

Abstract
Endogenous cannabinoids (endocannabinoids) are endogenous compounds that resemble the active ingredient of marijuana and activate the cannabinoid receptor in the brain. They mediate retrograde signaling from principal cells to both inhibitory [“depolarization-induced suppression of inhibition” (DSI)] and excitatory (“depolarization-induced suppression of excitation”) afferent fibers. Transient endocannabinoid release is triggered by voltage-dependent Ca2+ influx and is upregulated by group I metabotropic glutamate receptor activation. Here we show that muscarinic acetylcholine receptor (mAChR) activation also enhances transient endocannabinoid release (DSI) and induces persistent release. Inhibitory synapses in the rat hippocampal CA1 region of acute slices were studied using whole-cell patch-clamp techniques. We found that low concentrations (0.2–0.5 μm) of carbachol (CCh) enhanced DSI without affecting basal evoked IPSCs (eIPSCs) by activating mAChRs on postsynaptic cells. Higher concentrations of CCh (≥1 μm) enhanced DSI and also persistently depressed basal eIPSCs, mainly by releasing endocannabinoids. Persistent CCh-induced endocannabinoid release did not require an increase in [Ca2+]i but was dependent on G-proteins. Although they were independent at the receptor level, muscarinic and glutamatergic mechanisms of endocannabinoid release shared intracellular machinery. Replication of the effects of CCh by blocking acetylcholinesterase with eserine suggests that mAChR-mediated endocannabinoid release is physiologically relevant. This study reveals a new role of the muscarinic cholinergic system in mammalian brain.
Keywords: GABAergic IPSC, mAChR, DSI, retrograde messenger, retrograde signaling, mGluR
Activation of muscarinic acetylcholine receptors (mAChRs) affects individual neurons by influencing several types of ionic currents: the M-current (Adams et al., 1982; Halliwell and Adams, 1982), the K+ “leak” current (Madison et al., 1987), the slow Ca2+-activated K+ current (Cole and Nicoll, 1984), the Ca2+-activated nonselective cation current (Guérineau et al., 1995; Fraser and MacVicar, 1996), and voltage-dependent Ca2+ current (Beech et al., 1991; Howe and Surmeier, 1995; Yan and Surmeier, 1996; Shapiro et al., 1999). Muscarinic agonists also influence the firing behavior of neuronal populations by activating different mAChR subtypes on different classes of cells. For instance, “θ rhythms” (Fischer et al., 1999; Buzsaki, 2002) and “γ rhythms” (Fisahn et al., 1998) are initiated by mAChR agonists. Stimulation of cholinergic afferents activates a population of GABAergic interneurons (Pitler and Alger, 1992a; Behrends and ten Bruggencate, 1993), and the cholinergic induction of γ rhythms depends in large part on the activation of GABAergic interneurons (Fisahn et al., 1998, 2002).
The diverse phenomena caused by mAChR activation are generally thought to result directly from modulation of ionic currents. However, the existence of endogenous cannabinoids (endocannabinoids) (Di Marzo et al., 1998; Piomelli et al., 2000), lipid-derived messengers that resemble the psychoactive ingredient in marijuana (Ameri, 1999), and retrograde signaling systems that use endocannabinoids (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001) raises other possibilities. Normally, increases in [Ca2+]i in the principal cells (Llano et al., 1991; Pitler and Alger, 1992b;Ohno-Shosaku et al., 1998; Lenz and Alger, 1999) lead to the induction of “depolarization-induced suppression of inhibition” (DSI) (Alger and Pitler, 1995), which is caused by the release of endocannabinoids (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001) that selectively bind to and activate CB1-type cannabinoid receptors (CB1Rs). CB1Rs are selectively localized on the nerve terminals of a subset of GABAergic interneurons (Tsou et al., 1998; Katona et al., 1999), and CB1R activation causes a decrease in GABA release from the cells probably by reducing N-type Ca2+ current (Hoffman and Lupica, 2000; Wilson et al., 2001). Recent reports show that the activation of group I metabotropic glutamate receptors (mGluRs) can release endocannabinoids in the cerebellum (Maejima et al., 2001) and hippocampus (Varma et al., 2001; Ohno-Shosaku et al., 2002). In the hippocampus, low concentrations of mGluR agonists also enhance DSI without suppressing evoked IPSC (eIPSC) amplitudes, which suggests an interaction between mGluRs and Ca2+-dependent endocannabinoid release (Varma et al., 2001; Ohno-Shosaku et al., 2002).
mAChR agonists have two opposing effects on IPSCs (Pitler and Alger, 1992a; Behrends and ten Bruggencate, 1993): they depress eIPSCs and enhance spontaneous IPSC (sIPSC) activity. Not all interneurons are susceptible to DSI (Martin and Alger, 1999; Wilson et al., 2001) because they do not all possess CB1Rs (Tsou et al., 1998; Katona et al., 1999). mAChR activation enhances DSI of sIPSCs in part by selectively stimulating interneurons that are DSI susceptible (Martin and Alger, 1999; Martin et al., 2001). However, eIPSCs also undergo DSI, and DSI of eIPSCs is also very robust in the presence of an mAChR agonist. Hence, activation of susceptible interneurons is probably not the sole mechanism of DSI enhancement by mAChRs (Lenz and Alger, 1999). The depression of eIPSCs and the enhancement of DSI could be caused by direct modulation of ionic currents of interneurons. Alternatively, both effects could reflect increased endocannabinoid release.
In this study, we show that mAChR activation can enhance endocannabinoid release independently of the mGluR system. This is a new role of the cholinergic system in the brain and will broaden our understanding of interactions between neurotransmitter systems. Our results also show some similarities between the mAChR and mGluR systems. Endocannabinoids play diverse roles in retrograde signaling, neuroprotection (van der Stelt et al., 2001; Panikashvili et al., 2002), and analgesia (Calignano et al., 1998; Ameri, 1999). GABA reduction by endocannabinoid may be important in long-term potentiation induction (Carlson et al., 2002). Our study contributes to building a connection from the cholinergic system to these endocannabinoid-mediated phenomena.
MATERIALS AND METHODS
Preparation of slices. Hippocampal slices were obtained mainly from 4- to 6-week-old male Sprague Dawley rats. In some experiments, as noted, mice (25–40 gm) in which the gene for CB1R had been invalidated (Ledent et al., 1999) were used. All experiments were performed in accordance with the guidelines set forth by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine. After the animals were deeply anesthetized with halothane and decapitated, the hippocampi were removed and sectioned into 400-μm-thick slices in ice-cold saline using aVibratome (Technical Products International, St. Louis, MO). The slices were maintained at room temperature in an interface holding chamber in a humidified atmosphere saturated with 95% O2/5% CO2. The slices were used ≥1 hr after sectioning. The recording chamber warmed the submerged slice, and experiments were performed at 30 ± 1°C (Nicoll and Alger, 1981).
Electrophysiology. Whole-cell voltage-clamp recordings of CA1 pyramidal cells were done using the “blind” patch method (Blanton et al., 1989). Electrode resistances in the bath were 3–6 MΩ, and recordings with series resistance <35 MΩ were accepted. During experiments, series resistance was checked by −1 mV hyperpolarizing voltage steps, and data associated with obvious changes of series resistance or unstable current baseline were discarded. The holding potential was −70 mV in all experiments. Monosynaptic eIPSCs were elicited by 100 μsec extracellular stimuli delivered with concentric bipolar stimulating electrodes (David Kopf Instruments, Tujunga, CA) placed in stratum oriens between CA3 and CA1, 0.5–1 mm apart from the recording site. Data were collected using an Axopatch 1C amplifier (Axon Instruments, Union City, CA), filtered at 2 kHz, and digitized at 5 kHz using a Digidata 1200 and Clampex 8 software (Axon Instruments).
The intracellular recording solution contained (in mm): 90 CsCH3SO3, 50 CsCl, 1 MgCl2, 2 Mg-ATP, 0.3 Tris-GTP, 0.2 Cs4-BAPTA, 10 HEPES, and 5 QX-314 (pH 7.2 with CsOH and 295 mOsm). In some experiments (see Fig. 6), 0.3 mm Tris-GTP was replaced by 1 mmGTPγS-Li4 to block further activation of G-proteins, or 90 mmCsCH3SO3 was replaced by 35 mm Cs4-BAPTA (Molecular Probes, Eugene, OR) to block a rise in [Ca2+]i. The extracellular solution included (in mm): 120 NaCl, 3 KCl, 25 NaHCO3, 1 NaH2PO4, 2.5 CaCl2, 2 MgSO4, and 15 glucose (300 mOsm). The extracellular solution was oxygenated with 95% O2/5% CO2 gas and flowed continuously through the recording chamber at a rate of ∼1 ml/min.
Fig. 6.

