

Abstract
Transgenic mice expressing mutant amyloid precursor proteins (APPs) have provided important new information about the pathogenesis of Alzheimer's disease (AD) histopathology. However, the molecular basis of memory loss in these mice is poorly understood. One of the major impediments has been the difficulty of distinguishing between age-dependent and age-independent behavioral changes. To address this issue we studied in parallel two lines of APP transgenic mice expressing comparable levels of mutant and wild-type human APP. This enabled us to identify age-independent behavioral deficits that were not specifically related to mutant APP expression. When mice with age-independent deficits were eliminated, we detected memory loss in transgenic mice expressing mutant APP (Tg2576 mice) starting at ∼6 months, which coincided with the appearance of detergent-insoluble Aβ aggregates (Aβinsol). Genetically accelerating the formation of Aβinsol resulted in an earlier onset of memory decline. A facile interpretation of these results, namely that memory loss and Aβinsol were closely connected, was rejected when we extended our analysis to include older mice. No obvious correspondence between memory and Aβinsol was apparent in a combined group of old and young mice unless the mice were stratified by age, whereupon inverse correlations between memory and Aβinsol became evident. These results suggested that Aβinsol is a surrogate marker for small assemblies of Aβ that disrupt cognition and occur as intermediates during Aβinsol formation, and they are the first descriptive in vivo data supporting their role in impairing memory. These studies also provide a methodological framework within which to investigate these Aβ assemblies in vivo.
Keywords: Alzheimer's disease, transgenic, behavior, Aβ, SDS-soluble, insoluble, learning, memory
Memory loss is the cardinal manifestation of Alzheimer's disease (AD). The Aβ hypothesis of AD stipulates that Aβ aggregates form amyloid plaques that are neurotoxic, leading to neurodegeneration accompanied by dementia (Hardy and Allsop, 1991). The strongest evidence for this hypothesis comes from molecular genetic studies of amyloid precursor proteins (APPs) and presenilins in early-onset familial AD showing that all genetically linked mutations increase the propensity for Aβ to aggregate in vitro (for review, see Golde et al., 2000; Selkoe, 2001). The principal argument against Aβ stems from multiple studies showing little or no correlation between the quantity of amyloid deposition and dementia (Katzman et al., 1988; Delaere et al., 1990; Terry et al., 1991; Arriagada et al., 1992; Dickson et al., 1992; Berg et al., 1993).
Tg2576 mice overexpressing human APP695 with the “Swedish” mutation develop memory deficits and plaques with age (Hsiao et al., 1996), making them suitable for examining the relationship between Aβ and memory. Tg2576 mice show rapid increases in Aβ starting at ∼6 months and amyloid plaques beginning at 9–12 months (Kawarabayashi et al., 2001). Different forms of Aβ, biochemically distinguishable by their solubility properties, are present in varying amounts during the lifetime of Tg2576 mice (Kawarabayashi et al., 2001). Detergent-soluble Aβ is present throughout life, whereas Aβinsol is absent until ∼6 months.
One of the main challenges in cognitive studies of APP transgenic mice has been determining the onset and progression of memory deficits. These are important issues because identifying molecules causing memory loss depends on accurately determining when cognitive deficits first appear. In addition, when APP transgenic mice are used to evaluate potential therapies for AD, knowing how much memory loss has occurred at different stages of the disease is essential for setting up experimental designs. Two factors have made it difficult to establish definitive onsets of cognitive deficits. The first concerns the presence of age-independent behavioral deficits that might not be well distinguished from age-dependent cognitive deficits. The second concerns the subtlety of initial memory deficits, and whether the testing methods are sufficiently sensitive to detect small changes. Another significant problem in assessing memory in APP transgenic mice has been measuring the progression of memory decline across the entire life span of the mouse. This is critical for correlating memory loss with molecular markers appearing at different stages of disease. The main difficulty in this task is that the dynamic range for changes in molecular markers of AD is very large, whereas the dynamic range, or parametric space between the “ceiling” and “floor,” for many popular behavioral procedures may be quite small.
We identified age-independent behavioral deficits that were not specifically related to mutant APP expression in two lines of APP transgenic mice expressing comparable levels of wild-type APP and mutant APP. When these deficits were excluded, memory loss in Tg2576 coincided with the appearance of Aβinsol at ∼6 months. Accelerating Aβinsol production with mutant presenilin-1 (PS1) resulted in earlier loss of memory. These findings initially implicated Aβinsol in memory decline, but further analysis of the connection between Aβinsol and memory in older mice argued against a simple relationship. When memory and Aβinsolwere compared in young (5–6 months) and old (21–22 months) mice combined, no obvious correspondence was apparent, arguing against Aβinsol per se causing memory loss. However, when these mice were stratified by age, memory correlated inversely with Aβinsol, suggesting that Aβinsol is a surrogate marker for intermediate forms of Aβ that disrupt cognition and result in Aβinsol formation. Our findings challenge the classic amyloid cascade model but are in keeping with in vitro studies demonstrating neurotoxic properties of small Aβ assemblies (Roher et al., 1996; Lambert et al., 1998; Hartley et al., 1999; Wang et al., 1999) and are the first descriptive in vivo data supporting the hypothesis that these small Aβ assemblies cause learning and memory deficits.
MATERIALS AND METHODS
Mouse construction. The prion protein (PrP)-APP695 transgenes were generated by ligating Sall-flanked human APP695open reading frames into a hamster PrP cosmid vector (Scott et al., 1992), as described previously (Hsiao et al., 1995). Twenty-one Tg(Hacos.HuAPP695.WT) founders were generated in C57B6/SJL F3 mice. Tg5469 mice harbor ∼100 copies of Hacos.HuAPP695. WT, equivalent to that of HuAPP695.KM670/671NL (770-numbering) in Tg2576. Tg2576 and Tg5469 were all offspring of mice backcrossed successively to B6SJLF1 breeders, except for Tg2576 mice bred with transgenic mice expressing PS1 with the M146L mutation, Tg1 (Citron et al., 1997).
