

Abstract
Peroxynitrite (ONOO−) has been implicated as a causative factor in dopamine neuronal damage resulting from exposure to methamphetamine and1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), and it may be involved in the etiology of Parkinson's Disease. ONOO− causes a concentration-dependent and irreversible reduction in dopamine uptake by EM4 cells stably expressing the human dopamine transporter (hDAT). The effect of ONOO− is manifested as a reduction inVmax. Cysteine, dithiothreitol, glutathione, and N-acetyl-cysteine, reagents that interact directly with ONOO−, prevent this inhibition, whereas a scavenger of hydroxyl radical (dimethylsulfoxide), hydrogen peroxide (catalase), and superoxide (superoxide dismutase) did not. Dopamine in the extracellular medium protects the hDAT from ONOO−, whereas intracellular dopamine does not. Parachloromercuribenzoic acid and 2-aminoethyl methanethiosulfonate (MTSEA), which share with ONOO− the ability to modify cysteine sulfhydryls, also inhibit hDAT function. ONOO− treatment lowers cysteine-specific labeling of the hDAT by MTSEA-biotin, suggesting that ONOO− reacts with one or more cysteines in hDAT. A mutant of hDAT (X7C) in which all intracellular and extracellular loop cysteines were mutated was resistant to inhibition by ONOO−. Sensitivity to ONOO− was restored in mutants of hDAT in which reduced cysteines were present only in the first (C135) and third (C342) intracellular loops (CD-DAT), or in which C342 alone had been reintroduced into X7C (X7C-M342C). These results indicate that the hDAT is inhibited by ONOO− through oxidation of cysteine 342. Our studies also substantiate the possibility that drugs known to decrease DAT function in vivo (e.g., methamphetamine and MPTP) may exert their effects through ONOO−-mediated oxidative stress.
Keywords: dopamine transporter, peroxynitrite, cysteine residues, dopamine, Parkinson's disease, neurotoxic amphetamines
The dopamine transporter (DAT) is an integral membrane protein in dopamine (DA) nerve endings where it subserves the critical function of terminating the synaptic activity of DA through transport into the presynaptic process (Amara et al., 1998;Chen and Reith, 2000). Drugs or pathological conditions that disrupt the function of the DAT could have profound effects on behaviors and physiological processes that are mediated by DA (Jaber et al., 1997). Methamphetamine (Cubells et al., 1994; Sulzer et al., 1995) and MPP+ (Javitch and Snyder, 1984; Javitch et al., 1985; Gainetdinov et al., 1997) are substrates for inward transport by the DAT. Once inside the DA nerve terminal, these drugs displace DA from storage vesicles into the cytoplasm and eventually into the synapse through reverse outward transport (Cubells et al., 1994; Sulzer et al., 1995; Lotharius and O'Malley, 2000). In vitro studies have shown that reactive oxygen species (Fleckenstein et al., 1997b; Hanson et al., 1998;Haughey et al., 1999) have reversible inhibitory effects on the DAT that simulate closely the in vivo effects of methamphetamine on it (Haughey et al., 2000; Sandoval et al., 2001), leading to the conclusion that DAT function is acutely responsive to oxidative insult.
Methamphetamine (Davidson et al., 2001) and1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Blum et al., 2001; Schmidt and Ferger, 2001) are known to exert toxic effects on DA neurons, including persistent reductions in DAT function. Nitric oxide and superoxide have been implicated in the damaging effects of these drugs on DA neurons (Cadet et al., 1994; Schulz et al., 1995; Di Monte et al., 1996; Przedborski et al., 1996; Przedborski and Jackson-Lewis, 1998; Itzhak et al., 1999). However, the coincident production of nitric oxide and superoxide almost certainly results in the production of peroxynitrite (ONOO−) (Koppenol et al., 1992). ONOO− is a powerful oxidant that can modify proteins, nucleotides, lipids, and cell organelles, properties that are thought to underlie its cytotoxic potential (Beckman and Koppenol, 1996). Perhaps the best known posttranslational modification of proteins caused by ONOO− is the nitration of tyrosine residues (Ischiropoulos and al-Mehdi, 1995; Crow and Ischiropoulos, 1996). The appearance in brain of nitrotyrosine immunoreactivity after administration of methamphetamine (Imam et al., 1999; Imam and Ali, 2001) or MPTP (Ferrante et al., 1999; Pennathur et al., 1999) suggests that these drugs cause the production of ONOO−.
In light of evidence correlating ONOO−generation with drug-induced reductions in DAT function and neurotoxicity, we hypothesized that ONOO−might inactivate DAT. The DAT contains a total of 13 cysteine residues (Giros et al., 1991, 1992) that are arrayed throughout its transmembrane domains, and extracellular and intracellular loops, and these cysteines are potential targets for ONOO− reaction. The present studies use EM4 cells stably expressing the wild-type hDAT and selected cysteine mutants to establish that DA transport is inhibited by ONOO− via its action on cysteine 342 in the third cytoplasmic loop of the hDAT.
MATERIALS AND METHODS
Materials. The following materials were obtained from Sigma (St. Louis, MO): DA, N-acetyl-DA, dimethylsulfoxide (DMSO), cocaine, dithiothreitol (DTT), glutathione (GSH), cysteine, H2O2, sodium periodate,p-chloromercuribenzoic acid (pCMB),N-acetylcysteine, superoxide dismutase (SOD), anti-Flag monoclonal antibodies (M2), and Triton X-100. Catalase was a product of Boehringer Mannheim (Indianapolis, IN), and a monoclonal antibody against nitrotyrosine was from Cayman Chemical (Ann Arbor, MI). 3,4-[7-3H]dihydroxy-phenylethylamine ([3H]DA) (31.6 Ci/mmol) was obtained from NEN Life Science Products (Boston, MA). 2-aminoethyl methanethiosulfonate hydrobromide (MTSEA), [2-(trimethylammonium)ethyl] methanethiosulfonate bromide (MTSET), and MTSEA-biotin were obtained from Toronto Research Chemicals Inc. (Toronto, Canada). Nomifensine and 2-β-carbomethoxy-3-β-(4-fluorophenyl) tropane (β-CFT) were obtained from Research Biochemicals International (Natick, MA). Fetal bovine serum was purchased from HyClone (Logan, UT), and all cell culture media was from Invitrogen (Carlsbad, CA). Lactate dehydrogenase (LDH) assay kits were purchased from Promega (Madison, WI). Ultralink immobilized neutravidin plus agarose was from Pierce Endogen (Rockford, IL), and PVDF membranes were from Bio-Rad Laboratories (Hercules, CA).