Effect of CCh on eIPSC amplitude in cells loaded with GTPγS (1 mm) or high BAPTA (35 mm).A, With 1 mm GTPγS inside the postsynaptic cell, CCh (1 μm) did not reduce eIPSC amplitude, but WIN 55212-2 (2 μm) did. In all experiments, WIN 55212-2 was applied in the absence of CCh to prevent confounding of effects. Forty traces were averaged for this cell, and 30–60 traces were averaged for other cells. The stimulus artifacts were partially truncated graphically. We waited for ∼30 min for complete action of GTPγS. DSI had disappeared by this time because of inhibition of the Ca2+ current. B, With 1 mm intracellular GTPγS, 1 μm CCh did not affect eIPSC amplitude (n = 6; pairedt test; p > 0.1), but WIN55212-2 (2 μm) reduced it to 75 ± 6% (n = 5; paired t test; *p < 0.05).C, CCh (1 μm) reduced eIPSC amplitude in a reversible manner in the presence of 35 mm BAPTA inside the postsynaptic cell. In addition, high BAPTA did not prevent 50 μm ACPD from reducing eIPSC. Forty to 50 traces were averaged for this cell, and 40–60 traces were averaged for other cells. DSI was not observed with high BAPTA in either the presence or absence of CCh. D, In cells loaded with 35 mm BAPTA, 1 μm CCh reduced eIPSC amplitude to 61 ± 5% (n = 6; paired ttest; *p < 0.01), and 50 μm ACPD reduced it to 33 ± 8% (n = 5; pairedt test; *p < 0.05).E, In cells loaded with 1 mm GTPγS, AM251 (4 μm) increased eIPSC amplitude (filled circles; n = 5), indicating that GTPγS by itself stimulated endocannabinoid release. At 16 min of AM251 application, eIPSC amplitude was 143 ± 8% of the control amplitude. AM251 was also applied to control cells lacking GTPγS (open circles; n = 5). At 16 min, eIPSC amplitude in these cells was 113 ± 8% of the baseline. Each circle is mean value of five cells after averaging 15 traces (1 min) within a cell. *p < 0.05;t test for 16–17 min.
To isolate monosynaptic eIPSCs, ionotropic glutamate receptor blockers 10 μm1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide and 50 μm dl-2-amino-5-phosphonopentanoic acid were present in the bath solution throughout the experiments. For the experiments in Figures 3 and 7, slices were preincubated with 4 μm AM251 for 2–5 hr, except for four cells in Figure3Aa, which were treated with AM251 from the onset of the whole-cell configuration, i.e., for >20 min. Water-based stock solutions of carbachol (CCh),trans-[1S,3R]-1-amino-1,3-cyclopentanedicarboxylic acid (ACPD), atropine, LY341495 (Tocris, Ballwin, MO), or DMSO-based stock solutions of AM251 (Tocris) and (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate (WIN 55212–2) (Tocris) were added to the bath solution and perfused into the recording chamber when needed. The final concentration of DMSO was 0.01% (v/v). All other chemicals were purchased from Sigma (St. Louis, MO).
Fig. 3.
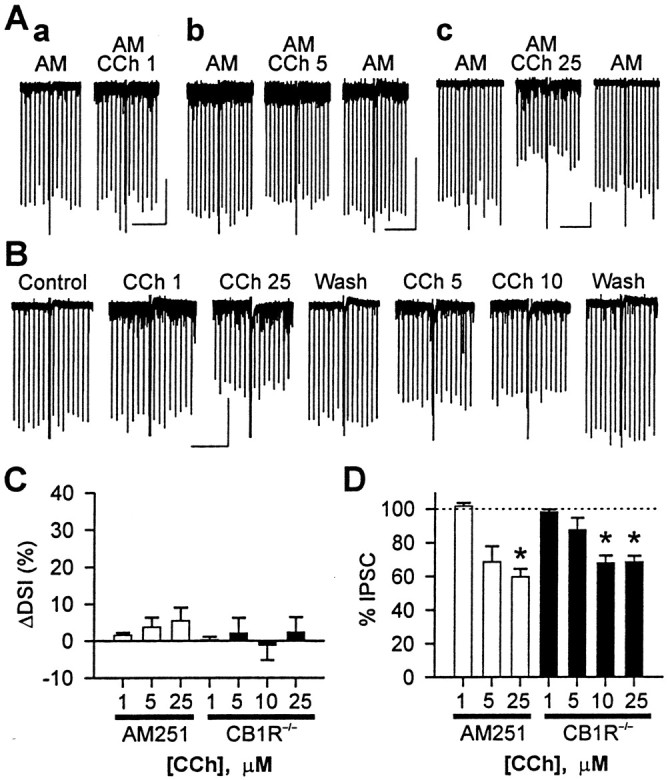
The effects of CCh on DSI and eIPSCs are reduced by AM251 and in CB1R−/− mice. A, Representative traces from three cells in rat slices treated with the CB1R antagonist AM251 (4 μm). One DSI trial per condition is shown. DSI was abolished by AM251, and CCh (1–25 μm) did not produce notable DSI. eIPSC amplitude was not changed by 1 μm CCh (a) but was reduced by 5 μm (b) or 25 μm CCh (c). Calibration: 200 pA, 30 sec.B, Representative traces of a cell from a CB1R−/− mouse. One DSI trial per condition is shown. DSI is absent in the presence or absence of CCh. eIPSCs were reduced by 5–25 μm CCh. Calibration: 200 pA, 30 sec.C, DSI was not changed significantly by 1–25 μm CCh in the presence of 4 μm AM251 (open bars) or in CB1R−/− mice (filled bars). In AM251 data,n = 5 for 1 or 25 μm CCh, andn = 4 for 5 μm CCh. For each concentration of CCh in AM251 data, p > 0.1 (paired t tests). In CB1R−/− data,n = 4, and p > 0.1 (repeated measures ANOVA). D, eIPSC amplitude was not changed by 1 μm CCh in rat slices with AM251 (open bar;p > 0.1; paired t test) or in slices from CB1R−/− mice (filled bar; p > 0.1; paired t test after repeated measures ANOVA). The effects of 5 μm CCh on eIPSC amplitude were variable from cell to cell and were not significant (p > 0.1; same tests as 1 μm). eIPSC was significantly reduced by 25 μm CCh (p < 0.05; same tests as 1 μm).
Fig. 7.
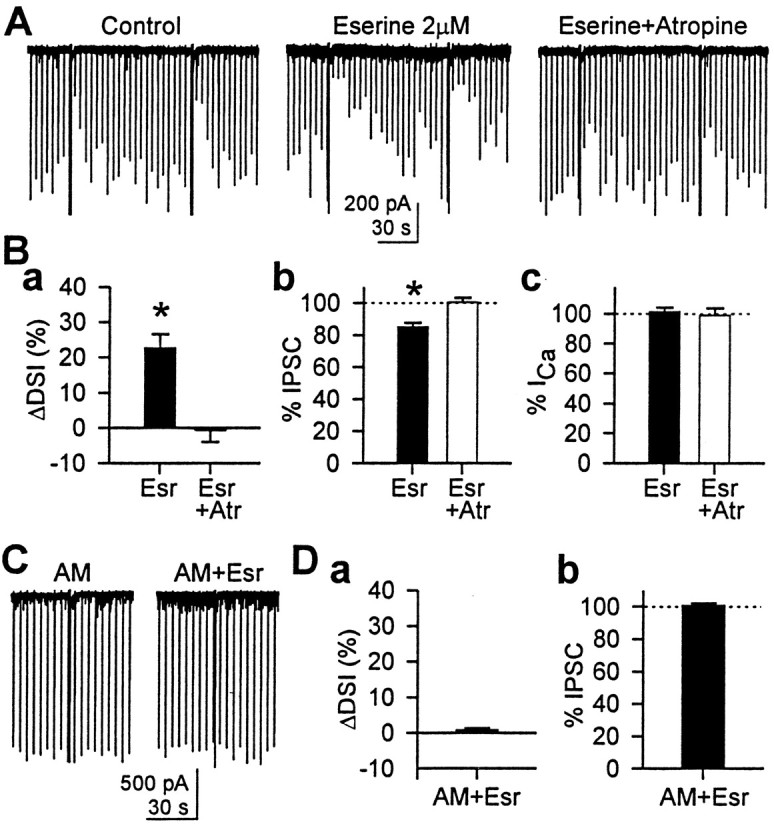
Eserine, the ACh esterase inhibitor, enhanced DSI.A, Sample traces showing that eserine (2 μm) enhanced DSI and reduced eIPSC. Both effects were reversed by 1 μm atropine. B, Group data (n = 5 cells) for the effects of eserine and atropine. a, DSI was significantly increased (ΔDSI = 23 ± 4%) by eserine (*p < 0.01; paired t test after repeated measures ANOVA).Filled bar, Eserine; open bar, eserine and atropine. b, eIPSC amplitude was significantly reduced by eserine to 85 ± 3% of control (*p< 0.05; paired t test after repeated measures ANOVA).c, Peak Ca2+ current, however, was not affected by 2 μm eserine (p > 0.1; repeated measures ANOVA).C, AM251 (4 μm) abolished the effects of eserine, indicating that the effects of eserine were mediated via CB1R.D, Group data (n = 5 cells) showing that AM251 blocked the enhancement of DSI (a) (p > 0.1; paired t test) and reduction of eIPSC amplitudes (b) (p > 0.1; paired t test) caused by eserine.