To generate mice bigenic for both PS1 and APP transgenes, we bred hemizygous Tg2576 mice congenic on 129S6 (129.Tg2576) with hemizygous Tg1 mice inbred in FVB (FVB.Tg1). All offspring were (FVB × 129)F1, which exhibits superior performance in the water maze (Crawley et al., 2000).
Analysis of transgene expression. Transgene products were examined in 3-month-old Tg5469 and 7-month-old Tg2576 mice. Whole brain homogenates (20% w/v) were prepared in TNE (50 mm Tris-Cl, pH 8.0, 150 mmNaCl, 5 mm EDTA with 1 mmphenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 1 mm benzamidine) buffer. An equal volume of TNE and detergents (1% IPEGAL, 1% deoxycholate, and 2% SDS) was added to the homogenate, sonicated, incubated on ice for 5–30 min, and boiled for 5 min. Homogenates were then centrifuged for 10 min at 14,000 rpm in a Beckman microcentrifuge. Protein content in the supernatant was measured (Bio-Rad Protein Assay). Loading dye (2×) with 10% β-mercaptoethanol was added to samples. After boiling for 5 min, 75 μg of homogenate was added per sample lane. Samples were fractionated using 7% SDS-PAGE. Proteins were electrophoretically transferred to Immobilon-P membranes (Millipore, Bedford, MA) and incubated with 22C11 (Chemicon, Temecula, CA) or 6E10 (Senetek) monoclonal APP antibodies. Blots were incubated with secondary rabbit anti-mouse antibodies (1:2500) (Sigma, St. Louis, MO) and visualized with 35S-protein A (Amersham Biosciences, Arlington, IL). Radioactivity was quantitated using a phosphorimager (Molecular Dynamics, Sunnyvale, CA).
Behavioral testing. We tested spatial reference learning and memory using a version of the conventional Morris water maze (Morris, 1984) in a cohort of 141 Tg+ Tg2576 mice and 149 Tg− littermates across their 2 year life span (see Tables 1, 2). Mice were selected from a closed colony created from successive matings with B6SJLF1 breeders. We also tested 60 Tg+ Tg5469 mice, overexpressing wild-type human APP695, and 62 Tg− littermates in the same strain background. Mice were grouped into four age ranges corresponding to four stages in Tg2576 plaque pathology and Aβinsol levels: (1) very young mice, 4–5 months, before the appearance of Aβinsol or plaques; (2) young mice, 6–11 months, during the initial appearance of Aβinsol and both amyloid plaques and punctate Aβ deposits; (3) middle-aged mice, 12–18 months, during the extensive deposition of plaques when Aβinsollevels are rising rapidly; and (4) old mice, 20–25 months, at a time when Aβ is leveling off and amyloid loads are comparable to those in Alzheimer's disease (see Tables 1, 2). Punctate Aβ deposits much smaller and sparser than mature plaques appear at 6–8 months, whereas amyloid plaques appear at 9–12 months and increase to numbers similar to those seen in Alzheimer's patients by 16–20 months (Irizarry et al., 1997; Kawarabayashi et al., 2001). Approximately equal numbers of male and female mice were tested. Mice homozygous for the retinal degeneration (rd) gene were not tested. All mice were naive and tested in a coded manner.
Table 1.
Profile of Tg2576 and Tg5469 mice tested in the Morris water maze
| Mice tested | Tg− mice | Tg+ mice | ||||
|---|---|---|---|---|---|---|
| Included | Excluded | Included | Excluded | |||
| Male | Female | n(%)1-a | Male | Female | n(%)1-a | |
| Tg5469 (wt) | ||||||
| 4–5 Months | 21 | 18 | 2 (5) | 15 | 18 | 3 (8) |
| 6–12 Months | 10 | 9 | 2 (10) | 13 | 7 | 4 (17) |
| Tg2576 (mutant) | ||||||
| Very young | 11 | 15 | 3 (10) | 9 | 11 | 5 (20) |
| Young | 15 | 19 | 4 (11) | 15 | 11 | 9 (26) |
| Middle-aged | 18 | 21 | 4 (9) | 21 | 16 | 7 (16) |
| Old | 22 | 13 | 4 (10) | 16 | 9 | 12 (32) |
Shown are ages, transgenotypes, genotypes, and performance competence of all mice tested. Tg2576 mice were grouped according to important stages in Aβ formation and plaque deposition: very young (4–5 months); young (6–11 months); middle-aged (12–18 months); and old (20–25 months).
Includes both genders. Number in parentheses represents percentage of all mice excluded in a given age group.
Table 2.
Profile of Tg2576 and Tg5469 mice tested in the Morris water maze
| Mice excluded | Tg5469 | Tg2576 | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4–5 Months | 6–11 Months | Very young | Young | Middle-aged | Old | |||||||||||||||||||
| Tg− | Tg+ | Tg− | Tg+ | Tg− | Tg+ | Tg− | Tg+ | Tg− | Tg+ | Tg− | Tg+ | |||||||||||||
| M | F | M | F | M | F | M | F | M | F | M | F | M | F | M | F | M | F | M | F | M | F | M | F | |
| Visible platform failure | 0 | 1 | 0 | 2 | 1 | 0 | 1 | 1 | 1 | 2 | 0 | 0 | 0 | 2 | 4 | 3 | 1 | 1 | 3 | 4 | 1 | 1 | 8 | 1 |
| Failure to orient to escape scoop | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 2 | 3 | 1 | 1 | 1 | 1 | 0 | 2 | 0 | 0 | 0 | 2 | 1 | 2 |
Performance-incompetent mice were excluded from the analysis of age-dependent changes in learning and memory. They were defined as outliers in the cued task or animals failing to orient to the escape scoop. Performance-incompetent mice exhibited one or more of the following traits: cork-screw circling, floating, gross motor abnormalities, poor attention, and apparent visual impairments. Thigmotaxis was not prevalent in either Tg+ or Tg− populations and was not a consistent feature of the performance-incompetent phenotype.
