

Abstract
Deprivation of afferent input in young animals results in transneuronal degeneration of postsynaptic sensory neurons in a variety of species and sensory pathways. Transneuronal degeneration is generally not seen in adult animals. The cellular and molecular basis for this dramatic developmental change in susceptibility is not understood. One possibility is that genes involved in the apoptotic process are involved in determining cell death or survival after afferent deprivation. To further investigate this possibility, we performed unilateral cochlear ablation on wild-type and bcl-2-overexpressing mice at a variety of ages. In postnatal day 5 (P5) or P8 wild-type mice, cochlea removal resulted in a 54% or 31% neuronal loss in the anteroventral cochlear nucleus (AVCN), respectively. When the same manipulation is performed on a P30 mouse, no loss of AVCN neurons occurs. This confirmed a rather abrupt change in the sensitivity to disruption of afferent input, a critical period. However, in littermates expressingbcl-2 under a neuron-specific enolase promoter, no significant loss of AVCN neurons was observed at any age after unilateral cochlear ablation. Furthermore, wild-type mice demonstrate rapid expression of activated caspase-3 in AVCN neurons within hours of deafferentation, whereas bcl-2-overexpressing mice do not. This suggests that bcl-2 can influence cell survival after removal of afferent input during the critical period and is consistent with the hypothesis that caspase-3 is one effector of cell death under these circumstances. These data are the first to indicate that known apoptotic mediators can play a role in central neuronal plasticity in models of afferent deprivation.
Keywords: deafferentation, bcl-2, caspase-3, apoptosis, cochlear nucleus, critical period
Removal of sensory receptors or changes in afferent activity as a function of environmental manipulations have profound effects on the maturation of neuronal structure and function (Levi-Montalcini, 1949; Hubel and Wiesel, 1970;Van der Loos and Woolsey, 1973; Brunjes, 1994). One dramatic example is deprivation-induced cell death, which is seen in a variety of systems when afferent activity is interrupted during development (Born and Rubel, 1988; Frazier and Brunjes, 1988; Catsicas et al., 1992;Galli-Resta et al., 1993; Linden, 1994; Sherrard and Bower, 1998; Baldi et al., 2000). A poorly understood property of deprivation-induced cell death, as well as other experience-induced changes in neural structure and function, is that alterations of the normal pattern of activity during a finite period early in life dramatically alter the CNS, whereas identical manipulations later in life have little or no effect (Wiesel and Hubel, 1963; Hubel and Wiesel, 1970; Berardi et al., 2000). The molecular bases underlying these so-called critical periods of heightened sensitivity to afferent manipulations are virtually unknown in any sensory system.
Studies in chicks and gerbils have shown that the rapid transneuronal changes in cochlear nucleus neurons after cochlea removal are attributable to elimination of afferent activity (Rubel et al., 1990; Lachica et al., 1996). We recently extended this work in the mouse (Mostafapour et al., 2000). Anteroventral cochlear nucleus (AVCN) neurons in young animals undergo an apoptotic-lke process that begins within 12 hr of sensory deprivation, peaks at ∼48 hr, and is complete by 96 hr. The same manipulation in older animals results in no cell death.
Programmed cell death and many pathological forms of cell death are regulated by a family of apoptotic genes, including thebcl-2 and caspase families (Miura et al., 1993; Yuan et al., 1993; Zheng and Flavell, 2000). We sought to determine whether these same families of molecules might mediate changes in the susceptibility of the nervous system to afferent deprivation during critical periods of development. We demonstrate that upregulation of bcl-2prevents loss of central auditory neurons after removal of the peripheral sense organ, the cochlea, during a critical period for development of the central auditory system. Furthermore, detection of activated caspase-3 within hours of deafferentation suggests that it may be an effector of cell death in this model. Surviving neurons appear healthy and continue normal expression of neuron-specific proteins.
