP are significantly more effective at releasing native BDNF from hippocampal neurons than low-frequency stimulation
The magnitude of native BDNF release from primary sensory neurons is regulated by the frequency and pattern of stimulation (Balkowiec and Katz, 2000; Lever et al., 2001). To determine whether similar mechanisms regulate activity-dependent secretion of BDNF from hippocampal neurons, we next examined the relationship between the pattern of stimulation and the magnitude of native BDNF release in hippocampal cultures. Because the magnitude of release of other peptides depends on the total number of pulses delivered (Whim and Lloyd, 1994), we designed five stimulation protocols in which the overall number of pulses, and consequently, average frequency, as well as the number of pulses in individual bursts, remained constant, whereas intraburst frequency and interburst interval were varied (Fig.2A): (1)continuous stimulation at 5 Hz, a frequency that corresponds to physiological levels of resting activity at hippocampal synapses (Bito et al., 1996); (2) bursts of 100 pulses at 10 Hz, delivered once every 20 sec, a frequency that does not produce significant changes of synaptic strength in hippocampal cultures (Deisseroth et al., 1996) and slices (Dudek and Bear, 1992); (3) bursts of 100 pulses at 25 Hz, delivered once every 20 sec, a “weak” LTP-inducing stimulus (Huber et al., 1998); (4) bursts of 100 pulses at 100 Hz, delivered once every 20 sec, a protocol that is commonly used to induce hippocampal LTP (Patterson et al., 1992;Dragunow et al., 1993; Korte et al., 1995, 1996; Figurov et al., 1996;Patterson et al., 1996; Kang et al., 1997); and (5) trains of 25 bursts, each burst of four pulses at 100 Hz, delivered with an interburst interval of 200 msec (at 5 Hz) and an intertrain interval of 20 sec, i.e., TBS, which mimics the typical firing pattern of hippocampal neurons during learning (O'Keefe and Recce, 1993) and is optimal for the induction of hippocampal LTP (Larson et al., 1986;Staubli and Lynch, 1987; Larson and Lynch, 1988; Staubli et al., 1999). The magnitude of BDNF release was compared among these stimulation protocols applied to newborn hippocampal neurons over a 30 min period.
Each stimulation protocol evoked detectable release of BDNF. However, 100 Hz tetanic stimulation and TBS resulted in significantly higher BDNF release compared with the other stimulation protocols (TBS, 44.32 ± 3.08 pg/ml, n = 30; 100 Hz, 39.27 ± 2.06 pg/ml, n = 42; 25 Hz, 25.13 ± 1.83 pg/ml,n = 40; 10 Hz, 11.76 ± 1.89 pg/ml,n = 14; 5 Hz, 2.36 ± 2.01 pg/ml,n = 16) (Fig. 2B). Interestingly, tetanic stimulation at 25 Hz, a protocol that has been shown to induce a lower magnitude of LTP compared with 100 Hz tetanic stimulation (Huber et al., 1998), resulted in an intermediate level of BDNF release that was significantly lower compared with BDNF release evoked by 100 Hz tetanus or TBS (Fig. 2B). These data indicate that there is a strong relationship between the pattern of stimulation and the magnitude of BDNF release from hippocampal neurons, such that stimulus protocols that are known to be most effective at inducing LTP are also most effective at evoking BDNF release.
To rule out the possibility that the increase in BDNF release detected in stimulated cultures was attributable to damage of cells by electrical activation, we compared cell survival between control and stimulated cultures, using the same stimulus protocols that were used for the analysis of BDNF release. Twenty-four hours after stimulation, there were no significant differences in the number of cells between control and stimulated cultures for any stimulation protocol tested (per well: 5 Hz, control, 8649 ± 483.98, stimulated, 10,287 ± 1163.21, n = 8, p = 0.2146; 25 Hz, control, 8343 ± 918.90, stimulated, 9432 ± 558.87,n = 8, p = 0.3285; 100 Hz, control, 11,241 ± 1101.61, stimulated, 10,026 ± 715.26,n = 8, p = 0.3706; TBS, control, 11,808 ± 904.36, stimulated, 11,700 ± 1043.91,n = 8, p = 0.9388).
Electrical stimulation-evoked release of native BDNF requires activation of TTX-sensitive sodium channels and can occur in the absence of glutamate receptor activation
We next examined whether native BDNF release from hippocampal neurons evoked by patterned electrical stimulation requires activation of voltage-gated sodium channels and, consequently, generation of action potentials. Regardless of the stimulation protocol used, evoked release of BDNF was abolished by treatment of cultures with 1.5 μm TTX, an inhibitor of voltage-dependent sodium channels, before stimulation (Fig. 3), indicating that action potentials are required for this release.
Fig. 3.

Activity-dependent release of native BDNF depends on action potentials. Mean levels of BDNF released in sister cultures of newborn hippocampal neurons during 30 min of electrical field stimulation [100 biphasic rectangular pulses of 4 msec, delivered at 25 Hz (n = 12), 100 Hz (n = 10), and TBS (n = 4), once every 20 sec] in the absence (black bars) or presence (gray bars) of 1.5 μm TTX, an inhibitor of voltage-dependent sodium channels. **p < 0.01; ***p < 0.001.