The water maze was tailored to Tg2576 mice in the B6/SJL strain background in a manner that enabled us to detect and distinguish all stages of memory loss. This protocol provided the sensitivity, specificity, and dynamic range needed to measure changes that are subtle early in life and gross late in life. Interpolation of probes during training provided sensitivity. Adoption of exclusion criteria for performance deficits gave specificity. Training extensively lent dynamic range. The assignment of mean probe scores (MPSs), which is the mean percentage time spent by a mouse in the target quadrant during the three probe trials, improved quantification of cognitive performance of individual mice for correlations with molecular markers and provided a single measure with a broad dynamic range. Protocols for Tg2576 mice in other strain backgrounds may need to be adjusted for strain-specific differences in rates of learning and performance deficits.
The water maze was a circular 1 or 1.2 m pool filled with water at 25–27°C and made opaque by the addition of nontoxic white paint. The pool was placed amid fixed spatial cues consisting of boldly patterned curtains and shelves containing distinct objects. Mice were placed in a beaker and gently lowered into the water facing the wall of the pool. Mice first underwent visible platform training for 3 consecutive days (eight trials per day), swimming to a raised platform (a square surface 12 × 12 cm2) marked with a black and white striped pole. Visible platform days were split into two training blocks of four trials for statistical analysis. During visible platform training, both the platform location (NE, SE, SW, or NW) and start position (N, NE, E, SE, S, SW, W, or NW, excluding the positions immediately adjacent to the platform) were varied pseudorandomly in each trial. Pseudorandomization ensured that all positions were sampled before a given position was repeated. Hidden-platform training was conducted over 9 consecutive days (four trials per day), wherein mice were allowed to search for a platform submerged 1.5 cm beneath the surface of the water. Mice failing to reach the platform within 60 sec were led to the platform with a metal escape scoop. During hidden-platform trials, the location of the platform remained constant (NE, SE, SW, or NW), and mice entered the pool in one of the seven pseudorandomly selected locations (N, NE, E, SE, S, SW, W, or NW, excluding the position immediately adjacent to the platform). After each hidden platform trial, mice remained on the platform for 30 sec and were removed from the platform and returned to their home cage with the escape scoop. Mice quickly learned to associate the scoop with escaping from the pool and consistently oriented to or followed the scoop on its appearance. The ability of mice to orient to or follow the escape scoop represented independent measures of vision and attention. At the beginning of the 4th, 7th, and 10th day of hidden platform training, a probe trial was conducted in which the platform was removed from the pool and mice were allowed to search for the platform for 60 sec. All trials were monitored by a camera mounted directly above the pool and were recorded and analyzed using a computerized tracking system (HVS image, Hampton UK). Further analysis was done using Wintrack (kindly provided by Dr. David Wolfer, University of Zurich, Switzerland).
The MPS was calculated for each mouse and used to assess retention of spatial information in the Morris water maze. By integrating information from the intercalated probes, the MPS represents a measurement of learning similar in concept to the previously described learning index (Gallagher et al., 1993), which samples memory at different stages of learning. Similar statistical results were found with MPS, the learning index and learning score (the weighted sum of percentage time spent in the target quadrant during probe trials), and we elected to represent our data using MPS because of ease of representation.
After testing, a subset of each group of mice was euthanized, and the right hemibrain was frozen in liquid nitrogen for Aβ measurements. All brains were analyzed in a coded manner.
Measurements of brain Aβ in Tg2576. Aβ was measured as described previously (Kawarabayashi et al., 2001). Frozen hemibrains were sequentially extracted in a two-step extraction [sonication in (1) 2% SDS and (2) 70% formic acid (FA)]. The latter fraction was designated Aβnsol. Measurements of Aβ in subregions of the brain, such as the hippocampus, produced unacceptably large variances. After sonication the samples were centrifuged at 100,000 × g for 1 hr at 4°C, the supernatant was recovered, and the pellet was sonicated with the next solution. Brain extracts were measured by sandwich ELISA as described previously (Suzuki et al., 1994; Gravina et al., 1995). The following systems were used: (1) BAN-50 capture and BA-27 or BC-05 detection or (2) 3160 capture and BA-27 or BC-05 detection, both of which detect Aβ1–40 and Aβ1–42, respectively. The direct comparison of many Tg2576 brains from mice of all ages showed that the amounts of Aβ40 and Aβ42 detected with 3160 capture ELISAs were essentially the same as when BAN-50 was used for capture. The 2% SDS extracts were diluted at least 1:40 so that the assay could be performed in 0.05% SDS. Greater dilutions were corrected for SDS so that they were also assayed in 0.05% SDS. The FA extract was neutralized by a 1:20 dilution into 1m Tris phosphate buffer, pH 8.0. The program Softmax was used to calculate femtomoles per milliliter by comparing the sample absorbance to the absorbance of known concentrations of synthetic Aβ1–40 and Aβ1–42 in identical solution as the samples, and these values were corrected with the wet weight of the original homogenate to be finally expressed as picomoles per gram wet weight. In all instances, nontransgenic tissues were processed identically in parallel with transgenic tissues.
RESULTS
Both wild-type and mutant human APP695 accentuate age-independent performance deficits in the water maze
Tg+ and Tg− littermates from Tg2576 and Tg5469 lines in a hybrid B6/SJL strain background at ages ranging from 4 to 25 months were tested in the Morris water maze (Tables1, 2). We identified a set of mice with performance problems or sensorimotor abnormalities common to both Tg2576 and Tg5469 lines that were designated performance-incompetent mice. The assignment of performance incompetence was done after the conclusion of testing by experimenters who were blind to transgene status. Performance-incompetent mice were identified by two criteria: (1) outliers in the last block of visible platform training, with escape latencies >2 SDs above the mean latency of Tg− mice; and (2) mice failing to orient to or follow the escape scoop. Animals with performance incompetence comprised 23% of Tg+ Tg2576 mice, 10% of Tg− Tg2576 littermates, 12% of Tg+ Tg5469 mice, and 6% of Tg− Tg5469 littermates. More Tg+ than Tg− mice were performance incompetent at every age tested in both Tg2576 and Tg5469 mice (χ2 = 9.96;p < 0.01), but the rates at which performance incompetence occurred in Tg2576 and Tg5469 mice did not differ significantly (χ2 = 3.6;p > 0.05). There was also no consistent increase with age in the proportion of Tg+ relative to Tg− mice with performance incompetence, suggesting that these behavioral deficits are age independent.