MATERIALS AND METHODS
Animals. Transgenic mice overexpressing the humanbcl-2 gene under the control of a neuron-specific enolase (NSE) promoter (NSE73a line) were the generous gift of Dr. D.-F. Chen (Harvard Medical School, Boston, MA) (Martinou et al., 1994). NSE73a male breeders were bred to wild-type C57BL/6 females. Litters were thus mixed of roughly equal numbers of transgenic and wild-type (control) mice. Mouse genotype was identified by tail-clip DNA analysis using PCR. All mouse cages were checked daily for new litters; new pups were considered 0 d of age on the day of birth [postnatal day 0 (P0)]. All animals were weaned at approximately P21. The numbers of animals used for each analysis are provided in Results. All procedures were approved by the University of Washington Animal Use Committee.
Surgical procedures. Litters born to heterozygous NSE73a breeders underwent surgery at P5, P8, or P30. Mice of all ages were anesthetized using inhaled methoxyflurane until they were areflexic; this level of anesthesia was maintained throughout the surgical procedure. In animals at age P8 and younger, an incision was made inferior to the pinna, and the tympanic membrane was identified. The middle-ear mesenchyme (if present) was aspirated, ossicles were removed, and the basal turn of the cochlea was visualized. Using a 30 gauge needle, the bony wall of the cochlea was penetrated and the contents were aspirated using a fine glass pipette. The modiolus was visualized and destroyed. The skin incision was closed using cyanoacrylic glue. Litters were returned to their parents within 2 hr. In P30 animals, a transtympanic approach was used. A small superior and posteriorly based flap of skin was raised in the external acoustic canal (this was later used for closure). The tympanic membrane and ossicles were visualized and removed. A 23 gauge needle was used to penetrate the bony cochlea, and its contents were aspirated. A pick was used to destroy the modiolus. The skin incision was closed with cyanoacrylic glue.
Histology. Litters of mice used to study the age dependence of afferent deprivation survived 7 d after unilateral ablation of the cochlea. A previous study (Mostafapour et al., 2000) shows that this survival period is sufficient to observe asymptotic transneuronal cell death. Animals were killed with CO2intoxication and perfused transcardially with phosphate-buffered 4% paraformaldehyde, pH 7.4. After perfusion, each brain was immediately dissected from the skull and placed in fresh fixative for 24–48 hr. Brains were serially dehydrated in graded alcohols, embedded in paraffin, and serially sectioned at 10 μm in the coronal plane. A one-in-five series of sections was mounted on gelatin-coated slides, dewaxed in xylene, stained with thionin, and coverslipped with DPX mounting medium (BDH Laboratories, Poole, UK). Photomicrocraphs were acquired by digital photography using a SPOT IIE camera (Digital Instruments, Sterling Heights, MI) and transferred to Adobe Photoshop 5.5 software (Adobe Systems, Inc., Seattle, WA).
Immunocytochemistry. Litters of mice used for immunocytochemical studies survived 1, 3, 6, 12, 24, or 48 hr or 3 weeks after unilateral ablation of the cochlea and were killed as described above. Brains were fixed, embedded in paraffin, sectioned, and mounted as noted above with the exception that the duration of fixation was 18 hr. Antigen retrieval was performed on the brainstem sections in preparation for immunohistochemistry (except for anti-NSE and anti-α-neurofilament staining). Slides were placed in a Coplin jar containing 10 mm citric acid solution, pH 6, steamed in a rice cooker for 25 min, and subsequently cooled in ice for 10 min. Slides were subsequently rinsed in buffered saline, incubated in 0.6% hydrogen peroxide in saline for 30 min, and rinsed again. Tissue was blocked for 1 hr with 5% normal goat serum and 1% Triton X-100 in PBS (anti-bcl-2) or 5% nonfat dry milk and 0.1% Triton X-100 in PBS [anti-NSE, anti-α-neurofilament, and anti-neuron-specific nuclear protein (NeuN)]. Sections were subsequently incubated overnight with primary antibody: anti-activated caspase-3 (rabbit polyclonal, 1:500; Cell Signaling Technology, Beverly, MA), anti-bcl-2 ΔC-21 (rabbit polyclonal, 1: 2000; Santa Cruz Biotechnology, Santa Cruz, CA), anti-α-neurofilament (1:20,000, provided by Dr. Virginia Lee, University of Pennsylvania School of Medicine, Philadelphia, PA), anti-NeuN (1:1000;Chemicon, Temecula, CA), or anti-NSE (1:500; Dako Inc., Glostrup, Denmark). Tissue was subsequently rinsed and incubated for 30 min with secondary antibody (Vector biotinylated goat anti-rabbit, 1:200; Vector Laboratories, Burlingame, CA) followed by rinsing and incubation in Vector avidin–biotin complex solution for 30 min at room temperature. HRP reaction product was visualized using diaminobenzidine as the chromagen.
