

Abstract
There is substantial evidence that bioenergetic defects and excitotoxicity may play a role in the pathogenesis of Huntington's disease (HD). Potential therapeutic strategies for neurodegenerative diseases in which there is reduced energy metabolism and NMDA-mediated excitotoxicity are the administration of the mitochondrial cofactor coenzyme Q10 and the NMDA antagonist remacemide. We found that oral administration of either coenzyme Q10 or remacemide significantly extended survival and delayed the development of motor deficits, weight loss, cerebral atrophy, and neuronal intranuclear inclusions in the R6/2 transgenic mouse model of HD. The combined treatment, using coenzyme Q10 and remacemide together, was more efficacious than either compound alone, resulting in a ∼32 and 17% increase in survival in the R6/2 and N171–82Q mice, respectively. Magnetic resonance imaging showed that combined treatment significantly attenuated ventricular enlargement in vivo. These studies further implicate defective energy metabolism and excitotoxicity in the R6/2 and N171–82Q transgenic mouse models of HD and are of interest in comparison with the outcome of a recent clinical trial examining coenzyme Q10 and remacemide in HD patients.
Keywords: Huntington's disease, excitotoxicity, coenzyme Q10, remacemide, mitochondria, transgenic
The mechanism of neuronal degeneration in Huntington's disease (HD) remains unknown, despite the identification of the disease-causing genetic defect as an expansion of a polyglutamine tract in the protein huntingtin (The Huntington's Disease Collaborative Research Group, 1993). The evidence suggests that the polyglutamine expansion confers a gain of function that in turn may be related to effects on gene transcription (Lin et al., 2000;Luthi-Carter et al., 2000). The effects on gene transcription may be linked to energy dysfunction and excitotoxicity, which are implicated in the pathogenesis of HD (Beal, 2000).
There is substantial evidence linking impaired energy metabolism to HD. Consistent with this, we and others found the following: (1) lactate is elevated in the cerebral cortex in basal ganglia of patients with HD (Jenkins et al., 1993); (2) the phosphocreatine to inorganic phosphate ratio is reduced in resting muscle of HD patients (Koroshetz et al., 1997); (3) mitochondrial toxins produce selective damage in the striatum of animals that closely resembles the pathology of HD (Beal et al., 1993a; Brouillet et al., 1995); and (4) there are reductions in mitochondrial electron transport enzymes in HD postmortem (Gu et al., 1996; Browne et al., 1997). Recent studies showed that mitochondria in HD lymphoblasts and fibroblasts show an increased susceptibility to depolarization that correlates directly with CAG repeat length (Sawa et al., 1999). The maximal rate of mitochondrial ATP generation in muscle is significantly reduced in both symptomatic HD patients and presymptomatic HD gene carriers, which correlates with CAG repeat length (Lodi et al., 2000).
In addition, there is also substantial evidence linking excitotoxicity to HD pathogenesis. Initial studies showed that kainic acid lesions replicated many pathologic features of HD (McGeer and McGeer, 1976;Schwarcz and Coyle, 1976). Subsequent studies with the NMDA agonist quinolinic acid showed that it could more accurately model HD because it produces relative sparing of NADPH-diaphorase neurons, which are spared in HD (Beal et al., 1986; Ferrante et al., 1993). The mitochondrial toxins malonate and 3-nitropropionic acid also produce striatal lesions that mimic HD and are mediated by excitotoxic mechanisms (Beal et al., 1993b; Greene et al., 1993; Brouillet et al., 1995). Both NMDA antagonists, as well as coenzyme Q10, block striatal lesions produced by malonate (Beal et al., 1993b, 1994; Greene et al., 1993). Furthermore, there are additive neuroprotective effects of NMDA antagonists with coenzyme Q10 (Schulz et al., 1996).
A major advance for studying disease pathogenesis and developing therapeutics has been the introduction of transgenic mouse models of HD. Transgenic mice expressing exon-1 of the human HD gene with an expanded CAG repeat and transgenic mice (N171–82Q) expressing a cDNA encoding a 171 amino acid N-terminal fragment of huntingtin containing 82 CAG repeats develop a progressive neurological disorder (Mangiarini et al., 1996; Schilling et al., 1999). At ∼6 weeks of age, the R6/2 mice develop loss of brain and body weight, and at 9–11 weeks they develop an irregular gait, abrupt shuttering stereotypic movements, resting tremors, and epileptic seizures. The brains of the R6/2 mice show striatal atrophy and neuronal intranuclear inclusions that are immunopositive for huntingtin and ubiquitin (Davies et al., 1997). The N171–82Q mice show similar findings, but these mice have a more delayed disease onset and longer survival (Schilling et al., 1999). In the present experiments, we examined whether oral administration of coenzyme Q10 and remacemide either alone or in combination could exert beneficial effects in the R6/2 and N171–82Q mouse models of HD.
MATERIALS AND METHODS
Animals. Male transgenic HD mice of the R6/2 strain were obtained from Jackson Laboratories (Bar Harbor, ME). The male R6/2 mice were bred with females from their background strain (B6CBAFI/J) for two generations. Male transgenic mice of the N171–82Q strain were originally obtained from Drs. Ross and Borchelt (The Johns Hopkins University) and backcrossed to the B6CBA background through 15 generations. The offspring were genotyped using a PCR assay on tail DNA. The mice were housed four to five in each cage under standard conditions with ad libitum access to water and food. All animal experiments were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by both the Veterans Administration and Boston University Animal Care Committees.
Treatment. At 21 d of age, R6/2 and littermate wild-type control mice were placed on either an unsupplemented diet or a diet supplemented with 0.2% coenzyme Q10(Vitaline, Ashland, OR) or 0.007% remacemide (Astra) or a combination of the two within the same pellet. Forty mice from the same generation were placed within each of the treatment groups. Groups were randomly pooled from multiple liters (six to eight) to ensure heterogeneity. N171–82Q mice were placed on the combination diet alone. In all, behavioral and survival data were obtained from ∼320 R6/2 mice and 160 N171–82Q mice. The diets were made into pelleted mouse chow (Purina Test Diets, Richmond, IN). The amount of food intake per mouse was found to be 4–5 gm/d, with no significant difference between treated and untreated mice. The calculated dose for coenzyme Q10 was 400 mg · kg−1 · d−1and 14 mg · kg−1 · d−1for remacemide. During the temporal progress of the disease, the food consumed per gram of mouse weight was stable until end stage (12–14 weeks) in both the R6/2 and N171–82Q mice.