Previous studies showed that BDNF release from hippocampal neurons can be evoked by stimulation with glutamate receptor agonists (Ghosh et al., 1994; Griesbeck et al., 1999) and by high-frequency activation of glutamatergic synapses (Hartmann et al., 2001). Because glutamate is the major excitatory neurotransmitter synthesized by hippocampal neurons and can be released in response to electrical stimulation, we tested whether activation of hippocampal neurons by glutamate played a role in BDNF release evoked by patterned electrical stimulation in our experiments. Pretreatment of hippocampal cultures with 20 μm CNQX, a non-NMDA receptor antagonist, and 100 μm APV, an NMDA receptor antagonist, had no significant effect on the magnitude of BDNF release evoked by 25 Hz, 100 Hz, or TBS patterns (Fig. 4A).
Fig. 4.

Release of native BDNF evoked by patterned electrical stimulation does not require glutamatergic synaptic activity. A, Mean levels of BDNF released in sister cultures of newborn hippocampal neurons during 30 min of electrical field stimulation [100 biphasic rectangular pulses of 4 msec, delivered at 25 Hz (n = 8), 100 Hz (n = 8), and TBS (n = 10), once every 20 sec] in the absence (black bars) or presence of CNQX (20 μm) and APV (100 μm), antagonists of, respectively, non-NMDA and NMDA glutamate receptors (CNQX-APV; gray bars). B, Mean levels of BDNF released in sister cultures during 30 min of either exposure to 100 μmtrans-[1S,3R]-ACPD, an agonist of mGluR I and II (t-ACPD; n= 12), or electrical field stimulation at the theta-burst pattern (TBS; n = 12) in the absence (black bars) or presence of metabotropic glutamate receptor antagonists AIDA (500 μm; gray bars) and LY341495 (100 μm; white bars); ***p < 0.001; n.s., not significant.
Canossa et al. (2001) demonstrated that activation of metabotropic glutamate receptors (mGluR) leads to secretion of native BDNF from adult hippocampal slices. Therefore, we next examined the role of mGluR in BDNF release. Thirty minute exposure to 100 μmtrans-[1S,3R]-ACPD, an agonist of mGluR I and II, evoked a significant increase in native BDNF release that was blocked by 100 μm LY341495, used as a general mGluR antagonist (Leslie et al., 2001), and by 500 μm AIDA, a specific mGluR I inhibitor (Canossa et al., 2001) (Fig. 4B). However, the magnitude of TBS-evoked BDNF release was not affected by pretreatment with these same mGluR antagonists (Fig. 4B). Together, these data indicate that glutamatergic receptor activation did not contribute to BDNF release evoked by patterned electrical stimulation in our study.
Release of native BDNF evoked by patterned electrical stimulation requires calcium influx through N-type voltage-activated calcium channels
To begin characterizing cellular mechanisms underlying activity-dependent release of native BDNF from hippocampal neurons, we next examined the role of extracellular calcium and specific voltage-activated calcium channels. Our initial experiments showed that removal of calcium from the extracellular solution completely abolishes release of native BDNF from hippocampal neurons evoked by 25 Hz, 100 Hz, and TBS stimulation (Fig.5A), indicating that extracellular calcium is required for this release.
Fig. 5.
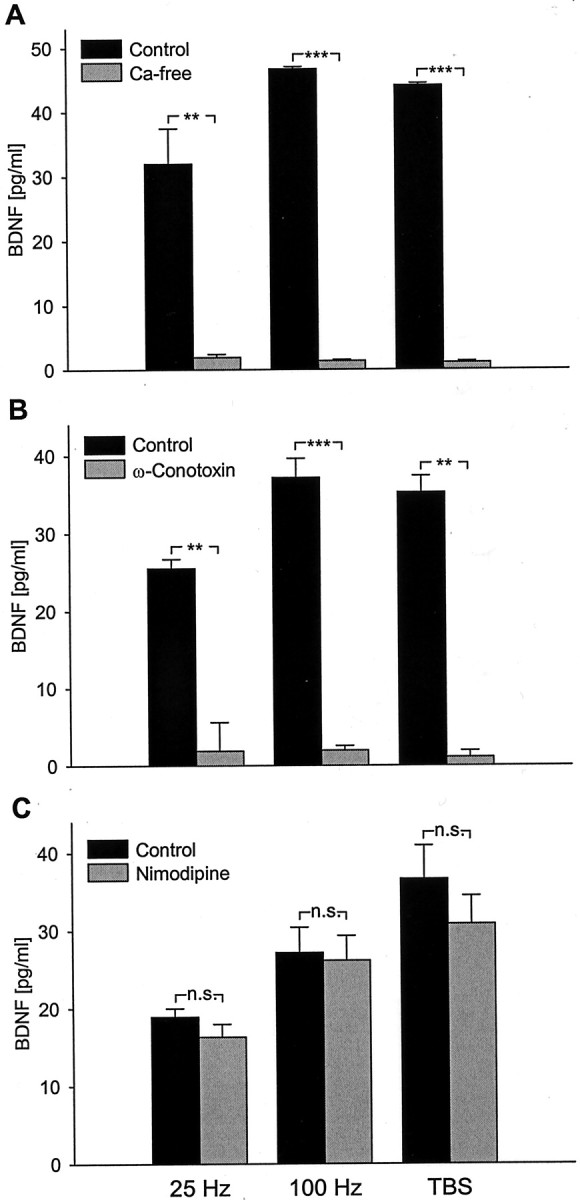
Release of native BDNF evoked by patterned electrical stimulation requires calcium influx through N-type voltage-activated calcium channels. Mean levels of BDNF released in sister cultures of newborn hippocampal neurons during 30 min of electrical field stimulation (100 biphasic rectangular pulses of 4 msec, delivered at 25 Hz, 100 Hz, and TBS, once every 20 sec):A, in the presence (Control; black bars) or absence (Ca-free; gray bars) of extracellular calcium (25 Hz, n = 6; 100 Hz, n = 4; TBS, n = 4);B, in the absence (Control; black bars) or presence of 1 μm ω-Conotoxin GVIA, an N-type channel antagonist (ω-Conotoxin; gray bars) (25 Hz, n = 4; 100 Hz,n = 10; TBS, n = 4);C, in the absence (Control; black bars) or presence of 2 μm nimodipine, an L-type channel antagonist (Nimodipine; gray bars) (25 Hz, n = 4; 100 Hz,n = 4; TBS, n = 6). **p < 0.01; ***p < 0.001;n.s., not significant.