Interestingly, we found no such deficits in 129S6 mice congenic for the Tg2576 transgene (129.Tg2576), leading us to conclude that performance deficits accentuated by APP695 overexpression may be strain specific. We concluded that overexpression of human APP695 accentuates performance deficits present in native B6/SJL mice.
Spatial reference learning and memory is normal until 6 months of age
The application of our exclusion criteria resulted in the generation of a cohort of mice in which sensorimotor performance deficits could be factored out of the interpretation of behavioral data, a conclusion supported by two observations: we found no significant differences in escape latencies in the last two blocks of training to the visible platform or in swim speeds during the third probe trial between Tg+ and Tg− mice at any age (data not shown). The impact of the performance-incompetent mice on the overall analysis was such that when these mice were not excluded, a trend showing Tg+ mice performing worse than Tg− mice appeared at all ages, because performance-incompetent mice were approximately twice as prevalent in Tg+ as in Tg− mice regardless of age. Therefore, all conclusions regarding cognitive function in Tg2576 were drawn on the basis of data gathered from the group of mice remaining after performance-incompetent mice were excluded (Tables 1, 2).
Spatial memory was assessed in probe trials three times during training, and the MPS was calculated for each mouse. No transgene effects were observed for MPSs (Fig.1a), escape latencies during hidden platform training (Fig. 2), or search biases in probe trials (Fig. 3) in mice <6 months old. We concluded that learning and memory in Tg2576 is normal until 6 months of age.
Fig. 1.

Age-dependent impairment in spatial reference memory occurs only in transgenic mice expressing mutant APP.a, The mean probe score (MPS), the mean percentage time spent in the target quadrant during all three probes, was used to assess retention of spatial information in the Morris water maze. Random swimming during all three probes would yield an MPS of 25%. There was a significant age-by-transgene interaction in Tg2576 mice (ANOVA; p < 0.05), with significantly lower scores in young (6–11 months), middle-aged (12–18 months), and old (20–25 months) Tg+ mice relative to Tg− mice (t test; *p < 0.05, **p < 0.01). MPSs of 6- to 12-month-old Tg5469 Tg+ mice were significantly higher than those of Tg− littermates (t test; **p < 0.01) and age-matched Tg+ Tg2576 mice (t test; ***p < 0.001), excluding APP overexpression as a cause of memory loss in Tg2576 mice. No significant decrease in MPS was observed in very young Tg2576 Tg+ mice devoid of Aβinsol. A significant drop in performance occurred in young Tg2576 Tg+ mice (t test; *p < 0.05), coincident with the appearance of Aβinsol. b, The learning curves of Tg+ and Tg− mice at different ages are represented, showing memory (percentage time in the target quadrant) assessed during the three probe trials,P1, P2, and P3, performed after the 12th, 24th, and 36th training trials. As expected, memory improves with training and appears to saturate. With age, there is a rightward shift of the curves. In Tg+ mice there is an apparent lowering of the saturation level of memory.
Fig. 2.

Acquisition of visible and hidden platform locations in Tg5469 and Tg2576 mice. a, Escape latencies of Tg5469 and Tg2576 mice in the visible platform version of the Morris water maze (2 blocks of 4 trials each day). No significant effect of transgene was found in the last two training blocks for either line of mouse at any age, mitigating against sensorimotor deficits as a potential explanation for impaired performance in acquisition or retention of the location of the hidden platform. Although middle-aged and old Tg2576 Tg+ mice showed an initial lag in performance, their performance was not significantly different from Tg− mice on the final day. b, Escape latencies of Tg5469 and Tg2576 mice swimming to the hidden platform in the Morris water maze. A significant age-by-transgene interaction for mean escape times during the last three training blocks was seen in Tg2576 mice (ANOVA;p < 0.005). Post hoc analysis showed significant effects of transgene in middle-aged and old Tg2576 mice (t test; **p < 0.0001).
Fig. 3.
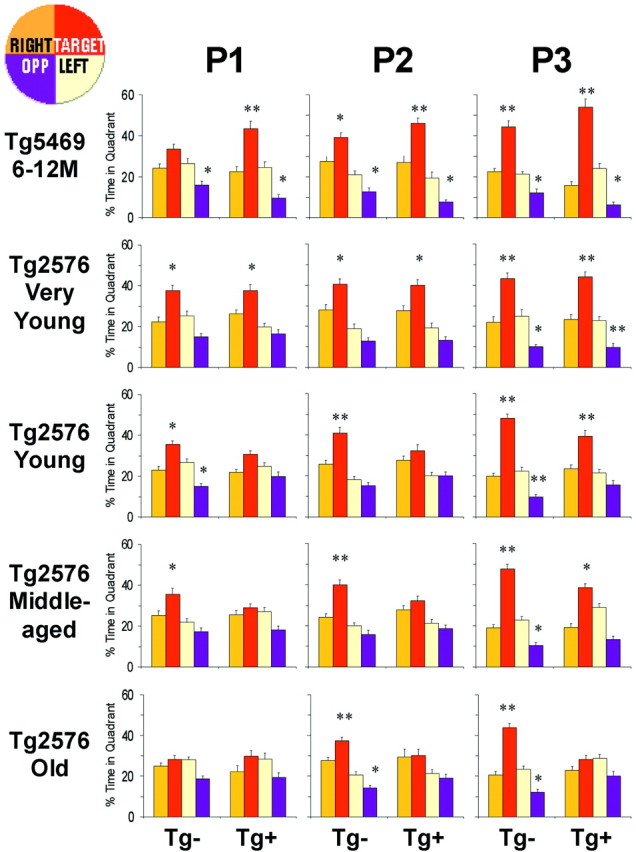
Retention of spatial reference information in Tg5469 and Tg2576 mice. Percentage time spent in each of four quadrants (inset diagram at top left) during three probe trials run at the beginning of the 4th, 7th, and 10th days of training in the Morris water maze in Tg5469 and Tg2576 mice. Tg5469 (6–12 months) and very young (4–5 months) Tg2576 mice spent a significantly greater proportion of time in the target quadrant in the first probe, regardless of transgene status. Search strategies of young (6–11 months) and middle-aged (12–18 months) Tg− mice were biased toward the target quadrant in the first probe, but those of Tg+ showed no significant bias until the third probe. Search strategies of old (20–25 months) Tg− mice were biased toward the target quadrant in the second probe, but Old Tg+ mice failed to exhibit a search bias in any probe. Differences between percentage time spent in the target and opposite quadrants compared with all other quadrants was determined using an ANOVA with Fischer's PLSD post hoc analysis (*p < 0.01; **p < 0.0001).P1, P2, and P3 are probe 1, probe 2, and probe 3, respectively.