Ca2+ imaging. For imaging pyramidal cell [Ca2+]i, 100 μm Fluo3 plus 0.1 mm EGTA was included in the recording pipette solution in place of 0.2 mm Cs4-BAPTA. After a whole-cell recording was established, the cell was held at −70 mV for 10 min before imaging to establish diffusion equilibrium of the dye. The dye was excited with a 480 nm wavelength light provided by filtering the output from a 100 W mercury bulb. Images were collected by a cooled CCD camera (Roper, Tucson, AZ) and analyzed by IPLab software (Scanalytics, Fairfax, VA). For each trial, eight baseline fluorescence images were acquired immediately before a DSI-inducing depolarization step was delivered. ΔF/F was calculated for a total of 100 images that were acquired sequentially every 130 msec, where F is the fluorescence intensity when the cell is at rest, and ΔF is the change in fluorescence during activity.
Data analysis. We evoked IPSCs at 4 sec intervals and depolarized the postsynaptic cell to 0 mV for 1 sec at 88 sec intervals. The mean amplitude of five eIPSCs just before the depolarization pulse was taken as the control amplitude, and the mean of four eIPSCs just after the pulse was taken as the eIPSC amplitude during the DSI period. The magnitude of DSI of eIPSCs was calculated by taking the ratio of these two mean amplitudes, i.e., DSI (%) = (1 − mean of four eIPSCs after depolarizing pulse/mean of five eIPSCs before depolarizing pulse) × 100.
A DSI value of 0% means no reduction of eIPSC, and a value of 100% means complete reduction. Values of three DSI trials were averaged to obtain a mean DSI in a given condition. In some experiments, we also studied DSI of sIPSCs. To calculate DSI of sIPSCs, we integrated the sIPSC waveforms for 20 sec before the depolarization pulse and 16 sec after the pulse. Before integration, we adjusted the baseline of the current waveform to 0. We took the ratio of the integrations (charge) after dividing them by time, i.e., DSI (%) = [1 − (charge through sIPSC for 16 sec after pulse/16 sec)/(charge through sIPSC for 20 sec before pulse/20 sec) × 100]. To quantify a change in DSI, we simply subtracted two values of DSI, i.e., ΔDSI = experimental DSI − control DSI.
Data analysis was done in Clampfit 8 (Axon Instruments), and graphs were drawn in SigmaPlot 2000 [Statistical Program for the Social Sciences (SPSS), Chicago, IL]. t tests were done in Excel XP (Microsoft Corporation, Redmund, WA), and ANOVA was done in Systat 10 (SPSS). All t tests were two-tailed tests, and thep value for significance was <0.05. For multiple comparisons, we used ANOVA (or repeated measures ANOVA, where appropriate), and only when p < 0.05 did we performt tests (or paired t tests).
RESULTS
High concentrations (≥1 μm) of the mAChR agonist, CCh, enhance DSI of sIPSCs indirectly by markedly enhancing the activity of the interneurons that are susceptible to DSI (Martin et al., 2001), but we wanted to determine whether mAChR activation could promote DSI process directly. Because DSI of sIPSCs changes with the number of DSI-sensitive interneurons that are active, we cannot determine the changes in the DSI process itself if the number of interneurons is changed. To circumvent this difficulty, we measured DSI of eIPSCs because the eIPSC is generated from the same number of interneurons before and after CCh application. At low concentrations (0.2 or 0.5 μm), CCh enhanced DSI of eIPSC significantly (paired t test; p < 0.01) (Fig.1A,B,E) without affecting eIPSC amplitudes (paired t test,p > 0.1) (Fig. 1F). The increase in DSI (ΔDSI) was 16 ± 4% (n = 7) in 0.2 μm CCh and 27 ± 5% (n = 7) in 0.5 μm (Fig. 1E). Note that, from the equation (see Materials and Methods), ΔDSI represents an absolute increase in %DSI, not a fractional increase over control DSI. Unlike submicromolar concentrations, higher concentrations of CCh (1 and 25 μm) not only enhanced DSI but also reduced eIPSC amplitudes (Fig. 1C,D). ΔDSI was 32 ± 6% (n = 11) in 1 μmCCh and 30 ± 5% (n = 5) in 25 μm CCh (paired t test with each control; p < 0.01) (Fig. 1E), although there was no significant difference among ΔDSIs at the four different concentrations (ANOVA; p > 0.1). One and 25 μm CCh significantly reduced eIPSC amplitude to 68 ± 4% and 29 ± 3% of control, respectively (pairedt test; p < 0.001) (Fig.1F). To test whether all effects of CCh are mediated by mAChRs, we applied 1 μm atropine, an mAChR antagonist, in the presence of CCh (Fig.1B,C). Atropine completely reversed the effects of 0.5 or 1 μm CCh on DSI and eIPSC amplitudes to the control level (paired t test with control;p > 0.1 after repeated measures ANOVA among control, CCh, and atropine groups).
Fig. 1.

CCh enhances DSI and depresses eIPSC amplitudes. eIPSCs were evoked every 4 sec, and cells were depolarized to 0 mV from the holding potential of −70 mV every 88 sec. A–D, Representative traces from four different cells. Two DSI trials per condition are shown. CCh enhanced DSI at 0.2–25 μm and reduced eIPSC amplitude as well at ≥1 μm. The antagonistic effect of atropine (1 μm) was tested with 0.5 and 1 μm CCh. E, Changes in DSI (ΔDSI) were calculated by subtracting control DSI from DSI with CCh (filled bars). DSI with CCh was greater than control DSI at 0.2, 0.5, 1, and 25 μmCCh (n = 7, 7, 11, and 5, respectively) (*p < 0.01; paired t test). At 0.5 μm (n = 5) and 1 μm(n = 5) CCh, 1 μm atropine reversed the effect of CCh (open bars; paired ttest after repeated measures ANOVA; p > 0.1).F, eIPSC amplitude was not changed by 0.2 and 0.5 μm CCh (paired t test;p > 0.1) but reduced by 1 and 25 μm(*p < 0.001; paired t test;filled bars). Atropine (1 μm) recovered the eIPSC amplitude reduced by 1 μm CCh (open bars; paired t test after repeated measures ANOVA; p > 0.1). G, Peak Ca2+ current activated by 0 mV pulse. In the presence of 0.2–1 μm CCh, the mean Ca2+ currents were 88–94% of control (filled bars). In the presence of 0.5 μm CCh plus atropine, the Ca2+ current showed more rundown (75 ± 4% of control; open bars). CCh (25 μm) reduced the eIPSC amplitude to 61 ± 8% of control. *p < 0.05; pairedt test between control and CCh.
It is unlikely that DSI enhancement and IPSC reduction by CCh was mediated through changes in ionic currents (e.g., M-current, K+ leak current, and Ca2+-activated K+ current). In the cells shown in Figure1, 0.5 μm CCh did not change the holding current significantly; it went from −163 ± 27 pA in control to −151 ± 22 pA in CCh (p > 0.1; pairedt test). CCh, 1 μm, increased inward holding current significantly, but only slightly, from −187 ± 18 pA in control to −198 ± 18 pA in CCh (p< 0.05; paired t test). The DSI enhancement or IPSC reduction by CCh cannot be attributed to these small changes in holding current.
The enhancement of DSI did not appear to be caused by an increase in the voltage-dependent Ca2+ current that initiates DSI. In the presence of 0.2–1 μm CCh, the mean Ca2+ currents were 88–94% of control, which could represent normal rundown of Ca2+ current (Fig. 1G), but in 25 μm CCh, the mean Ca2+ current was only 61 ± 8% of control. It is therefore unlikely that the increase in DSI caused by CCh is associated with a change in [Ca2+]i. Nevertheless, the Ca2+ current may be imperfectly voltage clamped, and CCh might have influenced depolarization-induced [Ca2+]itransients. To examine this issue further, we imaged [Ca2+]i from pyramidal cell somata simultaneously with measurements of DSI. We used brief voltage steps (0.25–0.5 sec) to induce a minimal degree of DSI and then bath applied 0.2 or 0.5 μm CCh. The low concentration of CCh enhanced DSI but did not affect the fluorescence signals for basal [Ca2+]i(n = 5; data not shown) or the transients in response to voltage steps, as measured by the ΔF/F ratio (Fig. 2). The lack of change in ΔF/F was not the result of dye saturation, because a larger Ca2+ response was detected when a longer voltage step was given (Fig.2B, inset). These results demonstrate that increases in depolarization-induced Ca2+transients are not necessary for CCh to enhance DSI.
