

Abstract
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) damages the nigrostriatal dopaminergic pathway as seen in Parkinson's disease (PD), a common neurodegenerative disorder with no effective protective treatment. Consistent with a role of glial cells in PD neurodegeneration, here we show that minocycline, an approved tetracycline derivative that inhibits microglial activation independently of its antimicrobial properties, mitigates both the demise of nigrostriatal dopaminergic neurons and the formation of nitrotyrosine produced by MPTP. In addition, we show that minocycline not only prevents MPTP-induced activation of microglia but also the formation of mature interleukin-1β and the activation of NADPH–oxidase and inducible nitric oxide synthase (iNOS), three key microglial-derived cytotoxic mediators. Previously, we demonstrated that ablation of iNOS attenuates MPTP-induced neurotoxicity. Now, we demonstrate that iNOS is not the only microglial-related culprit implicated in MPTP-induced toxicity because mutant iNOS-deficient mice treated with minocycline are more resistant to this neurotoxin than iNOS-deficient mice not treated with minocycline. This study demonstrates that microglial-related inflammatory events play a significant role in the MPTP neurotoxic process and suggests that minocycline may be a valuable neuroprotective agent for the treatment of PD.
Keywords: IL-1β, iNOS, minocycline, microglia, MPTP, NADPH–oxidase, neurodegeneration, Parkinson's disease
Parkinson's disease (PD) is a common neurodegenerative disorder whose cardinal clinical features include tremor, slowness of movement, stiffness, and postural instability (Fahn and Przedborski, 2000). These symptoms are primarily attributable to the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the consequent loss of their projecting nerve fibers in the striatum (Hornykiewicz and Kish, 1987;Pakkenberg et al., 1991). Although several approved drugs do alleviate PD symptoms, chronic use of these drugs is often associated with debilitating side effects (Kostic et al., 1991), and none seems to dampen the progression of the disease. So far, the development of effective neuroprotective therapies is impeded by our limited knowledge of the pathogenesis of PD. However, significant insights into the mechanisms by which SNpc dopaminergic neurons may die in PD have been achieved by the use of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which replicates in humans and nonhuman primates a severe and irreversible PD-like syndrome (Przedborski et al., 2000). In several mammalian species, MPTP reproduces most of the biochemical and pathological hallmarks of PD, including the dramatic neurodegeneration of the nigrostriatal dopaminergic pathway (Przedborski et al., 2000).
To elucidate PD pathogenic factors, and thus to develop therapeutic strategies aimed at halting its progression, we revisited the neuropathology of this disease in search of putative culprits. Aside from the dramatic loss of dopaminergic neurons, it appears that the SNpc is also the site of a robust glial reaction in PD and experimental models of PD (Vila et al., 2001b). Although gliosis and especially activated microglia may sometimes be associated with beneficial effects, often gliosis appears to be deleterious (Vila et al., 2001b). For instance, microglial cells, which are resident macrophages in the brain, have the ability to react promptly in response to insults of various natures (Kreutzberg, 1996) in that resting microglia quickly proliferate, become hypertrophic, and increase or express de novo a plethora of marker molecules (Banati et al., 1993;Kreutzberg, 1996). The multifunctional nature of activated microglia encompasses the upregulation of cell surface markers such as the macrophage antigen complex-1 (MAC-1), phagocytosis, and the production of cytotoxic molecules, including reactive oxygen species (ROS), nitric oxide (NO), and a variety of proinflammatory cytokines such as interleukin-1β (IL-1β) (Banati et al., 1993; Gehrmann et al., 1995;Hopkins and Rothwell, 1995). Given this, there is little doubt that activated microglia, through the actions of aforementioned factors, can inflict significant damage on neighboring cells.
Minocycline, a semisynthetic second-generation tetracycline, is an antibiotic that possesses superior penetration through the brain–blood barrier (Aronson, 1980). Minocycline has emerged as a potent inhibitor of microglial activation (Amin et al., 1996; Yrjanheikki et al., 1998,1999; Tikka and Koistinaho, 2001; Tikka et al., 2001a), an anti-inflammatory property completely separate from its antimicrobial action, and as an effective neuroprotective agent in experimental brain ischemia (Yrjanheikki et al., 1998, 1999), in the R6/2 mouse model of Huntington's disease (Chen et al., 2000), in traumatic brain injury (Sanchez Mejia et al., 2001), and in the 6-hydroxydopamine model of PD (He et al., 2001). In the present study, we report that, in the MPTP mouse model of PD, minocycline (1) mitigates, in a dose-dependent manner, the loss of dopaminergic cell bodies in the SNpc and of nerve terminals in the striatum, (2) reduces the levels of nitrotyrosine, a marker of protein nitrative modification, (3) prevents microglial activation with minimal effects on the astrocytic response, (4) reduces the formation of mature IL-1β and decreases activation of NADPH–oxidase and upregulation of inducible nitric oxide synthase (iNOS), two enzymes implicated in microglial-derived production of ROS and NO, respectively, and (5) protects against MPTP beyond the beneficial effect of iNOS ablation (Liberatore et al., 1999; Dehmer et al., 2000).
MATERIALS AND METHODS
Animals and treatment. All mice used in this study were 8-week-old male C57BL/6 mice from Charles River Laboratories (Wilmington, MA) and iNOS-deficient mice (C57BL/6-NOS2; The Jackson Laboratory, Bar Harbor, ME) and their wild-type littermates weighing 22–25 gm. For MPTP intoxication, mice received four intraperitoneal injections of MPTP-HCl (18 or 16 mg/kg of free base; Sigma, St. Louis, MO) in saline at 2 hr intervals. For minocycline treatment, mice received twice daily (12 hr apart) intraperitoneal injections of varying doses of minocycline-HCl ranging from 1.4 to 45 mg/kg (Sigma) in saline starting 30 min after the first MPTP injection and continuing through 4 additional days after the last injection of MPTP; control mice received saline only. Mice (n = 5–8 per group; saline–saline, saline–minocycline, MPTP–saline, and MPTP–minocycline) were killed at selected time points, and their brains were used for morphological and biochemical analyses. Procedures using laboratory animals were in accordance with the National Institutes of Health guidelines for the use of live animals and were approved by the institutional animal care and use committee of Columbia University. MPTP handling and safety measures were in accordance with our published recommendations (Przedborski et al., 2001b).
Immunoblots. Cytosolic and particulate fractions from selected mouse brain regions were prepared as described previously (Vila et al., 2001a) and used for either one-dimensional Western blot or dot-blot analyses. For Western blots, the following primary antibodies were used: monoclonal anti-p67phox (1:1000; Transduction Laboratories, Lexington, KY), polyclonal anti-calnexin (1:2000;Stressgen, Victoria, British Columbia, Canada). For dot-blot analyses, 25 μg of protein extracts were loaded onto the 0.2 μm nitrocellulose membrane in dot-blot apparatus (Bio-Rad, Hercules, CA), and blots were probed with an affinity-purified polyclonal antibody against nitrotyrosine (1:1000) (Przedborski et al., 2001a) that was preconjugated overnight at 4°C with 1:5000 dilution of horseradish-labeled donkey anti-rabbit IgG. For all blots, bound primary antibody was detected using a horseradish-conjugated antibody against IgG and a chemiluminescent substract (SuperSignal Ultra; Pierce, Rockford, IL). All films were quantified using the NIH Image analysis system.