Analysis of neuron number. Only cases in which every section through the AVCN on both sides of the brain was intact were included in this study. Every mounted section through the entire anteroposterior extent of the AVCN was examined using standard light microscopy. The posterior boundary of the AVCN was defined by the appearance of the dorsal cochlear nucleus. Neuron counts were performed on a Leitz Aristoplan microscope (Leitz, Wetzlar, Germany) with a 40× objective and a 10 × 10 reticule. Neuron counts were performed on a one-in-five series of thionin-stained sections from each brain. All AVCN neurons in a given section were counted. The criteria for a neuron to be counted were a well defined cytoplasm and nuclear outline and a clearly visible nucleolus. Neuron counts were obtained from the AVCN on both sides of the brainstem. The AVCN on the contralateral side provided a within-animal control. Total AVCN neuron number was defined as follows: total number of neurons = number counted × 5. The percentage of AVCN neuron loss on the lesioned side was calculated as follows: 100 × [1 − (number of neurons on ablated side/number of neurons on control side)]. Corrections for “double-counting” were not used because the nucleolus is small compared with section thickness. Stereological procedures were not used because our primary goal was the comparison of the two sides of the brainstem rather than the absolute numbers of neurons. Statistical comparisons were made using a paired Student's t test for comparison of neuron loss within a single age group.
RESULTS
bcl-2 overexpression removes sensitivity of AVCN neurons to deafferentation-induced cell death
Transgenic mice overexpressing bcl-2 from the NSE73a line (Martinou et al., 1994) were examined for expression ofbcl-2 in the AVCN. Immunohistochemical analysis of cochlear nucleus neurons revealed robust expression of bcl-2 in neurons throughout the AVCN (Fig.1a,b). Expression in the cytoplasm of AVCN neurons was demonstrated when examined at higher power. Minimal expression of native bcl-2 was detected in the AVCN of wild-type littermates (Fig. 1c).
Fig. 1.
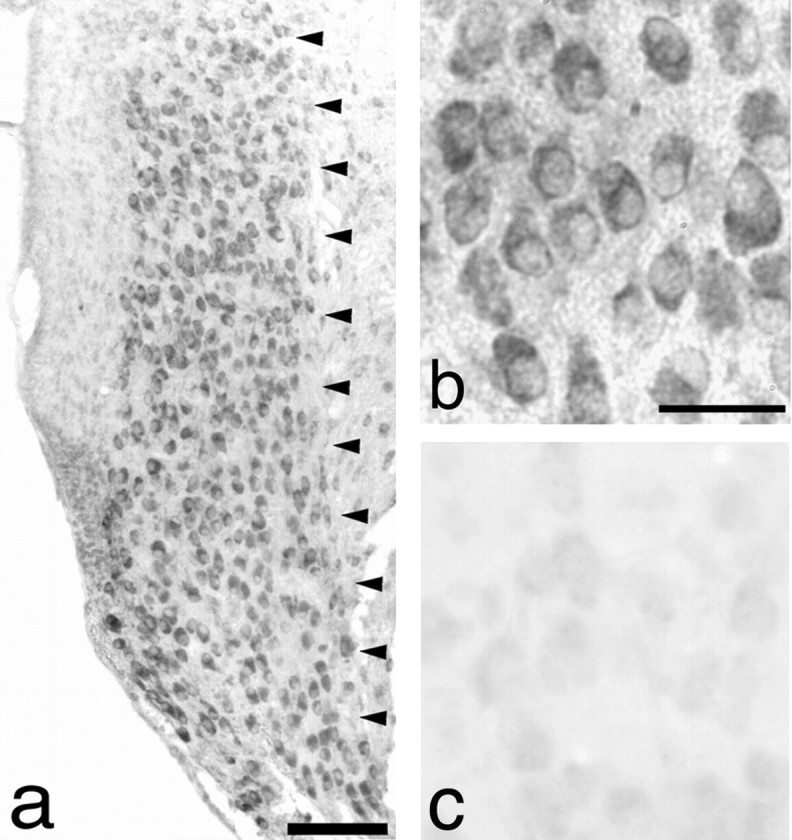
AVCN neurons in P5 mice overexpressbcl-2. Anti-bcl-2 ΔC-21 rabbit polyclonal antibody was used to examine expression ofbcl-2. a, Intense staining is noted at low power (10× magnification) throughout the AVCN (delineated byarrowheads) in an NSE73a mouse. b, Examination of the same section at 100× magnification reveals primarily cytoplasmic expression of bcl–bcl-2.c, Faint staining of native bcl-2 in a section through the AVCN of a wild-type littermate. Scale bars:a, 100 μm; b, 30 μm (also applies toc).