Fig. 2.

The enhancement of DSI by CCh is not associated with an increase in the depolarization-induced somatic Ca2+ transients. eIPSCs were evoked every 5 sec, and a 250-msec-long depolarizing voltage step to 0 mV was delivered at intervals of several minutes. A, The average magnitude of DSI in control solution was enhanced more than twofold in 0.2 μm CCh (*p < 0.05; Wilcoxon signed rank test; n = 5). B, Group data for the simultaneously measured ΔF/F ratio of the [Ca2+]i signals are shown for control conditions (left graph) and in the same cells in the presence of CCh (right graph). The DSI-inducing voltage step was given 1.5 sec after time 0. Note that the increase in DSI is not accompanied by an increase in ΔF/F. The inset on theright graph, showing an example of a larger ΔF/F change that was produced by a 1 sec voltage step in the same cells, demonstrates that the lack of measured changes in ΔF/F did not result from dye saturation.
Because DSI is mediated by the release of endocannabinoids, it was possible that the effects of mAChR activation on DSI and eIPSCs were mediated through an interaction with the endocannabinoid system. We tested this hypothesis by applying CCh either to cells in normal slices in the presence of the CB1R antagonist AM251 (4 μm) or to cells in slices from CB1R−/− mice (Fig.3). As reported (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001), CB1R antagonists blocked DSI in rat hippocampus (mean DSI in control condition = 1–3%) (Fig.3A), and DSI was absent in the CB1R−/− mouse (Varma et al., 2001;Wilson et al., 2001) (control DSI = 0 ± 1%;n = 4) (Fig. 3B). For comparison, DSI from rat slices in normal conditions (Fig. 1) was 30 ± 3% (n = 30 cells). In rat slices treated with AM251, application of 1–25 μm CCh did not produce significant DSI. ΔDSIs were 2 ± 1% (n = 5) for 1 μm CCh, 4 ± 3% (n = 4) for 5 μm CCh, and 6 ± 4% (n = 5) for 25 μm CCh (p > 0.1; paired t test in each group) (Fig. 3C). In CB1R−/−mouse slices, 1–25 μm CCh also did not cause DSI to appear (repeated measures ANOVA; p > 0.1) (Fig.3C). Mean ΔDSIs were in the range of −1 to 2% (n = 4). CCh at 1 μm did not reduce eIPSCs in rat slices treated with AM251 or in CB1R−/− mice (Fig. 3D). The mean eIPSC was 102 ± 2% of control in treated rat slices and 98 ± 2% in CB1R−/− mice. In control CB1R+/+ mice of the CD-1 strain, which is the background for CB1R−/− mice (Ledent et al., 1999), CCh (1 μm) increased DSI by 18 ± 4% (n = 3; p < 0.05; data not shown). This implies that the enhancement of DSI by 1 μm CCh (Fig. 1F) was mediated entirely by induced endocannabinoid release.
The mean eIPSC in 5 μm CCh was 69 ± 9% of control in rat slices treated with AM251 and 88 ± 7% in CB1R−/− slices, but these decreases were not significant (p > 0.1). CCh at 25 μm significantly reduced eIPSC amplitudes to 60 ± 5% of control in AM251-treated rat slices (p < 0.01; paired t test) and to 69 ± 4% in CB1R−/− slices (p < 0.05; paired t test after repeated measures ANOVA). Thus, 25 μm CCh reduced eIPSCs not only by a direct induction of persistent endocannabinoid release but by other mechanisms as well.
From the difference in the eIPSC depression caused by 25 μm CCh in control conditions (71%), and in the AM251-treated condition (40%), we infer that 25 μm CCh reduced eIPSC by 31% via the endocannabinoid pathway. Tests on the group data showed that this is statistically indistinguishable from the 32% eIPSC depression caused by 1 μm CCh (p > 0.1; t test). This leads to the conclusion that persistent, mAChR-induced release of endocannabinoids is saturated by 1 μm CCh. In the remaining experiments, we used 1 μm CCh, except where noted, to induce persistent endocannabinoid release maximally and to avoid the confounding effects of other mAChR-mediated mechanisms of eIPSC suppression that are produced by higher CCh concentrations.
mGluR activation can enhance release of endocannabinoids (Maejima et al., 2001; Varma et al., 2001; Ohno-Shosaku et al., 2002); therefore, it was necessary to determine whether mGluR activation could be involved in the action of CCh. For example, CCh might cause glutamatergic neurons to release glutamate, and this could activate mGluRs on the pyramidal cell and thereby stimulate endocannabinoid release. To test this possibility, we applied CCh together with 100 μm LY341495, an mGluR antagonist that at high concentration blocks all known mGluRs (Fitzjohn et al., 1998). We found that CCh at 1 μm was capable of increasing DSI and reducing eIPSC amplitudes in the presence of LY341495 (Fig.4A). ΔDSI was 24 ± 7% (n = 5; paired t test after repeated measures ANOVA; p < 0.05) (Fig. 4C). We confirmed that LY341495 had blocked mGluRs by observing that 10 μm ACPD had no effect on DSI (Fig.4A) (cf. Varma et al., 2001). ΔDSI was 1 ± 4% (n = 5; paired t test after repeated measures ANOVA; p > 0.5) (Fig. 4C). Therefore, mGluR activation is not necessary for CCh to enhance endocannabinoid release. To determine whether the mGluR and mAChR effects on DSI are independent, we also performed the converse experiment to rule out the possibility that activation of mGluRs on cholinergic neurons releases acetylcholine and thereby stimulates endocannabinoid release from pyramidal cells. Indeed, in the presence of 1 μm atropine, 5 or 10 μm ACPD enhanced DSI (Fig.4B). ΔDSI was 25 ± 4% (n = 5; paired t test after repeated measures ANOVA;p < 0.05) (Fig. 4D).
Fig. 4.
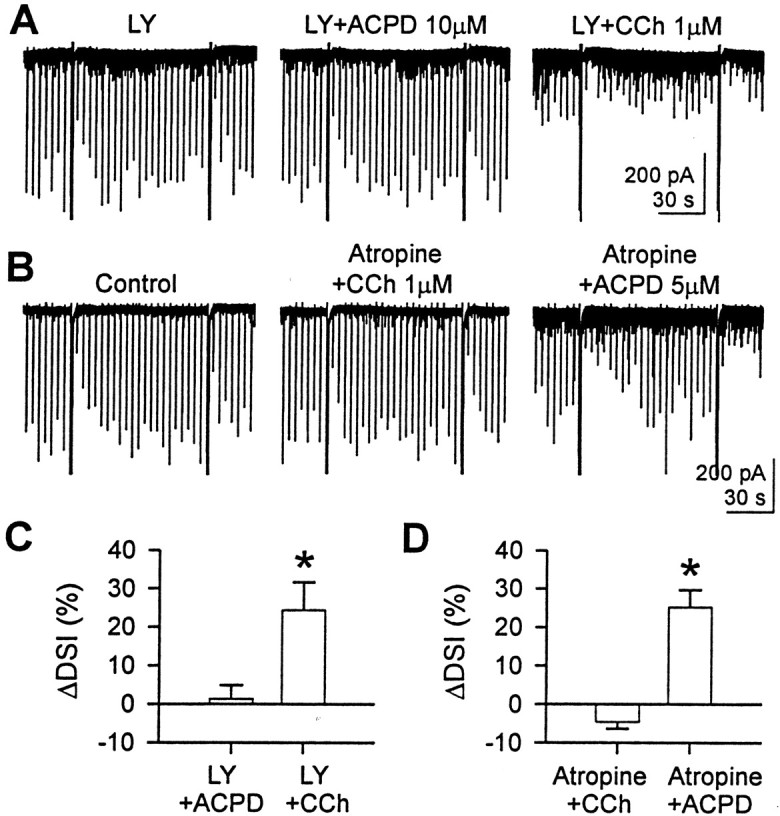
Activation of mAChR or mGluR can enhance DSI independently of each other. A, In the presence of LY341495 (100 μm), 1 μm CCh enhanced DSI and reduced the eIPSC amplitude, but the ability of 10 μmACPD to enhance DSI was antagonized by 100 μm LY341495.Traces are from one cell. B, In the presence of atropine (1 μm), 5 μm ACPD enhanced DSI, but 1 μm CCh did not. C, In five cells, ACPD (10 μm) and CCh (1 μm) were sequentially applied in the presence of 100 μmLY341495. CCh enhanced DSI significantly (*p < 0.05; paired t test after repeated measures ANOVA). No effect of ACPD (paired t test after repeated measures ANOVA; p > 0.5) indicates that LY341495 was effectively blocking mGluR. D, In five cells, CCh (0.5 or 1 μm) and ACPD (5 or 10 μm) were sequentially applied in the presence of 1 μm atropine. Data for two concentrations were pooled. ACPD enhanced DSI significantly (*p < 0.05; paired ttest after repeated measures ANOVA), whereas CCh did not (pairedt test after repeated measures ANOVA;p > 0.5 ).