RNA extraction and reverse transcription-PCR. Total RNA was extracted from midbrain, striatal, and cerebellar samples from all four groups of mice at selected time points and used for reverse transcription-PCR analysis as described previously (Vila et al., 2001a). The primer sequences used in this study were as follows: for mouse MAC-1, 5′-CAG ATC AAC AAT GTG ACC GTA TGG-3′ (forward) and 5′-CAT CAT GTC CTT GTA CTG CCG C-3′ (reverse); for mouse glial fibrillary acidic protein (GFAP), 5′-CAG GCA ATC TGT TAC ACT TG-3′ (forward) and 5′-ATA GCA CCA GGT GCT TGA AC-3′ (reverse); and for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-GTT TCT TAC TCC TTG GAG GCC AT-3′ (forward) and 5′-TGA TGA CAT CAA GAA GTG GTG AA-3′ (reverse). PCR amplification was performed for 26 cycles for MAC-1 and GFAP and 18 cycles for GADPH. After amplification, products were separated on a 5% PAGE. Gels were dried and exposed overnight to a phosphorimager screen, and then radioactivity was quantified using a computerized analysis system (Bio-Rad PhosphoImager system).
Immunohistochemistry and stereology. Brains were fixed and processed for immunostaining as described previously (Liberatore et al., 1999). Primary antibodies used in this study were as follows: rat anti-MAC-1 (1:200; Serotec, Raleigh, NC), mouse anti-GFAP (1:1000; Boehringer Mannheim, Indianapolis, IN), and a rabbit polyclonal anti-tyrosine hydroxylase (TH) (1:1000; Calbiochem, San Diego, CA). Immunostaining was visualized by using either 3,3′-diaminobenzine (brown) or SG substrate kit (gray blue; Vector Laboratories, Burlingame, CA). Sections were counterstained with thionin.
The total number of TH-positive SNpc neurons was counted in the various groups of animals at 7 d after the last MPTP or saline injection using the optical fractionator method as described previously (Liberatore et al., 1999). This is an unbiased method of cell counting that is not affected by either the volume of reference (SNpc) or the size of the counted elements (neurons). Striatal density of TH immunoreactivity was determined as described previously (Burke et al., 1990).
Assay of NOS catalytic activity. Ventral midbrain NOS activity was assessed by measuring both the calcium-dependent and calcium-independent conversion of [3H]arginine to [3H]citrulline as described previously (Liberatore et al., 1999).
Mature IL-1β measurement. Ventral midbrain content of mature murine IL-1β was done as described using an enzyme-linked immunosorbend assay kit specific for this cytokine (R & D Systems, Minneapolis, MN) (Li et al., 2000).
Measurement of striatal levels of 1-methyl-4-phenylpyridinium. This was done in MPTP–saline and MPTP–minocycline mice killed at 90 min after one intraperitoneal injection of 18 mg/kg MPTP using an HPLC method with ultraviolet detection (wavelength, 295 nm) as described previously (Przedborski et al., 1996).
Synaptosomal 1-methyl-4-phenylpyridinium uptake.Naïve mice were killed, and their striata were dissected out and processed for uptake experiments as described previously (Przedborski et al., 1992). The uptake of [3H]1-methyl-4-phenylpyridinium (MPP+) was assessed in the absence and presence of minocycline (concentration raging from 1 to 330 μm). The assay was repeated three times, each time using duplicate samples.
Mouse tissue slices and lactate measurement. Striatal slices (300 μm) were prepared and processed as described by Kindt et al. (1987) using 50 μmMPP+ and varying concentrations of minocycline (0–333 μm). At the end of the incubation (60 min; 37°C), media were collected and used for lactate quantification by enzymatic assay based on the formation of NADH, followed by 340 nm in a spectrophotometer. The assay was repeated three times, each time using duplicate samples.
Statistical analysis. All values are expressed as the mean ± SEM. Differences between means were analyzed using a two-tail Student's t test. Differences among means were analyzed using one-way ANOVA, with time, treatment, or genotype as the independent factors. When ANOVA showed significant differences, pairwise comparisons between means were tested by Newman–Keulspost hoc testing. In all analyses, the null hypothesis was rejected at the 0.05 level.
RESULTS
Minocycline attenuates MPTP-induced dopaminergic neurodegeneration
As illustrated in Figure1G, the numbers of SNpc TH-positive neurons varied significantly among the various groups of mice (F(9,71) = 7.045;p < 0.001). MPTP, 18 mg/kg for four injections over 8 hr, caused more than a 55% reduction in the number of SNpc dopaminergic neuron numbers, as evidenced by TH immunostaining (Fig.1C,G). In MPTP-treated mice, minocycline increased significantly the number of surviving SNpc TH-positive neurons in a dose-dependent manner (Fig.1D,G). Minocycline at a dose of 1.4 mg/kg twice daily had no effect on MPTP neurotoxicity, whereas at doses of 11.25 mg/kg twice daily and higher, there was significant neuroprotection (Fig. 1G). Even at the highest dose tested (45 mg/kg twice daily), minocycline was well tolerated and did not produce any behavioral abnormality. To test whether minocycline could provide complete neuroprotection, we examined another group of mice with less severe SNpc damage by injecting a lower dose of MPTP (16 mg/kg for four injections). In mice that received MPTP only, this lower regimen reduced numbers of SNpc TH-positive neurons by ∼30% compared with controls (Fig. 1E,G). Minocycline at 45 mg/kg twice daily produced >90% protection against MPTP at 16 mg/kg for four injections (Fig.1F,G).
Fig. 1.