Elevation of detergent-soluble Aβ in mice before 6 months is insufficient to disrupt spatial reference learning or memory
The Swedish mutation elevates Aβ levels substantially in Tg2576 mice as young as 2–3 months (Hsiao et al., 1996). With the exception of a small minority (10–20%) of 5 month-old mice in which Aβinsol is present, Aβ in all animals <6 months of age is soluble in aqueous or detergent buffers. Mere elevation of soluble Aβ in mice <6 months old does not disrupt learning and memory.
Spatial reference memory deteriorates progressively after 6 months of age
We observed a significant age-by-transgene interaction for MPSs (two-way ANOVA; p < 0.05) (Fig. 1) and also for three other measures of spatial information retention including percentage time in the target quadrant and proximity index during the third probe, as well as the learning index (data not shown). Post hocanalyses revealed no significant differences between MPSs of Tg+ and Tg− mice in very young (4–5 months) mice, but MPSs of young (6–11 months) Tg+ mice were significantly lower than those of Tg− mice and continued to decrease with increasing age. Very young Tg+ mice retained spatial reference information as well as Tg− littermates. Young and middle-aged (12–18 months) Tg+ mice were able to retain spatial reference information but required more reinforcement than Tg− littermates. Old (20–25 months) Tg+ mice, with some rare individual exceptions, were significantly impaired compared with younger Tg+ mice, showing no ability to acquire or retain any spatial information (Figs.2, 3). Examination of escape latencies revealed a similar age-related deterioration for spatial acquisition (Fig. 2). However, significant transgene effects were not observed until mice were middle-aged. No significant effects of gender were found for any cognitive measure.
MPSs of old Tg− mice were significantly lower than those of younger (4–18 months) Tg− mice (p < 0.05;t test), consistent with previous reports describing age-related deterioration in spatial memory in mice (Fordyce and Wehner, 1993; Bellush et al., 1996; Bach et al., 1999). However, compared with Tg+ mice, the decline in Tg− mice was smaller and did not occur until mice were much older.
The learning curves of Tg+ and Tg− mice at different ages are graphically represented in Figure 1b. These curves demonstrate that, with age, there is a rightward shift in the learning curves. In Tg+ mice, there also appears to be a lowering of the saturation level of learning.
Spatial memory deficits are not caused by APP overexpression
MPSs of Tg+ Tg5469 mice at 6–12 months were significantly higher than those of Tg− littermates or age-matched Tg2576 mice (Fig. 1), ruling out the possibility that deterioration of learning and memory in Tg2576 at 6–11 months was related to overexpression of human APP695. The enhancement of memory in Tg5469 may be related to overproduction of secreted APPa, which has been shown to improve memory when administered to rats (Roch et al., 1994; Meziane et al., 1998). This enhancement is unlikely to be caused by a nonspecific transgene insertional mutation, because it is associated with larger hippocampal long-term potentiation that can be blocked with antibodies to APPa (J. Steidl, L. Boland, and K. Ashe, unpublished observations). The facilitating effect of wild-type human APP695 is of less importance in Tg2576, in which cleavage at the β-secretase site is favored because of enhanced β-secretase activity in APP with the Swedish mutation (Sinha et al., 1999; Vassar et al., 1999). Aβ is produced five times more efficiently in Tg2576 than in Tg5469 mice (data not shown), consistent with previous experiments in vitroexamining the effect of the Swedish mutation on Aβ production (Cai et al., 1993; Citron et al., 1997). These results strongly implicate Aβ in the memory loss observed in 6- to 11-month-old Tg2576 mice.
Spatial memory loss coincides with the appearance of Aβinsol
Aβinsol measures aggregated Aβ and is a more sensitive measure of amyloid deposition than histopathological detection methods (Kawarabayashi et al., 2001). Brain detergent-soluble Aβ and Aβinsol levels were measured by ELISA in a subset of Tg2576 Tg+ mice after training in the Morris water maze (4–5 months, n = 13; 6 months, n = 15; 10 months, n = 11; >20 months, n = 12). Detergent-soluble Aβ was present at all ages. Aβinsol was present at the threshold of detection (>5 pmol/gm) in <10% of 4–5 month, 70% of 6 month, 100% of 10 month, and 100% of >20 month mice. To assess the effects of Aβinsol on learning and memory, we compared MPSs in a group of 4–6 month mice at an age before the appearance of well formed amyloid plaques, but during which time a subset have formed Aβinsol. MPSs assessed in 28 Tg+ mice, in which Aβinsol was above the threshold for detection (>5 pmol/gm) in 12 mice and not detectable (<2 pmol/gm) in 16 mice, were significantly lower in mice with Aβinsol(Fig. 4). We concluded that in Tg2576 the appearance of Aβinsol is associated with impaired memory. Supporting this view is the finding that 4- to 5-month-old animals devoid of Aβinsol showed no significant impairment.