Previous studies have shown that multiple calcium channels coexist in nerve terminals and control release of neurotransmitters and peptide neuromodulators from central neurons (Dunlap et al., 1995). Moreover, different voltage-gated calcium channels can be coupled to transmitter release during low- versus high-frequency electrical stimulation (Wright and Angus, 1996). Therefore, we next sought to determine the requirement for specific voltage-activated calcium channels in BDNF release evoked by different patterns of stimulation. Pretreatment of hippocampal cultures with 1 μm ω-Conotoxin GVIA, an N-type calcium channel antagonist, completely abolished BDNF release evoked by 25 Hz, 100 Hz, and TBS stimulation (Fig. 5B). On the other hand, treatment with an L-type channel antagonist, nimodipine (2 μm), had no significant effect on BDNF release evoked by any of the stimulation protocols tested (Fig.5C). These data indicate that native BDNF release from hippocampal neurons evoked by patterned electrical stimulation requires calcium entry specifically through N-type voltage-activated calcium channels.
Calcium entry through L-type channels has been shown to play an important role in activity-dependent BDNF expression in hippocampal neurons (Zafra et al., 1990, 1991; Ghosh et al., 1994; Tabuchi et al., 2000). Therefore, to determine whether calcium entry through L-type channels is capable of regulating release of native BDNF from hippocampal neurons, we exposed hippocampal cultures to 5 μm Bay K-8644, a selective agonist of L-type channels (Nowycky et al., 1985; Brosenitsch et al., 1998), in the presence of 15 mm KCl, a protocol that optimizes the efficacy of Bay K-8644 by slightly depolarizing the cells (Brosenitsch et al., 1998). Thirty minute exposure to Bay K-8644 and 15 mm KCl did not evoke detectable BDNF release (control, 2.48 ± 1.26 pg/ml; Bay K-8644, 2.50 ± 0.96 pg/ml; n = 8;p = 0.9896), whereas 3 d exposure led to a highly significant increase in extracellular BDNF concentration (control, 6.27 ± 1.66 pg/ml; Bay K-8644, 44.89 ± 10.76 pg/ml;n = 8; p = 0.0239). Exposure to 15 mm KCl alone, either for 30 min or 3 d, did not significantly affect BDNF release (data not shown). Considering the role of L-type channels in regulation of BDNF expression, this result indicates that calcium entry through L-type channels does not influence native BDNF release from hippocampal neurons in response to short-term stimulation but may increase the size of the releasable BDNF pool by increasing expression during prolonged chronic depolarization. In addition, exposure of hippocampal cultures to 1 μm ionomycin, a calcium ionophore, for 30 min had no significant effect on BDNF release (control, 4.34 ± 2.79 pg/ml; ionomycin, 5.96 ± 3.32 pg/ml; n = 6;p = 0.7219), further demonstrating that increasing calcium influx per se is not sufficient to increase native BDNF release in response to short-term stimulation.