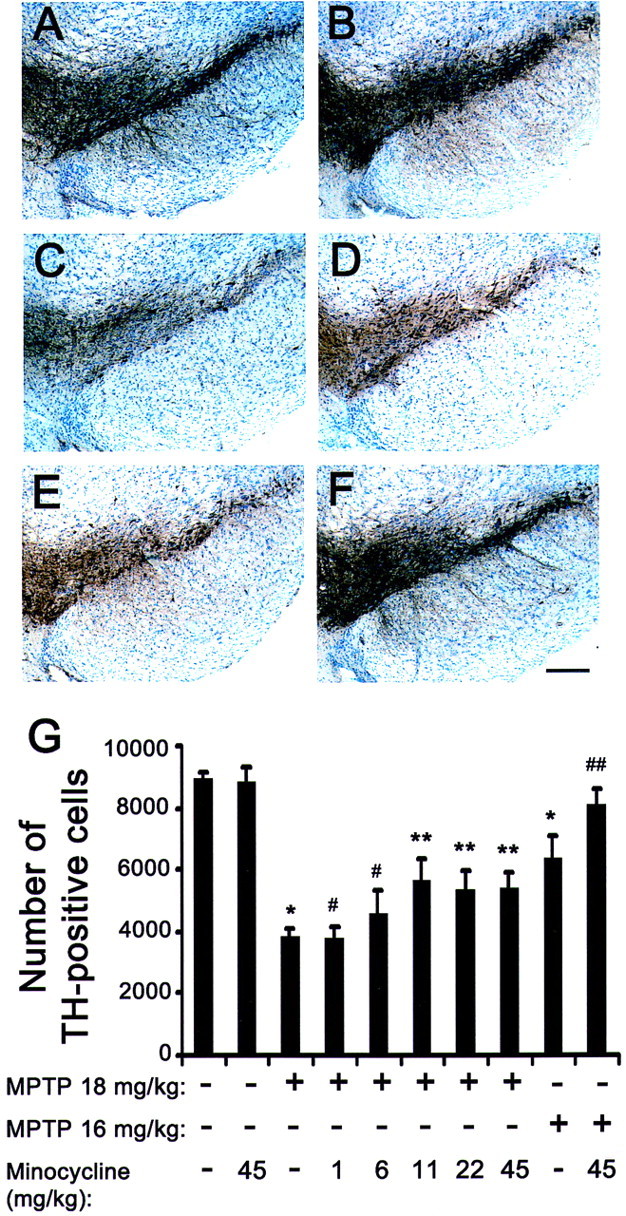
Effect of minocycline on MPTP-induced SNpc dopaminergic neuronal death. In saline-injected control mice treated without (A) or with (B; 45 mg/kg twice daily) minocycline, there are numerous SNpc TH-positive neurons (brown; A, B). MPTP (18 mg/kg for 4 injections) reduces the number of SNpc TH-positive neurons (C) 7 d after the last injection. In mice treated with both MPTP and minocycline, there is a noticeable attenuation of SNpc TH-positive neuronal loss (D). At a lower MPTP dosage (16 mg/kg for 4 injections), loss of TH-positive structures is less (E) and minocycline protection is more obvious (F). Scale bar, 50 μm. Bar graph shows SNpc TH-positive neuronal counts (G) assessed under the various experimental conditions. Minocycline 1,6, 11, 22,45, Mice injected with minocycline at 1.4, 6.1, 11.3, 22.5, and 45.0 mg/kg twice daily. *p < 0.05, fewer than saline-injected or minocycline-injected control mice. #p > 0.05, same as MPTP-injected mice. **p < 0.05, fewer than control mice but more than MPTP-injected mice. ##p < 0.05, more than MPTP-injected mice and not different from control mice. Values are means ± SEM (n = 6–8 per group).
Sparing of SNpc dopaminergic neurons does not always correlate with sparing of their corresponding striatal nerve fibers (Liberatore et al., 1999), which is essential for maintaining dopaminergic neurotransmission. To determine whether minocycline can prevent not only MPTP-induced loss of SNpc neurons but also the loss of striatal dopaminergic fibers, we assessed the density of TH immunoreactivity in striata from the different groups of mice (Fig.2). Four injections of MPTP at 18 and 16 mg/kg reduced striatal TH immunoreactivity compared with controls by 96 and 79%, respectively (Fig.2C,E,G). Mice that received minocycline (45 mg/kg twice daily) and four injections of 18 mg/kg MPTP (Fig. 2D,G) showed no protection of striatal dopaminergic fibers, whereas mice that received the same dose of minocycline and four injections of 16 mg/kg MPTP (Fig.2F,G) showed significant sparing of striatal TH-positive fibers. These findings indicate that minocycline protects the nigrostriatal pathway against the effects of the parkinsonian toxin MPTP.
Fig. 2.
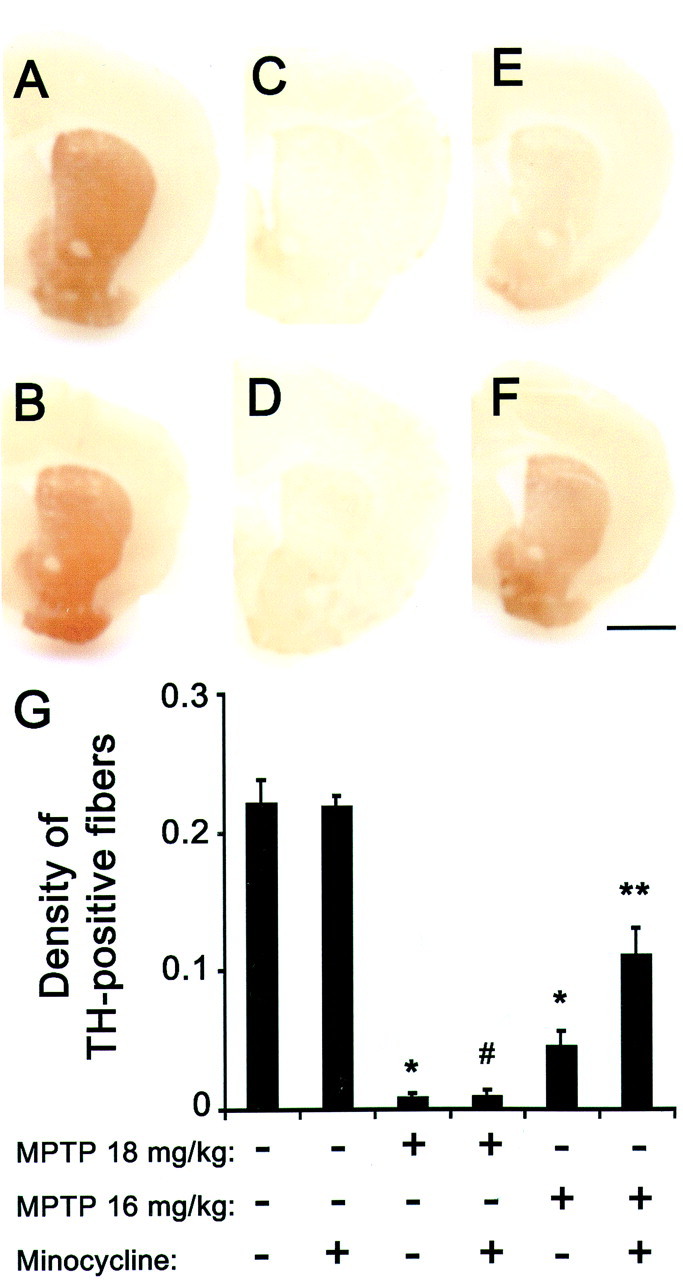
Effect of minocycline on MPTP-induced striatal dopaminergic fiber loss. In saline-injected control mice treated without (A) or with (B; 45 mg/kg twice daily) minocycline, there are a high density of striatal TH-positive fibers. MPTP (18 mg/kg for 4 injections) reduces the density of striatal TH-positive fibers (C) 7 d after the last injection. In mice treated with both MPTP and minocycline, there is also a noticeable striatal TH-positive fiber loss (D). At a lower MPTP dosage (16 mg/kg for 4 injections), loss of TH-positive structures is less (E) and minocycline protection is obvious (F). Scale bar, 1 mm. Bar graph shows striatal TH-positive optical density (G) assessed under the various experimental conditions (F(5,33) = 41.475;p < 0.001). *p < 0.05, fewer than saline-injected or minocycline-injected control mice. #p > 0.05, same as MPTP-injected mice. **p < 0.05, more than MPTP-injected mice but fewer than control mice. Values are means ± SEM (n= 6–8 per group).