Fig. 4.

Aβinsol in Tg2576 mice is associated with impaired water maze performance. Brain Aβinsol40 and Aβinsol42 levels were determined by ELISA in a group of 4- to 6-month-old Tg2576 Tg+ mice (n = 28). When mice were segregated according to the presence (>5 pmol/gm of either Aβinsol40 or Aβinsol42) (n = 12) or absence of Aβinsol(n = 16), MPSs were significantly lower in mice with Aβinsol (t test; *p < 0.05). When mice were segregated by age (≤5 months, n = 13; or 6 months, n= 15), no significant differences were observed (data not shown).
Mutant PS1 accelerates the conversion of detergent-soluble to insoluble Aβ in Tg2576 and produces an earlier onset of spatial memory deficits
Mutant PS1 increases the ratio of the more highly fibrillogenic Aβ42 to Aβ40 (Duff et al., 1996) and accelerates the appearance of amyloid plaques in mice bigenic for the Tg2576 transgene and mutant PS1 (Holcomb et al., 1998). In bigenic (PS1/APP) mice, amyloid deposits appear as early as 3 months and are abundant by 6–9 months. In contrast, mice with the Tg2576 transgene (APP) alone have few or no deposits at 6–9 months. Despite the dramatic difference in amyloid deposition between PS1/APP and APP mice at 6–9 months, no difference in acquisition or retention of spatial reference memory in the water maze between these two genotypes of mice has been detected in mice at these ages (Holcomb et al., 1999).
We speculated that the reason the effects of mutant PS1 on cognitive function have not yet been identified, despite its dramatic influence on Aβ metabolism, is because impaired memory is not associated with amyloid load but is related to the conversion of detergent-soluble to insoluble Aβ. We predicted that PS1/APP mice should exhibit an earlier onset of deficits than APP mice, because mutant PS1 would accelerate the formation of Aβinsol. We measured Aβinsol at 2 months in PS1 transgene-positive and -negative mice generated from crosses of 129.Tg2576 with FVB.Tg1 mice (Table 3). As predicted, mutant PS1 accelerated the conversion of SDS-soluble to insoluble Aβ, because Aβinsol was present above the threshold for detection (>5 pmol/gm) only in PS1/APP mice. Next we tested mice in the water maze at 4–5 months, when Aβinsol was present in all PS1/APP mice but in only ∼20% of APP mice (data not shown), and found significantly lower probe 1 scores in bigenic mice compared with all other genotypes (Fig. 5).
Table 3.
Expression of M146L mutant presenilin-1 transgenes accelerates formation of Aβinsol in (FVB × 129.Tg2576)F1 APP transgene-positive mice
| PS1(M146L) transgene3-a | Aβ40 (pmol/gm) | Aβ42(43) (pmol/gm) | ||
|---|---|---|---|---|
| Aβsol | Aβinsol | Aβsol | Aβinsol | |
| Positive | 59.7 | 15.7 | 9.9 | 47.1 |
| Positive | 57.3 | 15.7 | 9.5 | 61.5 |
| Positive | 68.4 | 171.4 | 9.4 | 200.8 |
| Negative | 41.8 | 1.1 | 3.5 | 0.7 |
| Negative | 41.6 | 1.4 | 3.6 | 0.5 |
| Negative | 50.2 | 1.3 | 4.0 | 0.5 |
Mice were 50–62 d old. APP transgene-negative mice are not comparable because they express only mouse APP, but values for APP transgene-negative mice in the same assay were Aβsol40 = 2.5 ± 0.5, Aβinsol40 = 0.3 ± 0.5, Aβsol42 = 0.37 ± 0.15, and Aβinsol42 = 0.12 ± 0.25. Aβsolis a SDS-soluble Aβ peptide; Aβinsol is a formic acid releasable Aβ peptide, after SDS extraction.
All mice were positive for the Tg2576 APP transgene array.
Fig. 5.

Mutant presenilin-1 accelerates memory loss in Tg2576 mice. At 4–5 months, Aβinsol was present in all PS1/APP mice but in only ∼20% of APP mice. PS1/APP mice (n = 21) showed significantly worse performance in probe 1 than APP (n = 11), PS1 (n = 14), or nontransgenic (n = 13) mice (*p < 0.05; **p < 0.01; ***p < 0.001).
Probe 1 scores were more sensitive than any other measure, including MPSs, in this experiment because (FVB × 129) F1 mice reached saturated performance early in this water maze protocol because of their superior learning ability relative to B6/SJL mice (Crawley et al., 2000), resulting in a “leftward” shift of their learning curves. MPSs generated for (FVB × 129) F1 mice showed the same trend but did not reach significance because probe 2 and probe 3 scores reached a plateau near the ceiling, obscuring the effects of probe 1 scores. This finding is consistent with our observations that earlier interpolated probes are more sensitive to subtle changes in learning and memory than MPSs, but MPSs represent a broader dynamic range than individual probe scores. Accuracy and dynamic range obtained through multiple measurements may be gained at the expense of sensitivity, and vice versa. This notwithstanding, it is likely that a compressed training protocol tailored to the superior learning ability of (FVB × 129) F1 mice (with probe scores interpolated along the ascending portion of the learning curve) would yield a more sensitive MPS. We concluded that mutant PS1 accelerates the onset of spatial memory deficits.
Spatial memory correlates inversely with Aβinsol in Tg2576 mice stratified by age
Our findings that the onset of memory loss coincided with the appearance of Aβinsol and that genetic acceleration of the formation of Aβinsol with PS1 resulted in an earlier onset of memory decline led us initially to surmise that Aβinsol directly or indirectly interfered with neural processes underlying spatial reference learning and memory. If this were true, then memory and Aβinsol would be inversely related. We therefore compared Aβinsol and MPSs in a pooled sample of 5- to 6-month-old, 10-month-old, and 21- to 22-month-old mice. Unexpectedly, we found no obvious relationship (Fig.6e). Total Aβinsol in the younger mice ranged from ∼1 to 20 pmol/gm, whereas Aβinsol in older mice was several orders of magnitude higher, ranging from ∼10,000 to 50,000 pmol/gm. Yet a subset of the older mice performed as well or better than average younger mice. On the basis of these observations, we rejected the idea that Aβinsol impaired cognition in Tg2576 mice and sought an alternative interpretation.