Electrical stimulation-evoked release of native BDNF from hippocampal neurons involves calcium mobilization from intracellular stores by a CICR mechanism
Previous studies have indicated that regulated secretion of neurotrophins requires calcium release from intracellular calcium stores, including caffeine–ryanodine-sensitive stores (Blöchl and Thoenen, 1995; Griesbeck et al., 1999; Canossa et al., 2001). To establish whether calcium release from caffeine–ryanodine-sensitive stores is capable of inducing release of native BDNF from hippocampal neurons, we initially examined the effects of caffeine alone. Thirty minute treatment of hippocampal cultures with 30 μmcaffeine, an agonist of ryanodine receptors, resulted in a significant increase in BDNF release that was blocked by 10 min pretreatment with 50 μm dantrolene, a ryanodine receptor antagonist (Blöchl and Thoenen, 1995) and 10 μm thapsigargin (Fig. 6A). These data show that activation of ryanodine-sensitive calcium stores is sufficient to evoke native BDNF release from hippocampal neurons. Because the caffeine–ryanodine-sensitive store is involved in CICR (Kuba, 1994), we next sought to examine the possibility that native BDNF release evoked by patterned electrical stimulation involves CICR. One of several criteria to establish the involvement of CICR is the presence of a caffeine-sensitive intracellular calcium store that is modulated by inhibitors of sarcoplasmic–endoplasmic reticulum Ca2+-ATPase, such as thapsigargin (Treiman et al., 1998). Therefore, we next examined the effect of blocking CICR with thapsigargin and dantrolene on release of native BDNF from hippocampal neurons in response to patterned electrical stimulation. Pretreatment with 10 μm thapsigargin, which by itself resulted in a significant release of BDNF (10.52 ± 2.45 pg/ml; n = 8), and 50 μmdantrolene significantly inhibited BDNF release evoked by TBS (Fig.6B). These data demonstrate that release of native BDNF from hippocampal neurons in response to electrical stimulation requires intact intracellular calcium stores, as well as calcium influx through N-type channels.
Fig. 6.

Release of native BDNF in response to patterned electrical stimulation requires calcium mobilization from ryanodine-sensitive stores. Mean levels of BDNF released in sister cultures of newborn hippocampal neurons during 30 min exposure to either 30 μm caffeine or electrical field stimulation at the theta-burst pattern (TBS) in the absence (black bars) or presence of dantrolene (50 μm), a selective antagonist of ryanodine receptor channels. Cultures were pretreated with thapsigargin (10 μm), which selectively inhibits the endoplasmic reticulum Ca2+-ATPase (gray bars);n = 8. ***p < 0.001.
DISCUSSION
The present study demonstrates for the first time the properties and cellular mechanisms of native BDNF release from hippocampal neurons in response to physiologically relevant patterns of electrical stimulation, including those known to induce LTP in the intact hippocampus. Our study shows that frequency-dependent release of native BDNF can be triggered directly by activation of voltage-gated sodium channels, in the absence of glutamate receptor activation. Moreover, this release requires both calcium influx through N-type channels and calcium mobilization from intracellular stores.
The most extensive studies on the role of BDNF in synaptic plasticity have focused on hippocampal LTP. Induction of LTP is a cooperative process that requires simultaneous activity of a large number of axons and a threshold of stimulus frequency and intensity (Bennett, 2000). Several studies indicate that, in fact, a critical level of BDNF is needed for LTP induction. For example, the postnatal increase in the ability of developing hippocampal synapses to undergo LTP (Jackson et al., 1993) is paralleled by an increase in the expression of BDNF and its receptor TrkB in the hippocampus (Maisonpierre et al., 1990). In addition, BDNF−/− and BDNF+/− animals show the same degree of impairment in LTP (Korte et al., 1995; Patterson et al., 1996). The present study demonstrates that TBS, which mimics the typical firing pattern of hippocampal neurons during learning, is highly effective at releasing BDNF from hippocampal neurons, whereas 5 Hz stimulation, which corresponds to resting activity at hippocampal synapses, is not. Our data, therefore, raise the possibility that the pattern dependence of hippocampal LTP is related to the fact that the magnitude of BDNF release from hippocampal neurons depends on the pattern and frequency of stimulation.
Several studies have indicated that, although 100 Hz tetanus and TBS are both effective at inducing hippocampal LTP, only TBS-induced LTP requires BDNF (Kang et al., 1997; Chen et al., 1999; Patterson et al., 2001) (but see Korte et al., 1995; Figurov et al., 1996). It has been suggested that this difference could be attributable to differential release of BDNF by TBS versus 100 Hz tetanus (Kang et al., 1997; Chen et al., 1999). Our data do not support this possibility, because 100 Hz tetanus and TBS were equally effective at releasing native BDNF in our model. Thus, although the frequency dependence of BDNF release could contribute to the relative effectiveness of TBS versus low-frequency stimulation at inducing LTP, other factors likely influence the relative importance of BDNF signaling in TBS-induced versus 100 Hz tetanus-induced LTP.
The role of calcium influx in activity-dependent release of neurotrophins has been a subject of considerable controversy. Most previous studies, in which chronic depolarizing agents were used to induce BDNF release, suggested a mechanism independent of extracellular calcium (Griesbeck et al., 1999) (but see Goodman et al., 1996). However, it has been demonstrated recently that extracellular calcium is required for BDNF-GFP release evoked by both KCl depolarization and high-frequency electrical stimulation of hippocampal neurons (Hartmann et al., 2001). Our results demonstrate that activity-dependent BDNF secretion requires both calcium influx, specifically through N-type channels, and calcium mobilization from intracellular stores. This dual requirement for extracellular and intracellular calcium strongly implicates a role for calcium-induced calcium release in activity-dependent secretion of BDNF.