Minocycline decreases MPTP-mediated nitrotyrosine formation
A significant part of the MPTP neurotoxic process is mediated by NO-related oxidative damage (Przedborski et al., 2000), the extent of which can be evaluated by assessing nitrotyrosine levels (Liberatore et al., 1999; Pennathur et al., 1999). In saline-injected mice, the levels of nitrotyrosine in ventral midbrain were similar between non-minocycline and minocycline-treated animals (Table1). In MPTP-injected mice (18 mg/kg for four injections), nitrotyrosine levels were significantly increased in ventral midbrain (brain region containing SNpc) and unchanged in cerebellum (brain region unaffected by MPTP) (Table 1). MPTP produced significantly smaller increases in nitrotyrosine levels in ventral midbrains of minocycline (45 mg/kg twice daily)-treated mice than in their non-minocycline-treated counterparts (Table 1). This confirms that minocycline not only attenuates the morphological but also the biochemical impacts of MPTP neurotoxicity.
Table 1.
Nitrotyrosine levels (pg/μg protein)
| Saline | Minocycline | MPTP | MPTP–minocycline | |
|---|---|---|---|---|
| Ventral midbrain | 16.2 ± 1.3 | 18.5 ± 1.7 | 32.2 ± 6.0* | 21.8 ± 1.81-160 |
| Cerebellum | 13.1 ± 0.8 | 14.0 ± 2.1 | 13.4 ± 1.0 | 11.7 ± 1.1 |
Nitrotyrosine levels are significantly different among groups in the ventral midbrain (F(3,23) = 4.56;p < 0.05) but not in cerebellum (F(3,23) = 0.618; p > 0.05).
p < 0.05, more than saline-injected and minocycline-injected control mice.
F1-160: p < 0.05, less than MPTP-injected mice but not different from both control groups. Saline, Mice injected with saline; Minocycline, mice injected with minocycline only (45 mg/kg twice daily); MPTP, mice injected with MPTP only (18 mg/kg MPTP for 4 injections in one day); MPTP–minocycline, mice injected with both MPTP and minocycline. Values are means ± SEM (n = 6–8 per group).
MPTP metabolism is unaffected by minocycline
The main determining factors of MPTP neurotoxic potency are its conversion in the brain to MPP+ followed by MPP+ entry into dopaminergic neurons and its subsequent blockade of mitochondrial respiration (Przedborski et al., 2000). To ascertain that resistance to the neurotoxic effects of MPTP provided by minocycline was not attributable to alterations in any of these three key MPTP neurotoxic steps, we measured striatal levels of MPP+ 90 min after injection of 18 mg/kg MPTP, striatal uptake of [3H]MPP+into synaptosomes, and striatal MPP+-induced lactate production, a reliable marker of mitochondrial inhibition (Kindt et al., 1987) (Table2). These investigations showed that striatal levels of MPP+ did not differ between MPTP-injected mice that either received or did not receive minocycline (45 mg/kg) 30 min after MPTP administration. In addition, minocycline up to 333 μm (maximal solubilizing concentration) did not affect striatal uptake of [3H]MPP+ or MPP+-induced lactate production (Table2).
Table 2.
Striatal MPTP metabolism
| MPP+ level | ||||
| Treatment | MPTP only | MPTP–minocycline pretreatment | MPTP–minocycline post-treatment | |
| μg/gm tissue | 6.42 ± 0.92 | 5.21 ± 0.66 | 6.52 ± 0.59 | |
| [3H]MPP+ uptake | ||||
| Minocycline (μm) | 0 | 10 | 100 | 333 |
| % of control | 100 | 98 ± 3 | 96 ± 3 | 82 ± 1 |
| MPP+-induced lactate | ||||
| Minocycline (μm) | 0 | 10 | 100 | 333 |
| μm/100 mg protein | 74 ± 4 | 71 ± 6 | 70 ± 6 | 67 ± 6 |
For MPP+ levels, minocycline (45 mg/kg) was given either 30 min before or after MPTP administration. Values are means ± SEM of either six mice per group (MPP+levels) or three independent experiments each performed in duplicate ([3H]MPP+ uptake and lactate levels). None of the presented values differ significantly (p > 0.05) from MPTP only (MPP+ levels) or from 0 μmminocycline ([3H]MPP+ uptake and lactate levels).
Minocycline inhibits MPTP-induced microglial activation
To determine whether neuroprotection by minocycline is associated with inhibition of MPTP-induced glial response, we examined the expression of MAC-1, a specific marker for microglia, and GFAP, a specific marker for astrocytes. As shown in Figure3B, MAC-1 mRNA contents (F(3,23) = 4.252; p < 0.05), but not GFAP mRNA contents (F(3,18) = 2.843; p > 0.05), varied significantly among the various group of mice. In saline-injected mice, ventral midbrain expression of MAC-1 and GFAP mRNA was minimal (Fig. 3A,B). In these animals, only a few faintly immunoreactive resting microglia and astrocytes were observed in SNpc and striatum by immunostaining (data no shown). In MPTP-injected mice (18 mg/kg for four injections) without treatment with minocycline, ventral midbrain expression of MAC-1 mRNA was significantly higher, whereas expression of GFAP mRNA, although also higher, was not significantly increased compared with saline controls (Fig. 3). Morphologically, numerous robustly immunoreactive MAC-1-positive activated microglia were observed 24 hr after the last injection of the toxin (Fig.4A–D). Although GFAP immunostaining appeared somewhat increased at 24 hr after the last MPTP injection (Fig.5A,B), the strongest GFAP reaction was noted 7 d after the last injection of MPTP (Fig. 5C,D). Conversely, in MPTP-injected mice treated with minocycline (45 mg/kg twice daily), ventral midbrain MAC-1 mRNA contents (Fig. 3) and SNpc and striatal immunostaining were similar to those seen in saline-injected mice (Fig.4E–H). In contrast, in MPTP-injected minocycline-treated mice, ventral midbrain GFAP mRNA content (Fig. 3) and SNpc immunostaining (Fig.5E,F) were almost as high and as intense as in MPTP-only mice. Staining with Isolectin B-4 (Sigma), another marker for microglia, gave results similar to that of MAC-1 (data not shown).