Behavior and weight assessment. Motor performance was assessed weekly from 21–63 d of age and twice weekly from 63–90 d of age in the R6/2 mice. The mice were given two training sessions to acclimate them to the rotarod apparatus (Columbus Instruments, Columbus, OH). During testing, the mice were placed on a rod rotated at 16 rpm. Each mouse had three separate trials at 60 sec each. The maximum score was 180 sec. The combined total length of time remaining on the rod was based as the measure of competency.
Survival. Mice were observed twice daily. The criterion for euthanization was the point in time at which the mice were unable to initiate movement after being gently prodded for 2 min. Mice had lost ∼40–50% of their body weight at this time point. Two independent observers confirmed the criterion for euthanization. This time point was identified as the time of death. Mice from both genders were equally included in the experimental paradigm. We have not experienced gender differences in survival in either transgenic HD mouse model. Mice dying prematurely (<70 d) were excluded from the study.
Nuclear magnetic resonance. Untreated and coenzyme Q10/ remacemide-treated R6/2 and littermate wild-type mice were anesthetized using halothane/N2O/O2 anesthesia (0.75–1% halothane; 2:1 O2/N2O). Body temperature was maintained using two water blankets surrounding the body at 38°C. Nuclear magnetic resonance (NMR)–T2-weighted magnetic resonance images were acquired with a repetition time/echo time of 3000/60 msec and a field of view of 32 mm (256 × 128 matrix size) and 1 mm slice thickness. A total of 5 untreated and 11 coenzyme Q10/remacemide-treated animals were evaluated. Mean age of the animals was 83.6 ± 4.2 versus 83.4 ± 6.1 d for the treated and untreated groups, respectively.
NMR data analysis. NMR-derived ventricular volumes were measured using home-written software. Mean signal intensity in the striatum and cortex surrounding the ventricles was measured in the T2-weighted images. Then the outline of the ventricles was segmented from the tissue using a threshold of 3 SDs above the mean signal intensity in the brain tissue. The ventricle size was measured in the lateral ventricle and dorsal third ventricles only.
Histology evaluation. At 21 d, R6/2 transgenic mice and negative littermate controls were fed diets containing 0.2% coenzyme Q10 or 0.007% remacemide, or the combination. Groups of 20 animals from each treatment paradigm were deeply anesthetized and then transcardially perfused with 4% buffered paraformaldehyde at 63 and 90 d. Approximately 250 mice were used for data collection in the neuropathological analysis. The brains were removed, post-fixed with the perfusant for 2 hr, weighed, cryoprotected in a graded series of 10 and 20% glycerol/2% DMSO solution, serially frozen sectioned at 50 μm, stored in six-well tissue collection clusters, and stained for Nissl substance (cresyl violet). Serially cut tissue sections were immunostained for huntingtin using an antibody (EM48; dilution, 1:1000) that recognizes the first 256 amino acids of human huntingtin lacking the polyglutamine and polypeptide stretches (Kuemmerle et al., 1999). The antibody reacts with N-terminal fragments of huntingtin expressed by transfection. An antibody to ubiquitin (dilution, 1:200; Dako, Carpinteria, CA) was also used in selected tissue sections to confirm the presence of aggregates.
Stereology and quantitation. Serial-cut coronal tissue sections from the rostral segment of the neostriatum at the level of the anterior commissure (interaural 5.34 mm/bregma 1.54 mm to interaural 3.7 mm/bregma −0.10 mm) (Franklin and Paxinos, 1997) were used for huntingtin aggregate analysis. Unbiased stereologic counts of huntingtin-positive aggregates (≥1.0 μm) were obtained from the neostriatum and layer 6 of the neocortex in 10 mice fed unsupplemented and supplemented diets containing the combination treatment of coenzyme Q10 and remacemide at 63 and 91 d, using Neurolucida Stereo Investigator software (Microbrightfield, Colchester, VT). The total areas of the neostriatum and motor cortex were defined from serial sections in which counting frames were sampled randomly. The dissector counting method was used in which huntingtin-positive aggregates were counted from an unbiased selection of serial sections in a defined volume of the neostriatum. Striatal neuron areas were analyzed by microscopic video capture using a Windows-based image analysis system for area measurement (Optimas, Bioscan Incorporated, Edmonds, WA). The software automatically identifies and measures profiles. All computer-identified cell profiles were manually verified as neurons and exported to Microsoft Excel. Cross-sectional areas were analyzed using Statview.
Statistics. The data are expressed as the mean ± SEM. Statistical comparisons of rotarod, weight data, and histological data were compared by ANOVA or repeated measures ANOVA. Survival data were analyzed using the Mantel–Cox log-rank test and Kaplan–Meier survival curves.
RESULTS
The effects of oral administration of coenzyme Q10, remacemide, and combined dietary supplement of coenzyme Q10 and remacemide in the diet on survival in the R6/2 and N171–82Q transgenic mice are shown in Figure1. All three treatment paradigms significantly improved survival. The mean survival in coenzyme Q10-treated R6/2 mice increased by 14.5% (coenzyme Q10: 112.9 ± 2.0 d; unsupplemented: 96.5 ± 1.8 d; p < 0.001). The percentage increase in survival using remacemide in the diet was 15.5% (remacemide: 114.2 ± 2.4 d; unsupplemented: 96.5 ± 1.8 d; p < 0.001). The combined treatment using both coenzyme Q10 and remacemide was significantly more efficacious than either compound alone (p < 0.001) and extended survival in the R6/2 mice by 31.8% (coenzyme Q10/remacemide: 127.2 ± 3.1 d; unsupplemented: 96.5 ± 1.8 d;p < 0.0001). This increase was more than twice that observed using either compound separately and is several times greater than other treatment strategies tested in R6/2 mice to date. Combined treatment in the N171–82Q mice also resulted in a significant extension of survival (coenzyme Q10/remacemide: 153.3 ± 13.7 d; unsupplemented: 127.4 ± 11.7 d;p < 0.05). These findings suggest that multiple compounds directed at different mechanistic targets may have additive effects on outcome measures.
Fig. 1.