It has been demonstrated previously that depolarization-induced calcium entry in hippocampal neurons can activate calcium release from intracellular stores (Sandler and Barbara, 1999). This observation is consistent with recent evidence that activity-dependent regulation of intracellular calcium levels is tightly controlled by interactions between calcium influx and activation of intracellular calcium stores. For example, decreasing extracellular calcium levels can prime activity-dependent calcium release from intracellular stores (Nohmi et al., 2000). Therefore, because high levels of neuronal activity can decrease the extracellular calcium concentration (Heinemann and Pumain, 1980; Borst and Sakmann, 1999; Stanley, 2000), it seems plausible that BDNF release from hippocampal neurons is potentiated during high-frequency firing, as occurs during hippocampal learning (O'Keefe and Recce, 1993), by increasing calcium mobilization from intracellular stores.
Our studies were conducted with 3-d-old cultures of newborn hippocampal neurons that lack excitatory glutamatergic synapses (Bito et al., 1996), as confirmed by our experiments with glutamate receptor antagonists. This model, therefore, allowed us to characterize in detail the requirement for calcium influx in BDNF release by using selective calcium channel antagonists without potentially confounding effects on glutamate release. Our results demonstrate that activity-dependent release of native BDNF from newborn hippocampal neurons requires calcium influx specifically through N-type channels, regardless of the pattern of stimulation. A similar requirement for N-type channels in release of neurotrophin-3 from retinotectal axon terminals has been reported recently (Wang et al., 2002). Our findings are supported by observations that blockade of N-type, but not L-type, channels also leads to a significant reduction of synaptic vesicle recycling in newborn hippocampal neurons (Pravettoni et al., 2000). Moreover, the fact that N-type calcium channels are located almost exclusively on axons of newborn hippocampal neurons grown for 3 d in culture (Pravettoni et al., 2000) suggests that our data primarily reflect BDNF release from axonal sites. Although it is a subject of debate whether BDNF is released presynaptically or postsynaptically or both (Hartmann et al., 2001; Kohara et al., 2001), findings from several laboratories indicate that BDNF can be released from axons (Altar et al., 1997; Conner et al., 1997; Fawcett et al., 1998; Fawcett et al., 2000; Kohara et al., 2001; Kojima et al., 2001). Our data are consistent with these findings but do not rule out the possibility that, in more mature neurons, release can occur from other sites as well.
Although there are limitations in extrapolating data from dissociate cultures to more intact systems, we believe that our results may help shed light on the role of BDNF in activity-dependent synaptic plasticity. For example, previous studies have indicated that BDNF, by promoting synaptic vesicle docking, enhances transmitter release in response to tetanic stimulation (Gottschalk et al., 1998; Pozzo-Miller et al., 1999). Moreover, highly active synapses are more susceptible to the potentiating effects of BDNF on presynaptic transmitter release (Gottschalk et al., 1998). Thus, based on the present findings, we propose that highly active synapses, in contrast to inactive or weakly active synapses, are more likely to meet the threshold conditions both for BDNF concentration and the potentiating effects of activity on BDNF-induced transmitter release and, therefore, induce LTP. This is essentially the model proposed by Gottschalk et al. (1998) based on their analysis of BDNF-stimulated glutamate release in hippocampal neurons. A similar mechanism could underlie the role of BDNF in activity-dependent synaptic plasticity leading to morphological changes in neuronal circuits, such as refinement of neuronal connections in developing visual cortex (Goodman and Shatz, 1993; Katz and Shatz, 1996; Schinder and Poo, 2000). Several studies indicate that BDNF can also act postsynaptically in hippocampal neurons by rapidly enhancing basal excitatory synaptic responses, including increases in postsynaptic NMDA receptor activity (Lessmann et al., 1994; Levine et al., 1995). Thus, one implication of our findings is that the increase in BDNF release with increasing frequency of presynaptic neuronal activity could partially compensate for the effects of synaptic fatigue by enhancing postsynaptic NMDA responses.
The duration of patterned electrical stimulation used in our study was significantly longer than standard LTP-inducing protocols because of the fact that the levels of native BDNF released during shorter stimulation periods were below the detectability limits of the ELISA assay (data not shown). This observation suggests that, in our model, release of native BDNF from hippocampal neurons occurs over a prolonged period of stimulation. Very recently, Gärtner and Staiger (2002)demonstrated that release of AdVBDNF from hippocampal neurons peaks within the first minute of stimulation and decays rapidly thereafter. On the other hand, Hartmann et al. (2001) demonstrated that tetanic stimulation-evoked release of BDNF-GFP from hippocampal neurons is characterized by a slow onset, in the range of minutes. These apparent discrepancies in the time course of BDNF secretion could reflect differences in the sorting and release of native BDNF, AdVBDNF, and BDNF-GFP (cf. Fawcett et al., 1997; Mowla et al., 1999) or other differences among the models used in these three studies.
Similar to primary sensory neurons, patterned electrical stimulation of hippocampal neurons is strikingly more effective than KCl depolarization at releasing native BDNF (present findings; Balkowiec and Katz, 2000). This result is in agreement with Griesbeck et al. (1999) who were unable to detect native BDNF release from primary cultures of hippocampal neurons briefly exposed to elevated KCl. The same treatment, however, led to a significant increase in release of AdVBDNF (Griesbeck et al., 1999). This result, together with our current findings, raises the possibility that, after adenovirus-mediated overexpression, at least some BDNF is shunted to a pool that is sensitive to KCl depolarization, whereas native BDNF is not.
