

Abstract
The present study uses the embryonic chick to examine in vivo the mechanisms and regulation of Schwann cell programmed cell death (PCD) in spinal and cranial peripheral nerves. Schwann cells are highly dependent on the presence of axons for survival because thein ovo administration of NMDA, which excitotoxically eliminates motoneurons and their axons by necrosis, results in a significant increase in apoptotic Schwann cell death. Additionally, pharmacological and surgical manipulation of axon numbers also affects the relative amounts of Schwann cell PCD. Schwann cells undergoing both normal and induced PCD display an apoptotic-like cell death, using a caspase-dependent pathway. Furthermore, axon elimination results in upregulation of the p75 and platelet-derived growth factor receptors in mature Schwann cells within the degenerating ventral root. During early development, Schwann cells are also dependent on axon-derived mitogens; the loss of axons results in a decrease in Schwann cell proliferation. Axon removal during late embryonic stages, however, elicits an increase in proliferation, as is expected from these more differentiated Schwann cells. In rodents, Schwann cell survival is regulated by glial growth factor (GGF), a member of the neuregulin family of growth factors. GGF administration to chick embryos selectively rescued Schwann cells during both normal PCD and after the loss of axons, whereas other trophic factors tested had no effect on Schwann cell survival. In conclusion, avian Schwann cells exhibit many similarities to mammalian Schwann cells in terms of their dependence on axon-derived signals during early and later stages of development.
Keywords: Schwann cells, programmed cell death, chick embryo, proliferation, peripheral nerves, neuregulin
During nervous system development, programmed cell death (PCD) is a process that entails the selective elimination of large numbers of initially overproduced neurons. The death of these neurons, primarily by apoptosis, has conventionally been attributed to a failure in competition for limited amounts of target-derived trophic factors. Consequently, PCD promotes construction of the functionally mature nervous system by establishing appropriate neuronal population sizes and proper matching of their connections (for review, see Burek and Oppenheim, 1996). Throughout the past several decades, extensive work has been done regarding the mechanisms and regulation of PCD in neurons. More recently, however, the study of PCD in glia of the CNS and in Schwann cells, the glia of the peripheral nervous system (PNS), has begun to receive attention in the context of establishing quantitative matching between neurons and ensheathing or myelinating glia.
Schwann cell PCD has primarily been studied in the rodent, in which it has been demonstrated that, in vitro, both embryonic precursors (Jessen et al., 1994; Gavrilovic et al., 1995; Dong et al., 1995, 1999), and early postnatal Schwann cells (Syroid et al., 1996,1999; Nakao et al., 1997) undergo apoptosis when deprived of growth factors. Complementary in vivo work has also shown the presence of early postnatal Schwann cell death and its regulation by axon-derived trophic factors (Grinspan et al., 1996; Trachtenberg and Thompson, 1996). These studies have proposed that, during development, early Schwann cells compete for limited supplies of trophic factors produced by their target axons, where the losers undergo PCD in an effort to establish proper Schwann cell numbers for achieving an optimal ratio of Schwann cells to axons.
The survival of developing mammalian Schwann cells is regulated by a variety of factors, but responsiveness depends on their developmental age. Whereas mature Schwann cells rely on the autocrine production of factors such as insulin-like growth factor-1, neurotrophin-3, and platelet-derived growth factor-BB (Meier et al., 1999; Syroid et al., 1999), Schwann cell survival during earlier stages of development is primarily regulated by growth factors known as the neuregulins. Through alternative splicing of the NRG-1 gene (Marchionni et al., 1993), many neuregulins, such as glia growth factor (GGF), can influence Schwann cell survival. Specifically, neuregulins increase the survival of Schwann cells in vitro when cultured in the absence of serum (Dong et al., 1995; Syroid et al., 1996), and in vivo when administered after neonatal axotomy (Grinspan et al., 1996;Trachtenberg and Thompson, 1996; Kopp et al., 1997).
In the present study we investigated Schwann cell PCD in the developing chick embryo, which, in contrast to mammals, provides a more advantageous model for studying embryonic Schwann cells in vivo during even the earliest stages of development and with the greater possibility of experimental manipulations. Previous studies of the chick embryo have described the existence of Schwann cell death in the spinal ventral roots (Chu-Wang and Oppenheim, 1978; Ciutat et al., 1996), and suggested that axons were necessary for survival. The current study further examines the mechanisms and regulation of Schwann cell PCD in the ventral root and oculomotor nerve. The results indicate that Schwann cells undergo an apoptotic-like programmed cell death that is regulated by axons and neuregulin signaling.
MATERIALS AND METHODS
Eggs and embryos
Fertilized eggs were purchased from local suppliers (COPAGA, Lleida, Catalonia, Spain; Tyson Farms, North Wilksboro, NC) and incubated in the laboratory at 37°C and 60% relative humidity until the desired embryonic age. The age of embryos was determined according to the Hamburger and Hamilton (1951) stage series.
Embryonic surgery and pharmacological experiments
Limb bud removal. In one group of embryos, the unilateral removal of the right hindlimb bud was performed on embryonic day 2 (E2) (stage 16), using a procedure described previously (Chu-Wang and Oppenheim, 1978; Caldero et al., 1998). Briefly, embryos were exposed through a window in the shell, and small strips of neutral red-agar were applied briefly over the area to be operated to obtain better visualization. The hindlimb bud was then removed with a flame-sharpened tungsten needle. Only embryos later determined to have a complete hindlimb deletion were used for further analysis. The side contralateral to the limb removal, together with sham limb bud removals (LBRs), were used as controls to compare the effects of the ablation on Schwann cell survival.
Neural tube removal. The neural tube was removed from embryos on E3–3.5 (stage 20–21) as previously described (Landmesser and Honig, 1986; Wang and Scott, 2000). Briefly, motoneurons were eliminated by aspirating the neural tube when neural crest migration was primarily complete, thereby leaving sensory ganglia intact. In some embryos only partial ablation of the neural tube was achieved, leaving one side of the ventral neural tube intact. These embryos were included in the analysis of Schwann cell death by using the intact ventral neural tube as a control.
Curare treatments. Some embryos were treated daily from E3 to E7 with d-tubocurarine (curare; Sigma, St. Louis, MO). The doses of curare were: 1 mg on E3 and E4, 1.5 mg on E5, and 2 mg on E6 and E7.
NMDA treatments. In another set of experiments, embryos of different ages were treated with a single dose of 1–2.5 mg of NMDA (Sigma). We have previously shown that 0.5–1 mg of NMDA is sufficient to produce massive axonal degeneration and necrotic motoneuron death (Ciutat et al., 1996). Embryos were killed at different times after NMDA application.
BAF treatments. Embryos were treated with a 40 μg dose of Boc-Asp-CH2F (BAF, Enzyme Systems Products, Livermore, CA) on E7 to inhibit caspase activation.
Trophic factor treatments. In some experiments, embryos were treated on E5 and E6 with a 5 μg dose of one of the following factors: (1) brain-derived neurotrophic factor (BDNF; Amgen, Inc., Thousand Oaks, CA), (2) insulin-like growth factor-1 (IGF-1; Cephalon, Inc., West Chester, PA), (3) human recombinant basic fibroblast growth factor (bFGF; California Biotechnology, Inc., Mountain View, CA), (4) nerve growth factor (NGF; a generous gift from Dr. Eugene Johnson), (5) neutralizing antibody to p75, a generous gift from Dr. Louis Reichardt), (6) neutralizing antibody to NGF (a generous gift from Dr. Yves Barde), and human recombinant GGF (a generous gift from Dr. Mark Marchionni, Cambridge Neuroscience, Boston, MA).
The neurotoxins and trophic factors were dissolved in saline and dropped directly onto the choroallantoic membrane (CAM; volumes of 100–200 μl) through a small window made in the shell. Embryos treated with identical volumes of physiological saline were used as controls. After treatments, the windows were sealed with adhesive tape, and the eggs were returned to the incubator.
Histology and cell counts
Light microscopy. Embryos were fixed overnight in Carnoy's solution and processed for paraffin embedding. Serial transverse sections of 6–12 μm were obtained through both the entire lumbosacral segments of spinal cord for ventral root analysis and the rostral hindbrain for oculomotor nerve analysis, and both were stained with thionin. By using either a 60× or 100× oil-immersion objective, all normal pyknotic cells and all normal nuclear profiles present in a segment of the L3 ventral nerve roots located between their exit from the spinal cord and their contact with the dorsal root ganglia were drawn with a camera lucida and then counted in alternate serial sections. In the hindbrain, the same quantification of pyknotic and normal nuclei was done in the oculomotor nerve using a 100× oil-immersion objective. For E6–E8 embryos, the entire length of both bilateral oculomotor nerves was quantified in every other section. For E9–E16 embryos, which had very large oculomotor nerves, pyknotic and normal nuclei within a single field of view at the exit point of the nerves from the hindbrain were quantified for both oculomotor nerves (E9–E10) or for a single nerve (E12–E14) within every other section. The criteria used to identify pyknotic cells were the same as those described by Clarke and Oppenheim (1995).
Electron microscopy. Embryos were fixed by immersion in 2.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, overnight at 4°C. After washing with phosphate buffer, ventral or dorsal roots or the most proximal part of the sciatic nerves were dissected, removed, and cut into small pieces. Samples were then post-fixed in 1% osmium tetroxide for 2 hr, dehydrated, and embedded in Durcupan ACM (Fluka, Buchs, Switzerland). Ultrathin sections were collected on copper grids, counterstained with uranyl acetate and lead citrate, and observed with a Zeiss EM 910 electron microscope (Zeiss, Oberkochen, Germany).
Immunohistochemistry
For immunohistochemistry, embryos were fixed by immersion in 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, overnight at 4°C. After cryoprotection with 30% sucrose in PB, embryos were embedded in Tissue-Tek OCT embedding medium (Miles, Elkhart, IN), frozen, and transversely sectioned (20 μm thick) with a cryostat. Sections were washed in PBS containing 0.1% Triton X-100 and incubated for 30 min with 10% normal goat serum (NGS; Sigma and Vector Laboratories, Burlingame, CA) in PBS to prevent nonspecific staining.