To investigate the effect of bcl-2 overexpression on central auditory neuron survival after afferent deprivation, transgenic mice and wild-type littermates underwent unilateral cochlea removal at ages P5, P8, and P30 followed by a 1 week survival period. Neuron counts in the AVCN were subsequently performed in the transgenic mice and littermate controls. The results of this analysis are shown in Figure2. We confirmed that in P5 wild-type animals at the time of cochlea removal, approximately one-half of AVCN neurons (mean 54%; p < 0.01) were lost within 1 week of afferent deprivation, confirming our previous results (Mostafapour et al., 2000). The same manipulation performed on P8 animals also results in significant loss of neurons (mean 31%; p < 0.01), although less than observed at age P5. In the AVCN neurons of P30 mice, no significant neuronal loss is seen after removal of input from the cochlea.
Fig. 2.

Neuron loss in the anteroventral cochlear nucleus after cochlear ablation. At age P5, wild-type mice demonstrate significant neuron loss (54 ± 10%; n = 5; mean ± SEM) versus transgenic mice, which showed no significant neuron loss (0 ± 1%; n = 4;p < 0.01). Similar results were obtained at age P8 (31 ± 6, n = 4 vs 5 ± 4,n = 7; p < 0.01). When cochlear ablation is performed at age P30, no significant neuron loss is observed in animals of either genotype.
When we examine the numbers of AVCN neurons after cochlea removal in littermate transgenic mice overexpressing bcl-2, the results are strikingly different. Unlike their wild-type littermates, mice overexpressing bcl-2 show no significant AVCN neuron loss after sensory deprivation at any age we examined (Fig. 2). To confirm that the surviving AVCN neurons ipsilateral to cochlea removal inbcl-2-overexpressing animals retained neuronal characteristics, we sought to detect neuronal markers in AVCN neurons at extended periods after deafferentation. An additional group of P5 pups underwent unilateral cochlea removal and were allowed to survive 3 weeks. Immunocytochemical examination of cochlear nucleus neurons revealed robust expression of α-neurofilament, NeuN, and NSE in neurons throughout the AVCN at comparable levels ipsilateral and contralateral to cochlea removal (Fig.3). Wild-type littermates underwent the same manipulation and showed similar staining characteristics, although fewer neurons were present in the ipsilateral AVCN (data not shown).
Fig. 3.