Fig. 3.

Minocycline prevents MPTP-induced MAC-1 transcription. A, B, Ventral midbrain MAC-1 mRNA levels but not GFAP mRNA levels are increased by 24 hr after MPTP injection compared with those of saline- or minocycline-injected mice. Minocycline prevents MPTP-induced MAC-1 mRNA increases. MAC-1 and GFAP mRNA values are normalized with GAPDH. Values are mean ± SEM ratios (n = 5–7 mice per group).Saline, Saline-treated; Mc, minocycline-treated;MPTP, MPTP-treated; M + Mc, MPTP plus minocycline-treated. *p < 0.05, higher than both saline- and minocycline-injected control groups. **p < 0.05, lower than MPTP-injected group and not different from both control groups.
Fig. 4.
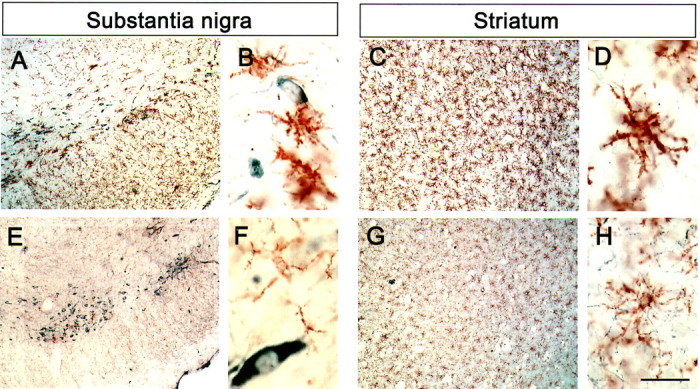
Minocycline prevents MPTP-induced microglia reaction. Microglia cells (brown) and TH-positive neurons (gray blue) are seen in both SNpc and striatum of all mice. One day after the last MPTP injection, numerous activated microglia (larger cell body, poorly ramified short and thick processes) are seen in SNpc (A, B) and striatum (C, D). Mice injected with both MPTP and minocycline show minimal microglial activation in SNpc (E) and striatum (G); here, microglial cell bodies are small and processes are thin and ramified (F, H). Scale bar:A, C, E, G, 1 mm; B, D, F,H, 100 μm.
Fig. 5.
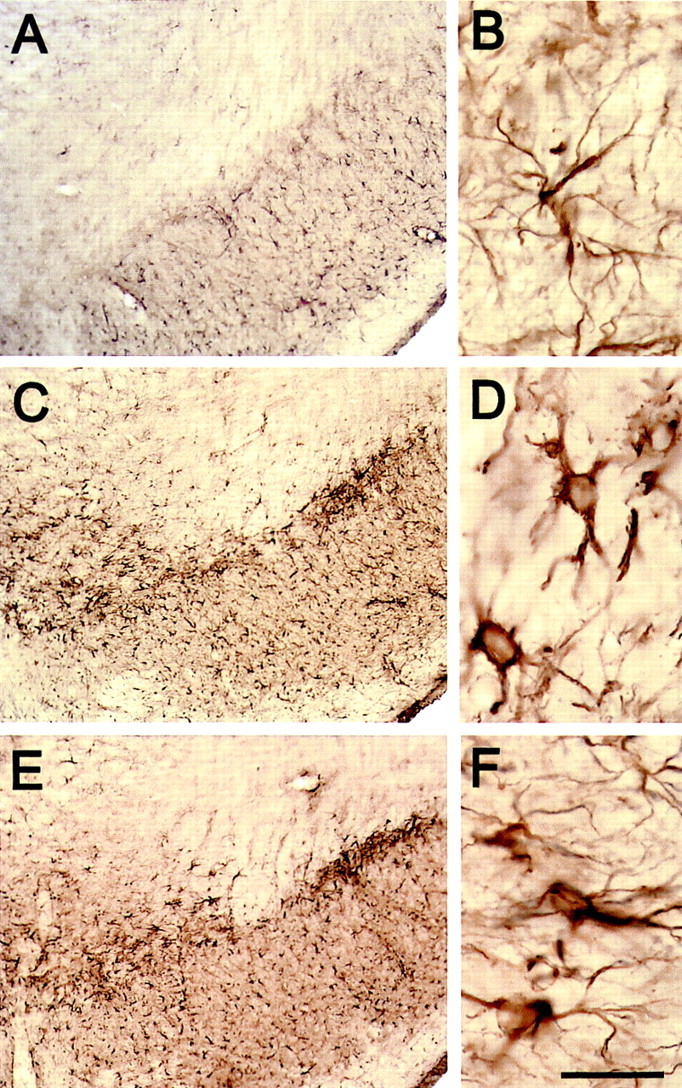
Minocycline does not affect MPTP-induced astrocytic reaction. One day after the last injection of MPTP, there is a mild astrocytic response (A, B), but 7 d after the last injection of MPTP, it becomes conspicuous (C, D). Minocycline does not affect the astrocytic response (E, F) 7 d after MPTP administration. Scale bar: A,C, E, 1 mm; B,D, F, 100 μm.
Minocycline prevents the production of microglial-derived deleterious mediators
Given the effect of minocycline on MPTP-induced microglial activation, we assessed whether the production of known microglial noxious mediators such as IL-1β, ROS, and NO will also be inhibited by minocycline (Fig. 6). The levels of ventral midbrain IL-1β differed significantly among the four group of mice (F(3,21) = 7.946;p < 0.001) (Fig. 6A). Ventral midbrain levels of the proinflammatory cytokine IL-1β in MPTP-injected mice (18 mg/kg for four injections) were significantly increased (Fig. 6A). However, MPTP produced significantly smaller increases in IL-1β levels in ventral midbrain of MPTP mice treated with minocycline (45 mg/kg twice daily) (Fig. 6A). iNOS activity (F(3,24) = 9.055; p < 0.001) and the ratio of membrane/total p67phox(F(3,23) = 4.336; p < 0.05) also varied significantly among the various groups. iNOS and NADPH–oxidase, two prominent enzymes of activated microglia that produce NO and ROS, respectively, exhibited induction patterns similar to those described for IL-1β in that ventral midbrain iNOS activity was increased by 200% (Fig. 6B) and NADPH–oxidase activation, evidenced by the translocation of its subunit p67phox from the cytosol to the plasma membrane, was increased by 80% 24 hr after the last injection of MPTP (Fig. 6C,D). MPTP-induced iNOS activity and NADPH–oxidase were both abolished by minocycline administration (Fig.6B–D).
Fig. 6.