To specifically immunolabel the Schwann cells, sections were incubated with the 1E8 monoclonal antibody (a generous gift from Dr. Nancy Ratner, University of Cincinnati Medical School and the Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), which recognizes chick P0 protein, an early marker of embryonic Schwann cells (Bhattacharyya et al., 1991) (diluted 1:10–1:100 in PBS-0.1% Triton X-100). Sections were then incubated sequentially with either (1) biotin-labeled anti-mouse IgG (diluted 1:200 in PBS) for 1 hr and rhodamine–avidin DCS (diluted 1:100 in PBS; Vector Laboratories), or (2) Cy3-conjugated AffiniPure goat anti-mouse IgG (H + L) (Jackson ImmunoResearch Laboratories, West Grove, PA). Some sections were further treated for simultaneous demonstration of P0 and DNA fragmentation by using terminal deoxynucleotidyl transferase-mediated dUTP-digoxigenin nick-end labeling (TUNEL) technique (fluorescent in situdetection kit for cell death from Boehringer Mannheim, Mannheim, Germany). For this procedure sections were incubated for 60 min at 37°C in a mixture containing terminal deoxynucleotidyl transferase and FITC-labeled dUTP according to the manufacturer's instructions. Sections were examined in a Zeiss LSM 310 confocal microscope (Zeiss, Oberkochen, Germany) using 543 nm helium–neon or 488 argon–ion excitation sources and appropriate selective barrier filters.
In other experiments, double label immunohistochemistry was performed with the 1E8 primary antibody (diluted 1:10) in conjunction with one of the following primary antibodies: (1) rabbit polyclonal IgG against c-Jun/sc45 (Santa Cruz Biotechnology; diluted 1:200) or (2) rabbit polyclonal IgG against cleaved–activated caspase 3 (New England Biolabs, Beverly, MA; diluted 1:200). Primary antibody dilutions were done in PBS-0.1% Triton X-100 and incubated at 4°C overnight. After washing in PBS, sections were incubated with the appropriate secondary antibody, either Cy3-conjugated AffiniPure goat anti-mouse IgG (H + L) (Jackson ImmunoResearch Laboratories, West Grove, PA) or Alexa 488 goat anti-rabbit IgG (H + L) conjugate (Molecular Probes, Eugene, OR; diluted 1:200) for 1 hr at room temperature. After staining, sections were washed, dehydrated, coverslipped, and analyzed on an Olympus America (Melville, NY) BX60 epifluorescent microscope with the appropriate fluorescent filters.
Some sections were also processed for immunocytochemistry by using the following primary antibodies: (1) monoclonal antibody against p75NGFR (a generous gift from Dr. Louis Reichardt; diluted 1:100), (2) rabbit polyclonal IgG against platelet-derived growth factor receptor-α (sc-338, Santa Cruz Biotechnology; diluted 1:200), (3) monoclonal antibody against microtubule-associated protein-5 (MAP-5; Boehringer Mannheim; diluted 1:50) or (4) rabbit polyclonal IgG against c-Jun/sc45 (Santa Cruz Biotechnology; diluted 1:200). Primary antibody dilutions were done in PBS–0.1% Triton X-100. Incubation was performed overnight at 4°C. After washing in PBS, sections were incubated with the appropriate secondary antibodies: fluorescein isothiocyanate (FITC)-labeled goat anti-mouse IgG (Southern Biotechnology Associates, Birmingham, AL; diluted 1:50) or FITC-labeled goat anti-rabbit IgG (Southern Biotechnology Associates; diluted 1:50). After washing, sections were counterstained with propidium iodide (5 μg/ml in PBS containing 100 μl/ml of ribonuclease type I-A; Sigma). Sections were examined in aZeiss LSM 310 confocal microscope (Zeiss, Oberkochen, Germany) using 543 nm helium–neon or 488 argon ion excitation sources and appropriate selective barrier filters.
Mitotic activity
To examine the proliferation of cells in the L3 ventral nerve roots of normal embryos and in embryos treated with either saline or NMDA, 5-bromo-2′-deoxyuridine (BrdU; Sigma) was used to label cells in S-phase of the mitotic cycle. A pulse of a nontoxic dose of BrdU (10 μg on E4–E10, 20 μg on E12 or 30 μg on E16) was administered onto the CAM. Two hours later embryos were killed, staged, fixed overnight in Carnoy's solution, embedded in paraffin, and serially sectioned at 6 μm (E4–E7), 8 μm (E8–E10), or 12 μm(E12 and E16). Sections were deparaffinized, treated with 0.07N NaOH for 2 min, rinsed in 0.1 m PBS, pH 8.5, and incubated with a mouse monoclonal anti-BrdU antibody (Becton Dickinson, Mountain View, CA) diluted 1:20 in 1% normal horse serum (NHS; Sigma)–PBS–0.1% Triton X-100. After washes in distilled H2O and PBS, sections were incubated with a biotin-conjugated goat anti-mouse IgG (Vector Laboratories) diluted 1:200 in 1% NHS–PBS–0.1% Triton X-100. After several washes in PBS, sections were incubated with the ABC complex (ABC elite; Vector Laboratories) according to manufacturer's instructions. Peroxidase activity was visualized by using 3,3′-diaminobenzidine–H2O2procedure. Sections were finally counterstained with hematoxylin. All heavily labeled BrdU cells and normal nuclei present in areas of 1 mm2 of the L3 ventral nerve root were counted in every other section with the help of a camera lucida. To analyze the BrdU incorporation in cells of the L3 ventral nerve roots of embryos treated with either saline or NMDA, a pulse of BrdU was administered 2 hr before killing the embryos. In some cases, paraffin sections from embryos fixed with 4% paraformaldehyde in 0.1m phosphate buffer, were double stained with TUNEL and BrdU. In this case, after the TUNEL procedure, sections were treated with 0.1% protease type XXIV (Sigma) in PBS at 37°C for 10 min. before incubation with the mouse monoclonal anti-BrdU antibody; a tetramethylrhodamine isothiocyanate-conjugated goat anti-mouse Ig (H + L) (Southern Biotechnology Associates) diluted 1:50 in 1% NHS–PBS–0.1% Triton X-100 was used as a secondary antibody.
In one experiment, E7.5 tissue containing sections of the hindbrain processed with thionin staining were examined, and mitotic figures were quantified in every other section throughout the entire length of both bilateral oculomotor nerves. Mitotic figures were present in all thionin-stained tissue and exhibit darkly stained chromatin arranged in formations characteristic of discrete phases of the mitotic cycle. The thionin-labeled mitotic figures were quantified instead of using BrdU analysis because this allowed for the identification of proliferating Schwann cells throughout many phases of the mitotic cycle, and not just the S-phase, which BrdU selectively labels. Additionally, mitotic figures were quantified in the oculomotor nerves in the same sections used for the analysis of pyknotic and normal cell numbers from the E7.5 embryos that received saline, NMDA, or GGF (see Fig. 11); processing tissue for BrdU precludes such an analysis.
Fig. 11.
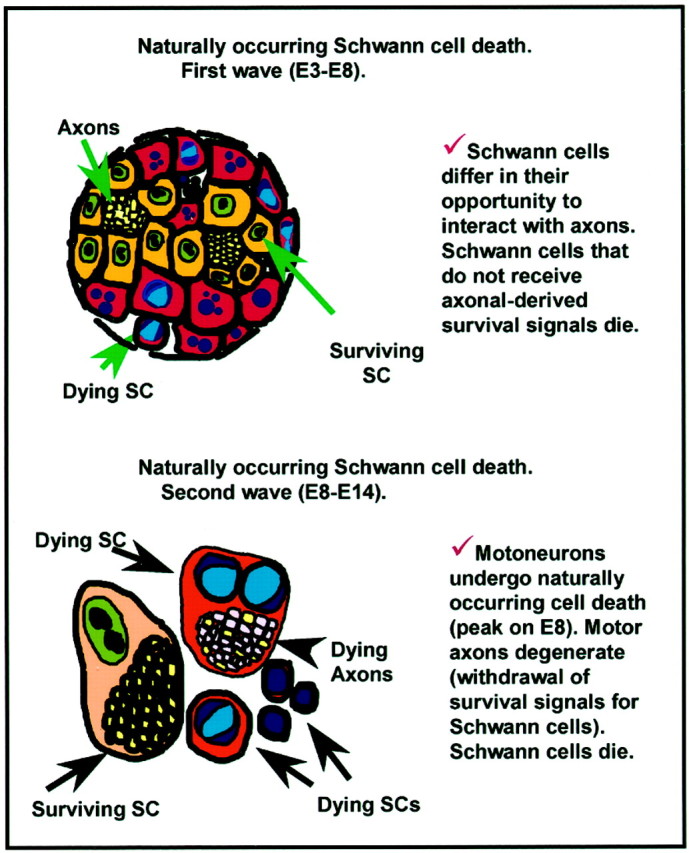
Possible models of axon regulation of Schwann cell PCD based on the present experiments and available literature.
RESULTS
NMDA treatment induces Schwann cell death in the ventral nerve root and oculomotor nerve
It has previously been shown that embryonic chick Schwann cells undergo a developmentally regulated period of PCD in the lumbar (L3) ventral nerve roots, exhibiting a biphasic profile with peaks of cell loss on E5–6 and E8.5 (Fig.1A, taken from Ciutat et al., 1996). Furthermore, Schwann cell death could be augmented byin ovo treatment of 1 mg of NMDA on E7, which excitotoxically eliminated spinal motoneurons and caused the degeneration of their respective axons (Ciutat et al., 1996). In the present study, the effects of NMDA on Schwann cell death were further examined to demonstrate the critical role of axons in regulating embryonic Schwann cell survival. First, in serially sectioned Nissl-stained tissue, the relative density of Schwann cells that displayed a pyknotic morphology was quantified after NMDA administration at several developmental stages. As shown in Figure1B, Schwann cell death in the L3 ventral roots is greatest at E10.5 after a single dose of NMDA (2.5 mg) given 12 hr earlier. Subsequent ages show a gradual decrease in Schwann cell death induced after NMDA with little, if any death occurring by E16.5 (Fig.1B), despite massive loss of motoneurons and their axons by necrosis (data not shown) at all ages. Additionally, the time course of Schwann cell death at E10 was also determined by quantifying pyknotic Schwann cells at several time intervals after NMDA treatment (2.5 mg) at E10. Relatively low levels of Schwann cell death were observed up to 9 hr after NMDA treatment. Subsequently a large increase in cell death occurred with a peak at 12 hr, followed by a decrease in cell death over the next 12 hr (Fig. 1C). The association of dying Schwann cells and degenerating ventral root axons can be seen in sections immunostained with an antibody to the axonal marker MAP-5 and counterstained with propidium iodine 7 hr after NMDA treatment on E7 (Fig. 1D,E). Dying Schwann cells, displaying condensed chromatin, can often be found associated with defasciculated axons after NMDA treatment (Fig. 1D). Embryos treated with saline, however, exhibit equivalent amounts of cell death to untreated nerves (Fig. 1E).