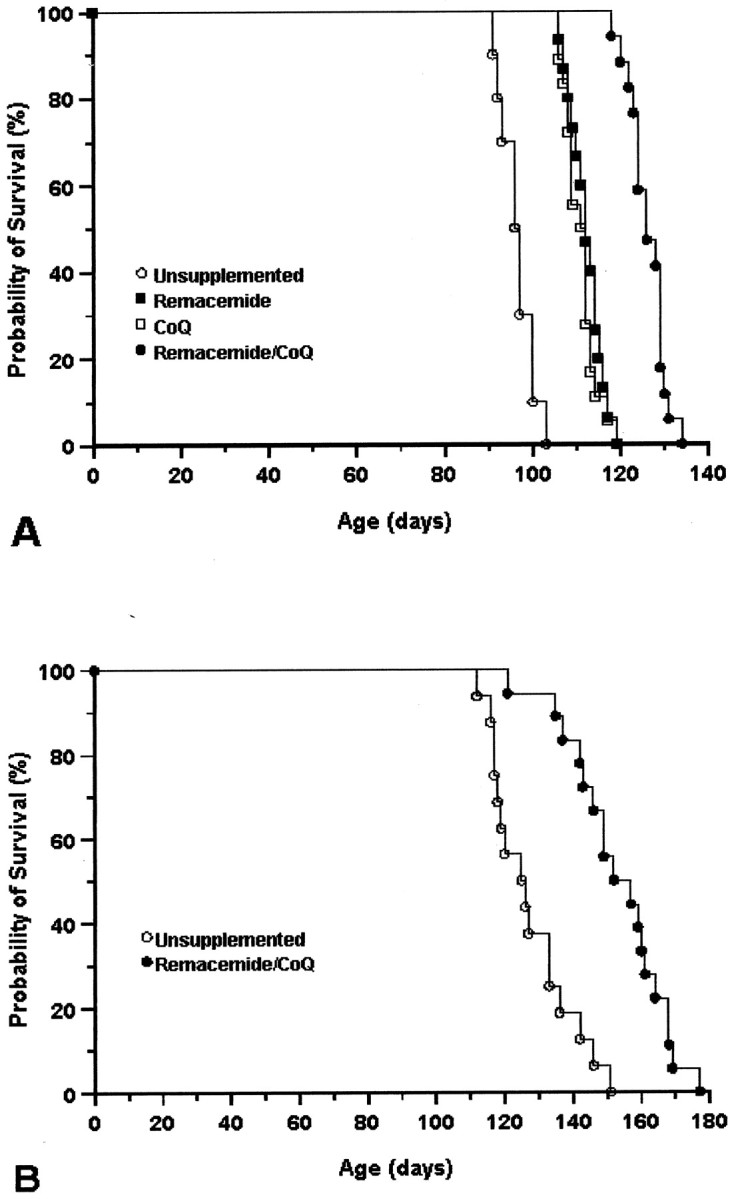
Kaplan–Meier survival curves showing the effects of coenzyme Q10, remacemide, and the combination of coenzyme Q10 and remacemide on cumulative survival in R6/2 transgenic mice (A) and the combined therapy in N171–82Q transgenic mice. Although both coenzyme Q10 and remacemide equally improved survival in the R6/2 mice (p < 0.001), the combination treatment was more than twice that of either compound alone (p < 0.0001). Although the combined therapy significantly extended survival in the N171–82Q mice (p < 0.05), the significance value was much lower because of marked variability in this murine model of Huntington's disease. CoQ, Coenzyme Q10.
Oral administration of coenzyme Q10, remacemide, and the combined coenzyme Q10/remacemide supplement significantly improved motor performance in the R6/2 mice (Fig. 2A). In contrast to unsupplemented R6/2 mice, dietary coenzyme Q10supplementation significantly improved rotarod performance throughout the measured life span of the R6/2 mice (coenzyme Q10: 134.1 ± 19.5 sec; unsupplemented: 92.8 ± 19.0 sec; p < 0.01; data represent combined means and SDs from 4–13 wks). Dietary supplementation with remacemide also resulted in significant motor improvement throughout the 4–13 week period, showing greater improved motor behavior than with coenzyme Q10 (remacemide: 143.6 ± 17 sec; unsupplemented: 92.8 ± 19.0 sec; p < 0.001; data represent combined means and SDs from 4–13 wks). In comparison with coenzyme Q10 or remacemide alone, the combined coenzyme Q10/remacemide treatment showed the greatest improvement in motor behavior (coenzyme Q10/remacemide: 150.5 ± 15.0 sec; unsupplemented: 92.8 ± 19.0 sec; p < 0.001; data represent combined means and SDs from 4–13 wks). Motor performance of the wild-type littermate control mice was ∼180 sec throughout the duration of the testing. The percentage increase in rotarod performance for coenzyme Q10, remacemide, and combined diet supplementation with both coenzyme Q10 and remacemide was 44.5, 54.7, and 62.2%, respectively, which is greater than the effect of 2% creatine supplementation (33%) that we reported previously (Ferrante et al., 2000).
Fig. 2.
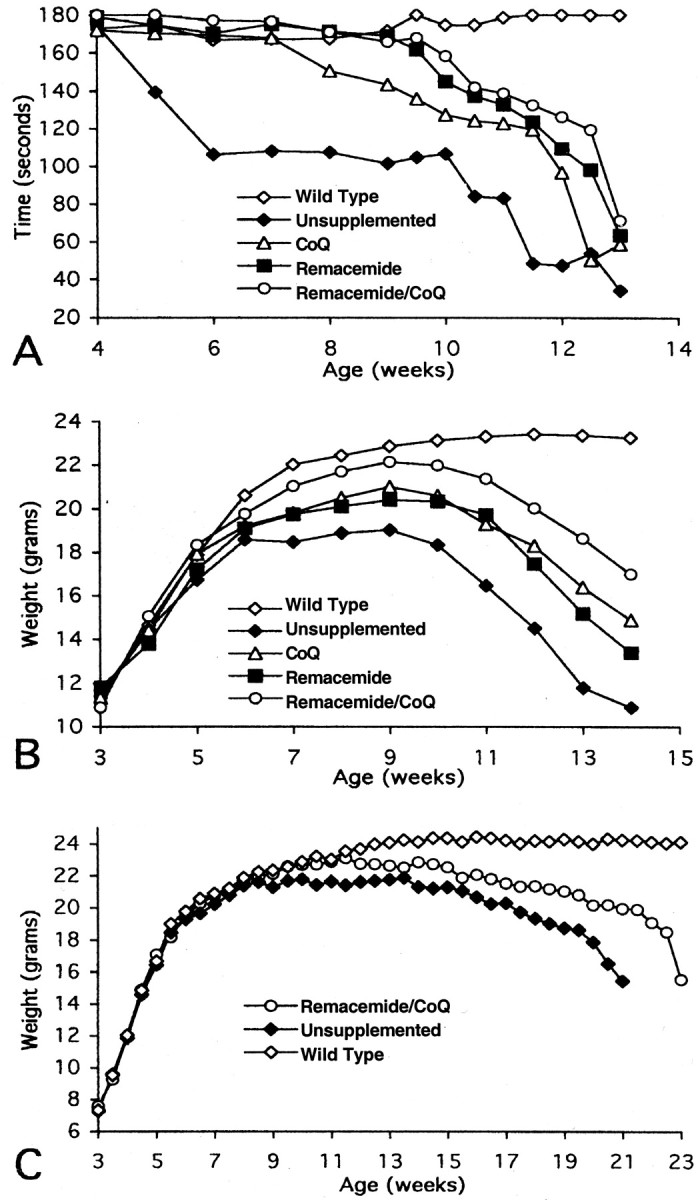
Effects of coenzyme Q10, remacemide, and the combination of coenzyme Q10 and remacemide on rotarod performance in R6/2 transgenic mice (A). All three treatments significantly improved motor performance throughout the extent of the lifespan of the mice. The combined treatment was more efficacious than either treatment alone. Effects of coenzyme Q10,remacemide, and the combination of coenzyme Q10 and remacemide resulted in significant attenuation of body weight loss in comparison with the unsupplemented R6/2 mice (B). The combined oral supplement resulted in less weight loss than either compound separately in the R6/2 transgenic mice. There was a significant reduction in weight loss from 9 and 7 weeks in the separate compound trials and combined treatment, respectively (p < 0.05; p < 0.01). The combined oral supplement resulted in a significant attenuation of weight loss in the N171–82Q mice at ∼15 weeks and continued throughout the remaining lifespan (C).CoQ, Coenzyme Q10.