Effects of MPTP and minocycline on microglial-derived deleterious factors IL-1β (A), iNOS (B), and NADPH–oxidase (C, D). MPTP (18 mg/kg for 4 injections) increases ventral midbrain mature IL-1β formation, iNOS catalytic activity, and NADPH–oxidase activation, as evidenced by the translocation of its subunit p67phox from the cytosol to the plasma membrane, 1 d after the last injection of MPTP. Minocycline (45 mg/kg twice daily) attenuates MPTP-related effects on mature IL-1β, iNOS, and NADPH–oxidase.Saline, Saline-treated; Mc, minocycline-treated; M, MPTP-treated;M+Mc, MPTP plus minocycline-treated. *p < 0.05, more than saline-injected or minocycline-injected control mice. **p < 0.05, less than MPTP-injected mice but not different from both control groups. Values are means ± SEM (n = 5–8 mice per group).
Minocycline confers resistance to MPTP beyond iNOS ablation
Previously, it has been demonstrated that iNOS ablation attenuates MPTP neurotoxicity (Liberatore et al., 1999; Dehmer et al., 2000). Thus, to demonstrate whether minocycline-mediated blockade of microglial activation protects solely because it inhibits iNOS induction, we compared the effect of MPTP (16 mg/kg for four injections) on the network of striatal dopaminergic nerve fibers between mutant iNOS-deficient mice that received or did not receive minocycline (45 mg/kg twice daily). As shown in Figure7, MPTP administration reduced by >80% the striatal density of TH-positive fibers both in wild-type and iNOS−/− mice; this is consistent with our previous data that ablation of iNOS protects against MPTP-induced SNpc dopaminergic neuronal loss but not against MPTP-induced striatal dopaminergic fiber destruction (Liberatore et al., 1999). In contrast, striatal TH-positive fiber densities were more than twofold higher in MPTP-treated wild-type and iNOS−/− mice that received minocycline compared with those that did not receive minocycline (Fig. 7). However, there was no difference in the magnitude of the minocycline beneficial effect between MPTP-treated iNOS−/− mice and their MPTP-treated wild-type counterparts (Fig. 7).
Fig. 7.

Minocycline attenuates MPTP-induced striatal damage by inhibiting microglia but not just by inhibiting iNOS. The optical density of striatal TH-positive fibers varied significantly among the various groups (F(7,47) = 83.576;p < 0.001). Minocycline, Mice injected with minocycline 45 mg/kg twice daily. MPTP, Mice injected with MPTP (4 injections of 16 mg/kg). *p < 0.05, fewer than saline-injected or minocycline-injected control mice. #p < 0.05, fewer than control mice but no different than wild-type mice injected with MPTP. **p < 0.05, fewer than control but more than MPTP-injected mice. # #p < 0.05, more than MPTP-injected mice but no different from wild-type mice injected with both MPTP and minocycline.
DISCUSSION
The main finding of this study is that inhibition of microglial activation by minocycline protects the nigrostriatal dopaminergic pathway against the noxious effects of the parkinsonian toxin MPTP. In mice that received minocycline, MPTP caused significantly less neuronal death in the SNpc, as evidenced by the greater number of TH-positive neurons, compared with those that received MPTP only (Fig. 1). Although less prominent, a similar observation was made for striatal dopaminergic nerve terminals (Fig. 2). The magnitude of resistance to MPTP in mice appears to result from a balance between the dose of minocycline and the dose of MPTP (Fig. 1), with the greatest neuroprotection observed in mice that received >11.25 mg/kg minocycline twice daily and MPTP at 16 mg/kg four times in 1 d and the least neuroprotection in mice that received the regimen of minocycline at 6.1 mg/kg twice daily and MPTP at 18 mg/kg four times in 1 d. In our study, minocycline was given twice daily beginning on the day of MPTP administration and continuing through 4 d thereafter because of its long half-life (>12 hr) and because we showed that, with this MPTP regimen, nigrostriatal degeneration occurs during the first 4 d after the last injection of MPTP (Jackson-Lewis et al., 1995). Therefore, we cannot exclude that greater protection could have been achieved if minocycline had been administered more frequently or for a longer period of time. Also, because we focused our assessment of nigrostriatal neurodegeneration at 7 d after MPTP administration, we cannot exclude with certainty that minocycline had delayed rather than prevented neuronal death. However, in light of what we know about how minocycline presumably mitigates cellular damage in a variety of experimental models (Tikka and Koistinaho, 2001; Tikka et al., 2001a), the aforementioned possibility appears unlikely. In addition, we did not pretreat mice with minocycline because we found that administration of minocycline before MPTP injection reduces striatal MPP+ levels by 20% (Table 2), which could complicate the interpretation of minocycline neuroprotection. Indeed, it is established that striatal contents of MPP+ correlate linearly with magnitudes of MPTP toxicity (Giovanni et al., 1991). Thus, to avoid this potential confounding factor in our study, all mice were injected first with MPTP and then with minocycline, which we found not to affect striatal MPP+ levels (Table 2). Along this line, it is also worth mentioning that minocycline, as used here, not only failed to alter MPP+ levels but also failed to interfere with other key aspects of MPTP metabolism (Przedborski et al., 2000), such as entry of MPP+ into dopaminergic neurons and inhibition of mitochondrial respiration at concentrations as high as 333 μm (Table 2).
Nitrotyrosine is a fingerprint of NO-derived modification of protein and has been documented as one of the main markers of oxidative damage mediated by MPTP (Schulz et al., 1995; Ara et al., 1998; Liberatore et al., 1999; Pennathur et al., 1999; Przedborski et al., 2001a). Consistent with our previous studies (Liberatore et al., 1999;Pennathur et al., 1999), nitrotyrosine levels increased substantially in brain regions affected by MPTP, such as ventral midbrain, but not in brain regions unaffected by MPTP, such as cerebellum (Table 1). As with the loss of SNpc neurons and striatal fibers, minocycline dramatically attenuated ventral midbrain increases in nitrotyrosine levels (Table1). Collectively, our data demonstrate that minocycline protects against morphological as well as biochemical abnormalities that arise from MPTP insult. That said, we now need to consider the nature of the mechanism underlying the beneficial effects of minocycline on MPTP neurotoxicity.
Previously, we demonstrated that, aside from a dramatic loss of dopaminergic neurons, gliosis is a striking neuropathological feature in the SNpc and the striatum in the MPTP mouse model as in PD (Liberatore et al., 1999). However, activated microglia appear in the SNpc earlier than reactive astrocytes (Liberatore et al., 1999) and at a time when only minimal neuronal death occurs (Jackson-Lewis et al., 1995). This supports the contention that the microglial response to MPTP arises early enough in the neurodegenerative process to contribute to the demise of SNpc dopaminergic neurons. Consistent with this is the demonstration that direct injection of the known microglial activator lipopolysaccharide into the rat SNpc causes a strong microglial response associated with significant dopaminergic neuronal death (Castano et al., 1998; Herrera et al., 2000; Kim et al., 2000). Given these data, the key to the minocycline neuroprotective effect in the MPTP mouse model may lie in the second main finding of our study, which is that minocycline prevented MPTP-induced microglial response in both the SNpc and the striatum (Figs. 3, 4). In contrast, minocycline did not alter MPTP-related astrocytic response (Fig. 5). These results suggest that minocycline acts on microglia specifically and not on all components of gliosis. Our data also support the view that reduction of MPTP-related microglial response seen after minocycline administration is not secondary to the attenuation of neuronal loss but rather the reverse. This interpretation does not rule out, however, that at least some of the neuroprotection of minocycline against MPTP is attributable to a direct action on neurons as suggested previously (Tikka et al., 2001b).