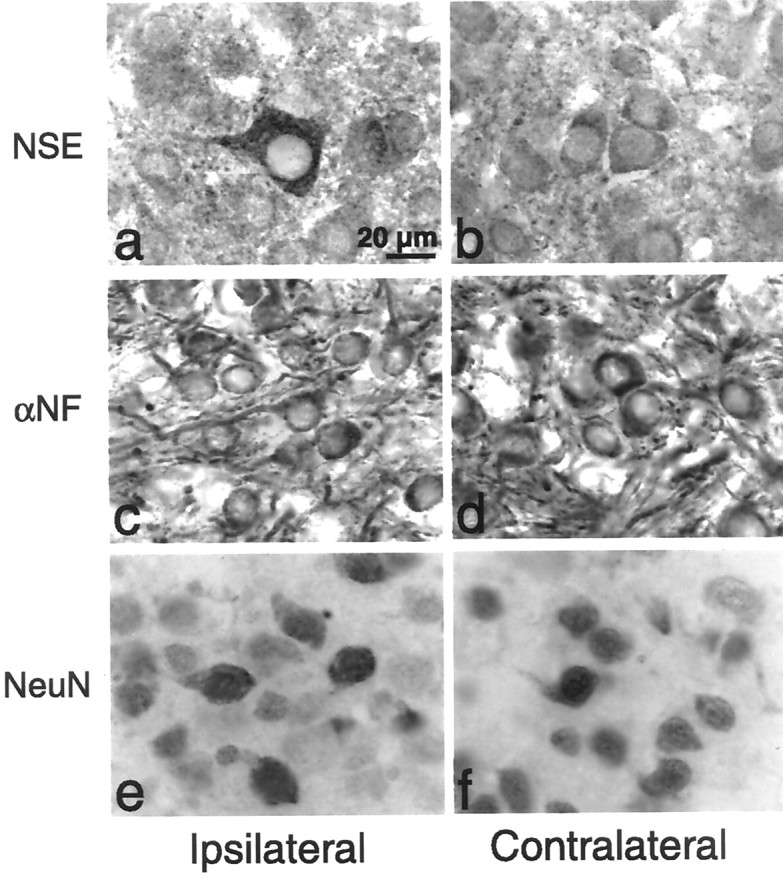
Expression of neuronal markers in the AVCN after cochlea removal in bcl-2-overexpressing animals. P5 animals underwent unilateral cochlea removal and survived 3 weeks.a, b, AVCN neurons demonstrate expression of NSE both ipsilateral and contralateral to cochlea removal. c, d, AVCN neurons demonstrate expression of α-neurofilament (α-NF) both ipsilateral and contralateral to cochlea removal. e, f, AVCN neurons demonstrate expression of NeuN both ipsilateral and contralateral to cochlea removal. All photomicrographs are at the same magnification.
Because the period of naturally occurring neuronal cell death may be affected by bcl-2 overexpression (Martinou et al., 1994), we also examined neuron number in the contralateral (i.e., unaffected) AVCN of transgenic animals and their littermate controls. Although there was a trend toward increased numbers of neurons in olderbcl-2-overexpressing animals, this was not statistically significant at any age examined. For example, the average ± SD total number of AVCN neurons contralateral to cochlea removal in P30 animals was 13,987 ± 1474 in bcl-2-overexpressing animals and 11,805 ± 1376 in wild-type littermates (p > 0.1).