Fig. 6.

Water maze performance correlates inversely with Aβinsol in young and old Tg2576 mice stratified by age. Brain Aβinsol40 and Aβinsol42 were determined by ELISA in 12 Tg2576 Tg+ mice at 5–6 months (a, b) and 12 Tg2576 Tg+ mice at 21–22 months (c, d). Significant correlations between MPSs and Aβinsol40 or Aβinsol42 were found in Tg2576 mice stratified by age, but not when the old and young mice were pooled together (e). Total Aβinsol ine is the sum of Aβinsol40 and Aβinsol42.
Remarkably, when the pooled mice were stratified by age (5- to 6-month-old and 21- to 22-month-old groups; there were too few mice in the 10-month-old group to obtain meaningful analyses), MPSs correlated inversely with Aβinsol in both age groups (Fig.6a–d). These results suggest a relationship between memory and Aβ that involves pathogenic Aβ assemblies formed when Aβ aggregates to form Aβinsol.
DISCUSSION
Since the creation of the first transgenic mice modeling AD over a decade ago (Quon et al., 1991), studies of these and subsequently generated mice have provided important information about the pathogenesis of AD histopathology. However, the pathogenesis of memory loss in these mice has been difficult to study. The major impediments to elucidating the relationship between cognitive function and molecular markers in APP transgenic mice have been the difficulty of distinguishing between age-dependent and age-independent changes in behavior and cognition and the inability to overcome several methodological problems inherent in testing mice with dynamic changes in cognition. We report here how we overcame these difficulties and the results of our investigations concerning the relationship between memory and Aβ.
Measuring memory in Tg2576 and other APP transgenic mice
Various reports about the onset of cognitive deficits in Tg2576 have indicated abnormalities appearing as early as 3 months, as late as 15 months, and at intermediate ages (Hsiao et al., 1996; Chapman et al., 1999; King et al., 1999; Pompl et al., 1999; Morgan et al., 2000). Although the involvement of Aβ in cognitive decline in Tg2576 and other APP transgenic mice has not been in dispute, disparate reports regarding the onset of cognitive impairment in Tg2576 mice have led researchers to various conclusions pertaining to the form of Aβ that is responsible. In one report, deficits were observed at 3 months, implicating soluble Aβ (King et al., 1999), whereas other studies have shown onsets at 9–11 or 15 months, invoking insoluble Aβ (Hsiao et al., 1996; Morgan et al., 2000). There are a number of possible explanations for these discrepancies that this study was designed to address.
We studied in parallel two lines of transgenic mice expressing similar levels of wild-type and mutant APP, which helped us identify and segregate behavioral effects that were not specifically related to mutant APP expression. We also tailored the Morris water maze to provide the sensitivity and dynamic range necessary to measure spatial memory during the life span of Tg2576 mice. Our protocol detected the onset of spatial memory deficits that map well onto post-translational modifications of Aβ. The earliest memory loss coincided with the appearance of Aβinsol at ∼6 months. Genetically accelerating the appearance of Aβinsol using mutant PS1 resulted in an earlier onset of memory decline. Hence, the onset of memory loss was associated with the appearance of Aβinsol in two different types of mice in which Aβinsol formed at distinct ages. This observation is supported by studies in TgCRND8 mice showing a rapid rise in Aβ at 10 weeks, probably indicative of the appearance of Aβinsol, before the demonstration of spatial memory deficits at 11 weeks (Chishti et al., 2001).
Our results contrast with other studies in Tg2576 and PDAPP mice that have not shown a close connection between the onset of memory deficits and the formation of Aβinsol. In PDAPP mice, Aβ rises rapidly between 4 and 8 months, probably signifying the appearance of Aβinsol (Games et al., 1995;Johnson-Wood et al., 1997), but no age-dependent deficits were detected at 6–9 months using a working memory version of the Morris water maze (Chen et al., 2000). Age-independent deficits, however, were already present (Chen et al., 2000). Age-dependent deficits in PDAPP mice were not detected until 13–15 months (Chen et al., 2000). It is possible that the age-dependent deficits present in younger PDAPP mice were obscured by age-independent deficits, or that the dynamic range of the test used to measure memory in these mice was more sensitive to larger changes present in older mice. In Tg2576 mice, deficits could not be detected at 11 months, 5 months after the appearance of Aβinsol, using the radial arm water maze to test spatial working memory (Morgan et al., 2000). One potential explanation could be that working memory is preserved longer than reference memory in Tg2576 mice. However, studies showing abnormal forced alternation in the T-maze as early as 7–8 months argue against this notion (P. Chapman, unpublished data). It is more likely that the testing parameters in the radial arm water maze task were optimized for detecting large deficits occurring in older mice and were less sensitive to smaller deficits emerging at younger ages.
Our conclusions relating the onset of memory loss to the appearance of Aβinsol depend on the validity of assigning the onset of decline in spatial reference memory to ∼6 months, a time point that differs from all other published results (Hsiao et al., 1996; Holcomb et al., 1999; King et al., 1999; Morgan et al., 2000). The discrepancies in the onset of memory loss in Tg2576 can be explained by methodological shortcomings, which we believe were resolved in the current study. The initial report indicated no significant differences between Tg+ and Tg− mice in the conventional Morris water maze at 3 or 6 months but a significant difference at 9–11 months (Hsiao et al., 1996). In this study, no systematic screen for performance-incompetent mice was used, which may explain why Tg+ mice lagged behind Tg− mice in the 9–11 month age group during two of the four training trials to the visible platform. Two possible explanations for the lack of a transgene effect at 6 months include the increased variance introduced by the inclusion of performance-incompetent mice that may have obscured potential differences and the possibility that the number of 6-month-old mice with Aβinsol, which was not measured in the initial study, was underrepresented. In another study, in which age-dependent cognitive deficits were not easily distinguished from age-independent performance deficits and in which exclusion criteria were not reported, a water maze retention deficit and abnormal performance in the Barnes maze were found in 3-month-old female Tg2576 mice (King et al., 1999). Two additional studies failed to detect spatial memory loss in 9-month-old Tg2576 mice (Holcomb et al., 1999;King et al., 1999). In both studies, single probes were performed after extensive training, equivalent to or more than that received by mice in our protocol during the third probe. We also found similar retention of spatial information in Tg+ and Tg− mice when we compared probe 3 scores in mice <20 months.