The effects of oral administration of coenzyme Q10, remacemide, and the combined dietary supplement of coenzyme Q10 and remacemide on body weight in R6/2 and N171–82Q transgenic mice are shown in Figure 2,B and C. Although all three dietary regimens resulted in significant attenuation of body weight loss in comparison with the unsupplemented R6/2 mice, the combined coenzyme Q10 and remacemide treatment resulted in less body weight loss than either coenzyme Q10 or remacemide treatment alone, again suggesting an additive effect. Significant body weight improvement began at 9 weeks for both coenzyme Q10 and remacemide and at 7 weeks for the combined coenzyme Q10 and remacemide treatment and continued throughout the course of the study until death. The total percentage increase in body weight from 4 to 14 weeks for coenzyme Q10, remacemide, and combined diet in R6/2 animals in comparison with unsupplemented R6/2 mice was 12.7, 10.1, and 20.3%, respectively. Although no significant differences were observed between coenzyme Q10 and remacemide, significance was found between combined coenzyme Q10 and remacemide treatment and either coenzyme Q10 or remacemide alone (p < 0.01). The combined treatment also resulted in a significant attenuation of body weight loss in N171–82Q mice (Fig. 2C) beginning at 110 d and continuing throughout the course of treatment until death. The total increase in body weight of treated mice from 3 to 21 weeks was 7.9% greater than that of unsupplemented N171–82Q mice.
We reported previously that the brain weight of R6/2 mice decreases significantly over time until death (Ferrante et al., 2000). Coenzyme Q10, remacemide, and the combined dietary supplement of coenzyme Q10 and remacemide delayed brain weight loss in the R6/2 mice in comparison with their littermate control mice (Table 1). A significant decrease in brain weight was delayed using each of the treatment paradigms until late in the disease process in the R6/2 mice. Although less brain weight loss was observed in the combined dietary supplement than in either coenzyme Q10 or remacemide alone, no significant differences were found between any of the three therapeutic strategies. At 13 weeks, there was a 16.1, 16.9, and 17.5% delayed brain weight loss in remacemide, coenzyme Q10, and the combined dietary supplement of coenzyme Q10 and remacemide, respectively, in the R6/2 mice as compared with unsupplemented R6/2 mice. The marked gross brain atrophy and ventricular enlargement in the unsupplemented R6/2 mice was attenuated using each treatment, especially with the coenzyme Q10 and remacemide combination (Fig.3). NMR in vivo imaging confirmed these findings in the unsupplemented and coenzyme Q10/remacemide-treated R6/2 mice (Fig.4). The average ventricle size in the coenzyme Q10/remacemide mice at 12 weeks of age was 13.6 ± 6.6 and 20.9 ± 3.5 mm3 in the untreated mice (p < 0.03) and 3.4 ± 0.2 mm3 in control mice.
Table 1.
Brain weights of R6/2 mice at 13 weeks
| R6/2 unsupplemented | Remacemide supplemented | Coenzyme Q supplemented | Remacemide/coenzyme Q supplemented | Control unsupplemented |
|---|---|---|---|---|
| 344 ± 19 | 410 ± 14* | 414 ± 18* | 417 ± 15* | 437 ± 5 |
Paraformaldehyde-fixed brain weights in milligrams from unsupplemented, remacemide-, coenzyme Q-, and remacemide/coenzyme Q-supplemented and littermate transgene negative R6/2 mice at 13 weeks of age.
p < 0.002.
Fig. 3.
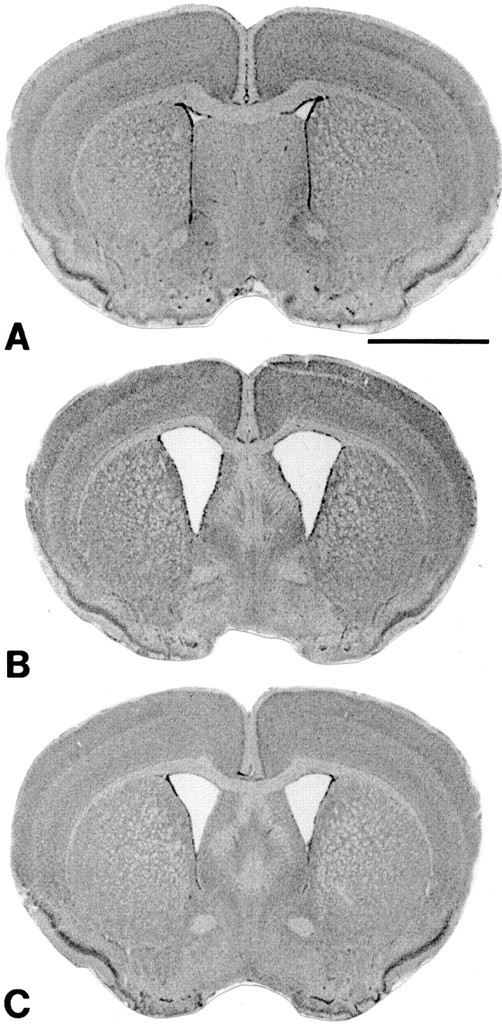
Effects of the combined treatment of coenzyme Q10 and remacemide on gross atrophy and ventricular enlargement in R6/2 mice at 13 weeks. In comparison with a littermate nonmutant transgene mouse (A), marked gross atrophy and enlarged lateral ventricles are evident in the unsupplemented R6/2 mouse (B) in coronal sections at the rostral level of the anterior commissure. Less atrophy and ventricular enlargement are seen in the supplemented mouse (C). Scale bar (shown in A): 2 mm.
Fig. 4.