Activated caspase-3 is expressed in AVCN neurons after deafferentation
To further investigate the mechanism underlying the elimination of the critical period for susceptibility to afferent deprivation bybcl-2 overexpression, we examined expression of the activated form of a downstream effector of apoptosis, caspase-3 (Zheng and Flavell, 2000). Our previous work showed that indirect evidence of apoptosis occurs as early as 12 hr after cochlea removal (Mostafapour et al., 2000). Thus, we sought to detect expression of activated caspase-3 at times leading up to and after this time point. Wild-type littermates and transgenic mice aged P5 underwent unilateral cochlear ablation and survived 1, 3, 6, 12, or 48 hr before immunohistochemical examination. These results are shown in Figures4 and 5. In wild-type mice, we found a dramatic increase in expression of activated caspase-3 in AVCN neurons ipsilateral to cochlea removal (Fig. 4a–c). Consistent with the cell death results, examination of AVCN neurons in mice overexpressingbcl-2 revealed no increase in caspase-3 activation (Fig.4d). The increased detection of activated caspase-3 in wild-type mice occurred as early as 6 hr after cochlea removal but was accentuated at 12 hr after cochlea removal (Fig. 5). Levels of activated caspase-3 decreased by 48 hr after cochlea removal. No increase in activated caspase-3 expression was detected inbcl-2-overexpressing mice at any time point after cochlea removal (Fig. 5).
Fig. 4.

Detection of activated caspase-3 in the AVCN after cochlea removal. a, Activated caspase-3-positive neurons can be detected in the ipsilateral AVCN (arrowheads) 12 hr after cochlea removal in C57BL/6 mice. b, Contralateral AVCN (arrowheads) from the same animal as shown in a demonstrates minimal detection of activated caspase-3. c, A higher-power photomicrograph of the same animal as in a demonstrates neurons containing activated caspase-3 ipsilateral to cochlea removal (arrows). d, No activated caspase-3 is detectable in the AVCN (arrowheads) ipsilateral to cochlear ablation in mice overexpressing bcl-2. Scale bars: a, 100 μm (also applies to b, d); c, 30 μm.
Fig. 5.

Time course of detection of activated caspase-3 in the AVCN after cochlea removal. Minimal activated caspase-3 was found in the AVCN of transgenic mice, and no difference was observed between sides of the brainstem. Activated caspase-3 was detected at higher levels in the ipsilateral AVCN of wild-type mice 12 hr after cochlea removal (146 ± 29, n = 7 for wild-type vs 6 ± 3, n = 4 forbcl-2-overexpressing mice; p < 0.05; mean ± SEM). Activated caspase-3 levels decreased by 48 hr after cochlea removal in wild-type animals. Where not shown, error bars are smaller than symbols.
DISCUSSION
The demonstration that bcl-2 overexpression in AVCN neurons removes the susceptibility of these neurons to afferent deprivation-induced cell death during the critical period provides evidence for a role for molecules that are considered mediators of apoptosis in modulating the response of an organism to sensory deprivation. It is well known that upregulation ofbcl-2 can prevent some forms of neuronal cell death bothin vitro and in vivo (Martinou et al., 1994;Allsopp et al., 1995). For example, retinal ganglion cells and facial motor neurons show increased survival during normal development in mice overexpressing bcl-2, and there is a reduced response to ischemic injury (Martinou et al., 1994). Furthermore, target-dependent neuronal cell death during development can be regulated by well conserved cell death pathways, as best evidenced by the dual role of the p75NTR receptor (Coulson et al., 1999). However, previous studies have not demonstrated a role for bcl-2 or other cell death mediators in the afferent activity-dependent CNS neuronal loss induced by sensory deprivation.
Several lines of evidence now support the hypothesis that the age dependence of neuronal loss after afferent deprivation (i.e., the critical period) is attributable to a change in the balance of proapoptotic versus antiapoptotic molecules expressed by the postsynaptic neurons. First, we show here that the age dependence of AVCN neuron survival to sensory input can be dramatically manipulated by altered expression of apoptotic regulatory genes. Increased expression of bcl-2 alone is sufficient to totally eliminate the sensitivity of AVCN neurons to removal of input at ages at which they are normally susceptible. Surviving neurons inbcl-2-overexpressing mice demonstrate neuronal characteristics immunocytochemically. In addition, homozygous deletion of bcl-2 causes increased loss of AVCN neurons in the adult mouse in a similar model, although not to levels of susceptibility comparable with those of P5 wild-type mice (Mostafapour et al., 2000).