Fig. 1.

Schwann cell death in L3 ventral roots of the embryonic chick after NMDA administration. Normally occurring Schwann cell death in the developing chick embryo displays a biphasic profile with peaks of death at E5–6 and E8.5 (p < 0.01 vs E8) (A, Ciutat et al., 1996). Induction of Schwann cell death in L3 ventral nerve root 12 hr after acute NMDA administration (2.5 mg) illustrates the developmentally regulated responsiveness of Schwann cells to NMDA (B). The time course of Schwann cell death induction after administration of NMDA (2.5 mg) on E10 (C); embryos taken at different time points after NMDA treatment show a peak of cell death after 12 hr, followed by a decrease over the next 12 hr. Ventral nerve root of E7 chick embryos immunostained with the axonal marker MAP-5 (green) and counterstained with propidium iodide (red) in embryos either treated with saline (E) or NMDA (D) 7 hr earlier. Below the ventral root there is a single degenerating axon segregated from the main ventral root fascicle associated with several dying Schwann cells (arrows). The circle delineates a dying Schwann cell with hemilunar caps of condensed chromatin, characteristic of apoptosis. SC, Spinal cord;VR, ventral root. In A, each point represents the mean ± SEM of 4–10 embryos, and inB and C, each point represents the mean ± SEM, with the number of embryos indicated.
Ultrastructural analysis provides additional evidence for the presence of embryonic Schwann cell PCD occurring both naturally during development, and experimentally induced by the loss of axons following NMDA treatment. At E3 and E6 healthy Schwann cells can be seen in close association with normal axons (Fig.2A,C). Schwann cells undergoing normal PCD are also observed at these ages, and they exhibit chromatin condensation, often in the form of peripheral crescents (Fig.2D), as well as an electron-dense cytoplasm and nuclear degradation (Fig. 2B). These naturally dying Schwann cells are often found to lack direct axon contact (Fig.2B). At E10.5, after a 1 mg dose of NMDA on E10, numerous dying Schwann cells, similar to those dying in normal or saline-treated embryos, could be found at the electron microscopic level. In contrast to the healthy Schwann cells associated with surviving axons in E10.5 saline-treated tissue (Fig.2E), NMDA-treated embryos exhibited dying Schwann cells, often in close association with degenerating axon bundles, which exhibit focal swellings, varicosities, and other regressive changes (Fig. 2F–H).
Fig. 2.
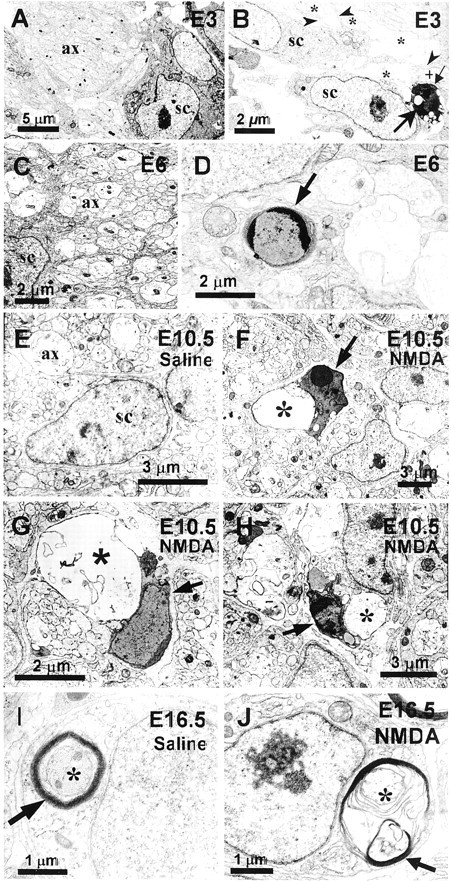
Ultrastructural analysis of Schwann cell death during normal development and after NMDA administration. Healthy Schwann cells (sc) are found associated with axons (ax) at E3 (A) and E6 (C). A Schwann cell undergoing PCD at E3 (large arrow) (B), exhibiting nuclear degradation and condensed chromatin, is separated from a healthy axon bundle (+) by a cellular process (small arrow), whereas, healthy Schwann cells (sc) are in close contact with normal bundles of axons (*) and in some cases extend processes between axons (arrowheads). Schwann cells undergoing PCD are also observed at E6 (D). Whereas E10.5 saline-treated embryos display healthy Schwann cells associated with normal axons bundles (ax) (E), dying Schwann cells (arrows) can often be found at E10.5, after NMDA treatment at E10, in close association with degenerating axons (*) (F–H). At E16.5 in saline-treated embryos, Schwann cells can be seen as ensheathing (arrow) normal axons (*) (I). However, at E16.5, after NMDA treatment on E16, Schwann cells exhibit the morphology of Wallerian degeneration, in which deteriorating myelin (arrow) can be seen around a degenerating axon (*) (J).
Although NMDA treatment on E16 also results in the elimination of motoneurons (Llado et al., 1999) and the degeneration of their axons by E16.5 (Fig. 2J), Schwann cells responded differently at this age (vs E10). Myelination begins in the chick around E14 (Saxod and Bouvet, 1982), and many Schwann cells at E16.5 in the L3 ventral root ensheath axons with multiple layers of myelin (Fig.2I). After axon loss by NMDA treatment, E16.5 Schwann cells exhibit myelin degeneration (Fig. 2J), but do not undergo cell death. This response by Schwann cells is characteristic of their response during Wallerian degeneration (Muller and Stoll, 1998) and supports the idea that these Schwann cells have differentiated sufficiently to maintain their own survival through autocrine mechanisms (Jessen and Mirsky, 1999; Meier et al., 1999).
To determine the generality of our observations regarding Schwann cell PCD, we have also examined a cranial motor nerve in the chick embryo, the oculomotor nerve (nIII). This nerve was chosen because of its large size and accessibility in the developing chick (Fig.3A). The glial cells in this nerve are of the Schwann cell lineage because they are reliably immunolabeled with the avian-specific 1E8 antibody (Fig.3B), which identifies the P0 protein expressed both in early and mature chick Schwann cells (Bhattacharyya et al., 1991). Initially, Schwann cells in the oculomotor nerve were analyzed in serial-sectioned Nissl-stained tissue at several developmental stages to determine the period of naturally occurring cell death. Here, as with ventral root Schwann cells, there are two distinguishable peaks of cell death, one occurring first around E6, and a smaller peak at E14 (Fig. 3C). In the developing chick, the normal period of oculomotoneuron PCD occurs between E10 and E15 (Steljes et al., 1999). Therefore, Schwann cell PCD in the oculomotor nerve, similar to the ventral root, occurs both before and during oculomotor neuron PCD.
Fig. 3.

Naturally occurring and NMDA-induced Schwann cell death in the embryonic chick oculomotor nerve. The bilateral chick oculomotor nerves (arrows) exit the hindbrain and are easily accessible at E8 (A). Schwann cells of the oculomotor nerve express the avian-specific homolog of the Schwann cell specific P0 protein (B), and display a developmentally regulated period of PCD with peaks at E6 and E14 (C). NMDA treatment induces a 75-fold increase in the relative density of dying Schwann cells (D). Each point in C and D indicates the mean ± SEM for three to seven embryos. Oculomotor nuclei in E7.5 saline-treated embryos (enclosed by dotted line, E) exhibit motoneuron PCD (arrow, F), as well as Schwann cell PCD in the nerve (arrowheads, G). NMDA treatment, however, induced an excitotoxic loss of motoneurons in all oculomotor nuclei except the parasympathetic preganglionic neurons of the accessory oculomotor nucleus (arrows, H), which exhibit characteristics of a necrotic death (I), such as hyperchromatic nuclei, cytoplasmic swelling and vacuolization, and loss of Nissl substance. After NMDA administration at E7, extensive numbers of pyknotic Schwann cells (arrowheads) can be seen throughout the E7.5 oculomotor nerve (J).
Similar to lumbar spinal motoneurons, the oculomotoneurons are also affected by NMDA administration, exhibiting massive cellular loss in all oculomotor nuclei, except in the preganglionic neurons within the chick accessory oculomotor nucleus, the avian homolog of the mammalian Edinger-Westphal nucleus (Fig. 3H). Compared with saline controls (Fig. 3E), which exhibit normal apoptotic oculomotoneuron PCD (Fig. 3F), E7.5 oculomotoneurons, after NMDA (1 mg) treatment at E7, display cytoplasmic swelling, loss of Nissl substance, and hyperchromatic nuclei, which are all characteristics of a necrotic cellular death (Fig.3I). Schwann cells of the oculomotor nerve are also affected indirectly by NMDA (via axon loss), demonstrating a 75-fold increase in the relative density of dying (pyknotic) Schwann cells (Fig. 3D,J), as compared with saline-treated controls (Fig. 3D,G).
Axon presence influences Schwann cell death
To further address the role of axons in regulating Schwann cell PCD, several experiments were done in which axonal number was manipulated in the embryonic chick ventral root. It has long been appreciated that the removal of the embryonic limb bud, before axon innervation, will induce massive amounts of motoneuron PCD from E6–E10, because of the removal of target-derived trophic signals (Hamburger, 1958; Chu-Wang and Oppenheim, 1978; Caldero et al., 1998). In the present study, LBR was used to reduce the number of axons, present during later stages of the Schwann cell death period (E5–E9). The hindlimb bud was removed on embryonic day 2, and afterwards Schwann cell death was quantified in both the ipsilateral and contralateral ventral roots in serial-sectioned Nissl stained tissue. Whereas a normal density of Schwann cell death was observed from E5 to E6, a large increase in Schwann cell PCD occurred on E7.5 and E8.5 in the ipsilateral ventral nerve roots after LBR (Fig.4A), as compared with both the contralateral side and normal controls. Thus, decreasing the number of axons, specifically during the time of heightened motoneuron PCD (E6–E10) results in an increase in Schwann cell death during that same period.