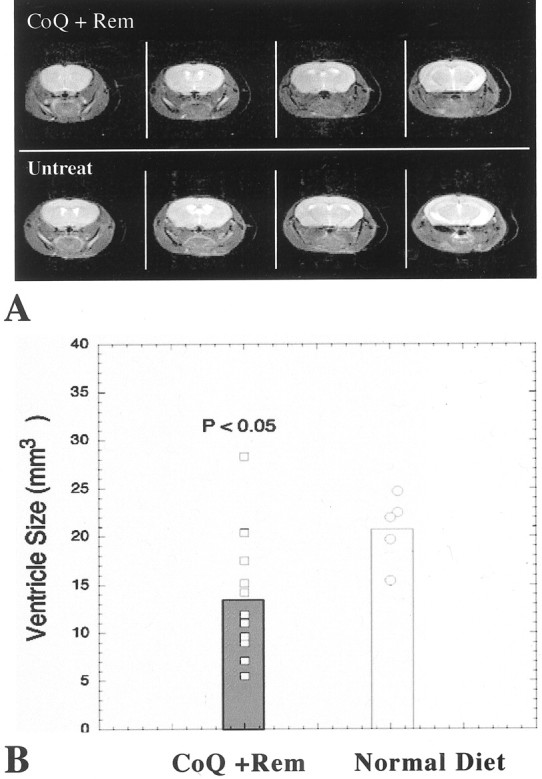
A, Selected T2-weighted images from four consecutive 1 mm slices in both a coenzyme Q10/remacemide-treated and an untreated animal. Note the much larger ventricles in the untreated animals. B,Bar graph of the effect of coenzyme Q10/remacemide treatment on ventricular size in the HD mice. CoQ, Coenzyme Q10;Rem, remacemide; Untreat, untreated.
Consistent with the brain weight loss and gross atrophy, striatal neuron atrophy is present in the R6/2 mice with a ∼38% overall decrease in area measurements (Ferrante et al., 2000). The neuroprotective effect of coenzyme Q10 and remacemide together significantly delayed striatal neuron atrophy at 91 d of age (wild-type littermate control: 120.9 ± 9.6 μm2; unsupplemented R6/2: 58.3 ± 14.7 μm2; coenzyme Q10: 89.1 ± 14.3 μm2; remacemide: 89.7 ± 12.3 μm2; coenzyme Q10/remacemide supplemented R6/2: 108.9 ± 12.4 μm2; p < 0.001) (Fig. 5). Neuronal atrophy was present in all the treated mice at end stage disease.
Fig. 5.
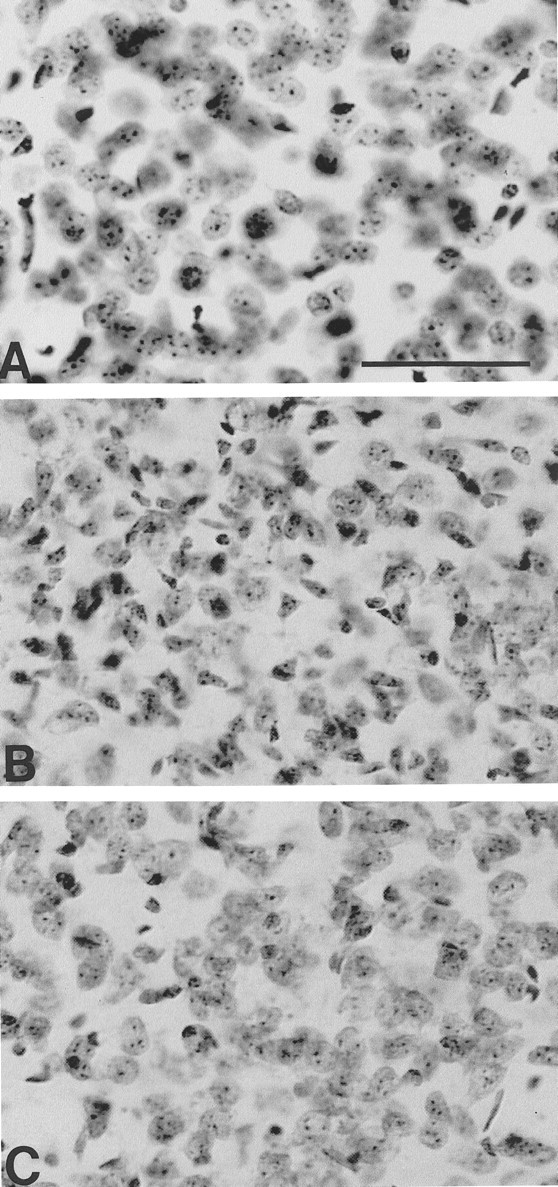
Photomicrographs of Nissl-stained tissue sections from the dorsomedial aspect of the neostriatum in a littermate nonmutant transgene mouse (A), unsupplemented R6/2 mouse (B), and a combined coenzyme Q10 and remacemide-treated R6/2 mouse (C) at 91 d of age. There is marked neuronal atrophy with small angulated neurons in the unsupplemented mouse (B), with relative preservation of neuronal size and number in the treated mouse (C). Scale bar (shown in A): 100 μm.
Huntingtin-positive aggregate formation is an early and progressive process throughout the brains of R6/2 mice. These aggregates increase in both size and number with age. Dietary supplementation with combined treatment of coenzyme Q10 and remacemide resulted in significant reductions of striatal aggregate number. Aggregate counts within the striatum at 9 and 13 week time points were significantly reduced in comparison with untreated R6/2 mice (p < 0.01) (Figs.6, 7). There was an 8.2 and 15.7% decrease in striatal volume loss at 9 and 13 weeks, respectively, in the combined treatment as compared with the untreated R6/2 mice in the estimated area of rostral striatum measured (striatal volumes, untreated 9 and 13 weeks: 12.8 and 10.9 mm3). The percentage decrease in aggregate number in treated mice was 32 and 36% at those same time points [estimated striatal huntingtin (htt) aggregates, untreated 9 and 13 weeks: 363 × 103 and 530 × 103; combined coenzyme Q10 and remacemide, 9 and 13 weeks: 275 × 103 and 390 × 103].
Fig. 6.

Graph of the number of huntingtin-positive aggregates in the neostriatum at 9 and 13 weeks in unsupplemented (unsup) and combined coenzyme Q10 and remacemide-treated (CoQ/Rem) R6/2 mice. There was a significant (p < 0.01) delay in the formation of aggregates within the neostriatum in the combined treatment mice at both time points, in comparison with the unsupplemented mice.
Fig. 7.