In conclusion, the present study shows that activity-dependent secretion of native BDNF from hippocampal neurons is highly frequency dependent, indicating that BDNF release can encode temporal features of neuronal activity. Moreover, our data provide support for a novel mechanism of activity-dependent BDNF release involving calcium influx specifically through N-type channels, as well as calcium-induced calcium release from intracellular stores.
Footnotes
This work was supported by United States Public Health Service grants (National Heart, Lung, and Blood Institute) (D.M.K.).
Correspondence should be addressed to Dr. David M. Katz, Department of Neurosciences, Case Western Reserve University School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106. E-mail:dmk4@po.cwru.edu.
A. Balkowiec's present address: Department of Biological Structure and Function, Oregon Health and Science University, 611 Southwest Campus Drive, Portland, OR 97239.
REFERENCES
- 1.Akaneya Y, Tsumoto T, Kinoshita S, Hatanaka H. Brain-derived neurotrophic factor enhances long-term potentiation in rat visual cortex. J Neurosci. 1997;17:6707–6716. doi: 10.1523/JNEUROSCI.17-17-06707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- 3.Androutsellis-Theotokis A, McCormack WJ, Bradford HF, Stern GM, Pliego-Rivero FB. The depolarisation-induced release of [125I]BDNF from brain tissue. Brain Res. 1996;743:40–48. doi: 10.1016/s0006-8993(96)00981-x. [DOI] [PubMed] [Google Scholar]
- 4.Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balkowiec A, Katz DM. Cellular mechanisms underlying activity-dependent release of endogenous BDNF from hippocampal neurons. Soc Neurosci Abstr. 2001;27:801.14. [Google Scholar]
- 6.Bennett MR. The concept of long term potentiation of transmission at synapses. Prog Neurobiol. 2000;60:109–137. doi: 10.1016/s0301-0082(99)00006-4. [DOI] [PubMed] [Google Scholar]
- 7.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 8.Black IB. Trophic regulation of synaptic plasticity. J Neurobiol. 1999;41:108–118. [PubMed] [Google Scholar]
- 9.Blöchl A, Thoenen H. Characterization of nerve growth factor (NGF) release from hippocampal neurons: evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur J Neurosci. 1995;7:1220–1228. doi: 10.1111/j.1460-9568.1995.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 10.Borst JGG, Sakmann B. Depletion of calcium in the synaptic cleft of a calyx-type synapse in the rat brainstem. J Physiol (Lond) 1999;521:123–133. doi: 10.1111/j.1469-7793.1999.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brady R, Zaidi SIA, Mayer C, Katz DM. BDNF is a target-derived survival factor for arterial baroreceptor and chemoafferent primary sensory neurons. J Neurosci. 1999;19:2131–2142. doi: 10.1523/JNEUROSCI.19-06-02131.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brevet A, Pinto E, Peacock J, Stockdale FE. Myosin synthesis increased by electrical stimulation of skeletal muscle cell cultures. Science. 1976;193:1152–1154. doi: 10.1126/science.959833. [DOI] [PubMed] [Google Scholar]
- 13.Brosenitsch TA, Salgado-Commissariat D, Kunze DL, Katz DM. A role for L-type calcium channels in developmental regulation of transmitter phenotype in primary sensory neurons. J Neurosci. 1998;18:1047–1055. doi: 10.1523/JNEUROSCI.18-03-01047.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 15.Canossa M, Gärtner A, Campana G, Inagaki N, Thoenen H. Regulated secretion of neurotrophins by metabotropic glutamate group I (mGluRI) and Trk receptor activation is mediated via phospholipase C signalling pathways. EMBO J. 2001;20:1640–1650. doi: 10.1093/emboj/20.7.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci. 1999;19:7983–7990. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 19.Dragunow M, Beilharz E, Mason B, Lawlor P, Abraham W, Gluckman P. Brain-derived neurotrophic factor expression after long-term potentiation. Neurosci Lett. 1993;160:232–236. doi: 10.1016/0304-3940(93)90420-p. [DOI] [PubMed] [Google Scholar]
- 20.Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 22.Fawcett JP, Aloyz R, McLean JH, Pareek S, Miller FD, McPherson PS, Murphy RA. Detection of brain-derived neurotrophic factor in a vesicular fraction of brain synaptosomes. J Biol Chem. 1997;272:8837–8840. doi: 10.1074/jbc.272.14.8837. [DOI] [PubMed] [Google Scholar]
- 23.Fawcett JP, Bamji SX, Causing CG, Aloyz R, Ase AR, Reader TA, McLean JH, Miller FD. Functional evidence that BDNF is an anterograde neuronal trophic factor in the CNS. J Neurosci. 1998;18:2808–2821. doi: 10.1523/JNEUROSCI.18-08-02808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fawcett JP, Alonso-Vanegas MA, Morris SJ, Miller FD, Sadikot AF, Murphy RA. Evidence that brain-derived neurotrophic factor from presynaptic nerve terminals regulates the phenotype of calbindin-containing neurons in the lateral septum. J Neurosci. 2000;20:274–282. doi: 10.1523/JNEUROSCI.20-01-00274.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 26.Gärtner A, Staiger V. Neurotrophin secretion from hippocampal neurons evoked by long-term-potentiation-inducing electrical stimulation patterns. Proc Natl Acad Sci USA. 2002;99:6386–6391. doi: 10.1073/pnas.092129699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 28.Goodman CS, Shatz CJ. Developmental mechanisms that generate precise patterns of neuronal connectivity. Cell [Suppl] 1993;72:77–98. doi: 10.1016/s0092-8674(05)80030-3. [DOI] [PubMed] [Google Scholar]