Site-directed mutagenesis and stable transfection of hDAT.EM4 cells, which are HEK 293 cells stably transfected with the macrophage embryonic receptor to increase their adherence to tissue culture plastic (Robbins and Horlick, 1998), were used as host cells for stable expression of hDAT (Ferrer and Javitch, 1998; Chen et al., 2000; Hastrup et al., 2001). All forms of hDAT used presently were tagged at the N terminus with the tandem Flag-HA epitope (18 amino acids) as previously described (Hastrup et al., 2001). Introduction of this epitope tag removed the first 22 amino acids of the hDAT from the amino terminus of all constructs, including C6, and did not significantly affect expression or transport (Saunders et al., 2000;Hastrup et al., 2001). In addition to wild-type hDAT, the following constructs, with mutations specified, were used: X7C (C90A, C135A, C306A, C319M, C342M, C581L); cysteine-depleted (CD)-DAT (C90A, C243A, C306A, C319M, C463S, C523S, C530A, C581L); and X7C-M342C (C342 reintroduced into X7C). In descriptive terms, X7C lacks cysteines in all of its putative intracellular and extracellular loops and in the sixth transmembrane domain (C319); CD-DAT lacks all putative extracellular and transmembrane free sulfhydryls, and contains disulfide-bridged C180-C189, C135 in the first intracellular loop and C342 in the third intracellular loop; and X7C-M342C is X7C (above) into which C342 has been re-introduced. Stably transfected cells were grown in DMEM supplemented with 10% fetal bovine serum (HyClone) at 37°C and 5% CO2.
Treatment of DAT with ONOO− and cysteine sulfhydryl reagents. Cells were grown to 90% confluence (∼0.8 × 106 cells) on 24 well plates. Media was removed, and cells were washed three times with Krebs'–Ringer's–HEPES (KRH) containing (in mm): 150 NaCl, 4.7 KCl, 2.2 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 10 HEPES, and 10 glucose, pH 7.4. ONOO− was synthesized by the quenched-flow method of Beckman et al. (1994), and its concentration was determined by the extinction coefficient ε302 = 1670m/cm-1. The hydrogen peroxide contamination of ONOO− was removed by manganese dioxide chromatography and filtration (Beckman et al., 1994). ONOO− was added to hDAT-expressing cells with rapid mixing and cells were incubated at 25°C for 10 min. Cells were washed three times with KRH, and uptake was assessed as described below. Concentrated solutions of ONOO− were allowed to decompose at room temperature in phosphate buffer at pH 7.4 (confirmed spectrophotometrically) before addition to cells and this reverse-order addition served as a control for the ONOO− solvents. LDH activity was measured in cells and culture media in some experiments to ensure that ONOO− was not causing cell lysis. For experiments aimed at assessing the specificity of ONOO− actions on the hDAT, antioxidants and/or reducing agents (cysteine, DTT, GSH, andN-acetyl-cysteine) or scavengers of reactive oxygen species (DMSO, catalase, and SOD) were added 5 min before ONOO−.
The ability of DA to modulate the effects of ONOO− on hDAT was assessed by two different methods. First, hDAT-expressing cells were incubated with various concentrations of DA (10–100 μm) for 10 min at 37°C after which they were washed and exposed to ONOO−. Second, cells were incubated with DA as described above, but were exposed to ONOO− without washing. These experiments were designed to determine the ability of intracellular or extracellular DA, respectively, to modulate the effects of ONOO− on hDAT function. Treatments with cysteine sulfhydryl reagents (pCMB, MTSEA, MTSET) or dopamine quinone were performed as described for ONOO−. Stable o-quinones were formed by oxidizing DA orN-acetyl-DA with one equivalent of NaIO4 immediately before addition to cells (Graham, 1978; Graham et al., 1978).
[3H] DA uptake. Cells were incubated with 3 μm DA containing 25 nm [3H] DA for 2 min at room temperature. Nonspecific uptake was defined in the presence of 10 μm nomifensine and was typically ∼5% of total uptake. Cells were washed and solubilized in 1% Triton X-100, and tritium was counted by liquid scintillation spectrometry. Kinetic analysis of hDAT (control and ONOO−-treated) was performed by incubating cells with increasing concentrations of unlabeled DA and a constant concentration of [3H] DA. Apparent Vmax andKm values were estimated using nonlinear regression curve fitting with a one-site binding equation (GraphPad Prism).
MTSEA-biotin labeling of hDAT. The wild-type hDAT used in the present studies has a Flag-HA epitope-tag (18 amino acids) replacing the first 22 amino acids of the N terminus (Saunders et al., 2000; Hastrup et al., 2001), and this tag was used for immunoblotting. MSTEA-biotin was used to label cysteine residues in hDAT as previously described (Daniels and Amara, 1998; Javitch, 1998), after treatment of intact cells with ONOO− or other sulfhydryl reagents. Reductions in MTSEA-biotin labeling of hDAT caused by ONOO− serves as a nonquantitative index of cysteine modification. Intact cells were treated with ONOO− or other sulfhydryl reagents and washed three times with KRH. Cells were homogenized in PBS, and washed membranes were solubilized in 1% Triton X-100. Biotinylated proteins were isolated from solubilized membranes by adsorption to neutravidin agarose for 60 min at room temperature, and after three washes, agarose beads were eluted with an SDS-stop solution. Proteins were resolved by SDS-PAGE (Laemmli, 1970), blotted to PVDF membranes, and probed with anti-Flag M2 antibodies (to measure hDAT). In some experiments, membranes from ONOO−-treated cells were immunoprecipitated with an antibody against nitrotyrosine (MacMillan-Crow and Thompson, 1999) and subsequently probed with anti-Flag antibodies to determine if the hDAT had been tyrosine-nitrated.
RESULTS
Effects of ONOO− on hDAT function
Exposure of intact EM4 cells stably expressing the hDAT to ONOO− for 10 min caused a concentration-dependent decrease in DA uptake, as shown in Figure1. DAT function was reduced by ∼10% at a concentration of 0.5 mm, and concentrations of 1.0–1.5 mm reduced DA uptake by 50–90%, respectively. At 2.0 mm ONOO−, hDAT function was completely inhibited. If ONOO− was allowed to decompose before addition to cells, this reverse order addition at a reagent concentration of 2 mm did not inhibit DA uptake. The overall effect of ONOO−concentration on DAT function was significant (p< 0.01; ANOVA). The inhibitory effects of 1–2 mm ONOO− were also significant (p < 0.05; Bonferroni's test). ONOO− did not cause lysis of cells as measured by LDH release into the cell culture media (data not shown). If cells were preloaded with 3H-DA and then exposed to ONOO−, we did not observe a release of DA (data not shown). The kinetic parameters of DA uptake were determined after exposure to 1 mmONOO− and revealed a reduction inVmax from 22.9 pmol/min per well to 10.5 pmol/min per well (46% control; p < 0.05, Bonferroni's test). The apparent Kmfor DA uptake was not significantly altered by 1 mm ONOO− (1.35 μm for control compared with 1.25 μm after ONOO−).