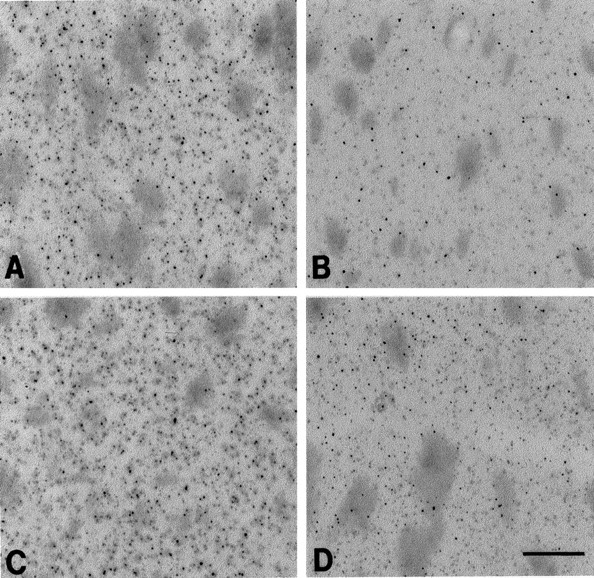
Photomicrographs of immunostained huntingtin tissue sections from the dorsomedial aspect of the neostriatum at the rostral level of the anterior commissure in combined coenzyme Q10 and remacemide-treated and unsupplemented R6/2 mice at 9 weeks (A and B, respectively) and at 13 weeks (C and D, respectively). There are significantly fewer huntingtin-positive aggregates in treated mice at 9 and 13 weeks (A, C) in comparison with the unsupplemented mice at the same time points (B, D). Scale bar (shown inD): 100 μm.
DISCUSSION
Previous work has shown that both coenzyme Q10 and remacemide exert neuroprotective effects. Coenzyme Q10 is an essential cofactor of electron transport chain where it accepts electrons from complexes I and II (Beyer, 1992). Coenzyme Q10 also serves as an important antioxidant in both mitochondria and lipid membranes (Beyer, 1992; Noack et al., 1994). Coenzyme Q10administration significantly increases brain mitochondrial concentrations of coenzyme Q10 in mature and older animals (Matthews et al., 1998). Administration of coenzyme Q10 also increases α-tocopherol concentrations in mitochondria, consistent with a sparing effect (Lass et al., 1999). Coenzyme Q10 is neuroprotective against experimental ischemia in rats (Ostrowski, 2000), and it protects against striatal lesions produced by administration of the mitochondrial toxins malonate and 3-nitropropionic acid (Beal et al., 1994; Matthews et al., 1998). Coenzyme Q10improves survival in a transgenic animal model of amyotrophic lateral sclerosis (Matthews et al., 1998). Some patients with mitochondrial disorders treated with coenzyme Q10 show clinical and biochemical improvement (Ihara et al., 1989; Nishikawa et al., 1989; Abe et al., 1991; Chariot et al., 1999).
Remacemide is an NMDA receptor channel blocker that has a lower affinity for the receptor than several other NMDA channel blockers (Porter and Greenamyre, 1995). This may be related to better tolerability and fewer behavioral side effects than those seen with agents such as MK-801, which dissociates from the channel slowly, and may therefore block normal synaptic activity. Remacemide attenuates excitotoxicity in vitro and focal ischemic lesions in vivo (Bannan et al., 1994; Black et al., 1995) and is effective in blocking malonate lesions (Greene et al., 1996). Remacemide has been tolerated well in clinical trials for cerebral ischemia and in HD patients (Kieburtz et al., 1996; Dyker and Lees, 1999).
In the present experiments, we examined whether oral administration of coenzyme Q10 or remacemide or a combination of both could exert beneficial effects in transgenic mouse models of HD. We found that oral administration of both coenzyme Q10 and remacemide produces a significant improvement of survival in a transgenic mouse model of HD. The increase in survival in the R6/2 model was 14.5% using coenzyme Q10, 15.5% using remacemide, and 32% using the combined coenzyme Q10/remacemide treatment. We also found that administration of coenzyme Q10and remacemide produces significant improvement in motor performance, a delay in loss of body weight, a delay in gross brain and striatal neuron atrophy, and attenuation of the development of neuronal intranuclear inclusions in the striatum. Both coenzyme Q10 and remacemide therefore produce significant benefit, and the combination was more efficacious than oral administration of creatine (Ferrante et al., 2000), minocycline (Chen et al., 2000), or intracerebroventricular administration of caspase inhibitors (Ona et al., 1999). These findings strongly suggest that combined therapies, targeting different mechanisms of cell death, may have cumulative beneficial effects.
Both a delay of onset and an altered course in the progression of disease can be identified in the behavioral and neuropathological analyses by comparison of the slopes of the curves throughout the course of treatment. Although there is a significant extension of survival in the treated R6/2 mice, there does not appear to be an effect on disease progression. The slopes of the Kaplan–Meier curves are similar. However, these mice have a particularly severe phenotype, which makes this difficult to assess. In the N171–82Q mice, the slope of the survival curve is more gradual with treatment, suggesting a treatment effect on disease progression. The body weight curve of the combined therapy reflects a delay in onset. This is also observed in the motor performance profiles, such that remacemide and the combined therapies delay motor dysfunction, whereas coenzyme Q10 presents with a more gradual course of dysfunction from the onset until end stage disease. In addition, htt aggregate counts in the combined treatment paradigm have a more gradual slope, suggesting again a less rapid disease progression.
The present results extend those found previously with mitochondrial toxin models of HD to transgenic mouse models, showing significant improvements on survival, motor performance, weight loss, and neuropathological features. The present findings are of great interest in the context of a recent clinical trial of coenzyme Q10 and remacemide in HD patients (Huntington Study Group, 2002). The CARE–HD trial of the Huntington Study Group randomized 340 patients to coenzyme Q10 or remacemide or the combination using a 2 × 2 factorial design (Kieburtz, 1999). The patients were treated for 30 months, and the primary outcome measure was the total functional capacity (TFC) scale of the unified Huntington's disease rating scale. The results of this trial showed no significant effects of either agent on the primary outcome measure (Huntington Study Group, 2001). Treatment with coenzyme Q10 showed a trend toward slowing in TFC decline (13%) over 30 months (p = 0.15); however, the study was not powered to be able to detect an effect of this magnitude. There was a nominally significant effect in slowing functional decline (22% slowing; p = 0.02) and a trend toward slowing functional decline (18% slowing; p < 0.06) on the independence scale. Coenzyme Q10 also showed trends toward a beneficial effect on two cognitive tests and on behavior.