Fig. 4.

Manipulation of axon number influences Schwann cell PCD. A reduction in the number of ventral root axons, after induced motoneuron PCD by limb bud removal at E2, results in an increase in Schwann cell PCD in the ipsilateral ventral roots at E7.5 (p < 0.001) and E8.5 (p < 0.01), as compared with Schwann cell PCD in the contralateral ventral roots of the same embryos (A). Conversely, an increased in the number of ventral root axons, following the prevention of motoneuron PCD by activity blockade with daily curare treatment from E3 to E7, results in a decrease in Schwann cell PCD on E9 (p < 0.01) and E10 (p < 0.03), as compared with age-matched saline-treated control embryos (B). Each point represents the mean ± SEM of 4–11 embryos.
To determine if an increase in the number of axons could influence Schwann cell PCD, neuronal activity in embryos was blocked with curare, a nicotinic acetylcholine receptor-blocking agent. Curare treatment has been shown to prevent normal motoneuron PCD from E6 to E10, thereby increasing the number of axons in the ventral root (Pittman and Oppenheim, 1978; Oppenheim et al., 2000). Embryos were treated daily with curare from E3 to E7, and the relative density of Schwann cell death was determined in the ventral roots in Nissl-stained tissue. Compared with control levels, normal amounts of Schwann cell death occurred from E5 to E8, however, on E9 and E10 when motoneuron and axon numbers are greatly increased, Schwann cell death was significantly decreased (Fig. 4B). Therefore, increasing the number of axons, by preventing motoneuron cell death from E6 to E10 results in a reduction of normal Schwann cell death during this time period. The differences in the timing of altered Schwann cell death after limb bud removal versus activity blockade (Fig. 4, compare A,B) are likely caused by the different methods used to perturb axon numbers.
Evidence that axons are critical for Schwann cell survival was also demonstrated by the removal of axons during the early peak period of ventral root Schwann cell PCD (E3–E6). To accomplish this, lumbar neural tubes were carefully removed from embryos on E3, resulting in the loss of ventral horn motoneurons and their axons, while leaving sensory ganglia intact (Fig.5B,F). Embryos with neural tube removal, as well as control embryos with intact neural tubes and ventral roots (Fig. 5A,E), were examined for changes in Schwann cell PCD. Other embryos in the analysis included those with incomplete neural tube removal, resulting in an intact contralateral ventral horn (Fig. 5C) and an ablated ipsilateral ventral horn (Fig. 5D). An increase in the number of pyknotic Schwann cells in the ipsilateral ventral roots could be seen within 1–2 hr after neural tube removal (Fig. 5). An even greater amount of Schwann cell death was observed 6–8 hr after neural tube removal (Fig. 5F,H,J), as compared with either control or contralateral ventral roots (Fig. 5E,G,J). Thus, removing axons during the early phase of Schwann cell PCD by way of neural tube removal also results in a significant increase in Schwann cell death.
Fig. 5.
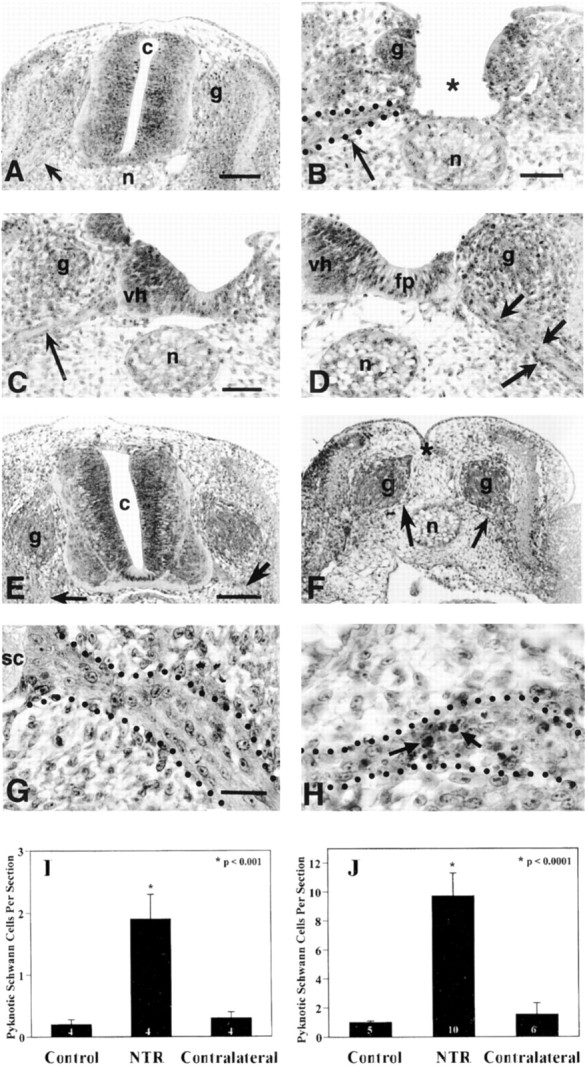
Neural tube removal at E3 in the chick embryo results in an increase in Schwann cell death in the spinal ventral roots. Compared with intact neural tubes displaying normal ventral roots (arrow, A), neural tubes surgically removed at E3, leave only the sensory ganglia (g) and ventral roots intact (arrow,B) by 2 hr after surgery. In some embryos, neural tube removal was incomplete and often resulted in one intact contralateral ventral horn with associated nerve root (arrow,C) and one ablated ipsilateral ventral horn (D), containing many pyknotic cells within its nerve root (arrows). Similar results were observed 6–8 hr (vs 1–2 hr) after neural tube removal on E3 (E–H). A control embryo with an intact neural tube (E) displays intact ventral roots (arrows), and an embryo 6–8 hr after complete neural tube removal exhibits intact sensory ganglia (g) and ventral roots (arrows,F). Little, if any, Schwann cell death is found in control embryo ventral roots (G), whereas 6–8 hr after complete neural tube removal there is a large increase in pyknotic Schwann cells (arrows,H) in the ventral roots. Quantification of pyknotic Schwann cells revealed a significant increase in the ipsilateral ventral roots as compared with both control and contralateral roots at 1–2 hr (I) and 6–8 hr (J) after neural tube removal. c, Central canal;g, sensory ganglia; n, notochord;vh, ventral horn; fp, floor plate;sc, spinal cord. In I andJ, the number in the barsrepresents sample size.
Schwann cells display an apoptotic-like cell death
To further examine if Schwann cell death is apoptotic, several experiments were conducted to identify apoptotic-related protein expression and morphological characteristics of dying Schwann cells during both normal development and after induction by NMDA administration. As previously described, E7.5 Schwann cells, immunolabeled with the 1E8 antibody, a Schwann cell marker, display DNA fragmentation, a hallmark of apoptosis (Gavrieli et al., 1992), when examined with the TUNEL technique after NMDA treatment on E7 (Fig.6A) (Ciutat et al., 1996). Many 1E8-labeled Schwann cells on E7.5 can also be identified by the sc45 antibody, which appears to label a specific downstream caspase-3 cleavage substrate, providing additional evidence for an apoptotic type of cell death (Fig. 6B) (Casas et al., 2001). Dying Schwann cells also exhibit an apoptotic morphology when conventionally stained sections are examined with the light microscope (Fig. 6C). Schwann cells in the oculomotor nerve of normal control embryos also undergo an apoptotic form of PCD. Many Schwann cells throughout the nerve at E8 are labeled with an antibody to the activated form of caspase-3, which is a common effector caspase activated in later stages of apoptosis (Fig. 6D) (Harvey and Kumar, 1998). Dying Schwann cells in the oculomotor nerve at E8 can also be identified by the sc45 antibody (Fig.6E). Finally, dying oculomotor nerve Schwann cells in Nissl-stained tissue also display a pyknotic morphology (Fig.6F).
Fig. 6.
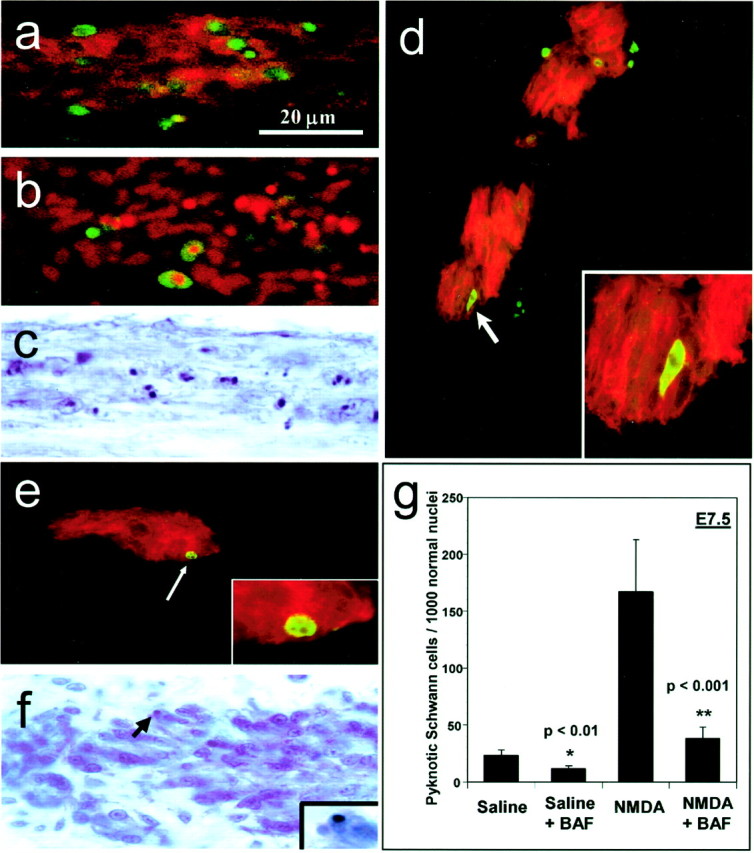
Schwann cells display characteristics of an apoptotic death in normal and NMDA-treated embryos and use a caspase-dependent pathway. Ventral root Schwann cells labeled with the 1E8 antibody (red) at E7.5, after treatment with NMDA on E7, display numerous TUNEL-positive cells (green), indicating DNA fragmentation (a). Also after NMDA treatment, dying Schwann cells are labeled with the sc45 antibody (green), which labels a downstream caspase-3 cleavage protein (b). Pyknotic Schwann cells in the ventral root, displaying darkly stained condensed nuclei are abundant in Nissl-stained tissue after NMDA treatment (c). In the oculomotor nerve, dying Schwann cells labeled with 1E8 (red) are colabeled with an antibody to activated caspase-3 (green) (d), as well as with the antibody to sc45 (green) (e). Normally dying Schwann cells in the oculomotor nerve in Nissl-stained tissue display a pyknotic morphology with darkly stained, condensed nuclear chromatin (arrow,f). Inset in fshows a pyknotic Schwann cell at higher magnification. Embryos were treated at E7 with either saline or NMDA, followed by a 40 μg dose of BAF to prevent caspase activation. Analysis at E7.5 showed significant reductions in Schwann cell death after BAF treatment in both saline-treated (p < 0.01) and NMDA-treated (p < 0.001) embryos (g). Bars represent the mean ± SEM for four to seven embryos. The scale ina is for a–f.