Inhibition of microglial activation using minocycline has also been demonstrated in vitro (Tikka et al., 2001b) and in other experimental models of acute and chronic brain insults (Yrjanheikki et al., 1998, 1999; Tikka and Koistinaho, 2001; Tikka et al., 2001a) and results, presumably, from the blockade of p38 mitogen-activated protein kinase (Tikka et al., 2001a). It is believed that activated microglia exerts cytotoxic effects in the brain through two very different and yet complementary processes (Banati et al., 1993). First, they can act as phagocytes, which involve direct cell-to-cell contact. Second, they are capable of releasing a large variety of potentially noxious substances (Banati et al., 1993). Consistent with the notion that minocycline inhibits the ability of microglia to respond to injury, we show that minocycline not only prevents the microglial morphological response to MPTP but also the microglial production of cytotoxic mediators such as IL-1β and the induction of critical ROS- and NO-producing enzymes such as NADPH–oxidase and iNOS (Fig. 6). Although we did not test this, it is quite relevant to mention that minocycline may also prevent the induction of cyclooxygenase-2, a key enzyme in the production of potent proinflammatory prostanoids, either directly or indirectly via the blockade of IL-1β formation (Yrjanheikki et al., 1999). Little is known about the actual role of IL-1β in either MPTP or PD neurodegenerative process, except that IL-1β immunoreactivity is found in glial cells from postmortem PD SNpc samples (Hunot et al., 1999) and that blockade of interleukin converting enzyme, the known activator of IL-1β, attenuates MPTP-induced neurodegeneration in mice (Klevenyi et al., 1999). As for ROS, oxidative stress is a prominent pathogenic hypothesis in both MPTP and PD (Przedborski and Jackson-Lewis, 2000). However, many of the microglial-derived ROS, such as superoxide, cannot readily transverse cellular membranes (Halliwell and Gutteridge, 1991), making it unlikely that these extracellular reactive species gain access to dopaminergic neurons and trigger intraneuronal toxic events. Alternatively, superoxide can react with NO in the extracellular space to form the highly reactive tissue-damaging species peroxynitrite, which can cross the cell membrane and injure neurons. Therefore, microglial-derived superoxide, by contributing to peroxynitrite formation, may be significant in this model. As for NO in both MPTP and PD, the pivotal pathogenic role for microglial-derived NO is supported by the demonstration that ablation of iNOS attenuates SNpc dopaminergic neuronal death (Liberatore et al., 1999; Dehmer et al., 2000) and the production of ventral midbrain nitrotyrosine after MPTP administration (Liberatore et al., 1999). In this context, it is worth mentioning that minocycline, which protects in global brain ischemia (Yrjanheikki et al., 1998) and in a mouse model of Huntington's disease (Chen et al., 2000), appears to do so by abating iNOS expression and activity. Remarkably, iNOS ablation does protect SNpc neurons from MPTP toxicity but does not protect striatal nerve terminals and does not prevent microglial activation (Liberatore et al., 1999). This is in striking contrast to the effect of minocycline treatment, which protects both dopaminergic cell bodies and nerve fibers and inhibits the entire microglial response. This strongly suggests that microglial-associated deleterious factors other than iNOS are involved in the demise of the nigrostriatal pathway in the MPTP mouse model of PD and possibly in PD itself. Consistent with this interpretation are our data in iNOS−/−mice (Fig. 7), which show that minocycline protects striatal dopaminergic fibers regardless of the presence or absence of iNOS expression. Therefore, our study provides strong support to the idea that activated microglia are important contributors to the overall demise of SNpc dopaminergic neurons in the MPTP mouse model of PD and, possibility, in PD itself. It also suggests that therapeutic interventions aimed at preventing the loss of striatal dopaminergic fibers, which is essential to maintaining dopaminergic neurotransmission, must target microglial-derived factors other than iNOS.
Footnotes
This study was supported by National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke Grants R29 NS37345, RO1 NS38586, RO1 NS42269, and P50 NS38370, NIH/National Institute on Aging Grant RO1 AG13966, United States Department of Defense Grant DAMD 17-99-1-9471, the Lowenstein Foundation, the Lillian Goldman Charitable Trust, the Parkinson's Disease Foundation, the Muscular Dystrophy Association, the Amyotrophic Lateral Sclerosis (ALS) Association, and Project-ALS. M.V. is the recipient of a fellowship from the Human Frontier Science Program Organization, and P.T. is the recipient of German Research Foundation Grant TE 343/1-1.
Correspondence should be addressed to Dr. Serge Przedborski, Departments of Neurology and Pathology, BB-307, Columbia University, 650 West 168th Street, New York, NY 10032. E-mail: SP30@columbia.edu.