There is discordance between the outcome we observed in the transgenic mice and the outcome of the human clinical trial. The results with coenzyme Q10 alone in the R6/2 mice were an improved survival of 14.5%, which is similar to the magnitude of improvement seen in HD patients (Huntington Study Group, 2002); however, there was no effect of remacemide in HD patients, whereas it was efficacious in the mice. This raises an important issue of whether therapeutic testing in the HD mice will be useful in predicting efficacy in humans. There are a number of potential explanations for the observed discrepancy. One is that HD patients are much more heterogeneous than genetically modified mice. Most importantly, the dose of remacemide used in the HD patients was 5.7 mg · kg−1 · d−1, which was at the limit of tolerability, whereas a dose of 14 mg · kg−1 · d−1was used in the mice (a 2.5-fold difference). The lack of efficacy of remacemide in HD patients therefore may be the result of too low a dose to adequately block the NMDA receptor. Alternative NMDA-antagonists may prove more efficacious. Another important issue is that the HD transgenic mice used in the experiments have a high expression of an N-terminal fragment of huntingtin, rather than full-length huntingtin. These mice show little cell loss and are resistant to excitotoxins, whereas mice expressing full-length huntingtin show enhanced vulnerability to excitotoxicity (Hansson et al., 1999; Zeron et al., 2001). It is possible that the pathophysiology of neurodegeneration in the HD transgenic mice that we studied may not be entirely reminiscent of that occurring in the human illness. Finally, the disease stage in which the therapeutic trials were initiated was markedly different. The mice were started on treatment at day 21, well before the onset of clinical symptoms, whereas the human trials were initiated in patients with symptomatic HD. Therefore, it will be of interest to determine whether remacemide shows efficacy in the mice if administered after the onset of symptoms, as in human clinical trials.
Transgenic mouse models of HD hold great promise for the screening of novel therapeutics that may then be successfully translated to the treatment of HD patients. The present results, however, suggest that issues of dosing and timing of drug administration will need to be carefully considered to predict efficacy in humans. The findings that remacemide and coenzyme Q10 improve the clinical and neuropathological phenotype in transgenic mice provides further evidence that mitochondrial dysfunction and excitotoxicity may contribute to HD pathogenesis. The present results also suggest that multicombination therapies with agents targeting differing pathogenic mechanisms may exert additive therapeutic effects.
Footnotes
This work was supported by The Department of Defense and National Institutes of Health Grants NS38180 (M.F.B.), NS35255 (S.M.H., R.J.F.), NS37102 and AG13846 (R.J.F.), AG12992 (M.F.B., R.J.F.), and AT00613 (S.M.H., R.J.F., M.F.B.), the Veterans Administration (R.J.F.), the Hereditary Disease Foundation (R.J.F., S.M.H., M.F.B.), the Huntington's Disease Society of America (R.J.F., S.M.H., M.F.B.), and the Norwegian Research Council (O.A.A.). The secretarial assistance of Sharon Melanson is gratefully acknowledged. Photographic assistance and histology preparation were provided by James Kubilus, Kerry Cormier, and Karen Smith.
Correspondence should be addressed to Dr. M. Flint Beal, Neurology Department, New York Hospital–Cornell Medical Center, 525 East 68th Street, New York, NY 10021, E-mail:fbeal@mail.med.cornell.edu, or Dr. Robert J. Ferrante, GRECC Unit, 182B, Bedford VA Medical Center, 200 Springs Road, Bedford, MA 01730, E-mail: rjferr@bu.edu.
REFERENCES
- 1.Abe K, Fujimura H, Nishikawa Y, Yorifuki S, Mezaki T, Hirono N, Nishitani N, Kameyama M. Marked reduction in CSF lactate and pyruvate levels after CoQ therapy in a patient with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS). Acta Neurol Scand. 1991;83:356–359. doi: 10.1111/j.1600-0404.1991.tb03962.x. [DOI] [PubMed] [Google Scholar]
- 2.Bannan PE, Graham DI, Lees KR, McCulloch J. Neuroprotective effect of remacemide hydrochloride in focal cerebral ischemia in the cat. Brain Res. 1994;664:271–275. doi: 10.1016/0006-8993(94)91984-4. [DOI] [PubMed] [Google Scholar]
- 3.Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000;23:294–300. doi: 10.1016/s0166-2236(00)01584-8. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:4181–4192. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- 5.Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993a;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beal MF, Brouillet E, Jenkins B, Henshaw R, Rosen B, Hyman BT. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J Neurochem. 1993b;61:1147–1150. doi: 10.1111/j.1471-4159.1993.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 7.Beal MF, Henshaw R, Jenkins BG, Rosen BR, Schulz JB. Coenzyme Q10 and nicotinamide block striatal lesions produced by the mitochondrial toxin malonate. Ann Neurol. 1994;36:882–888. doi: 10.1002/ana.410360613. [DOI] [PubMed] [Google Scholar]
- 8.Beyer RE. An analysis of the role of coenzyme Q in free radical generation and as an antioxidant. Biochem Cell Biol. 1992;70:390–403. doi: 10.1139/o92-061. [DOI] [PubMed] [Google Scholar]
- 9.Black MA, Tremblay R, Mealing G, Ray R, Durkin JP, Whitfield JF, Blosser J, Morley P. N-methyl-d-aspartate- or glutamate-mediated toxicity in cultured rat cortical neurons is antagonized by FPL 15896AR. J Neurochem. 1995;65:2170–2177. doi: 10.1046/j.1471-4159.1995.65052170.x. [DOI] [PubMed] [Google Scholar]
- 10.Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MMK, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 12.Chariot P, Brugieres P, Eliezer-Vanerot MC, Geny C, Binaghi M, Cesaro P. Choreic movements and MRI abnormalities in the subthalamic nuclei reversible after administration of coenzyme Q10 and multiple vitamins in a patient with bilateral optic neuropathy. Mov Disord. 1999;14:855–859. doi: 10.1002/1531-8257(199909)14:5<855::aid-mds1023>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 13.Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of huntington disease. Nat Med. 2000;6:797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- 14.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiari L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 15.Dyker AG, Lees KR. Remacemide hydrochloride: a double-blind, placebo-controlled, safety and tolerability study in patients with acute ischemic stroke. Stroke. 1999;30:1796–1801. doi: 10.1161/01.str.30.9.1796. [DOI] [PubMed] [Google Scholar]