To further demonstrate that Schwann cells use an apoptotic pathway for cell death, embryos were treated in ovo with Boc-Asp-CH2F (BAF; 40 μg), a general caspase inhibitor. Embryos were initially treated with either saline or NMDA together with BAF on E7, and Schwann cell death was then examined at E7.5 in the spinal ventral roots. As assessed in serial sections of Nissl-stained tissue, BAF treatment significantly reduced the density of dying Schwann cells in both saline- and NMDA-treated embryos compared with controls without BAF treatment (Fig. 6G). Taken together, these data indicate that Schwann cells undergo PCD by an apoptotic, caspase-dependent pathway.
Schwann cells upregulate the PDGF receptor and the p75 receptor in response to degenerating axons
Early postnatal Schwann cells in the rat have been found to express both PDGF and the PDGF receptor (PDGF-R), which has been implicated as a potential mechanism for autocrine-mediated survival (Meier et al., 1999; Lobsiger et al., 2000). Additionally, after loss of axon contact, Schwann cells upregulate the low-affinity nerve growth factor receptor, p75, (Taniuchi et al., 1986; Lemke and Chao, 1988), which may also be involved in Schwann cell death postnatally (Soilu-Hänninen et al., 1999; Syroid et al., 2000). In the present study, the expression of the p75 receptor and the PDGF receptor were examined by immunolabeling in Schwann cells in the ventral root after NMDA treatment on E16. Embryos were given a 1 mg dose of NMDA on E16, and tissue from the ventral nerve roots was examined 48 and 96 hr later. The tissue was immunostained using either an antibody to PDGF-R, or an antibody to p75, and counterstained with propidium iodide to identify cell nuclei. Schwann cells associated with degenerating motor axons exhibit a cytoplasmic upregulation of the PDGF-R within 48 hr after NMDA treatment (Fig.7A,B) but do not show increased expression within the intact sensory fibers, which are not affected by NMDA (Fig. 7C). The p75 receptor exhibits a similar response, in which upregulation occurs in Schwann cells adjacent to the degenerating motor fibers 96 hr after NMDA treatment, but not in Schwann cells ensheathing normal sensory fibers (Fig.7D). Confocal analysis of the general pattern of immunostaining for both PDGF-R and p75 indicate that Schwann cells are the source of these receptors versus degenerating axons. These data indicate that, in response to axon loss, Schwann cells of late embryonic ages upregulate the PDGF and p75 receptors, which may be involved in an autocrine-mediated survival response, as previously shown in rodents (Meier et al., 1999; Syroid et al., 2000).
Fig. 7.

Mature Schwann cells upregulate the PDGF receptor and the p75 receptor in response to axon loss induced by NMDA treatment. Schwann cells, labeled with propidium iodine (red), upregulate the PDGF receptor (green) in the ventral root 48 hr after a 1 mg dose of NMDA on E16 (A). PDGF receptor immunoreactivity is increased in the cytoplasmic regions of elongated Schwann cells, often delineating nodes of Ranvier (arrow), adjacent to degenerating motor axons in the ventral root (B), but not in the dorsal root, which contain normally ensheathed sensory axons unaffected by NMDA (C). Schwann cells also exhibit upregulation of the p75 receptor (green) in the ventral root, but not in the dorsal root 96 hr after NMDA treatment at E16 (D). VR, Ventral root;DR, dorsal root; DRG, dorsal root ganglia.
Axons provide Schwann cells with a mitogenic signal
The following experiments were conducted to determine if Schwann cell proliferation, similar to PCD, is also regulated by axon-derived signals. Initially, the developmental profile of Schwann cell proliferation was established by using the mitotic marker, BrdU. The relative density of Schwann cells heavily labeled with BrdU, compared with quiescent nuclei, is greatest on E4 and E5, and followed by a gradual decrease throughout the remainder of incubation (Fig.8A). To investigate the role of axons in regulating Schwann cell proliferation, NMDA was administered at different developmental stages to eliminate ventral root axons, and the density of heavily labeled BrdU Schwann cells was quantified 12 hr after NMDA treatment at each specific age. On E10, NMDA treatment resulted in a decrease in the density of heavily BrdU-labeled Schwann cells, however, NMDA treatment on E16 increased the density of proliferating Schwann cells when examined 24 hr later (Fig. 8B).
Fig. 8.

Axons regulate Schwann cell proliferation during development. Schwann cell proliferation in the lumbar ventral root is greatest at E4–E5, followed by a gradual reduction throughout the remainder of incubation (A). Although NMDA treatment reduces Schwann cell proliferation at E10, an increase in proliferation at E16 is evident by 24 hr after NMDA treatment (*p < 0.05) (B). The absence of BrdU (red) and TUNEL (green) double labeling at E10.5 indicates that mitotic Schwann cells do not undergo cell death after NMDA treatment at E10 (C). In A, each point represents the mean ± SEM for three to six embryos, and inB, numbers in barsindicate sample size.
To determine whether proliferating Schwann cells are susceptible to cell death, BrdU labeling was done in conjunction with the TUNEL technique. In embryos treated with NMDA on E10, followed by BrdU administration 10 hr later, and analyzed at E10.5 we have been unable to detect double labeling of BrdU-positive and TUNEL-positive Schwann cells (Fig. 8C). This finding suggests that the decrease in proliferating Schwann cells after NMDA treatment may be attributable directly to the loss of an axon-derived mitogen, rather than to the loss of mitotically active Schwann cells by PCD.
Glial growth factor selectively regulates Schwann cell death
Because axons of the developing chick appear to regulate several aspects of Schwann cell development, we next asked whether specific axon-derived trophic factors are involved in Schwann cell PCD. In the first experiments, various trophic factors were administered to developing chick embryos in ovo on both E5 and E6. To determine the effects of these trophic factors on normal Schwann cell PCD, the relative density of pyknotic Schwann cells was quantified at E6.5 in the L3 ventral roots in serial sectioned Nissl-stained tissue. Most of the trophic factors tested had no effect on Schwann cell PCD as compared with controls, including BDNF, IGF-1, bFGF, and NGF (Fig.9B). Additionally, neutralizing antibodies against p75 and NGF both failed to affect Schwann cell PCD (Fig. 9A). Because early Schwann cell precursors in mammals have been shown to be highly dependent on neuregulins for survival (Dong et al., 1995), chick embryos were treated with GGF, a member of the neuregulin family of proteins. GGF treatment resulted in a significant decrease in the relative density of dying Schwann cell in the ventral root compared with controls (Fig.9A).
Fig. 9.

GGF selectively reduces Schwann cell death in the ventral root in both normal and NMDA-treated embryos. In controls, doses of GGF (5 μg) on E5 and E6 significantly decrease Schwann cell PCD, whereas other growth factors or neutralizing antibodies to p75 and NGF are ineffective (A, B). After NMDA (1 mg) treatment in ovo on E7 plus GGF (5 μg), Schwann cell death is not significantly reduced on E7.5 (C). However, after administration of a second dose of GGF at E7.25, ∼50% of Schwann cells are rescued from NMDA-induced cell death (D). Bars represent the mean ± SEM and the number of embryos is indicated within bars.
To further examine the role of GGF in regulating Schwann cell PCD, embryos were treated at E7 with both NMDA (1 mg), to induce Schwann cell death, and GGF (5 μg). These treatments were then followed by administration of either saline or a second dose of GGF 6 hr later (E7.25). Analysis was performed on Schwann cell PCD in both the ventral roots and the oculomotor nerve in serial sectioned Nissl-stained tissue at E7.5. The relative density of pyknotic Schwann cells in the ventral root did not change after a single treatment with NMDA plus GGF at E7 followed by saline treatment at E7.25 (Fig. 9C). However, embryos treated twice with GGF (E7.0 and E7.25), exhibited a significant decrease in the relative density of dying Schwann cells (Fig. 9C).
In the oculomotor nerve, the PCD of Schwann cells after NMDA was reduced by either a single GGF treatment on E7.0 or double GGF treatment on E7.0 and E7.25 (Fig.10A). The GGF double-treatment group exhibited a larger reduction in the density of pyknotic Schwann cells compared with both the single GGF treatment group and the NMDA-only group (Fig. 10A).
Fig. 10.

GGF regulates Schwann cell death, but not proliferation, in the oculomotor nerve after NMDA administration. Schwann cell death was significantly reduced at E7.5 after NMDA treatment (1 mg) on E7 by either a single dose of GGF (5 μg) on E7, or two doses of GGF on E7 and E7.25 (A). GGF treatment did not significantly affect Schwann cell proliferation, as assessed by quantification of mitotic figures (B). Examples of mitotic figures of proliferating Schwann cells found in S-phase (C), metaphase (D), and anaphase (E) that were included in the mitotic counts in (B). All bars represent mean ± SEM for four to seven embryos.