- 29.Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- 30.Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griesbeck O, Canossa M, Campana G, Gärtner A, Hoener MC, Nawa H, Kolbeck R, Thoenen H. Are there differences between the secretion characteristics of NGF and BDNF? Implications for the modulatory role of neurotrophins in activity-dependent neuronal plasticity. Microsc Res Tech. 1999;45:262–275. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<262::AID-JEMT10>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 32.Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinemann U, Pumain R. Extracellular calcium activity changes in cat sensorimotor cortex induced by iontophoretic application of amino acids. Exp Brain Res. 1980;40:247–250. doi: 10.1007/BF00237788. [DOI] [PubMed] [Google Scholar]
- 34.Heymach JVJ, Krüttgen A, Suter U, Shooter EM. The regulated secretion and vectorial targeting of neurotrophins in neuroendocrine and epithelial cells. J Biol Chem. 1996;271:25430–25437. doi: 10.1074/jbc.271.41.25430. [DOI] [PubMed] [Google Scholar]
- 35.Hoener MC. Role played by sodium in activity-dependent secretion of neurotrophins—revisited. Eur J Neurosci. 2000;12:3096–3106. doi: 10.1046/j.1460-9568.2000.00190.x. [DOI] [PubMed] [Google Scholar]
- 36.Huber KM, Sawtell NB, Bear MF. Brain-derived neurotrophic factor alters the synaptic modification threshold in visual cortex. Neuropharmacology. 1998;37:571–579. doi: 10.1016/s0028-3908(98)00050-1. [DOI] [PubMed] [Google Scholar]
- 37.Jackson PS, Suppes T, Harris KM. Stereotypical changes in the pattern and duration of long-term potentiation expressed at postnatal days 11 and 15 in the rat hippocampus. J Neurophysiol. 1993;70:1412–1419. doi: 10.1152/jn.1993.70.4.1412. [DOI] [PubMed] [Google Scholar]
- 38.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 39.Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- 40.Katz DM, Balkowiec A. Frequency-dependent release of endogenous BDNF from hippocampal neurons in culture. Soc Neurosci Abstr. 2001;27:801.15. [Google Scholar]
- 41.Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- 42.Kohara K, Kitamura A, Morishima M, Tsumoto T. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science. 2001;291:2419–2423. doi: 10.1126/science.1057415. [DOI] [PubMed] [Google Scholar]
- 43.Kojima M, Takei N, Numakawa T, Ishikawa Y, Suzuki S, Matsumoto T, Katoh-Semba R, Nawa H, Hatanaka H. Biological characterization and optical imaging of brain-derived neurotrophic factor-green fluorescent protein suggest an activity-dependent local release of brain-derived neurotrophic factor in neurites of cultured hippocampal neurons. J Neurosci Res. 2001;64:1–10. doi: 10.1002/jnr.1080. [DOI] [PubMed] [Google Scholar]
- 44.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci USA. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuba K. Ca2+-induced Ca2+ release in neurones. Jpn J Physiol. 1994;44:613–650. doi: 10.2170/jjphysiol.44.613. [DOI] [PubMed] [Google Scholar]
- 47.Larson J, Lynch G. Role of N-methyl-d-aspartate receptors in the induction of synaptic potentiation by burst stimulation patterned after the hippocampal theta-rhythm. Brain Res. 1988;441:111–118. doi: 10.1016/0006-8993(88)91388-1. [DOI] [PubMed] [Google Scholar]
- 48.Larson J, Wong D, Lynch G. Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long-term potentiation. Brain Res. 1986;368:347–350. doi: 10.1016/0006-8993(86)90579-2. [DOI] [PubMed] [Google Scholar]
- 49. Leslie KR, Nelson SB, Turrigiano GG. Postsynaptic depolarization scales quantal amplitude in cortical pyramidal neurons. J Neurosci 21 2001. RC170(1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 51.Lever IJ, Bradbury EJ, Cunningham JR, Adelson DW, Jones MG, McMahon SB, Marvizon JC, Malcangio M. Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J Neurosci. 2001;21:4469–4477. doi: 10.1523/JNEUROSCI.21-12-04469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu B, Gottschalk W. Modulation of hippocampal synaptic transmission and plasticity by neurotrophins. Prog Brain Res. 2000;128:231–241. doi: 10.1016/S0079-6123(00)28020-5. [DOI] [PubMed] [Google Scholar]