Second, caspase-3, a so-called “effector” caspase in neurons (Zheng and Flavell, 2000), is detected in activated form at increased levels within hours of deafferentation in susceptible mice. This is reduced in the presence of bcl-2 overexpression. It is possible thatbcl-2 overexpression masks or overrides the effects of other antiapoptotic or proapoptotic mediators that are more functionally important in the wild-type mouse. However, our detection of caspase-3 activation in wild-type animals suggests that the pathway leading to cell death in this model goes through caspase-3 and is sensitive to expression of members of the bcl-2 family of cell death mediators (Adams and Cory, 1998). The expression of upstream “initiator” caspases after deafferentation and the possible effect of bcl-2 overexpression on these caspases remains to be examined.
The sequence of events occurring in AVCN neurons after deafferentation in this model agrees with contemporary models of apoptosis. The earliest event we have detected after deafferentation in mice thus far is activation of caspase-3, demonstrated here, which occurs within 6–12 hr of removal of afferent input to the AVCN in juvenile animals. Our previous work using the same model demonstrated detection of terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)-labeled AVCN neurons in C57BL/6 mice as early as 12 hr, peaking at 48 hr after cochlea removal (Mostafapour et al., 2000). Detection of TUNEL, in turn, preceded loss of AVCN neurons, which was first detectable at 48 hr and was complete by 96 hr after cochlea removal (Mostafapour et al., 2000). Thus, caspase-3 activation precedes TUNEL detection and neuron loss in the AVCN after deprivation in susceptible animals. Overexpression of bcl-2 halts both activation of caspase-3 and neuron loss in these animals.
These results suggest that apoptotic pathway molecules may also play a role in more subtle neuronal changes resulting from altered afferent input such as those that mediate learning and memory (Mattson, 2000). The central effects of neuron loss have profound implications for functional rehabilitation with auditory prostheses such as cochlear or auditory brainstem implants and support early intervention in cases of early hearing loss. The role of apoptosis in central neuronal loss in other sensory systems has yet to be demonstrated.
Footnotes
This work was supported by National Institutes of Health Grant DC03829. We thank Dr. Dong-Feng Chen for providingbcl-2-overexpressing mice and Dr. Virginia Lee for anti-α-neurofilament antibody.
Correspondence should be addressed to Edwin W Rubel, Virginia Merrill Bloedel Hearing Research Center, Box 357923, CHDD Building, Room CD176, University of Washington, Seattle, WA 98195. E-mail:rubel@u.washington.edu.
REFERENCES
- 1.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 2.Allsopp TE, Kiselev S, Wyatt S, Davies AM. Role of Bcl-2 in the brain-derived neurotrophic factor survival response. Eur J Neurosci. 1995;7:1266–1272. doi: 10.1111/j.1460-9568.1995.tb01116.x. [DOI] [PubMed] [Google Scholar]
- 3.Baldi A, Calia E, Ciampini A, Riccio M, Vetuschi A, Persico AM, Keller F. Deafferentation-induced apoptosis of neurons in thalamic somatosensory nuclei of the newborn rat: critical period and rescue from cell death by peripherally applied neurotrophins. Eur J Neurosci. 2000;12:2281–2290. doi: 10.1046/j.1460-9568.2000.00119.x. [DOI] [PubMed] [Google Scholar]
- 4.Berardi N, Pizzorusso T, Maffei L. Critical periods during sensory development. Curr Opin Neurobiol. 2000;10:138–145. doi: 10.1016/s0959-4388(99)00047-1. [DOI] [PubMed] [Google Scholar]