Because Schwann cell death was measured as a relative density compared with healthy nuclei, the present results cannot establish whether GGF has reduced cell death, or increased proliferation, a previously described role of GGF (Minghetti et al., 1996; Cheng et al., 1998; Maurel and Salzer, 2000). Therefore, proliferation in the oculomotor nerve was assessed by quantifying the relative density of Schwann cells that displayed mitotic profiles in Nissl-stained tissue. These mitotic figures exhibit chromatin condensation morphologies consistent with mitosis (Bergman and Afifi, 1974), such as large, darkly stained nuclei containing balls of heterochromatin (Fig.10C) and chromatin separation resembling distinct phases of mitosis, such as metaphase and anaphase (Fig. 10D,E). Quantification of these mitotic figures in the different treatment groups resulted in no significant differences between the groups (Fig.10B). Because there was no change in the relative density of mitotic figures after GGF treatment, it seems likely that GGF in this situation acts directly to reduce Schwann cell PCD without affecting proliferation.
DISCUSSION
In the present series of experiments we have examined the mechanisms and regulation of Schwann cell death in the embryonic chick. This avian model provides an advantage over mammalian models in that it allows for in vivo experimentation and analysis of individual embryos during the earliest time periods of Schwann cell development. Consistent with the previous studies in mammals, our results demonstrate that axons are major regulators of Schwann cell survival and suggest that the neuregulin family of peptide growth factors (GGFs) may be an important axon-derived signal for regulating Schwann cell survival.
Axons regulate Schwann cell survival
In the present study we have shown that Schwann cells in the embryonic chick are highly dependent on the presence of axons for their survival, which is a finding in accord with our previous report (Ciutat et al., 1996). The pharmacological or surgical manipulation of motoneuron and axon numbers resulted in complementary results for Schwann cell survival, in that (1) increases in Schwann cell PCD occurred after axon elimination and (2) decreases in Schwann cell PCD occurred when axon number was increased by preventing motoneuron PCD by activity blockade. Additionally, observations at the ultrastructural level revealed that dying Schwann cells were often found in association with degenerating axons rather than with healthy axons. Induced Schwann cell PCD, as well as normal Schwann cell PCD, displays many characteristics of an apoptotic-like death including TUNEL-positive staining, immunolabeling of c-Jun/sc45, and cleaved caspase-3, rescue by caspase inhibition, as well as morphological features highly characteristic of apoptotic PCD (Kerr et al., 1995). These experiments demonstrate that axons are key regulators of embryonic Schwann cell survival and suggest that a major function of Schwann cell death is for establishing optimal Schwann cell numbers for ensheathment and myelination.
After treatment with NMDA, all dying Schwann cells were found to be in close contact with degenerating axons, however, in rare cases seemingly healthy Schwann cells were in close proximity with degenerating axons. In these cases it is assumed that these normal appearing Schwann cells are likely to degenerate and die later. Additionally, the elimination of axons by NMDA treatment does not appear to have a direct effect on Schwann cell survival. Although it is not currently known whether embryonic Schwann cells of the ventral root or oculomotor nerve at these ages express functional NMDA receptors, this seems unlikely. For example, as we have shown previously (Ciutat et al., 1996), Schwann cells display apoptotic-like cell death in response to NMDA, whereas the motoneurons undergo a necrotic cell death. Additionally, although spinal motoneurons exhibit developmental differences in their susceptibility to NMDA, in that even very large doses fail to affect motoneurons before E7 (Caldero et al., 1997), with the doses and ages examined in the present study virtually all motoneurons underwent necrotic cell death.
The fact that Schwann cells are rescued from both naturally occurring and NMDA-induced cell death by treatment with BAF, a general caspase inhibitor, has the potential caveat that motoneuron survival may also be affected by caspase inhibition. This seems unlikely, however, because NMDA-induced motoneuron death is necrotic (Ciutat et al., 1996), and BAF or other caspase inhibitors fail to prevent necrotic motoneuron death (Y.-M. Chan, W. Wu, H. K. Yip, K.-F. So, and R. W. Oppenheim, unpublished observations).
Axons also play a critical role in mediating the survival of developing Schwann cells in the rodent. However, because of the limitations ofin utero experimentation on mammalian embryos, much of the evidence concerning axon regulation of Schwann cell survival in vivo is restricted to early postnatal ages. For example, postnatal Schwann cells undergo normal PCD, which can be significantly increased in both the distal stumps and nerve terminal upon sciatic nerve transection (Grinspan et al., 1996; Syroid et al., 1996; Trachtenberg and Thompson, 1996). Additional evidence has come from work donein vitro where both embryonic Schwann cell precursors (Jessen et al., 1994; Gavrilovic et al., 1995; Dong et al., 1995, 1999) and neonatal Schwann cells (Syroid et al., 1996, 1999; Nakao et al., 1997; Maurel and Salzer, 2000) undergo rapid apoptosis if cultured without axons or trophic support. These studies support our findings that Schwann cells in the embryonic chick in vivo are dependent on axons for survival during early development.
It is currently thought that Schwann cell precursors compete for trophic support from outgrowing axons and that failure to achieve axon contact results in PCD (Marchionni et al., 1993; Grinspan et al., 1996;Syroid et al., 1996). Our results support this model and provide evidence for two distinct phases of normal Schwann cell PCD in the chick (Fig. 11). In normal embryos, Schwann cell PCD displays a biphasic developmental profile in both the ventral root (E5.5 and E8.5) and in the oculomotor nerve (E6–7 and E14). The first peak, or wave, of Schwann cell PCD likely occurs because Schwann cell precursors fail to associate with and acquire adequate trophic support from axons. This wave of Schwann cell PCD occurs before the onset of PCD in both the spinal (E6-E10) and oculomotor motoneurons (E10-E15). It is followed by a transient decrease in Schwann cell death because the remaining Schwann cells have now obtained trophic support. Subsequently, as motor axons begin to reach their target areas and motoneuron PCD ensues, those Schwann cells in contact with the axons of the sub-population of motoneurons that degenerate lose their trophic support and exhibit a second wave of Schwann cell PCD, which peaks shortly after the onset of motoneuron PCD.
The current study has discovered many similarities between avian and mammalian Schwann cell survival regulation, including the comparable roles of axons and GGF, presumably acting as a limiting axon-derived trophic molecule. The major differences between these species, however, are the timing of both Schwann cell death and maturation. Whereas much of chicken Schwann cell death and differentiation occurs in ovo, Schwann cell development in the mouse and rat occurs both prenatally and postnatally. In vitro analysis has shown that early Schwann cell precursors are most susceptible to cell death at E12 in the mouse (Dong et al., 1999) and at E14 in the rat (Jessen et al., 1994), and that cell death of immature Schwann cells extends into early postnatal ages as well (Syroid et al., 1996). Although quantification of in vivo Schwann cell death throughout development has not yet been performed in mammals, the in vitro results suggest that much Schwann cell death occurs in coincidence with the PCD periods of their motoneuron counterparts, which occurs from E13 to E18 in the mouse spinal cord (Lance-Jones, 1982; Oppenheim et al., 1986) and between E15 and E19 in the rat (Harris and McCaig, 1984; Oppenheim et al., 1986). Whether a distinct earlier phase of cell death occurs in mammals as in the chick is not known.
Schwann cell proliferation during development
The results of our experiments examining Schwann cell proliferation indicate that similarities exist between avian and mammal species concerning the regulation of Schwann cell proliferation. During early embryonic stages in the chick, Schwann cell proliferation is initially high followed by a gradual decrease in mitosis throughout incubation. An analogous pattern of Schwann cell proliferation has been described in the rat sciatic nerve, where cell division is highest during late embryonic ages and declines substantially during the first two postnatal weeks (Stewart et al., 1993). We have also demonstrated that axons regulate early Schwann cell proliferation. When examined 2 hr after BrdU administration, the absence of TUNEL and BrdU double labeling suggest that axon elimination results in a decrease in mitosis caused by the loss of a putative axon-derived mitogen. Although it is possible that Schwann cells could undergo cell death later at the time of G0/G1 arrest, these data are consistent with observations of axon-driven Schwann cell proliferation in the rat (Wood and Bunge, 1975) and in cultured chick Schwann cells in the presence or absence of neurons (McCarthy and Partlow, 1976). More recent work has shown that the proliferation of human Schwann cells is also regulated by neuregulin signaling derived from cocultured axons (Morrissey et al., 1995).
Avian and mammalian Schwann cell proliferation is also comparable during later stages of Schwann cell development. We have shown that older avian Schwann cells (i.e., E16.5) respond to the loss of axons by increased proliferation. Mature mammalian Schwann cells respond to axon degeneration by undergoing morphological transformations (e.g., the breakdown of myelin) and altered gene expression, followed by rapid proliferation and the establishment of an environment favorable for axon regeneration (Fawcett and Keynes, 1990). Our results suggest that at E16.5 many avian Schwann cells are relatively mature and respond to axon degeneration by initiating proliferation as part of the regeneration process. Support for this conclusion comes from our observations of Wallerian degeneration at the ultrastructural level in which E16.5 Schwann cells display myelin sheath breakdown as they surround degenerating axons. Therefore, Schwann cell proliferation in the developing chick appears to follow rules similar to dividing mammalian Schwann cells, providing further support for the chick embryo as a useful model for the in vivo study of Schwann cell development.
Trophic factor regulation of Schwann cell survival
Our data demonstrate that the survival of developing Schwann cells in the chick embryo can be selectively regulated by GGF, which is likely to be one of the axon-derived factors mediating the previously described regulation of Schwann cell survival by axons. The in ovo administration of exogenous GGF, but not other trophic factors tested by us, rescued dying chick Schwann cells during both normal development and in response to axon degeneration. Additionally, we found that two separate doses of GGF, given after NMDA treatment, had a greater effect on Schwann cell survival then a single dose. This effect is likely a dose-dependent response, but we cannot exclude the possibility that the second dose is available at a time when Schwann cells are more responsive to the survival effects of GGF. Although the dose of GGF used here rescued Schwann cells without significantly affecting proliferation, it is possible that a higher dose could also induce proliferation as has been found to occur in cultured rat Schwann cell precursors (Dong et al., 1995).
Previous studies have confirmed that neuregulins are survival factors for developing mammalian Schwann cells both in vitro (Dong et al., 1995; Syroid et al., 1996) and in vivo (Grinspan et al., 1996; Trachtenberg and Thompson, 1996). The importance of neuregulin signaling for Schwann cell survival has also been demonstrated in mice with mutations in either the NRG-1 gene (Meyer and Birchmeier, 1995) or in the neuregulin ErbB receptors (Riethmacher et al., 1997; Morris et al., 1999). In these mutants, peripheral axons are devoid of Schwann cells by early embryonic ages. Furthermore, the expression and anterograde transport of neuregulin isoforms in motor and sensory neurons have been shown in both avian and mammalian species (Marchionni et al., 1993; Loeb et al., 1999), indicating that neuregulin signaling can occur at the juxtaposition of axon–Schwann cell association.