Thus, mGluRs and mAChRs independently act on the endocannabinoid system; however, they may share intracellular machinery for endocannabinoid release. We addressed this issue by determining whether the two pathways would occlude each other when they were maximally stimulated. In the presence of a high concentration (25 μm) of CCh, we applied 50 μm ACPD, which at this concentration reduces eIPSC amplitudes and largely occludes DSI (Morishita et al., 1998) (Fig.5A,B). In 25 μm CCh, DSI of eIPSC or sIPSC is very clear, but it did not change when 50 μm ACPD was applied (paired t test; p > 0.5). ΔDSI was −2 ± 4% for eIPSC (n = 5) and −1 ± 2% for sIPSC (n = 7) (Fig. 5D). In addition, neither the amplitude of the eIPSC (107 ± 9% of control) nor the magnitude of the charge carried by sIPSC (102 ± 9% of control) was changed by 50 μm ACPD (paired t test; p > 0.1) (Fig.5E). This could imply that the endocannabinoid release system was already saturated by 25 μm CCh, such that no more endocannabinoids were available for release by the mGluR pathway. However, significant DSI (60 ± 1%, n = 5, for eIPSCs; 71 ± 3%, n = 7, for sIPSCs) could still be obtained in the presence of high CCh (Fig.5A,B), showing that despite apparent inability to stimulate the persistent release pathway further, the transient release of endocannabinoids by the Ca2+-dependent pathway that initiates DSI could still be activated. Therefore, endocannabinoid release in general was not saturated. This result implies the existence of two independent pathways for endocannabinoid release.
Fig. 5.

A high concentration of either CCh or ACPD prevented the other from enhancing DSI and reducing eIPSC amplitude.A, ACPD (50 μm), which normally reduces eIPSC amplitude and occludes DSI at this concentration, did not affect DSI or eIPSC when applied with 25 μm CCh. Note that 50 μm ACPD had little effect on either eIPSC or sIPSC.B, In this cell, only sIPSCs were measured. DSI and eIPSC amplitudes were unaffected by 50 μm ACPD in the presence of 25 μm CCh. Ca2+ current and associated transient current were blanked for clarity. For clear comparison of DSI, a fast inward current activated by depolarization was subtracted from the baseline. The two DSI trials in each column are consecutive. C, In this cell, 50 μm ACPD was applied before 1 μm CCh. CCh slightly enhanced DSI but had no effect on eIPSC amplitude. D, Group data of ΔDSI. When 25 μm CCh was applied first (open bars), 50 μm ACPD had no effect on DSI of eIPSC (n = 5; paired t test;p > 0.5) or DSI of sIPSC (n = 7; paired t test; p > 0.5). When 50 μm ACPD was applied first (filled bar), 1 μm CCh slightly increased DSI of eIPSC, but it was not significant (n = 5; pairedt test; p > 0.1). In the experiments in which CCh was applied first, both sIPSC and eIPSC were measured in four cells, only sIPSC was measured in three cells, and only eIPSC was measured in one cell. E, When 25 μm CCh was applied first (open bars), 50 μm ACPD had no effect on amplitude of eIPSC (n = 5; paired t test;p > 0.1) or charge of sIPSC (n= 7; paired t test; p > 0.5). When 50 μm ACPD was applied first (filled bar), 1 μm CCh did not change the amplitude of eIPSC (n = 5; paired t test;p > 0.1). Charge carried by sIPSC was measured by integrating the area under sIPSC for 20 sec before and 16 sec after the 0 mV pulse. “Charge per second” was used as a magnitude of sIPSC.
To determine whether maximal mGluR activation was similarly capable of occluding endocannabinoid release stimulated by mAChRs, we tested the reverse order of agonist application. When ACPD (50 μm) was applied first, DSI of the eIPSC was partly occluded (DSI was 29 ± 5% in control and 21 ± 3% in ACPD; n= 5) (Fig. 5C), and 1 μm CCh did not significantly enhance it (n = 5; ΔDSI = 7 ± 4%; paired t test; p > 0.1) (Fig.5D). Furthermore, the eIPSC amplitude was not changed by 1 μm CCh in the presence of 50 μm ACPD (99 ± 5% of control; pairedt test; p > 0.1) (Fig. 5E). Evidently, 50 μm ACPD induced the persistent release of endocannabinoid to such an extent that GABA release from DSI-susceptible interneurons was largely inhibited, thus occluding DSI and additional effects of CCh on eIPSCs. We conclude that the mAChR and mGluR pathways share intracellular machinery for cannabinoid release, but they do not activate them equivalently (see Discussion).
Do DSI and the persistent release pathways for endocannabinoid release rely only on Ca2+ or also G-proteins? To investigate this issue, we included 1 mm GTPγS, which locks G-proteins into a persistently active state and thus inhibits further activation by an agonist, in the pipette solution (Fig.6). We waited for ∼30 min for complete dialysis of the cell with GTPγS. The voltage-dependent Ca2+ current was gradually inhibited as expected (Dolphin, 1995) and finally abolished during this time; consequently, DSI was abolished. Every cell, however, showed DSI until ∼15 min after the onset of whole-cell configuration. This means that all pyramidal cells tested in the GTPγS experiment were able to release endocannabinoids and that the presynaptic interneurons possessed CB1R. Nevertheless, with GTPγS inside the postsynaptic cell, bath application of 1 μm CCh did not reduce the eIPSC amplitude (100 ± 3% of control; n = 6; paired t test; p > 0.1) (Fig.6A,B). The reduction of eIPSC amplitude by subsequent application of 2 μm WIN 55212-2 confirmed that the eIPSCs could be reduced by CB1R activation, thus showing that the interneurons possessing CB1Rs were still active (Fig. 6A,B). WIN 55212-2 (2 μm) reduced the eIPSC amplitude to 75 ± 6% of control (n = 5; paired t test;p < 0.05). We attempted to test the hypothesis further by including 1 mm GDPβS in the pipette because it can block G-proteins. However, in four of six cells, GDPβS did not block DSI or the ability of CCh to enhance it; in fact, DSI was enhanced by GDPβS (data not shown). Similar anomalous enhancing effects of GDPβS on presumed G-protein-mediated responses have been reported (Andrade et al., 1986; Paris and Pouyssegur, 1990). In any event, this complication meant that we could not use GDPβS to study the role of G-proteins. The results in Figure 6 show that the CCh effect on persistent endocannabinoid release was mediated by postsynaptic G-proteins.
Because GTPγS is an activator of G-proteins, GTPγS itself could induce G-protein-dependent endocannabinoid release directly. If this is true, an alternative interpretation of the GTPγS results (Fig.6A,B) is possible. Stimulation of the G-protein pathway by GTPγS might occlude the ability of CCh to release more endocannabinoids. Thus, in GTPγS-loaded cells, CCh would appear to have no effect (Fig.6A,B), but this would not be attributable to inhibition of the CCh pathway by GTPγS.
To test this possibility, we checked whether GTPγS by itself can release endocannabinoids. After allowing 1 mm GTPγS to diffuse into the postsynaptic cell for 20–30 min, we bath-applied 4 μm AM251 (Fig. 6E). When applied to cells loaded with 1 mm intracellular GTPγS, AM251 increased eIPSC amplitudes to 143 ± 8% at 16 min of application (n = 5; p < 0.01; pairedt test), suggesting that persistent release of endocannabinoids caused by GTPγS had been suppressing the eIPSCs. Before accepting this conclusion, it was necessary to rule out another possible interpretation. The CB1R antagonist AM251, like SR141716A, is actually an inverse agonist (Bouaboula et al., 1997; Pan et al., 1998). Moreover, CB1R is constitutively active and can bind GTP in the absence of ligand (Pan et al., 1998). Because an inverse agonist binds to and stabilizes the receptor in a GDP-bound, inactive state, it can produce opposite effects of those produced by ligand-bound receptor. To determine whether the increase of eIPSC produced by AM251 reflected this inverse agonist effect, we applied 4 μmAM251 to cells recorded with a normal pipette solution (i.e., without GTPγS). AM251 slightly increased eIPSC amplitudes to 113 ± 8% at 16 min of application, but the effect was variable from cell to cell and not significant (n = 5; p > 0.1; paired t test). Therefore, the inverse agonist effect does not play a substantial role in the actions of AM251, although we cannot rule out a slight contribution. The difference between the eIPSC amplitudes in cells loaded with GTPγS and control cells was significant (comparison made at 16–17 min of AM251 application;p < 0.05; t test). We infer that the enhancement of eIPSCs caused by AM251 is attributable to its blockade of CB1Rs that have been activated by a persistent, GTPγS-induced release of endocannabinoids. This action of GTPγS occludes the ability of CCh to suppress eIPSCs.