REFERENCES
- 1.Amin AR, Attur MG, Thakker GD, Patel PD, Vyas PR, Patel RN, Patel IR, Abramson SB. A novel mechanism of action of tetracyclines: effects on nitric oxide synthases. Proc Natl Acad Sci USA. 1996;93:14014–14019. doi: 10.1073/pnas.93.24.14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ara J, Przedborski S, Naini AB, Jackson-Lewis V, Trifiletti RR, Horwitz J, Ischiropoulos H. Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Proc Natl Acad Sci USA. 1998;95:7659–7663. doi: 10.1073/pnas.95.13.7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aronson AL. Pharmacotherapeutics of the newer tetracyclines. J Am Vet Med Assoc. 1980;176:1061–1068. [PubMed] [Google Scholar]
- 4.Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7:111–118. doi: 10.1002/glia.440070117. [DOI] [PubMed] [Google Scholar]
- 5.Burke RE, Cadet JL, Kent JL, Karanas AL, Jackson-Lewis V. An assessment of the validity of densitometric measures of striatal tyrosine hydroxylase-positive fibers: relationship to apomorphine-induced rotations in 6-hydroxydopamine lesioned rats. J Neurosci Methods. 1990;35:63–73. doi: 10.1016/0165-0270(90)90095-w. [DOI] [PubMed] [Google Scholar]
- 6.Castano A, Herrera AJ, Cano J, Machado A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J Neurochem. 1998;70:1584–1592. doi: 10.1046/j.1471-4159.1998.70041584.x. [DOI] [PubMed] [Google Scholar]
- 7.Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6:797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- 8.Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB. Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J Neurochem. 2000;74:2213–2216. doi: 10.1046/j.1471-4159.2000.0742213.x. [DOI] [PubMed] [Google Scholar]
- 9.Fahn S, Przedborski S. Parkinsonism. In: Rowland LP, editor. Merritt's neurology. Lippincott Williams & Wilkins; New York: 2000. pp. 679–693. [Google Scholar]
- 10.Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Rev. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- 11.Giovanni A, Sieber BA, Heikkila RE, Sonsalla PK. Correlation between the neostriatal content of the 1-methyl-4-phenylpyridinium species and dopaminergic neurotoxicity following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration to several strains of mice. J Pharmacol Exp Ther. 1991;257:691–697. [PubMed] [Google Scholar]
- 12.Halliwell B, Gutteridge JM. Free radicals in biology and medicine. Clarendon; Oxford: 1991. [Google Scholar]
- 13.He Y, Appel S, Le W. Minocycline inhibits microglial activation and protects nigral cells after 6-hydroxydopamine injection into mouse striatum. Brain Res. 2001;909:187–193. doi: 10.1016/s0006-8993(01)02681-6. [DOI] [PubMed] [Google Scholar]
- 14.Herrera AJ, Castano A, Venero JL, Cano J, Machado A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis. 2000;7:429–447. doi: 10.1006/nbdi.2000.0289. [DOI] [PubMed] [Google Scholar]
- 15.Hopkins SJ, Rothwell NJ. Cytokines and the nervous system. I. Expression and recognition. Trends Neurosci. 1995;18:83–88. [PubMed] [Google Scholar]
- 16.Hornykiewicz O, Kish SJ. Biochemical pathophysiology of Parkinson's disease. In: Yahr M, Bergmann KJ, editors. Parkinson's disease. Raven; New York: 1987. pp. 19–34. [PubMed] [Google Scholar]
- 17.Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, Agid Y, Dugas B, Hirsch EC. FceRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-α in glial cells. J Neurosci. 1999;19:3440–3447. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson-Lewis V, Jakowec M, Burke RE, Przedborski S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration. 1995;4:257–269. doi: 10.1016/1055-8330(95)90015-2. [DOI] [PubMed] [Google Scholar]
- 19.Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kindt MV, Heikkila RE, Nicklas WJ. Mitochondrial and metabolic toxicity of 1-methyl-4-(2′-methylphenyl)-1,2,3,6-tetrahydropyridine. J Pharmacol Exp Ther. 1987;242:858–863. [PubMed] [Google Scholar]
- 21.Klevenyi P, Andreassen O, Ferrante RJ, Schleicher JRJ, Friedlander RM, Beal MF. Transgenic mice expressing a dominant negative mutant interleukin-1beta converting enzyme show resistance to MPTP neurotoxicity. NeuroReport. 1999;10:635–638. doi: 10.1097/00001756-199902250-00035. [DOI] [PubMed] [Google Scholar]
- 22.Kostic V, Przedborski S, Flaster E, Sternic N. Early development of levodopa-induced dyskinesias and response fluctuations in young-onset Parkinson's disease. Neurology. 1991;41:202–205. doi: 10.1212/wnl.41.2_part_1.202. [DOI] [PubMed] [Google Scholar]
- 23.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 24.Li M, Ona VO, Guegan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000;288:335–339. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- 25.Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- 26.Pakkenberg B, Moller A, Gundersen HJG, Mouritzen A, Pakkenberg H. The absolute number of nerve cells in substantia nigra in normal subjects and in patients with Parkinson's disease estimated with an unbiased stereological method. J Neurol Neurosurg Psychiatry. 1991;54:30–33. doi: 10.1136/jnnp.54.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pennathur S, Jackson-Lewis V, Przedborski S, Heinecke JW. Mass spectrometric quantification of 3-nitrotyrosine, ortho-tyrosine, and o,o′-dityrosine in brain tissue of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice, a model of oxidative stress in Parkinson's disease. J Biol Chem. 1999;274:34621–34628. doi: 10.1074/jbc.274.49.34621. [DOI] [PubMed] [Google Scholar]
- 28.Przedborski S, Jackson-Lewis V. ROS, Parkinson's disease: a view to a kill. In: Poli G, Cadenas E, Packer L, editors. Free radicals in brain pathophysiology. Dekker; New York: 2000. pp. 273–290. [Google Scholar]
- 29.Przedborski S, Kostic V, Jackson-Lewis V, Naini AB, Simonetti S, Fahn S, Carlson E, Epstein CJ, Cadet JL. Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J Neurosci. 1992;12:1658–1667. doi: 10.1523/JNEUROSCI.12-05-01658.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Przedborski S, Jackson-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM. Role of neuronal nitric oxide in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci USA. 1996;93:4565–4571. doi: 10.1073/pnas.93.10.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Przedborski S, Jackson-Lewis V, Djaldetti R, Liberatore G, Vila M, Vukosavic S, Almer G. The parkinsonian toxin MPTP: action and mechanism. Restor Neurol Neurosci. 2000;16:135–142. [PubMed] [Google Scholar]
- 32.Przedborski S, Chen Q, Vila M, Giasson BI, Djaldatti R, Vukosavic S, Souza JM, Jackson-Lewis V, Lee VM, Ischiropoulos H. Oxidative post-translational modifications of alpha-synuclein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson's disease. J Neurochem. 2001a;76:637–640. doi: 10.1046/j.1471-4159.2001.00174.x. [DOI] [PubMed] [Google Scholar]
- 33.Przedborski S, Jackson-Lewis V, Naini A, Jakowec M, Petzinger G, Miller R, Akram M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. J Neurochem. 2001b;76:1265–1274. doi: 10.1046/j.1471-4159.2001.00183.x. [DOI] [PubMed] [Google Scholar]
- 34.Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48:1393–1399. doi: 10.1097/00006123-200106000-00051. [DOI] [PubMed] [Google Scholar]
- 35.Schulz JB, Matthews RT, Muqit MMK, Browne SE, Beal MF. Inhibition of neuronal nitric oxide synthase by 7-nitroindazole protects against MPTP-induced neurotoxicity in mice. J Neurochem. 1995;64:936–939. doi: 10.1046/j.1471-4159.1995.64020936.x. [DOI] [PubMed] [Google Scholar]
- 36.Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-d-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166:7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- 37.Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation, proliferation of microglia. J Neurosci. 2001a;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tikka T, Usenius T, Tenhunen M, Keinanen R, Koistinaho J. Tetracycline derivatives and ceftriaxone, a cephalosporin antibiotic, protect neurons against apoptosis induced by ionizing radiation. J Neurochem. 2001b;78:1409–1414. doi: 10.1046/j.1471-4159.2001.00543.x. [DOI] [PubMed] [Google Scholar]
- 39.Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D, Korsmeyer SJ, Przedborski S. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Proc Natl Acad Sci USA. 2001a;98:2837–2842. doi: 10.1073/pnas.051633998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vila M, Jackson Lewis V, Guégan C, Wu DC, Teismann P, Choi D-K, Tieu K, Przedborski S. The role of glial cells in Parkinson's disease. Curr Opin Neurol. 2001b;14:483–489. doi: 10.1097/00019052-200108000-00009. [DOI] [PubMed] [Google Scholar]
- 41.Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]