Fig. 1.

Effects of ONOO− on hDAT activity. Intact EM4 (∼0.8 × 106) cells stably expressing wild-type hDAT were exposed to the indicated concentrations of ONOO− for 10 min at 25°C in 24 well tissue culture plates. Cells were washed 3× with KRH, and the uptake of 3H-DA was determined immediately as described in Materials and Methods. The open circle above the 2 mm ONOO− concentration marker represents the effects of decomposed ONOO− on DA uptake. Data represent means ± SEM for four experiments run in triplicate. The effect of ONOO− on hDAT activity is reported as percentage of control (control levels of DA uptake were 13.5 pmol/min per well). The overall effect of ONOO− on DA uptake was significant (p < 0.01; ANOVA). Concentrations of ONOO− of 1 mm and higher were also significantly different from control levels of DA uptake (*p < 0.05; Bonferroni's test).
Specificity of the effect of ONOO− on hDAT
Reagents known to react directly with ONOO−, including cysteine, DTT, GSH, andN-acetyl-cysteine (Radi et al., 1991; Halliwell et al., 1999), provided almost complete protection against the inhibitory effects of ONOO− on hDAT. Figure2A shows that 1 mm concentrations of each reactant, with the exception of GSH, reduced the effect of ONOO− from a 50% inhibition to ∼20% inhibition. GSH provided complete protection against ONOO−. These reagents did not alter hDAT activity in the absence of ONOO−. Attempts to reverse the effect of ONOO−on hDAT with these same reagents were not successful (data not shown). Figure 2B shows that a scavenger of hydroxyl radical (DMSO), hydrogen peroxide (catalase), or superoxide (SOD), was ineffective in preventing ONOO−-induced inhibition of hDAT. DMSO, catalase, and SOD were devoid of effects on hDAT activity in the absence of ONOO−(data not shown).
Fig. 2.
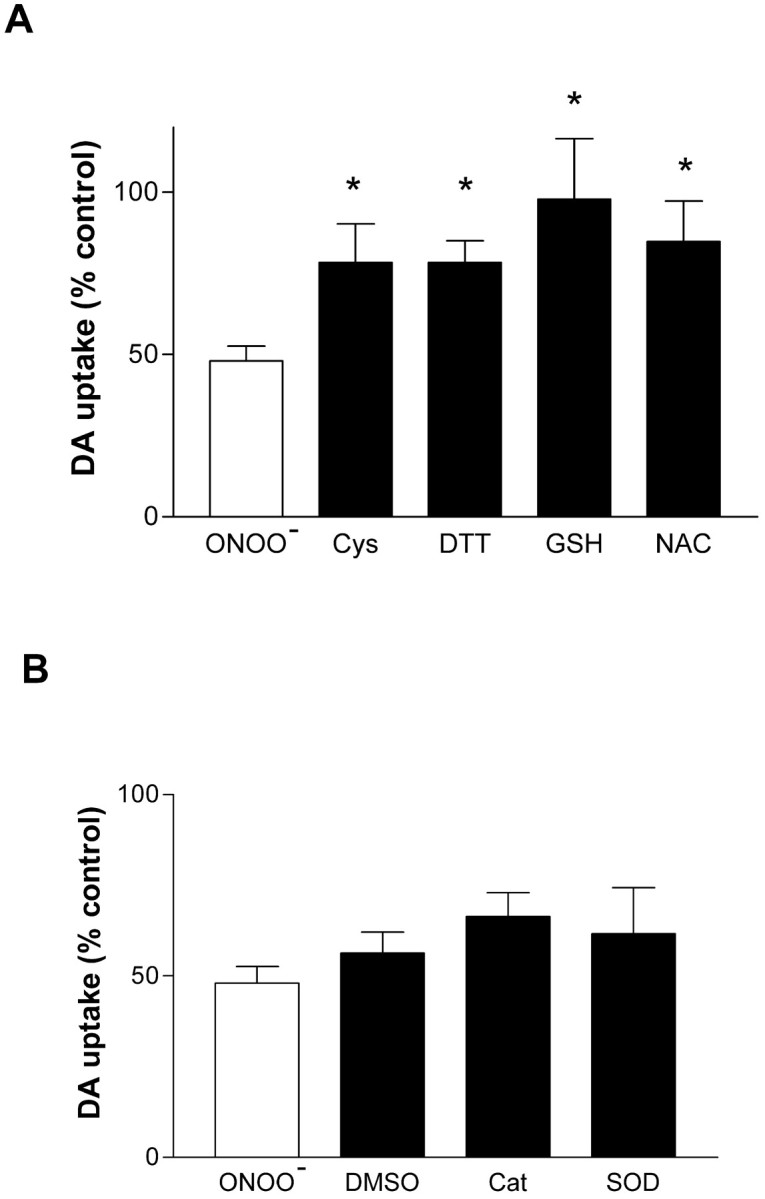
Effects of antioxidants and radical scavengers on ONOO−-induced inhibition of the hDAT. Intact EM4 cells expressing wild-type hDAT were exposed to ONOO− (1 mm) for 10 min at 25°C. The indicated reagents were added to cells 5 min before ONOO− and remained present during treatment.A shows the effects of antioxidants and reducing agents (1 mm concentrations for all reagents) on the ONOO−-induced inhibition of hDAT activity.B shows the effects of a scavenger of hydroxyl radical (DMSO, 50 mm), hydrogen peroxide (catalase [Cat], 10 U/ml), and superoxide (SOD, 10 U/ml) on the ONOO−-induced (1 mm) inhibition of the hDAT. The results represent mean ± SEM of four or five experiments run in triplicate for each condition. *Indicates that the effects of that treatment were significantly different from ONOO− alone (p < 0.05; Bonferroni's test).
Effects of DA on ONOO−-induced inhibition of hDAT
The hDAT is known to undergo a substrate-induced conformational change that increases the accessibility of cysteine 342 to sulfhydryl reagents (Chen et al., 2000). Therefore, we tested the effects of extracellular and intracellular DA on ONOO−-induced inactivation of hDAT. Figure 3 shows that extracellular DA provided almost complete protection of hDAT from inactivation by ONOO−. Concentrations of DA from 50–100 μm reduced the inhibitory effect of 1 mmONOO− from 52% to 10–15%. By contrast, when cytoplasmic DA levels were increased via hDAT-mediated uptake, and extracellular DA was removed by rapid washing just before the addition of ONOO−, the effect of ONOO− was only slightly altered. Thus, preincubation of intact cells with 50–100 μm DA followed by washing reduced the inhibitory effects of ONOO− by only ∼10%. The effect of extracellular DA on ONOO− inactivation of the hDAT was significant (p < 0.05; ANOVA), whereas the effect of intracellular DA was not. Nomifensine, β-CFT, and cocaine, antagonists of the hDAT, did not prevent ONOO−-induced inactivation of the hDAT (data not shown).