- 16.Ferrante RJ, Kowall NW, Cipolloni PB, Beal MF. Excitotoxin lesions in primates as a model of Huntington's disease: histopathologic and neurochemical characterization. Exp Neurol. 1993;119:46–71. doi: 10.1006/exnr.1993.1006. [DOI] [PubMed] [Google Scholar]
- 17.Ferrante RJ, Andreassen OA, Jenkins BG, Dedeoglu A, Kuemmerle S, Kubilus JK, Kaddurah-Daouk R, Hersch SM, Beal MF. Neuroprotective effects of creatine in a transgenic mouse model of Huntington's disease. J Neurosci. 2000;20:4389–4397. doi: 10.1523/JNEUROSCI.20-12-04389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; New York: 1997. [Google Scholar]
- 19.Greene JG, Porter RH, Eller RV, Greenamyre JT. Inhibition of succinate dehydrogenase by malonic acid produces an “excitotoxic” lesion in rat striatum. J Neurochem. 1993;61:1151–1154. doi: 10.1111/j.1471-4159.1993.tb03634.x. [DOI] [PubMed] [Google Scholar]
- 20.Greene JG, Porter RH, Greenamyre JT. ARL-15896, a novel N-methyl-d-aspartate receptor ion channel antagonist: neuroprotection against mitochondrial metabolic toxicity and regional pharmacology. Exp Neurol. 1996;137:66–72. doi: 10.1006/exnr.1996.0007. [DOI] [PubMed] [Google Scholar]
- 21.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AHV. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 22.Hansson O, Petersen A, Leist M, Nicotera P, Castilho RF, Brundin P. Transgenic mice expressing a Huntington's disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci USA. 1999;96:8727–8732. doi: 10.1073/pnas.96.15.8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huntington Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington's disease. Neurology. 2001;57:401–404. doi: 10.1212/wnl.57.3.397. [DOI] [PubMed] [Google Scholar]
- 24.Ihara Y, Namba R, Kuroda S, Sato T, Shirabe T. Mitochondrial encephalomyopathy (MELAS): pathological study and successful therapy with coenzyme Q10 and idebenone. J Neurol Sci. 1989;90:263–271. doi: 10.1016/0022-510x(89)90112-3. [DOI] [PubMed] [Google Scholar]
- 25.Jenkins BG, Koroshetz WJ, Beal MF, Rosen BR. Evidence for impairment of energy metabolism in vivo in Huntington's disease using localized 1H NMR spectroscopy. Neurology. 1993;43:2689–2695. doi: 10.1212/wnl.43.12.2689. [DOI] [PubMed] [Google Scholar]
- 26.Kieburtz K. Antiglutamate therapies in Huntington's disease. J Neural Transm Suppl. 1999;55:97–102. doi: 10.1007/978-3-7091-6369-6_9. [DOI] [PubMed] [Google Scholar]
- 27.Kieburtz K, Feigin A, McDermott M, Como P, Abwender D, Zimmerman C, Hickey C, Orme C, Claude K, Sotack J, Greenamyre JT, Dunn C, Shoulson I. A controlled trial of remacemide hydrochloride in Huntington's disease. Mov Disord. 1996;11:273–277. doi: 10.1002/mds.870110310. [DOI] [PubMed] [Google Scholar]
- 28.Koroshetz WJ, Jenkins BG, Rosen BR, Beal MF. Energy metabolism defects in Huntington's disease and effects of coenzyme Q10. Ann Neurol. 1997;41:160–165. doi: 10.1002/ana.410410206. [DOI] [PubMed] [Google Scholar]
- 29.Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntingtin aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- 30.Lass A, Forster MJ, Sohal RS. Effects of coenzyme Q10 and α-tocopherol administration on their tissue levels in the mouse: elevation of mitochondrial α-tocopherol by coenzyme Q10. Free Radic Biol Med. 1999;26:1375–1382. doi: 10.1016/s0891-5849(98)00330-x. [DOI] [PubMed] [Google Scholar]
- 31.Lin X, Antalffy B, Kang D, Orr HT, Zoghbi HY. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci. 2000;3:157–163. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- 32.Lodi R, Schapira AH, Manners D, Styles P, Wood NW, Taylor DJ, Warner TT. Abnormal in vivo skeletal muscle energy metabolism in Huntington's disease and dentatorubropallidoluysian atrophy. Ann Neurol. 2000;48:72–76. [PubMed] [Google Scholar]
- 33.Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- 34.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 35.Matthews RT, Yang S, Browne S, Baik M, Beal MF. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci USA. 1998;95:8892–8897. doi: 10.1073/pnas.95.15.8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGeer EG, McGeer PL. Duplication of biochemical changes of Huntington's chorea by intrastriatal injections of glutamic and kainic acids. Nature. 1976;263:517–519. doi: 10.1038/263517a0. [DOI] [PubMed] [Google Scholar]
- 37.Nishikawa Y, Takahashi M, Yorifuji S, Nakamura Y, Ueno S, Tarui S, Kozuka T, Nishimura T. Long-term coenzyme Q10 therapy for a mitochondrial encephalomyopathy with cytochrome c oxidase deficiency: a 31P NMR study. Neurology. 1989;39:399–403. doi: 10.1212/wnl.39.3.399. [DOI] [PubMed] [Google Scholar]
- 38.Noack H, Kube U, Augustin W. Relations between tocopherol depletion and coenzyme Q during lipid peroxidation in rat liver mitochondria. Free Radic Res. 1994;20:375–386. doi: 10.3109/10715769409145637. [DOI] [PubMed] [Google Scholar]
- 39.Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature. 1999;399:263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- 40.Ostrowski RP. Effect of coenzyme Q10 on biochemical and morphological changes in experimental ischemia in the rat brain. Brain Res Bull. 2000;53:399–407. doi: 10.1016/s0361-9230(00)00406-8. [DOI] [PubMed] [Google Scholar]
- 41.Porter RH, Greenamyre JT. Regional variations in the pharmacology of NMDA receptor channel blockers: implications for therapeutic potential. J Neurochem. 1995;64:614–623. doi: 10.1046/j.1471-4159.1995.64020614.x. [DOI] [PubMed] [Google Scholar]
- 42.Sawa A, Wiegand GW, Cooper J, Margolis RL, Sharp AH, Lawler JF, Jr, Greenamyre JT, Snyder SH, Ross CA. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat Med. 1999;5:1194–1198. doi: 10.1038/13518. [DOI] [PubMed] [Google Scholar]
- 43.Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 44.Schulz JB, Matthews RT, Henshaw DR, Beal MF. Neuroprotective strategies for treatment of lesions produced by mitochondrial toxins: implications for neurodegenerative diseases. Neuroscience. 1996;71:1043–1048. doi: 10.1016/0306-4522(95)00527-7. [DOI] [PubMed] [Google Scholar]
- 45.Schwarcz R, Coyle JT. Adenylate cyclase activity in chick retina. Gen Pharmacol. 1976;7:349–354. doi: 10.1016/0306-3623(76)90019-7. [DOI] [PubMed] [Google Scholar]
- 46.The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 47.Zeron MM, Wellington CL, Krebs C, Leavin BR, Baimbridge K, Hayden MR, Raymond LA. Enhanced sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a transgenic mouse model of Huntington's disease. Soc Neurosci. 2001;27:1150. [Google Scholar]