As a check on this interpretation, we can estimate the magnitude of the endocannabinoid-dependent eIPSC suppression caused by loading cells with GTPγS. AM251 increased the eIPSC 43% above control (100%) in these cells. This suggests that GTPγS-released endocannabinoids caused a 30% reduction in eIPSC [1 − (100/143%)]. Even if this is corrected for a possible AM251 inverse-agonist effect (we took 13% as a conservative estimate; see preceding paragraph), the net increase in GTPγS-loaded cells would be 30% (43 − 13%), and GTPγS would have suppressed eIPSCs by 23% [1 − (100/130%)]. These estimates are similar to but perhaps smaller than the 32% reduction in eIPSC caused by 1 μm CCh (Fig. 1). It is possible that GTPγS totally occludes the effects of 1 μm CCh by maximally activating the G-protein-dependent, endocannabinoid release pathway or, alternatively, that the occlusion is only partial, and some other consequence of GTPγS loading helps prevent the CCh effects. In any case, the absence of CCh effects on eIPSCs in the GTPγS-loaded cells implies that CCh acts on the postsynaptic mAChRs, because postsynaptic treatment interfered with the effect of CCh.
We then tested whether the persistent release of cannabinoid induced by CCh is initiated by intracellular Ca2+, because DSI is. To suppress increases in intracellular Ca2+, we included 35 mm BAPTA in the pipette solution (Fig.6C,D), which abolished DSI, as expected. However, even with 35 mm intracellular BAPTA, CCh (1 μm) reduced eIPSC amplitudes to 61 ± 5% of control (paired t test; p< 0.01) in a reversible manner. This did not differ significantly from the depression (to 68%) that we observed in control conditions (Fig.1) (t test; p > 0.1). In addition, 50 μm ACPD depressed eIPSC amplitude to 33 ± 8% of control in the high internal BAPTA condition, which did not differ significantly from the suppression to 45 ± 14% eIPSC that we observed in control conditions (data not shown; n = 5). Thus, evidently, neither the mAChR nor the mGluR pathways require intracellular Ca2+ to release endocannabinoids persistently.
To determine whether the effects of mAChR activation on endocannabinoid release may be physiologically relevant, we asked whether ambient ACh can contribute to endocannabinoid release by applying eserine, an ACh esterase inhibitor. Bath application of 2 μm eserine enhanced DSI and reduced eIPSC amplitude, and the effects were maximal at 10–15 min of treatment (Fig.7A). Reversal of the eserine effects by 1 μm atropine indicated that the effects were mediated by mAChR. The mimicry of CCh effects by eserine itself shows that a small amount of ACh is constitutively released into extracellular space. ΔDSI induced by 2 μmeserine was 23 ± 4% (n = 5; p < 0.01; paired t test after repeated measures ANOVA) (Fig.7B). The amplitude of eIPSC was significantly reduced by eserine to 85 ± 3% of control (p < 0.05; paired t test after repeated measures ANOVA), and this was completely reversed by 1 μm atropine [ΔDSI = −1 ± 3% (p > 0.1), and eIPSC = 100 ± 3% of control (p > 0.1)]. The effects of eserine were not secondary to changes in Ca2+ current because eserine did not affect Ca2+ current (p > 0.1; repeated measures ANOVA) (Fig.7B). Pretreatment of slices with AM251 (4 μm) abolished the effects of eserine (Fig.7C), showing that they were mediated by endocannabinoids. The absence of DSI in the control condition (−1 ± 1%;n = 5) showed that AM251 was effective. With AM251, ΔDSI induced by 2 μm eserine was 1 ± 1% (n = 5; p > 0.1; pairedt test), and eIPSC amplitude was 101 ± 2% of control (p > 0.1; paired t test) (Fig.7D). These results indicate that ambient levels of physiological ACh may be sufficient to induce endocannabinoid release
DISCUSSION
This is the first study showing that mAChR activation enhances the release of endocannabinoids. The enhancement appeared in two forms. Low levels of mAChR activation increased the transient release of endocannabinoids that we record as DSI. At higher levels, mAChR activation directly induced the release of endocannabinoids that suppress baseline eIPSCs outside of the DSI period. Endocannabinoid release induced by high concentrations of CCh persisted for as long as the agonist was applied. The enhancement of DSI was fully blocked by an antagonist of CB1R and was absent in CB1R−/− mice, and we conclude that at low concentrations (≤0.5 μm), CCh only affected eIPSCs by enhancing the transient release process. The persistent effect was prominent when CCh levels were ≥1 μm. This was also blocked by CB1R antagonist and absent in CB1R−/− mice but was contaminated by a CB1R-independent effect when CCh concentrations were >5 μm. Although, in principle, enhancement of transient endocannabinoid release could be mediated by an increase in Ca2+ current, or more generally by an increase in [Ca2+]i, we observed neither. Enhancement of DSI via mGluR activation is also not associated with an increase in [Ca2+]i(Ohno-Shosaku et al., 2002). Application of the AChE inhibitor eserine mimicked all the effects of CCh. Showing that increases in endogenous levels of ACh can induce persistent endocannabinoid release implies that such release may be physiologically relevant.
The mGluR and mAChR effects are initiated independently, because antagonists of one receptor do not affect responses mediated by the other. mGluR and mAChR effects on endocannabinoid release do co-occlude, as expected if they share common intracellular signaling systems. Nevertheless, despite numerous similarities, the mGluR and mAChR effects on endocannabinoid release are not identical. Although maximal activation of mGluRs can suppress eIPSCs to the extent that DSI is almost completely occluded, maximal activation of mAChRs causes less occlusion, and in fact, pronounced DSI is often present in high CCh concentrations. High concentrations of ACPD suppress eIPSCs an average of 67%, and this is entirely mediated by endocannabinoids (Varma et al., 2001). In comparison, high concentrations of CCh (25 μm) suppress eIPSCs an average of 71%, but only approximately half of this was mediated by endocannabinoids (Fig.3D). Most intriguingly, high ACPD is unable to suppress eIPSCs, or further occlude DSI, in the presence of a high concentration of CCh. The existence of DSI in the presence of high CCh clearly implies that endocannabinoids can still be released by the transient, Ca2+-dependent pathway. mAChRs seem to activate the persistent release pathway with only approximately half the efficiency of mGluRs, and high concentrations of CCh prevent full activation of persistent release. Working out the details of the intracellular regulation of endocannabinoid release will clearly be an important task for the future.
Our data suggest that distinct biochemical pathways are involved in the two forms of endocannabinoid release. The persistent release of endocannabinoids either by mAChR (Fig. 6) or by mGluR activation (Maejima et al., 2001; Ohno-Shosaku et al., 2002) is clearly G-protein dependent. We found that persistent, mAChR-mediated endocannabinoid release could not be prevented by loading the cells with BAPTA (35 mm), a very high concentration that is fully effective in blocking Ca2+-dependent processes in these cells. This agrees with findings on the persistent, mGluR-mediated release (Maejima et al., 2001). We therefore conclude that the persistent release pathway is not Ca2+dependent. Because DSI was abolished by 35 mm BAPTA, we are unable to say whether the enhancement of transient endocannabinoid release is Ca2+ dependent. We had thought that this enhancement might be caused by an increase in the Ca2+ signal associated with DSI, but neither voltage-dependent Ca2+ currents nor [Ca2+]i was increased during enhancement of DSI. The enhancement process itself could still be Ca2+ dependent; for example, it might be initiated by mAChR activation only when [Ca2+]i has been elevated by the Ca2+ influx that initiates DSI.
Despite several similarities between our work and previous work, there are some differences. It has been reported that GTPγS blocks mGluR-induced endocannabinoid release in cerebellar Purkinje cells (Maejima et al., 2001), whereas we find that GTPγS strongly activates mAChR-induced endocannabinoid release in hippocampal CA1 cells (Fig.6). The differences in cell or receptor types may be responsible, although it is not clear whether persistent induction of endocannabinoid release has been ruled out in the cerebellum. GDPβS reduced, but did not fully block, DSI in cultured hippocampal cells (Ohno-Shosaku et al., 2002), and DSI was still enhanced by mGluR agonists, although to a lesser extent than in the control condition. In our hands, however, GDPβS did not inhibit DSI and may have enhanced it. A possible explanation is that GDPβS can be converted in cells to GTPβS, an activator of G-proteins, via the enzyme nucleoside diphosphate kinase (Eckstein et al., 1979; Paris and Pouyssegur, 1990; Piacentini and Niroomand, 1996). If this effect occurs more readily in adult cells in slices than in the cultured cells, then it could account for the different results obtained with GDPβS.