Fig. 3.

Effects of DA on the ONOO−-induced inhibition of hDAT. Intact EM4 cells stably expressing the hDAT were treated with ONOO−(1 mm) in the presence of extracellular or intracellular DA. Cells were incubated for 10 min at 25°C with the indicated concentrations of DA. ONOO− was added to cells with (intracellular DA) or without (extracellular DA) removal of exogenous DA from the incubation medium and treatment was continued for 10 min. Cells were then washed three times with KRH, and uptake of3H-DA was determined. The data are means ± SEM of four or five experiments run in triplicate. Controls for intracellular and extracellular DA on hDAT activity omitted ONOO−treatment. The overall effect of extracellular DA on the ONOO−-induced inhibition of hDAT was significant (p < 0.05; ANOVA), whereas the effect of intracellular DA was not.
Effects of cysteine-sulfhydryl reagents on hDAT
Various reagents that react specificity with cysteine residues in membranes (van Iwaarden et al., 1992) were tested for their effects on hDAT. Figure 4 shows that pCMB caused significant reductions in DA uptake. At a concentration of 100 μm, pCMB was more inhibitory (95% inhibition of hDAT) than ONOO− (55% inhibition). In agreement with previous results (Ferrer and Javitch, 1998; Chen et al., 2000), the cell membrane-permeable cysteine reactant MTSEA (1 mm) lowered DA uptake to 30% of control, whereas equimolar concentrations of the impermeable MTSET had no effect on DA uptake.
Fig. 4.

Effects of cysteine sulfhydryl reagents on hDAT activity. Intact EM4 cells stably expressing wild-type hDAT were exposed to ONOO− (1 mm), pCMB (100 μm), MTSEA (1 mm), or MTSET (1 mm) for 10 min at 25°C for 10 min. Cells were washed three times with KRH, and the uptake of 3H-DA was determined. Results represent the mean ± SEM of four or five experiments run in triplicate for each treatment. *Indicates that the treatment was significantly different from untreated controls (p < 0.05; Bonferroni's test).
Effect of ONOO− on cysteine labeling with MTSEA-biotin
MTSEA-biotin labels reduced cysteines in proteins and we used it presently to determine if the reduction in hDAT activity caused by ONOO− was related to oxidation of cysteine residues. Intact cells were exposed to 1 mmONOO− and washed membranes were isolated and labeled with MTSEA-biotin. The results are presented in Figure5 and show that untreated hDAT was readily detected by MTSEA-biotin labeling. ONOO− treatment caused a substantial reduction in labeling of the hDAT by MTSEA-biotin, indicative of cysteine modification. Treatment of cells with pCMB or MTSEA reduced MTSEA-biotin labeling of hDAT much like ONOO− (Fig. 5). Digital scans of the data in Figure 5 establish that ONOO− reduced MTSEA-biotinylation of the hDAT by ∼80%. The reductions in labeling caused by pCMB (70%) and MTSEA (45%) were somewhat smaller in magnitude when compared with ONOO−. We did not observe evidence of ONOO−-induced tyrosine nitration of the hDAT under the same conditions where it reduced MTSEA-biotin labeling (data not shown).
Fig. 5.

The effect of ONOO− on MTSEA-biotin labeling of cysteine residues in hDAT. Intact EM4 cells stably expressing wild-type hDAT were exposed to ONOO− (1 mm), pCMB (100 μm), or MTSEA (1 mm) for 10 min at 25°C. Controls were treated with decomposed ONOO−. Cells were washed three times with KRH and then homogenized in PBS. Membranes pelleted by a 15 min centrifugation step at 40,000 ×g were washed three times with PBS and exposed to 1 mm MTSEA-biotin for 30 min at 4°C. Unreacted MTSEA-biotin was removed by washing, and membranes were solubilized in 1% Triton X-100. Biotinylated proteins were separated from unlabeled proteins by adsorption to neutravidin agarose beads for 60 min at room temperature. Beads were washed, and adsorbed proteins were eluted with SDS-stop solution. Proteins were resolved by SDS-PAGE, blotted to PVDF membranes, and probed for hDAT with the use of antibodies against the Flag-epitope tag (8 μg/ml). The hDAT was visualized with enhanced chemiluminescence. This experiment was repeated on four other occasions with the same result.
Identification of the cysteine residues in hDAT that are targeted by ONOO−
The data in Figure 5 suggested a relative loss of reduced cysteines in hDAT after ONOO− treatment, but stopped short of identifying the most critical cysteine target or targets. Therefore, we used mutant forms of the hDAT to identify the cysteine targets for ONOO− attack. The results in Figure 6 show that X7C (a mutant of hDAT in which all intracellular and extracellular cysteines and one transmembrane cysteine were mutated), was relatively resistant to inhibition by ONOO−. Wild-type hDAT was inhibited by 50% after treatment with 1 mmONOO−, but the X7C mutant was inhibited by only 11% after the same treatment. The effect of ONOO− on X7C was significantly different from its effect on wild-type hDAT (p < 0.001; Bonferroni's test). The CD-DAT mutant, which only contains reduced cysteines in its first (C135) and third (C342) intracellular loops, was inhibited by ONOO− as was a mutant in which C342 alone had been reintroduced into X7C (X7C-M342C). ONOO− reduced DA uptake mediated by CD-DAT and X7C-M342C to ∼50% of control, the same extent as seen in wild-type hDAT.
Fig. 6.

The effect of ONOO− on cysteine mutants of hDAT. Intact EM4 cells stably expressing wild-type hDAT or the indicated cysteine mutants were exposed to ONOO− (1 mm) for 10 min at 25°C. Cells were washed three times with KRH, and the uptake of3H-DA was determined. X7C is a mutant of the wild-type (WT) hDAT in which all intracellular and extracellular loop cysteines were mutated (see Materials and Methods). CD-DAT is a cysteine-deficient mutant that contains reduced cysteines only in its first (C135) and third (C342) intracellular loops. X7C-M342C is a mutant into which only cysteine 342 has been reintroduced into X7C. Data are expressed as a percentage of control, where each mutant (untreated) served as its own control for ONOO− treatment. The results represent mean ± SEM of five to seven experiments run in triplicate. *Indicates that the effect of ONOO− was significantly different from its effect on WT hDAT (p < 0.05; Bonferroni's test).