Although many of the trophic factors we examined (IGF-1, BDNF, NGF, and bFGF) did not display survival-promoting activity in the chick, many of these factors have been shown to affect mammalian Schwann cell survival (Mirsky and Jessen, 1999). Although species differences may be a factor in the apparent discrepancies, it seems more likely that they may also reflect Schwann cell maturation. Whereas early Schwann cell precursors in rodents rely heavily on axon-derived neuregulin signaling for survival (Mirsky and Jessen, 1999), developing mammalian Schwann cells eventually become axon-independent and use IGF-1, IGF-2, PDGF-BB, and NT-3 as part of an autocrine mechanism to regulate their own survival (Meier et al., 1999; Syroid et al., 1999). We have found that after axon elimination in the chick at E16, Schwann cells upregulate the PDGFα receptor, which is likely to be a component of autocrine-mediated survival in these more mature Schwann cells, similar to the situation in rodents. Other factors such as BDNF have been shown to be mainly involved in the response of mature mammalian Schwann cells to regenerating axons (Zhang et al., 2000). Additionally, although bFGF (FGF-2) has been shown to exhibit a survival effect on Schwann cell precursors in vitro (Jessen et al., 1994), it does so transiently and only in the presence of IGF-1 (Gavrilovic et al., 1995) and does not have the same potent survival-promoting abilities as the neuregulins (Dong et al., 1995).
We have also demonstrated that neutralizing antibodies against the p75 receptor and NGF do not affect Schwann cell PCD in the embryonic chick. During mammalian Schwann cell development NGF–p75 signaling has been linked to the induction of apoptosis in cultured postnatal Schwann cells (Soilu-Hänninen et al., 1999). Additional evidence from the examination of p75-deficient mice, however, has shown that NGF–p75 signaling is more involved in injury-induced Schwann cell death than in PCD during normal development (Ferri and Bisby, 1999; Syroid et al., 2000). Furthermore, the survival of embryonic Schwann cells from p75-deficient mice is comparable with wild-type mice, both in vitro and in vivo (Syroid et al., 2000), indicating that p75 is not required for survival regulation during early development. Therefore, we conclude that NGF–p75 interactions in embryonic chick Schwann cells are not involved in the regulation of normal PCD. However, we have found that p75 is upregulated in Schwann cells in response to axon elimination in older embryos (at E16). Comparable upregulation of p75 has been shown in mature mammalian Schwann cells after axon degeneration (Taniuchi et al., 1986). At present the role of p75 in mature Schwann cells after axon degeneration is not clearly understood. The p75 receptor may be involved in Schwann cell migration (Anton et al., 1994; Bentley and Lee, 2000), in the presentation of growth factors to regenerating nerves (Heumann et al., 1987; Zhou et al., 1996), or in the regulation of Schwann cell death during regeneration (Syroid et al., 2000).
In summary, we conclude that early developing Schwann cells in the chick embryo are highly dependent on the presence of axons to regulate both their survival and proliferation. Based on our present findings in the chick, as well as on previous work with mammals, the neuregulin family of glial growth factors are likely to be major axon-derived molecules that regulate Schwann cell survival and ultimately control the optimal number of Schwann cells that are available for myelination and axon ensheathment.
Footnotes
This work was supported by National Institutes of Health Grants NS 36945 and NS20402 (R.W.O.) and grants from Ministerio de Ciencia y Tecnología (SAF 2000–0168) and Fundació la Caixa (J.E.E.).
Correspondence should be addressed to Ronald W. Oppenheim, Department of Neurobiology and Anatomy, Wake Forest University School of Medicine, One Medical Center Boulevard, Winston-Salem, NC 27157. E-mail: roppenheim@wfubmc.edu.
REFERENCES
- 1.Anton ES, Weskamp G, Reichardt LF, Matthew WD. Nerve growth factor and its low-affinity receptor promote Schwann cell migration. Proc Natl Acad Sci USA. 1994;91:2795–2799. doi: 10.1073/pnas.91.7.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bentley CA, Lee KF. p75 is important for axon growth and Schwann cell migration during development. J Neurosci. 2000;20:7706–7715. doi: 10.1523/JNEUROSCI.20-20-07706.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergman RA, Afifi AK. Atlas of microscopic anatomy: a companion to histology and neuroanatomy. W.B. Saunders; Philadelphia: 1974. [Google Scholar]
- 4.Bhattacharyya A, Frank E, Ratner N, Brackenbury R. P0 is an early marker of the Schwann cell lineage in chickens. Neuron. 1991;7:831–844. doi: 10.1016/0896-6273(91)90285-8. [DOI] [PubMed] [Google Scholar]
- 5.Burek MJ, Oppenheim RW. Programmed cell death in the developing nervous system. Brain Pathol. 1996;6:427–446. doi: 10.1111/j.1750-3639.1996.tb00874.x. [DOI] [PubMed] [Google Scholar]
- 6.Caldero J, Ciutat D, Llado J, Castan E, Oppenheim RW, Esquerda JE. Effects of excitatory amino acids on neuromuscular development in the chick embryo. J Comp Neurol. 1997;387:73–95. [PubMed] [Google Scholar]
- 7.Caldero J, Prevette D, Mei X, Oakley RA, Li L, Milligan C, Houenou L, Burek M, Oppenheim RW. Peripheral target regulation of the development and survival of spinal sensory and motor neurons in the chick embryo. J Neurosci. 1998;18:356–370. doi: 10.1523/JNEUROSCI.18-01-00356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casas C, Ribera J, Esquerda JE. Antibodies against c-Jun N-terminal peptide cross-react with neo-epitopes emerging after caspase-mediated proteolysis during apoptosis. J Neurochem. 2001;77:904–915. doi: 10.1046/j.1471-4159.2001.00314.x. [DOI] [PubMed] [Google Scholar]
- 9.Cheng L, Esch FS, Marchionni MA, Mudge AW. Control of Schwann cell survival and proliferation: autocrine factors and neuregulins. Mol Cell Neurosci. 1998;12:141–156. doi: 10.1006/mcne.1998.0706. [DOI] [PubMed] [Google Scholar]
- 10.Chu-Wang I-W, Oppenheim RW. Cell death of motoneurons in the chick embryo spinal cord. I. A light and electron microscopic study of naturally occurring and induced cell loss during development. J Comp Neurol. 1978;177:33–58. doi: 10.1002/cne.901770105. [DOI] [PubMed] [Google Scholar]
- 11.Ciutat D, Caldero J, Oppenheim RW, Esquerda JE. Schwann cell apoptosis during normal development and after axonal degeneration induced by neurotoxins in the chick embryo. J Neurosci. 1996;16:3979–3990. doi: 10.1523/JNEUROSCI.16-12-03979.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clarke PGH, Oppenheim RW. Neuron death in vertebrate development: in vivo methods. In: Schwartz LM, Osborne BA, editors. Methods in cell biology: cell death, Vol 46. Academic; New York: 1995. pp. 277–321. [PubMed] [Google Scholar]
- 13.Dong Z, Brennan A, Liu N, Yarden Y, Lefkowitz G, Mirsky R, Jessen KR. Neu differentiation factor is a neuron-glia signal and regulates survival, proliferation, and maturation of rat Schwann cell precursors. Neuron. 1995;15:585–596. doi: 10.1016/0896-6273(95)90147-7. [DOI] [PubMed] [Google Scholar]
- 14.Dong Z, Sinanan A, Parkinson D, Parmentier E, Mirsky R, Jessen KR. Schwann cell development in embryonic mouse nerves. J Neurosci Res. 1999;56:334–348. doi: 10.1002/(SICI)1097-4547(19990515)56:4<334::AID-JNR2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 15.Fawcett JW, Keynes RJ. Peripheral nerve regeneration. Annu Rev Neurosci. 1990;13:43–60. doi: 10.1146/annurev.ne.13.030190.000355. [DOI] [PubMed] [Google Scholar]
- 16.Ferri CC, Bisby MA. Improved survival of injured sciatic nerve Schwann cells in mice lacking the p75 receptor. Neurosci Lett. 1999;272:191–194. doi: 10.1016/s0304-3940(99)00618-7. [DOI] [PubMed] [Google Scholar]
- 17.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gavrilovic J, Brennan A, Mirsky R, Jessen KR. Fibroblast growth factors and insulin growth factors combine to promote survival of rat Schwann cell precursors without induction of DNA synthesis. Eur J Neurosci. 1995;7:77–85. doi: 10.1111/j.1460-9568.1995.tb01022.x. [DOI] [PubMed] [Google Scholar]
- 19.Grinspan JB, Marchionni MA, Reeves M, Coulaloglou M, Scherer SS. Axonal interactions regulate Schwann cell apoptosis in developing peripheral nerve: neuregulin receptors and the role of neuregulins. J Neurosci. 1996;16:6107–6118. doi: 10.1523/JNEUROSCI.16-19-06107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamburger V. Regression versus peripheral control of differentiation in motor hypoplasia. Am J Anat. 1958;102:365–410. doi: 10.1002/aja.1001020303. [DOI] [PubMed] [Google Scholar]
- 21.Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- 22.Harris AJ, McCaig CD. Motoneuron death and motor unit size during embryonic development of the rat. J Neurosci. 1984;4:13–24. doi: 10.1523/JNEUROSCI.04-01-00013.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harvey NL, Kumar S. The role of caspases in apoptosis. Adv Biochem Eng Biotechnol. 1998;62:107–128. doi: 10.1007/BFb0102307. [DOI] [PubMed] [Google Scholar]