Our findings are relevant to the issue of “spread” of endocannabinoids from one cell to another. Because GTPγS induced endocannabinoid release, the failure of CCh to decrease eIPSCs further might have been caused by saturation of CB1R by endocannabinoid that was released by GTPγS. In that case, we would not detect the additional release induced by CCh. However, this possibility can be excluded because WIN 55212-2 remained capable of depressing eIPSCs in GTPγS-loaded cells; thus, the CB1Rs on presynaptic terminals cannot have been saturated. A more likely explanation for the absence of CCh effects in GTPγS-loaded cells is that both GTPγS and CCh cause a persistent release of endocannabinoids by activating a common G-protein-dependent pathway. Previous activation of this pathway by GTPγS would occlude the ability of CCh to release more endocannabinoids from that cell. However, nearby cells were not filled with GTPγS, and if endocannabinoids from these cells could diffuse to the interneuronal synapses on the GTPγS-loaded cell, bath-applied CCh would have reduced the eIPSC amplitude. We conclude that failure of CCh to decrease eIPSCs recorded in GTPγS-loaded cells means that spillover of cannabinoid from nearby cells is unlikely. This agrees with work from the cerebellum (Maejima et al., 2001) and striatum (Gerdeman et al., 2002) that suggests that endocannabinoids do not normally spread from one cell to another, but is at odds with the finding of Wilson and Nicoll (2001) that endocannabinoids released from one cell can act on other cells within a radius of ≤20 μm. The reason for the discrepancy is not known, although it is possible that the release of endocannabinoids from cells stimulated under whole-cell voltage-clamp conditions (Wilson and Nicoll, 2001) is more pronounced than that from intact cells. Pitler and Alger (1994) and Morishita and Alger (2001) saw no evidence for spread of DSI from neighboring cells in most experiments in which the population of intact neighboring cells was stimulated antidromically. In any case, all of these studies agree that retrograde signaling mediated by endocannabinoids is a very localized process.
To date, four of the five mAChR subtypes (M1–M5) have been identified in the hippocampus (Levey et al., 1995). They can be divided into two families, with the odd-numbered members being coupled to phospholipase C through Gq and the even-numbered members being coupled to adenylate cyclase through Gi/Go (Caulfield, 1993;Caulfield and Birdsall, 1998). Group I mGluRs, in particular mGluR5 (Morishita et al., 1998; Morishita and Alger, 1999), are effective in mimicking and occluding DSI and do so by releasing endocannabinoids (Varma et al., 2001; Ohno-Shosaku et al., 2002; our unpublished observations). In the cerebellum, mGluR1 activation releases endocannabinoids (Maejima et al., 2001). The similarity between this previous work and our present results strongly suggests that M1, M3, or, conceivably, M5 will be found to mediate CCh-induced endocannabinoid release. Interestingly, the M1 subtype drives population γ rhythms in the hippocampus (Fisahn et al., 2002), and activation of CB1Rs can affect rhythmic firing behavior as well (Hajos et al., 2000). We propose that some of the mAChR effects on rhythmic firing behavior are mediated by CB1Rs.
These results may contribute to the reconciliation of contradictory published data. We reported that mGluR antagonists could often reduce DSI (Morishita et al., 1998; Morishita and Alger, 1999; Varma et al., 2001), but other reports did not replicate this observation (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). However, we had studied DSI of eIPSCs in the absence of muscarinic agonists, whereas in other experiments (Wilson and Nicoll, 2001), high concentrations of CCh were used to enhance sIPSC activity so that DSI of sIPSCs could be studied. Therefore, our demonstration that high CCh can obscure the effects of ACPD on eIPSCs (Fig. 5) can account in part for the discrepant observations, in addition to the likelihood that variations in ambient levels of glutamate play a role (Varma et al., 2001).
Activation of nicotinic AChRs in conjunction with NMDA receptor activation increases the levels of endocannabinoids synthesized in cultured neocortical cells (Stella and Piomelli, 2001), and activation of dopamine D2 receptors increases endocannabinoid synthesis and release in the striatum (Giuffrida et al., 1999). The present work, together with the recent discoveries that mGluR activation enhances endocannabinoid release, greatly expands the scope of these systems. Endocannabinoids must now be suspected to be involved in a wider variety of neuronal activities than thought previously, and conversely, understanding the workings of the conventional transmitter systems will not be possible without a full appreciation of their interactions with the endocannabinoids.
Footnotes
The work was supported by United States Public Health Service Grants RO1 DA14725 and RO1 NS30219 (B.E.A.) J.K. was supported by the Training Program in Neuroscience T32 DE1474. Most of this work is contained in the PhD thesis of J.K. We thank Scott Thompson and Darrin Brager for their comments on a draft of this manuscript.
Correspondence should be addressed to Dr. Bradley E. Alger, Department of Physiology, University of Maryland School of Medicine, 655 West Baltimore Street, Baltimore, MD 21201. E-mail:balger@umaryland.edu.
REFERENCES
- 1.Adams PR, Brown DA, Constanti A. Pharmacological inhibition of the M-current. J Physiol (Lond) 1982;332:223–262. doi: 10.1113/jphysiol.1982.sp014411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alger BE, Pitler TA. Retrograde signaling at GABAA-receptor synapses in the mammalian CNS. Trends Neurosci. 1995;18:333–340. doi: 10.1016/0166-2236(95)93923-l. [DOI] [PubMed] [Google Scholar]
- 3.Ameri A. The effects of cannabinoids on the brain. Prog Neurobiol. 1999;58:315–348. doi: 10.1016/s0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- 4.Andrade R, Malenka RC, Nicoll RA. A G protein couples serotonin and GABAB receptors to the same channels in hippocampus. Science. 1986;234:1261–1265. doi: 10.1126/science.2430334. [DOI] [PubMed] [Google Scholar]
- 5.Beech DJ, Bernheim L, Mathie A, Hille B. Intracellular Ca2+ buffers disrupt muscarinic suppression of Ca2+ current and M current in rat sympathetic neurons. Proc Natl Acad Sci USA. 1991;88:652–656. doi: 10.1073/pnas.88.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Behrends JC, ten Bruggencate G. Cholinergic modulation of synaptic inhibition in the guinea pig hippocampus in vitro: excitation of GABAergic interneurons and inhibition of GABA-release. J Neurophysiol. 1993;69:626–629. doi: 10.1152/jn.1993.69.2.626. [DOI] [PubMed] [Google Scholar]
- 7.Blanton MG, Lo Turco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- 8.Bouaboula M, Perrachon S, Milligan L, Canat X, Rinaldi-Carmona M, Portier M, Barth F, Calandra B, Pecceu F, Lupker J, Maffrand J-P, Le Fur G, Casellas P. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. J Biol Chem. 1997;54:1064–1072. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- 9.Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- 10.Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- 11.Carlson G, Wang Y, Alger BE. Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci. 2002;5:723–724. doi: 10.1038/nn879. [DOI] [PubMed] [Google Scholar]
- 12.Caulfield MP. Muscarinic receptors–characterization, coupling and function. Pharmacol Ther. 1993;58:319–379. doi: 10.1016/0163-7258(93)90027-b. [DOI] [PubMed] [Google Scholar]
- 13.Caulfield MP, Birdsall NJM. International Union of Pharmacolgy. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- 14.Cole AE, Nicoll RA. Characterization of a slow cholinergic post-synaptic potential recorded in vitro from rat hippocampal pyramidal cells. J Physiol (Lond) 1984;352:173–188. doi: 10.1113/jphysiol.1984.sp015285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21:521–528. doi: 10.1016/s0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- 16.Dolphin AC. Voltage-dependent calcium channels and their modulation by neurotransmitters and G proteins. Exp Physiol. 1995;80:1–36. doi: 10.1113/expphysiol.1995.sp003825. [DOI] [PubMed] [Google Scholar]
- 17.Eckstein F, Cassel D, Levkovitz H, Lowe M, Selinger Z. Guanosine 5′-O-(2-thiodiphosphate), an inhibitor of adenylate cyclase stimulation by guanine nucleotides and fluoride ions. J Biol Chem. 1979;254:9829–9834. [PubMed] [Google Scholar]
- 18.Fisahn A, Pike FG, Buhl EH, Paulsen O. Cholinergic induction of network oscillations at 40 Hz in the hippocampus in vitro. Nature. 1998;394:186–189. doi: 10.1038/28179. [DOI] [PubMed] [Google Scholar]