DISCUSSION
ONOO− is a powerful oxidant and cytotoxin whose production has been associated with conditions that result in damage to DA neurons. For example, the neurotoxic amphetamines (Imam et al., 1999, 2001a,b; Imam and Ali, 2001) and MPTP (Schulz et al., 1995; Ferrante et al., 1999; Pennathur et al., 1999) are thought to exert toxicity to DA neurons, at least in part, through the production of ONOO−. The appearance of nitrotyrosine immunoreactivity in postmortem brain from individuals with Parkinson's disease and other neurodegenerative conditions (Beckman and Koppenol, 1996; Good et al., 1996, 1998; Heales et al., 1999) also serves as indirect evidence of ONOO− production. Methamphetamine (Fleckenstein et al., 1997a,c; Bennett et al., 1998; Hanson et al., 1998; Kokoshka et al., 1998; Metzger et al., 1998; Sandoval et al., 2000) and MPTP (Kilbourn et al., 2000; Poyot et al., 2001) cause reductions in DAT function, and the levels of hDAT are reduced in Parkinson's disease as well (Kaufman and Madras, 1991; Guttman et al., 1997; Marek et al., 2001; Varrone et al., 2001). Although the DAT is known to be sensitive to inhibition by certain reactive oxygen species (Fleckenstein et al., 1997b; Hanson et al., 1998; Davidson et al., 2001; Sandoval et al., 2001), the effects of ONOO− on DAT have not been investigated directly.
Our results establish that ONOO− causes a selective and irreversible reduction in hDAT function. DA uptake into intact EM4 cells stably expressing the hDAT was diminished by ONOO− through a mechanism that reflected a reduction in Vmax. ONOO− did not cause cell lysis, and it did not provoke release of DA from preloaded cells, suggesting that its effects were mediated by a direct effect on the inward transport properties of the hDAT. The hDAT has a total of 13 cysteine residues arrayed throughout its 12 transmembrane domains and intracellular and extracellular loops (Amara et al., 1998; Chen and Reith, 2000). Recent molecular studies using a number of mutants of the hDAT have revealed the importance of its cysteines in DA transport and ligand binding (Ferrer and Javitch, 1998; Chen et al., 2000; Hastrup et al., 2001;Whitehead et al., 2001). In particular, cysteine 342, located in the third intracellular loop, is critical to hDAT function. These results provided ample precedence to focus attention on cysteine residues as targets for the inhibitory effects of ONOO−.
Several lines of evidence establish that ONOO− inactivates the hDAT through modification of cysteine residues. First, ONOO− is a powerful cysteine oxidant (Radi et al., 1991), and cysteine residues in proteins are among the most reactive amino acids with ONOO−(Alvarez et al., 1999). Second, cysteine residues in the hDAT are known to be critical determinants of its function (Ferrer and Javitch, 1998;Javitch, 1998; Chen et al., 2000; Hastrup et al., 2001; Whitehead et al., 2001). We found that the membrane-permeable sulfhydryl reagents pCMB and MTSEA inhibited hDAT function like ONOO−. MTSET, a membrane-impermeable sulfhydryl reagent, had no effect on hDAT activity, in agreement with previous results (Chen et al., 2000). Third, MTSEA-biotin labeling of hDAT was substantially reduced by a concentration of ONOO− that significantly inhibits hDAT function. Taken together, these findings support the conclusion that ONOO− modifies hDAT function through its effects on cysteine residues.
Intact cell membranes are not thought to present barriers to ONOO− (Beckman et al., 1994; Beckman and Koppenol, 1996), so we tested a series of cysteine mutants of hDAT to determine the potential inhibitory site of action of ONOO−. The X7C mutant, in which all intracellular and extracellular loop cysteines were mutated, was relatively resistant to inhibition by ONOO−. The CD-DAT mutant, which only contains reduced cysteines 135 and 342, was sensitive to inhibition to ONOO−. The X7C-M342C, a mutant into which C342 alone had been reintroduced into X7C, was also inhibited by ONOO− to the same extent as wild-type hDAT. These results point to cysteine 342 as the site in hDAT at which ONOO− acts to lower its activity. Cysteine 342 is not the only cysteine residue modified by ONOO− (Fig. 5) or other oxidants, but it appears that modification of cysteine 342 has the most severe consequences on hDAT function.
DA and other substrates that are transported by the DAT are known to cause a conformational alteration in hDAT that makes cysteine 342 more accessible to sulfhydryl reagents (Chen et al., 2000). Therefore, we predicted initially that intracellular DA would make hDAT more susceptible to inhibition by ONOO−. It was found, however, that when cells were loaded with DA before ONOO− treatment (and extracellular DA was removed by washing), the effect of ONOO−on hDAT was neither increased nor decreased. On the other hand, when cells remained exposed to external DA during ONOO− treatment, inhibition of hDAT was completely prevented. The ability of extracellular DA to prevent ONOO−-induced inactivation of hDAT appears to be the result of a detoxification of ONOO− through its reaction with DA. It has recently been observed that DA can react with ONOO− to produce the o-quinone of DA (Pannala et al., 1997; Kerry and Rice-Evans, 1999).We did not find evidence that the DA quinone is a substrate for uptake by the hDAT (our unpublished observations), so we conclude that the reaction of DA-quinone with external hDAT cysteines, like that shown by MTSET, would not lead to inhibition of uptake. It may seem paradoxical that 10 μm DA could block the effects of 1 mm ONOO−. However, ONOO− is extremely unstable at neutral pH, where it has an apparent half-life of 1.9 sec (Beckman et al., 1990). Taking this into account, the bolus addition of 250 μm ONOO− is approximately equivalent to a steady-state level of 1 μm maintained for 7 min (Beckman et al., 1994). Therefore, it is likely that cells were exposed to an effective concentration of ONOO− that is in the same range as that of DA (i.e., micromolar). The situation is different for intracellular cysteine residues in hDAT. DA-quinone, synthesized by electrochemical oxidation of DA, is known to prevent ligand binding to hDAT as a result of DA-quinone-mediated modification of cysteine 342 (Whitehead et al., 2001). Thus, our results are entirely consistent with those of Whitehead et al. (2001) and suggest that ONOO− reacts with intracellular DA to create the DA-quinone, which in turn modifies cysteine 342 and causes partial loss of transport function.
The reduced transport Vmax caused by ONOO− treatment is most likely mediated by a direct effect on uptake of modification of Cys342. DAT activity can also be altered by changes in its membrane trafficking. For example, amphetamine leads to a reduction in surface expression of the hDAT and diminished DA uptake (Saunders et al., 2000). Nonetheless, the time of exposure to ONOO− was relatively brief (10 min), and cell surface labeling of the hDAT remained strong after treatment with ONOO− (our unpublished observations). These results represent the first characterization of the effects of ONOO−on the hDAT, and they establish the possibility that drug- and disease-induced oxidative stress can alter hDAT function. ONOO− has been implicated as a causative factor under conditions in which DA neurons are damaged, including the neurotoxic amphetamines and in Parkinson's disease.