- 54.Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 55.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 56.McDonough PM, Stella SL, Glembotski CC. Involvement of cytoplasmic calcium and protein kinases in the regulation of atrial natriuretic factor secretion by contraction rate and endothelin. J Biol Chem. 1994;269:9466–9472. [PubMed] [Google Scholar]
- 57.Moore AN, Waxham MN, Dash PK. Neuronal activity increases the phosphorylation of the transcription factor cAMP response element-binding protein (CREB) in rat hippocampus and cortex. J Biol Chem. 1996;271:14214–14220. doi: 10.1074/jbc.271.24.14214. [DOI] [PubMed] [Google Scholar]
- 58.Mowla SJ, Pareek S, Farhadi HF, Petrecca K, Fawcett JP, Seidah NG, Morris SJ, Sossin WS, Murphy RA. Differential sorting of nerve growth factor and brain-derived neurotrophic factor in hippocampal neurons. J Neurosci. 1999;19:2069–2080. doi: 10.1523/JNEUROSCI.19-06-02069.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nohmi M, Hua SY, Liu C, Kuba K. Ryanodine- and thapsigargin-insensitive Ca2+-induced Ca2+ release is primed by lowering external Ca2+ in rabbit autonomic neurons. Pflügers Arch. 2000;440:588–599. doi: 10.1007/s004240000326. [DOI] [PubMed] [Google Scholar]
- 60.Nowycky MC, Fox AP, Tsien RW. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature. 1985;316:440–443. doi: 10.1038/316440a0. [DOI] [PubMed] [Google Scholar]
- 61.O'Keefe J, Recce ML. Phase relationship between hippocampal place units and the EEG theta rhythm. Hippocampus. 1993;3:317–330. doi: 10.1002/hipo.450030307. [DOI] [PubMed] [Google Scholar]
- 62.Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- 63.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 64.Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- 65.Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- 66.Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knock-out mice. J Neurosci. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pravettoni E, Bacci A, Coco S, Forbicini P, Matteoli M, Verderio C. Different localizations and functions of L-type and N-type calcium channels during development of hippocampal neurons. Dev Biol. 2000;227:581–594. doi: 10.1006/dbio.2000.9872. [DOI] [PubMed] [Google Scholar]
- 68.Sandler VM, Barbara JG. Calcium-induced calcium release contributes to action potential-evoked calcium transients in hippocampal CA1 pyramidal neurons. J Neurosci. 1999;19:4325–4336. doi: 10.1523/JNEUROSCI.19-11-04325.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23:639–645. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- 70.Stanley EF. Presynaptic calcium channels and the depletion of synaptic cleft calcium ions. J Neurophysiol. 2000;83:477–482. doi: 10.1152/jn.2000.83.1.477. [DOI] [PubMed] [Google Scholar]
- 71.Staubli U, Lynch G. Stable hippocampal long-term potentiation elicited by “theta” pattern stimulation. Brain Res. 1987;435:227–234. doi: 10.1016/0006-8993(87)91605-2. [DOI] [PubMed] [Google Scholar]
- 72.Staubli U, Scafidi J, Chun D. GABAB receptor antagonism: facilitatory effects on memory parallel those on LTP induced by TBS but not HFS. J Neurosci. 1999;19:4609–4615. doi: 10.1523/JNEUROSCI.19-11-04609.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tabuchi A, Nakaoka R, Amano K, Yukimine M, Andoh T, Kuraishi Y, Tsuda M. Differential activation of brain-derived neurotrophic factor gene promoters I and III by Ca2+ signals evoked via L-type voltage-dependent and N-methyl-d-aspartate receptor Ca2+ channels. J Biol Chem. 2000;275:17269–17275. doi: 10.1074/jbc.M909538199. [DOI] [PubMed] [Google Scholar]
- 74.Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends Pharmacol Sci. 1998;19:131–135. doi: 10.1016/s0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]
- 75.Wang X, Butowt R, Vasko MR, von Bartheld CS. Mechanisms of the release of anterogradely transported neurotrophin-3 from axon terminals. J Neurosci. 2002;22:931–945. doi: 10.1523/JNEUROSCI.22-03-00931.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whim MD, Lloyd PE. Differential regulation of the release of the same peptide transmitters from individual identified motor neurons in culture. J Neurosci. 1994;14:4244–4251. doi: 10.1523/JNEUROSCI.14-07-04244.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wright CE, Angus JA. Effects of N-, P- and Q-type neuronal calcium channel antagonists on mammalian peripheral neurotransmission. Br J Pharmacol. 1996;119:49–56. doi: 10.1111/j.1476-5381.1996.tb15676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D. Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 1990;9:3545–3550. doi: 10.1002/j.1460-2075.1990.tb07564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zafra F, Castrén E, Thoenen H, Lindholm D. Interplay between glutamate and gamma-aminobutyric acid transmitter systems in the physiological regulation of brain-derived neurotrophic factor and nerve growth factor synthesis in hippocampal neurons. Proc Natl Acad Sci USA. 1991;88:10037–10041. doi: 10.1073/pnas.88.22.10037. [DOI] [PMC free article] [PubMed] [Google Scholar]