- 19.Fisahn A, Yamada M, Duttaroy A, Gan J-W, Deng C-X, McBain CJ, Wess J. Muscarinic induction of hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents. Neuron. 2002;33:615–624. doi: 10.1016/s0896-6273(02)00587-1. [DOI] [PubMed] [Google Scholar]
- 20.Fischer Y, Gahwiler BH, Thompson SM. Activation of intrinsic hippocampal theta oscillations by acetylcholine in rat septo-hippocampal cocultures. J Physiol (Lond) 1999;519:405–413. doi: 10.1111/j.1469-7793.1999.0405m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzjohn SM, Bortolotto ZA, Palmer MJ, Doherty AJ, Ornstein PL, Schoepp DD, Kingston AE, Lodge D, Collingridge GL. The potent mGlu receptor antagonist LY341495 identifies roles for both cloned and novel mGlu receptors in hippocampal synaptic plasticity. Neuropharmacology. 1998;37:1445–1458. doi: 10.1016/s0028-3908(98)00145-2. [DOI] [PubMed] [Google Scholar]
- 22.Fraser DD, MacVicar BA. Cholinergic-dependent plateau potential in hippocampal CA1 pyramidal neurons. J Neurosci. 1996;16:4113–4128. doi: 10.1523/JNEUROSCI.16-13-04113.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- 24.Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 1999;2:358–363. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- 25.Guérineau NC, Bossu J-L, Gahwiler BH, Gerber U. Activation of a nonselective cationic conductance by metabotropic glutamatergic and muscarinic agonists in CA3 pyramidal neurons of the rat hippocampus. J Neurosci. 1995;15:4395–4407. doi: 10.1523/JNEUROSCI.15-06-04395.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hajos N, Katona I, Naiem SS, Mackie K, Ledent C, Mody I, Freund TF. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci. 2000;12:3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- 27.Halliwell JV, Adams PR. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res. 1982;250:71–92. doi: 10.1016/0006-8993(82)90954-4. [DOI] [PubMed] [Google Scholar]
- 28.Hoffman AF, Lupica CR. Mechanisms of cannabinoid inhibition of GABAA synaptic transmission in the hippocampus. J Neurosci. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howe AR, Surmeier DJ. Muscarinic receptors modulate N-, P-, and L-type Ca2+ currents in rat striatal neurons through parallel pathways. J Neurosci. 1995;15:458–469. doi: 10.1523/JNEUROSCI.15-01-00458.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ledent C, Valverde O, Cossu G, Petitet F, Aubert J-F, Beslot F, Bohme GA, Imperato A, Pedrazzini T, Roques BP, Vassart G, Fratta W, Parmentier M. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science. 1999;283:401–404. doi: 10.1126/science.283.5400.401. [DOI] [PubMed] [Google Scholar]
- 32.Lenz RA, Alger BE. Calcium dependence of depolarization-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J Physiol (Lond) 1999;521:147–157. doi: 10.1111/j.1469-7793.1999.00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levey AI, Edmunds SM, Koliatsos V, Wiley RG, Heilman CJ. Expression of m1–m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation. J Neurosci. 1995;15:4077–4092. doi: 10.1523/JNEUROSCI.15-05-04077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Llano I, Leresche N, Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- 35.Madison DV, Lancaster B, Nicoll RA. Voltage clamp analysis of cholinergic action in the hippocampus. J Neurosci. 1987;7:733–741. doi: 10.1523/JNEUROSCI.07-03-00733.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maejima T, Hasimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- 37.Martin LA, Alger BE. Muscarinic facilitation of the occurrence of depolarization-induced suppression of inhibition in rat hippocampus. Neuroscience. 1999;92:61–71. doi: 10.1016/s0306-4522(98)00745-3. [DOI] [PubMed] [Google Scholar]
- 38.Martin LA, Wei D-S, Alger BE. Heterogeneous susceptibility of GABAA receptor-mediated IPSCs to depolarization-induced suppression of inhibition in rat hippocampus. J Physiol (Lond) 2001;532:685–700. doi: 10.1111/j.1469-7793.2001.0685e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morishita W, Alger BE. Evidence for endogenous excitatory amino acids as mediators in DSI of GABAAergic transmission in hippocampal CA1. J Neurophysiol. 1999;82:2556–2564. doi: 10.1152/jn.1999.82.5.2556. [DOI] [PubMed] [Google Scholar]
- 40.Morishita W, Alger BE. Direct depolarization and antidromic action potentials transiently suppress dendritic IPSPs in hippocampal CA1 pyramidal cells. J Neurophysiol. 2001;85:480–484. doi: 10.1152/jn.2001.85.1.480. [DOI] [PubMed] [Google Scholar]
- 41.Morishita W, Kirov SA, Alger BE. Evidence for metabotropic glutamate receptor activation in the induction of depolarization-induced suppression of inhibition in hippocampal CA1. J Neurosci. 1998;18:4870–4882. doi: 10.1523/JNEUROSCI.18-13-04870.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicoll RA, Alger BE. A simple chamber for recording from submerged brain slices. J Neurosci Methods. 1981;4:153–156. doi: 10.1016/0165-0270(81)90049-2. [DOI] [PubMed] [Google Scholar]
- 43.Ohno-Shosaku T, Sawada S, Yamamoto C. Properties of depolarization-induced suppression of inhibitory transmission in cultured rat hippocampal neurons. Pflügers Arch. 1998;435:273–279. doi: 10.1007/s004240050512. [DOI] [PubMed] [Google Scholar]
- 44.Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 45.Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M. Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci. 2002;15:953–961. doi: 10.1046/j.1460-9568.2002.01929.x. [DOI] [PubMed] [Google Scholar]
- 46.Pan X, Ikeda SR, Lewis DL. SR 141716A acts as an inverse agonist to increase neuronal voltage-dependent Ca2+ currents by reversal of tonic CB1 cannabinoid receptor activity. Mol Pharmacol. 1998;54:1064–1072. doi: 10.1124/mol.54.6.1064. [DOI] [PubMed] [Google Scholar]
- 47.Panikashvili D, Simeondiou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shomani E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2002;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- 48.Paris S, Pouyssegur J. Guanosine 5′-O-(3-thiotriphosphate) and guanosine 5′-O-(2-thiodiphosphate) activate G proteins and potentiate fibroblast growth factor-induced DNA synthesis in hamster fibroblasts. J Biol Chem. 1990;265:11567–11575. [PubMed] [Google Scholar]
- 49.Piacentini L, Niroomand F. Phosphotransfer reactions as a means of G protein activation. Mol Cell Biochem. 1996;157:59–63. doi: 10.1007/BF00227881. [DOI] [PubMed] [Google Scholar]
- 50.Piomelli D, Giuffrida A, Calignano A, Rodriguez de Fonseca F. The endocannabinoid system as a target for therapeutic drugs. Trends Pharmacol Sci. 2000;21:218–224. doi: 10.1016/s0165-6147(00)01482-6. [DOI] [PubMed] [Google Scholar]
- 51.Pitler TA, Alger BE. Cholinergic excitation of GABAergic interneurons in the rat hippocampal slice. J Physiol (Lond) 1992a;450:127–142. doi: 10.1113/jphysiol.1992.sp019119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pitler TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci. 1992b;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pitler TA, Alger BE. Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G protein involvement in a presynaptic mechanism. Neuron. 1994;13:1447–1455. doi: 10.1016/0896-6273(94)90430-8. [DOI] [PubMed] [Google Scholar]
- 54.Shapiro MS, Loose MD, Hamilton SE, Nathanson NM, Gomeza J, Wess J, Hille B. Assignment of muscarinic receptor subtypes mediating G-protein modulation of Ca2+ channels by using knockout mice. Proc Natl Acad Sci USA. 1999;96:10899–10904. doi: 10.1073/pnas.96.19.10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stella N, Piomelli D. Receptor-dependent formation of endogenous cannabinoids in cortical neurons. Eur J Pharmacol. 2001;425:189–196. doi: 10.1016/s0014-2999(01)01182-7. [DOI] [PubMed] [Google Scholar]
- 56.Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- 57.van der Stelt M, Veldhuis WB, van Haaften GW, Fezza F, Bisogno T, Bar PR, Veldink GA, Vliegenthart JFG, Di Marzo V, Nicolay K. Exogenous anandamide protects rat brain against acute neuronal injury in vivo. J Neurosci. 2001;21:8765–8771. doi: 10.1523/JNEUROSCI.21-22-08765.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Varma N, Carlson GC, Ledent C, Alger BE. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci 21 2001. RC188(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- 60.Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- 61.Yan Z, Surmeier DJ. Muscarinic (m2/m4) receptors reduce N- and P-type Ca2+ currents in rat neostriatal cholinergic interneurons through a fast, membrane-delimited, G-protein pathway. J Neurosci. 1996;16:2592–2604. doi: 10.1523/JNEUROSCI.16-08-02592.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]