Footnotes
This work was supported by National Institutes of Health Grants DA06067 (S.U.P.), DA10756 (D.M.K.), DA11495 (J.A.J.), DA14942 (J.A.J.), and MH57324 (J.A.J.), and by a Veterans Affairs Merit Award (D.M.K.).
Correspondence should be addressed to Dr. Donald M. Kuhn, Department of Psychiatry and Behavioral Neurosciences, Wayne State University School of Medicine, 2125 Scott Hall, 540 East Canfield, Detroit, MI 48201. E-mail: donald.kuhn@wayne.edu.
REFERENCES
- 1.Alvarez B, Ferrer-Sueta G, Freeman BA, Radi R. Kinetics of peroxynitrite reaction with amino acids and human serum albumin. J Biol Chem. 1999;274:842–848. doi: 10.1074/jbc.274.2.842. [DOI] [PubMed] [Google Scholar]
- 2.Amara SG, Sonders MS, Zahniser NR, Povlock SL, Daniels GM. Molecular physiology and regulation of catecholamine transporters. Adv Pharmacol. 1998;42:164–168. doi: 10.1016/s1054-3589(08)60718-8. [DOI] [PubMed] [Google Scholar]
- 3.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 4.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beckman JS, Chen J, Ischiropoulos H, Crow JP. Oxidative chemistry of peroxynitrite. Methods Enzymol. 1994;233:229–240. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- 6.Bennett BA, Hollingsworth CK, Martin RS, Harp JJ. Methamphetamine-induced alterations in dopamine transporter function. Brain Res. 1998;782:219–227. doi: 10.1016/s0006-8993(97)01281-x. [DOI] [PubMed] [Google Scholar]
- 7.Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, Verna JM. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson's disease. Prog Neurobiol. 2001;65:135–172. doi: 10.1016/s0301-0082(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 8.Cadet JL, Ali S, Epstein C. Involvement of oxygen-based radicals in methamphetamine-induced neurotoxicity: evidence from the use of CuZnSOD transgenic mice. Ann NY Acad Sci. 1994;738:388–391. doi: 10.1111/j.1749-6632.1994.tb21827.x. [DOI] [PubMed] [Google Scholar]
- 9.Chen N, Reith ME. Structure and function of the dopamine transporter. Eur J Pharmacol. 2000;405:329–339. doi: 10.1016/s0014-2999(00)00563-x. [DOI] [PubMed] [Google Scholar]
- 10.Chen N, Ferrer JV, Javitch JA, Justice JB., Jr Transport-dependent accessibility of a cytoplasmic loop cysteine in the human dopamine transporter. J Biol Chem. 2000;275:1608–1614. doi: 10.1074/jbc.275.3.1608. [DOI] [PubMed] [Google Scholar]
- 11.Crow JP, Ischiropoulos H. Detection and quantitation of nitrotyrosine residues in proteins: in vivo marker of peroxynitrite. Methods Enzymol. 1996;269:185–194. doi: 10.1016/s0076-6879(96)69020-x. [DOI] [PubMed] [Google Scholar]
- 12.Cubells JF, Rayport S, Rajendran G, Sulzer D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J Neurosci. 1994;14:2260–2271. doi: 10.1523/JNEUROSCI.14-04-02260.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daniels GM, Amara SG. Selective labeling of neurotransmitter transporters at the cell surface. Methods Enzymol. 1998;296:307–318. doi: 10.1016/s0076-6879(98)96023-2. [DOI] [PubMed] [Google Scholar]
- 14.Davidson C, Gow AJ, Lee TH, Ellinwood EH. Methamphetamine neurotoxicity: necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res Brain Res Rev. 2001;36:1–22. doi: 10.1016/s0165-0173(01)00054-6. [DOI] [PubMed] [Google Scholar]
- 15.Di Monte DA, Royland JE, Jakowec MW, Langston JW. Role of nitric oxide in methamphetamine neurotoxicity: protection by 7-nitroindazole, an inhibitor of neuronal nitric oxide synthase. J Neurochem. 1996;67:2443–2450. doi: 10.1046/j.1471-4159.1996.67062443.x. [DOI] [PubMed] [Google Scholar]
- 16.Ferrante RJ, Hantraye P, Brouillet E, Beal MF. Increased nitrotyrosine immunoreactivity in substantia nigra neurons in MPTP treated baboons is blocked by inhibition of neuronal nitric oxide synthase. Brain Res. 1999;823:177–182. doi: 10.1016/s0006-8993(99)01166-x. [DOI] [PubMed] [Google Scholar]
- 17.Ferrer JV, Javitch JA. Cocaine alters the accessibility of endogenous cysteines in putative extracellular and intracellular loops of the human dopamine transporter. Proc Natl Acad Sci USA. 1998;95:9238–9243. doi: 10.1073/pnas.95.16.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fleckenstein AE, Metzger RR, Gibb JW, Hanson GR. A rapid and reversible change in dopamine transporters induced by methamphetamine. Eur J Pharmacol. 1997a;323:R9–R10. doi: 10.1016/s0014-2999(97)00148-9. [DOI] [PubMed] [Google Scholar]
- 19.Fleckenstein AE, Metzger RR, Beyeler ML, Gibb JW, Hanson GR. Oxygen radicals diminish dopamine transporter function in rat striatum. Eur J Pharmacol. 1997b;334:111–114. doi: 10.1016/s0014-2999(97)01175-8. [DOI] [PubMed] [Google Scholar]
- 20.Fleckenstein AE, Metzger RR, Wilkins DG, Gibb JW, Hanson GR. Rapid and reversible effects of methamphetamine on dopamine transporters. J Pharmacol Exp Ther. 1997c;282:834–838. [PubMed] [Google Scholar]
- 21.Gainetdinov RR, Fumagalli F, Jones SR, Caron MG. Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J Neurochem. 1997;69:1322–1325. doi: 10.1046/j.1471-4159.1997.69031322.x. [DOI] [PubMed] [Google Scholar]
- 22.Giros B, el Mestikawy S, Bertrand L, Caron MG. Cloning and functional characterization of a cocaine-sensitive dopamine transporter. FEBS Lett. 1991;295:149–154. doi: 10.1016/0014-5793(91)81406-x. [DOI] [PubMed] [Google Scholar]
- 23.Giros B, el Mestikawy S, Godinot N, Zheng K, Han H, Yang-Feng T, Caron MG. Cloning, pharmacological characterization, and chromosome assignment of the human dopamine transporter. Mol Pharmacol. 1992;42:383–390. [PubMed] [Google Scholar]