- 24.Heumann R, Korsching S, Bandtlow C, Thoenen H. Changes of nerve growth factor synthesis in nonneuronal cells in response to sciatic nerve transection. J Cell Biol. 1987;104:1623–1631. doi: 10.1083/jcb.104.6.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jessen KR, Brennan A, Morgan L, Mirsky R, Kent A, Hashimoto Y, Gavrilovic J. The Schwann cell precursor and its fate: a study of cell death and differentiation during gliogenesis in rat embryonic nerves. Neuron. 1994;12:509–527. doi: 10.1016/0896-6273(94)90209-7. [DOI] [PubMed] [Google Scholar]
- 26.Jessen KR, Mirsky R. Why do Schwann cells survive in the absence of axons? Ann NY Acad Sci. 1999;883:109–115. [PubMed] [Google Scholar]
- 27.Kerr JF, Gobe GC, Winterford CM, Harmon BV. Anatomical methods in cell death. Methods Cell Biol. 1995;46:1–27. doi: 10.1016/s0091-679x(08)61921-4. [DOI] [PubMed] [Google Scholar]
- 28.Kopp DM, Trachtenberg JT, Thompson WJ. Glial growth factor rescues Schwann cells of mechanoreceptors from denervation-induced apoptosis. J Neurosci. 1997;17:6697–6706. doi: 10.1523/JNEUROSCI.17-17-06697.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lance-Jones C. Motoneuron cell death in the developing lumbar spinal cord of the mouse. Dev Brain Res. 1982;256:473–479. doi: 10.1016/0165-3806(82)90192-4. [DOI] [PubMed] [Google Scholar]
- 30.Landmesser L, Honig MG. Altered sensory projections in the chick hind limb following the early removal of motoneurons. Dev Biol. 1986;118:511–531. doi: 10.1016/0012-1606(86)90023-0. [DOI] [PubMed] [Google Scholar]
- 31.Lemke G, Chao M. Axons regulate Schwann cell expression of the major myelin and NGF receptor genes. Development. 1988;102:499–504. doi: 10.1242/dev.102.3.499. [DOI] [PubMed] [Google Scholar]
- 32.Llado J, Caldero J, Ribera J, Tarabal O, Oppenheim RW, Esquerda JE. Opposing effects of excitatory amino acids on chick embryo spinal cord motoneurons: excitotoxic degeneration or prevention of programmed cell death. J Neurosci. 1999;19:10803–10812. doi: 10.1523/JNEUROSCI.19-24-10803.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lobsiger CS, Schweitzer B, Taylor V, Suter U. Platelet-derived growth factor-BB supports the survival of cultured rat Schwann cell precursors in synergy with neurotrophin-3. Glia. 2000;30:290–300. doi: 10.1002/(sici)1098-1136(200005)30:3<290::aid-glia8>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 34.Loeb JA, Khurana TS, Robbins JT, Yee AG, Fischbach GD. Expression patterns of transmembrane and released forms of neuregulin during spinal cord and neuromuscular synapse development. Development. 1999;126:781–791. doi: 10.1242/dev.126.4.781. [DOI] [PubMed] [Google Scholar]
- 35.Marchionni MA, Goodearl AD, Chen MS, Bermingham-McDonogh O, Kirk C, Hendricks M, Danehy F, Misumi D, Sudhalter J, Kobayashi K. Glial growth factors are alternatively spliced erbB2 ligands expressed in the nervous system. Nature. 1993;362:312–318. doi: 10.1038/362312a0. [DOI] [PubMed] [Google Scholar]
- 36.Maurel P, Salzer JL. Axonal regulation of Schwann cell proliferation and survival and the initial events of myelination requires PI 3-kinase activity. J Neurosci. 2000;20:4635–4645. doi: 10.1523/JNEUROSCI.20-12-04635.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCarthy KD, Partlow LM. Neuronal stimulation of (3H)thymidine incorporation by primary cultures of highly purified non-neuronal cells. Brain Res. 1976;114:415–426. doi: 10.1016/0006-8993(76)90963-x. [DOI] [PubMed] [Google Scholar]
- 38.Meier C, Parmantier E, Brennan A, Mirsky R, Jessen KR. Developing Schwann cells acquire the ability to survive without axons by establishing an autocrine circuit involving insulin-like growth factor, neurotrophin-3, and platelet-derived growth factor-BB. J Neurosci. 1999;19:3847–3859. doi: 10.1523/JNEUROSCI.19-10-03847.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–390. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- 40.Minghetti L, Goodearl AD, Mistry K, Stroobant P. Glial growth factors I-III are specific mitogens for glial cells. J Neurosci Res. 1996;43:684–693. doi: 10.1002/(SICI)1097-4547(19960315)43:6<684::AID-JNR5>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 41.Mirsky R, Jessen KR. The neurobiology of Schwann cells. Brain Pathol. 1999;9:293–311. doi: 10.1111/j.1750-3639.1999.tb00228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morris JK, Lin W, Hauser C, Marchuk Y, Getman D, Lee KF. Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron. 1999;23:273–283. doi: 10.1016/s0896-6273(00)80779-5. [DOI] [PubMed] [Google Scholar]
- 43.Morrissey TK, Levi AD, Nuijens A, Sliwkowski MX, Bunge RP. Axon-induced mitogenesis of human Schwann cells involves heregulin and p185erbB2. Proc Natl Acad Sci USA. 1995;92:1431–1435. doi: 10.1073/pnas.92.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muller HW, Stoll G. Nerve injury and regeneration: basic insights and therapeutic interventions. Curr Opin Neurol. 1998;11:557–562. doi: 10.1097/00019052-199810000-00019. [DOI] [PubMed] [Google Scholar]
- 45.Nakao J, Shinoda J, Nakai Y, Murase S, Uyemura K. Apoptosis regulates the number of Schwann cells at the premyelinating stage. J Neurochem. 1997;68:1853–1862. doi: 10.1046/j.1471-4159.1997.68051853.x. [DOI] [PubMed] [Google Scholar]
- 46.Oppenheim RW, Houenou L, Pincon-Raymond M, Powell JA, Rieger F, Standish LJ. The development of motoneurons in the embryonic spinal cord of the mouse mutant, muscular dysgenesis (mdg/mdg): survival, morphology, and biochemical differentiation. Dev Biol. 1986;114:426–436. doi: 10.1016/0012-1606(86)90207-1. [DOI] [PubMed] [Google Scholar]
- 47.Oppenheim RW, Prevette D, D'Costa A, Wang S, Houenou LJ, McIntosh JM. Reduction of neuromuscular activity is required for the rescue of motoneurons from naturally occurring cell death by nicotinic-blocking agents. J Neurosci. 2000;20:6117–6124. doi: 10.1523/JNEUROSCI.20-16-06117.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pittman RH, Oppenheim RW. Neuromuscular blockade increases motoneuron survival during normal cell death in the chick embryo. Nature. 1978;271:364–366. doi: 10.1038/271364a0. [DOI] [PubMed] [Google Scholar]
- 49.Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature. 1997;389:725–730. doi: 10.1038/39593. [DOI] [PubMed] [Google Scholar]
- 50.Saxod R, Bouvet J. Quantitative analysis of growth and myelination of cutaneous nerve fibers in the chick. Dev Neurosci. 1982;5:143–155. doi: 10.1159/000112671. [DOI] [PubMed] [Google Scholar]
- 51.Soilu-Hänninen M, Ekert P, Bucci T, Syroid D, Bartlett PF, Kilpatrick TJ. Nerve growth factor signaling through p75 induces apoptosis in Schwann cells via a Bcl-2-independent pathway. J Neurosci. 1999;19:4828–4838. doi: 10.1523/JNEUROSCI.19-12-04828.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steljes TP, Kinoshita Y, Wheeler EF, Oppenheim RW, von Bartheld CS. Neurotrophic factor regulation of developing avian oculomotor neurons: differential effects of BDNF and GDNF. J Neurobiol. 1999;41:295–315. [PubMed] [Google Scholar]
- 53.Stewart HJ, Morgan L, Jessen KR, Mirsky R. Changes in DNA synthesis rate in the Schwann cell lineage in vivo are correlated with the precursor–Schwann cell transition and myelination. Eur J Neurosci. 1993;5:1136–1144. doi: 10.1111/j.1460-9568.1993.tb00968.x. [DOI] [PubMed] [Google Scholar]
- 54.Syroid DE, Maycox PR, Burrola PG, Liu N, Wen D, Lee KF, Lemke G, Kilpatrick TJ. Cell death in the Schwann cell lineage and its regulation by neuregulin. Proc Natl Acad Sci USA. 1996;93:9229–9234. doi: 10.1073/pnas.93.17.9229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Syroid DE, Zorick TS, Arbet-Engels C, Kilpatrick TJ, Eckhart W, Lemke G. A role for insulin-like growth factor-I in the regulation of Schwann cell survival. J Neurosci. 1999;19:2059–2068. doi: 10.1523/JNEUROSCI.19-06-02059.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Syroid DE, Maycox PJ, Soilu-Hanninen M, Petratos S, Bucci T, Burrola P, Murray S, Cheema S, Lee KF, Lemke G, Kilpatrick TJ. Induction of postnatal Schwann cell death by the low-affinity neurotrophin receptor in vitro and after axotomy. J Neurosci. 2000;20:5741–5747. doi: 10.1523/JNEUROSCI.20-15-05741.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taniuchi M, Clark HB, Johnson EM. Induction of nerve growth factor receptor in Schwann cells after axotomy. Proc Natl Acad Sci USA. 1986;83:4094–4098. doi: 10.1073/pnas.83.11.4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trachtenberg JT, Thompson WJ. Schwann cell apoptosis at developing neuromuscular junctions is regulated by glial growth factor. Nature. 1996;379:174–177. doi: 10.1038/379174a0. [DOI] [PubMed] [Google Scholar]
- 59.Wang G, Scott SA. The “waiting period” of sensory and motor axons in early chick hindlimb: its role in axon pathfinding and neuronal maturation. J Neurosci. 2000;20:5358–5366. doi: 10.1523/JNEUROSCI.20-14-05358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wood PM, Bunge RP. Evidence that sensory axons are mitogenic for Schwann cells. Nature. 1975;256:662–664. doi: 10.1038/256662a0. [DOI] [PubMed] [Google Scholar]
- 61.Zhang JY, Luo XG, Xian CJ, Liu ZH, Zhou XF. Endogenous BDNF is required for myelination and regeneration of injured sciatic nerve in rodents. Eur J Neurosci. 2000;12:4171–4180. [PubMed] [Google Scholar]
- 62.Zhou XF, Rush RA, McLachlan EM. Differential expression of the p75 nerve growth factor receptor in glia and neurons of the rat dorsal root ganglia after peripheral nerve transection. J Neurosci. 1996;16:2901–2911. doi: 10.1523/JNEUROSCI.16-09-02901.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]