- 5.Born DE, Rubel EW. Afferent influences on brain stem auditory nuclei of the chicken: presynaptic action potentials regulate protein synthesis in nucleus magnocellularis neurons. J Neurosci. 1988;8:901–919. doi: 10.1523/JNEUROSCI.08-03-00901.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunjes PC. Unilateral naris closure and olfactory system development. Brain Res Brain Res Rev. 1994;19:146–160. doi: 10.1016/0165-0173(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 7.Catsicas M, Pequignot Y, Clarke PG. Rapid onset of neuronal death induced by blockade of either axoplasmic transport or action potentials in afferent fibers during brain development. J Neurosci. 1992;12:4642–4650. doi: 10.1523/JNEUROSCI.12-12-04642.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coulson EJ, Reid K, Bartlett PF. Signaling of neuronal cell death by the p75NTR neurotrophin receptor. Mol Neurobiol. 1999;20:29–44. doi: 10.1007/BF02741363. [DOI] [PubMed] [Google Scholar]
- 9.Frazier LL, Brunjes PC. Unilateral odor deprivation: early postnatal changes in olfactory bulb cell density and number. J Comp Neurol. 1988;269:355–370. doi: 10.1002/cne.902690304. [DOI] [PubMed] [Google Scholar]
- 10.Galli-Resta L, Ensini M, Fusco E, Gravina A, Margheritti B. Afferent spontaneous electrical activity promotes the survival of target cells in the developing retinotectal system of the rat. J Neurosci. 1993;13:243–250. doi: 10.1523/JNEUROSCI.13-01-00243.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hubel DH, Wiesel TN. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol (Lond) 1970;206:419–436. doi: 10.1113/jphysiol.1970.sp009022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lachica EL, Zirpel L, Rubel EW. Auditory system plasticity and regeneration. Thieme; New York: 1996. Intracellular mechanisms involved in the afferent regulation of neurons in the avian cochlear nucleus. pp. 333–353. [Google Scholar]
- 13.Levi-Montalcini R. The development of the acoustico-vestibular center in the chick embryo in the absence of the afferent root fibers and of descending fiber tracts. J Comp Neurol. 1949;91:209–242. doi: 10.1002/cne.900910204. [DOI] [PubMed] [Google Scholar]
- 14.Linden R. The survival of developing neurons: a review of afferent control. Neuroscience. 1994;58:671–682. doi: 10.1016/0306-4522(94)90447-2. [DOI] [PubMed] [Google Scholar]
- 15.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, Huarte J. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 16.Mattson MP. Apoptotic and anti-apoptotic synaptic signaling mechanisms. Brain Pathol. 2000;10:300–312. doi: 10.1111/j.1750-3639.2000.tb00264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miura M, Zhu H, Rotello R, Hartwieg EA, Yuan J. Induction of apoptosis in fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- 18.Mostafapour SP, Cochran SL, Del Puerto NM, Rubel EW. Patterns of cell death in mouse anteroventral cochlear nucleus neurons after unilateral cochlea removal. J Comp Neurol. 2000;426:561–571. doi: 10.1002/1096-9861(20001030)426:4<561::aid-cne5>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 19.Rubel EW, Hyson PL, Durham D. Afferent regulation of neurons in the brain stem auditory system. J Neurobiol. 1990;21:169–196. doi: 10.1002/neu.480210112. [DOI] [PubMed] [Google Scholar]
- 20.Sherrard RM, Bower AJ. Role of afferents in the development and cell survival of the vertebrate nervous system. Clin Exp Pharmacol Physiol. 1998;25:487–495. doi: 10.1111/j.1440-1681.1998.tb02241.x. [DOI] [PubMed] [Google Scholar]
- 21.Van der Loos H, Woolsey TA. Somatosensory cortex: structural alterations following early injury to sense organs. Science. 1973;179:395–398. doi: 10.1126/science.179.4071.395. [DOI] [PubMed] [Google Scholar]
- 22.Wiesel TN, Hubel DH. Single-cell responses in striate cortex of kittens deprived of vision in one eye. J Neurophysiol. 1963;26:1003–1017. doi: 10.1152/jn.1963.26.6.1003. [DOI] [PubMed] [Google Scholar]
- 23.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 24.Zheng TS, Flavell RA. Divinations and surprises: genetic analysis of caspase function in mice. Exp Cell Res. 2000;256:67–73. doi: 10.1006/excr.2000.4841. [DOI] [PubMed] [Google Scholar]