- 24.Good PF, Werner P, Hsu A, Olanow CW, Perl DP. Evidence of neuronal oxidative damage in Alzheimer's disease. Am J Pathol. 1996;149:21–28. [PMC free article] [PubMed] [Google Scholar]
- 25.Good PF, Hsu A, Werner P, Perl DP, Olanow CW. Protein nitration in Parkinson's disease. J Neuropathol Exp Neurol. 1998;57:338–342. doi: 10.1097/00005072-199804000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- 27.Graham DG, Tiffany SM, Bell WR, Jr, Gutknecht WF. Autoxidation versus covalent binding of quinones as the mechanism of toxicity of dopamine, 6-hydroxydopamine, and related compounds toward C1300 neuroblastoma cells in vitro. Mol Pharmacol. 1978;14:644–653. [PubMed] [Google Scholar]
- 28.Guttman M, Burkholder J, Kish SJ, Hussey D, Wilson A, DaSilva J, Houle S. [11C]RTI-32 PET studies of the dopamine transporter in early dopa-naive Parkinson's disease: implications for the symptomatic threshold. Neurology. 1997;48:1578–1583. doi: 10.1212/wnl.48.6.1578. [DOI] [PubMed] [Google Scholar]
- 29.Halliwell B, Evans P, Whiteman M. Assessment of peroxynitrite scavengers in vitro. Methods Enzymol. 1999;301:333–342. doi: 10.1016/s0076-6879(99)01097-6. [DOI] [PubMed] [Google Scholar]
- 30.Hanson GR, Gibb JW, Metzger RR, Kokoshka JM, Fleckenstein AE. Methamphetamine-induced rapid and reversible reduction in the activities of tryptophan hydroxylase and dopamine transporters: oxidative consequences? Ann NY Acad Sci. 1998;844:103–107. [PubMed] [Google Scholar]
- 31.Hastrup H, Karlin A, Javitch JA. Symmetrical dimer of the human dopamine transporter revealed by cross-linking Cys-306 at the extracellular end of the sixth transmembrane segment. Proc Natl Acad Sci USA. 2001;98:10055–10060. doi: 10.1073/pnas.181344298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haughey HM, Brown JM, Fleckenstein AE, Hanson GR. Oxygen radicals differentially affect Na+/Cl(-)-dependent transporters. Eur J Pharmacol. 1999;379:107–110. doi: 10.1016/s0014-2999(99)00487-2. [DOI] [PubMed] [Google Scholar]
- 33.Haughey HM, Brown JM, Wilkins DG, Hanson GR, Fleckenstein AE. Differential effects of methamphetamine on Na(+)/Cl(-)-dependent transporters. Brain Res. 2000;863:59–65. doi: 10.1016/s0006-8993(00)02094-1. [DOI] [PubMed] [Google Scholar]
- 34.Heales SJ, Bolanos JP, Stewart VC, Brookes PS, Land JM, Clark JB. Nitric oxide, mitochondria and neurological disease. Biochim Biophys Acta. 1999;1410:215–228. doi: 10.1016/s0005-2728(98)00168-6. [DOI] [PubMed] [Google Scholar]
- 35.Imam SZ, Ali SF. Aging increases the susceptibility to methamphetamine-induced dopaminergic neurotoxicity in rats: correlation with peroxynitrite production and hyperthermia. J Neurochem. 2001;78:952–959. doi: 10.1046/j.1471-4159.2001.00477.x. [DOI] [PubMed] [Google Scholar]
- 36.Imam SZ, Crow JP, Newport GD, Islam F, Slikker W, Jr, Ali SF. Methamphetamine generates peroxynitrite and produces dopaminergic neurotoxicity in mice: protective effects of peroxynitrite decomposition catalyst. Brain Res. 1999;837:15–21. doi: 10.1016/s0006-8993(99)01663-7. [DOI] [PubMed] [Google Scholar]
- 37.Imam SZ, Newport GD, Itzhak Y, Cadet JL, Islam F, Slikker W, Ali SF. Peroxynitrite plays a role in methamphetamine-induced dopaminergic neurotoxicity: evidence from mice lacking neuronal nitric oxide synthase gene or overexpressing copper-zinc superoxide dismutase. J Neurochem. 2001a;76:745–749. doi: 10.1046/j.1471-4159.2001.00029.x. [DOI] [PubMed] [Google Scholar]
- 38.Imam SZ, el-Yazal J, Newport GD, Itzhak Y, Cadet JL, Slikker W, Jr, Ali SF. Methamphetamine-induced dopaminergic neurotoxicity: role of peroxynitrite and neuroprotective role of antioxidants and peroxynitrite decomposition catalysts. Ann NY Acad Sci. 2001b;939:366–380. doi: 10.1111/j.1749-6632.2001.tb03646.x. [DOI] [PubMed] [Google Scholar]
- 39.Ischiropoulos H, al-Mehdi AB. Peroxynitrite-mediated oxidative protein modifications. FEBS Lett. 1995;364:279–282. doi: 10.1016/0014-5793(95)00307-u. [DOI] [PubMed] [Google Scholar]
- 40.Itzhak Y, Martin JL, Ali SF. Methamphetamine- and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurotoxicity in inducible nitric oxide synthase- deficient mice. Synapse. 1999;34:305–312. doi: 10.1002/(SICI)1098-2396(19991215)34:4<305::AID-SYN6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 41.Jaber M, Jones S, Giros B, Caron MG. The dopamine transporter: a crucial component regulating dopamine transmission. Mov Disord. 1997;12:629–633. doi: 10.1002/mds.870120502. [DOI] [PubMed] [Google Scholar]
- 42.Javitch JA. Probing structure of neurotransmitter transporters by substituted-cysteine accessibility method. Methods Enzymol. 1998;296:331–346. doi: 10.1016/s0076-6879(98)96025-6. [DOI] [PubMed] [Google Scholar]
- 43.Javitch JA, Snyder SH. Uptake of MPP(+) by dopamine neurons explains selectivity of parkinsonism-inducing neurotoxin, MPTP. Eur J Pharmacol. 1984;106:455–456. doi: 10.1016/0014-2999(84)90740-4. [DOI] [PubMed] [Google Scholar]
- 44.Javitch JA, D'Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci USA. 1985;82:2173–2177. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaufman MJ, Madras BK. Severe depletion of cocaine recognition sites associated with the dopamine transporter in Parkinson's-diseased striatum. Synapse. 1991;9:43–49. doi: 10.1002/syn.890090107. [DOI] [PubMed] [Google Scholar]
- 46.Kerry N, Rice-Evans C. Inhibition of peroxynitrite-mediated oxidation of dopamine by flavonoid and phenolic antioxidants and their structural relationships. J Neurochem. 1999;73:247–253. doi: 10.1046/j.1471-4159.1999.0730247.x. [DOI] [PubMed] [Google Scholar]
- 47.Kilbourn MR, Kuszpit K, Sherman P. Rapid and differential losses of in vivo dopamine transporter (DAT) and vesicular monoamine transporter (VMAT2) radioligand binding in MPTP-treated mice. Synapse. 2000;35:250–255. doi: 10.1002/(SICI)1098-2396(20000315)35:4<250::AID-SYN2>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 48.Kokoshka JM, Vaughan RA, Hanson GR, Fleckenstein AE. Nature of methamphetamine-induced rapid and reversible changes in dopamine transporters. Eur J Pharmacol. 1998;361:269–275. doi: 10.1016/s0014-2999(98)00741-9. [DOI] [PubMed] [Google Scholar]
- 49.Koppenol WH, Moreno JJ, Pryor WA, Ischiropoulos H, Beckman JS. Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem Res Toxicol. 1992;5:834–842. doi: 10.1021/tx00030a017. [DOI] [PubMed] [Google Scholar]
- 50.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 51.Lotharius J, O'Malley KL. The parkinsonism-inducing drug 1-methyl-4-phenylpyridinium triggers intracellular dopamine oxidation. A novel mechanism of toxicity. J Biol Chem. 2000;275:38581–38588. doi: 10.1074/jbc.M005385200. [DOI] [PubMed] [Google Scholar]
- 52.MacMillan-Crow LA, Thompson JA. Immunoprecipitation of nitrotyrosine-containing proteins. Methods Enzymol. 1999;301:135–145. doi: 10.1016/s0076-6879(99)01076-9. [DOI] [PubMed] [Google Scholar]
- 53.Marek K, Innis R, van Dyck C, Fussell B, Early, Eberly S, Oakes D, Seibyl J. [(123)I]beta-CIT SPECT imaging assessment of the rate of Parkinson's disease progression. Neurology. 2001;57:2089–2094. doi: 10.1212/wnl.57.11.2089. [DOI] [PubMed] [Google Scholar]
- 54.Metzger RR, Hanson GR, Gibb JW, Fleckenstein AE. 3–4-Methylenedioxymethamphetamine-induced acute changes in dopamine transporter function. Eur J Pharmacol. 1998;349:205–210. doi: 10.1016/s0014-2999(98)00196-4. [DOI] [PubMed] [Google Scholar]
- 55.Pannala AS, Rice-Evans CA, Halliwell B, Singh S. Inhibition of peroxynitrite-mediated tyrosine nitration by catechin polyphenols. Biochem Biophys Res Commun. 1997;232:164–168. doi: 10.1006/bbrc.1997.6254. [DOI] [PubMed] [Google Scholar]
- 56.Pennathur S, Jackson-Lewis V, Przedborski S, Heinecke JW. Mass spectrometric quantification of 3-nitrotyrosine, ortho-tyrosine, and O,O′-dityrosine in brain tissue of 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine-treated mice, a model of oxidative stress in Parkinson's disease. J Biol Chem. 1999;274:34621–34628. doi: 10.1074/jbc.274.49.34621. [DOI] [PubMed] [Google Scholar]
- 57.Poyot T, Conde F, Gregoire MC, Frouin V, Coulon C, Fuseau C, Hinnen F, Dolle F, Hantraye P, Bottlaender M. Anatomic and biochemical correlates of the dopamine transporter ligand 11C-PE2I in normal and parkinsonian primates: comparison with 6-[18F]fluoro-l-dopa. J Cereb Blood Flow Metab. 2001;21:782–792. doi: 10.1097/00004647-200107000-00003. [DOI] [PubMed] [Google Scholar]
- 58.Przedborski S, Jackson-Lewis V. Mechanisms of MPTP toxicity. Mov Disord. 1998;13:35–38. [PubMed] [Google Scholar]
- 59.Przedborski S, Jackson-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci USA. 1996;93:4565–4571. doi: 10.1073/pnas.93.10.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 61.Robbins AK, Horlick RA. Macrophage scavenger receptor confers an adherent phenotype to cells in culture. Biotechniques. 1998;25:240–244. doi: 10.2144/98252st04. [DOI] [PubMed] [Google Scholar]
- 62.Sandoval V, Hanson GR, Fleckenstein AE. Methamphetamine decreases mouse striatal dopamine transporter activity: roles of hyperthermia and dopamine. Eur J Pharmacol. 2000;409:265–271. doi: 10.1016/s0014-2999(00)00871-2. [DOI] [PubMed] [Google Scholar]
- 63.Sandoval V, Riddle EL, Ugarte YV, Hanson GR, Fleckenstein AE. Methamphetamine-induced rapid and reversible changes in dopamine transporter function: an in vitro model. J Neurosci. 2001;21:1413–1419. doi: 10.1523/JNEUROSCI.21-04-01413.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saunders C, Ferrer JV, Shi L, Chen J, Merrill G, Lamb ME, Leeb-Lundberg LM, Carvelli L, Javitch JA, Galli A. Amphetamine-induced loss of human dopamine transporter activity: an internalization-dependent and cocaine-sensitive mechanism. Proc Natl Acad Sci USA. 2000;97:6850–6855. doi: 10.1073/pnas.110035297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmidt N, Ferger B. Neurochemical findings in the MPTP model of Parkinson's disease. J Neural Transm. 2001;108:1263–1282. doi: 10.1007/s007020100004. [DOI] [PubMed] [Google Scholar]
- 66.Schulz JB, Matthews RT, Muqit MM, Browne SE, Beal MF. Inhibition of neuronal nitric oxide synthase by 7-nitroindazole protects against MPTP-induced neurotoxicity in mice. J Neurochem. 1995;64:936–939. doi: 10.1046/j.1471-4159.1995.64020936.x. [DOI] [PubMed] [Google Scholar]
- 67.Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Iwaarden PR, Driessen AJ, Konings WN. What we can learn from the effects of thiol reagents on transport proteins. Biochim Biophys Acta. 1992;1113:161–170. doi: 10.1016/0304-4157(92)90037-b. [DOI] [PubMed] [Google Scholar]
- 69.Varrone A, Marek KL, Jennings D, Innis RB, Seibyl JP. [(123)I]beta-CIT SPECT imaging demonstrates reduced density of striatal dopamine transporters in Parkinson's disease and multiple system atrophy. Mov Disord. 2001;16:1023–1032. doi: 10.1002/mds.1256. [DOI] [PubMed] [Google Scholar]
- 70.Whitehead RE, Ferrer JV, Javitch JA, Justice JB. Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J Neurochem. 2001;76:1242–1251. doi: 10.1046/j.1471-4159.2001.00125.x. [DOI] [PubMed] [Google Scholar]