Tracing the molecular basis of memory deficits in Tg2576 mice
Our investigations have helped us understand better the molecular basis of memory decline in Tg2576 mice. King and colleagues (1999)proposed that soluble Aβ caused memory loss occurring at 3 months. We showed that before 6 months, behavioral abnormalities were age independent and related to APP overexpression, and soluble Aβ had no deleterious effect on memory. We next considered the possibility that memory decline is caused by Aβinsol, which is a biochemical measure of aggregated Aβ that is related to amyloid load (Kawarabayashi et al., 2001). Inverse correlations between memory and amyloid load have been shown in both PDAPP mice (Chen et al., 2000) and PS1/APP mice (Gordon et al., 2001), suggesting a relationship between memory loss and plaque deposition. In these studies the age ranges of mice analyzed were 6 and 2 months, respectively.
In contrast, when we studied mice across a broader age range, 17 months, we showed no correlation between memory and Aβinsol and concluded that the major cause of memory loss in Tg2576 is not Aβinsol. However, when mice were stratified by age, inverse correlations between memory and Aβinsol appeared similar to those in PDAPP and PS1/APP mice (Chen et al., 2000; Gordon et al., 2001). Importantly, Aβinsol in some cognitively intact old mice was 100–1000 times higher than in impaired young mice. We considered whether this effect was caused by individual variations in susceptibility of old mice to Aβinsol, but reasoned that if this were the case, then the relationship between memory and Aβinsol in old mice would have been obliterated. Similarly, if there were a floor effect inherent in the water maze test that could prevent the detection of a decrease in performance proportional to the increase in Aβinsol in old mice, this would not explain how the inverse relationship between memory and Aβinsol was preserved.
To explain our findings, we propose that both amyloid load and Aβinsol may be surrogate measures for one or more small Aβ assemblies that disrupt learning and memory (Fig.7). An extension of this hypothesis is that these Aβ assemblies may also be involved in the conversion of detergent-soluble to insoluble Aβ. Although it is possible that these Aβ assemblies comprise a subset of the insoluble Aβ species, it would be difficult to explain how some old mice with very high levels of Aβinsol could have relatively normal cognitive function if this were the case. Therefore, we believe that these Aβ assemblies reside in the soluble Aβ fraction.
Fig. 7.

Proposed model of small Aβ assemblies disrupting learning and memory. a, Tg2576 mice <6 months have soluble Aβ (circles) but no memory loss. This indicates that the soluble Aβ species present in mice <6 months do not disrupt cognitive function. Mice >6 months show memory loss coinciding with the appearance of detergent-insoluble Aβ aggregates (large starbursts), also referred to as Aβinsol. However, when memory and Aβinsolwere studied in mice across a broad age range, no correlation was found, arguing against Aβinsol being the major cause of memory loss in Tg2576 mice. Yet inverse correlations between memory and Aβinsol became apparent when mice were stratified by age. To explain these findings, we propose that amyloid load and Aβinsol are both surrogate measures for one or more small Aβ assemblies (stars) that disrupt learning and memory. An extension of this hypothesis is that these Aβ assemblies may also be involved in the conversion of soluble to insoluble Aβ. Although it is possible that these Aβ assemblies comprise a subset of the insoluble Aβ species, the difficulty of reconciling in some old mice very high levels of Aβinsol with relatively normal cognitive function mitigates against this possibility. It is more likely that the Aβ assemblies reside among the soluble Aβ species.b, The hypothetical cascade involving small Aβ assemblies contrasts with the classic amyloid cascade hypothesis, in which cognitive dysfunction and dementia are caused by the accumulation of insoluble Aβ aggregates resulting in neuronal destruction. In the alternate cascade involving Aβ assemblies, memory loss and dementia are caused by a subset of soluble Aβ species that are present in the subpopulation of mice that possess Aβinsol, the presence of which reflects the Aβ assemblies.
Relevance to human AD studies
This idea might also explain a puzzling inconsistency in the relationship between amyloid load and memory in AD. Several early reports showed little or no correlation between amyloid load and dementia (Terry et al., 1991; Arriagada et al., 1992; Berg et al., 1993). More recently, when more sensitive antibody-based methods were used to measure Aβ deposits and subject pools that represented a broad range of cognitive impairment were examined, highly significant and robust correlations were found (Cummings et al., 1996; Bartoo et al., 1997; Naslund et al., 2000). Despite these methodological improvements, however, it has remained difficult to explain why some individuals with high plaque loads are cognitively normal (Katzman et al., 1988; Delaere et al., 1990; Dickson et al., 1992). We observed a similar phenomenon in a subset of old Tg2576 mice. Some possibilities are that these individuals have greater cognitive reserve or are less susceptible to amyloid plaques. The results of our investigations in Tg2576 mice suggest another possibility. If cognitive decline is caused by small Aβ assemblies formed during the conversion of detergent-soluble to insoluble Aβ, then certain individuals with low levels of these Aβ assemblies could be cognitively intact but would nevertheless accumulate large amounts of deposits or Aβinsol over time.
Although the intermediate form or forms of Aβ disrupting memory have not yet been isolated from brain tissue, they may be related to one or more Aβ species that have recently been synthesized and studiedin vitro. The idea that small Aβ oligomers or protofibrils cause neural toxicity or dysfunction and are linked to the conversion of detergent-soluble to insoluble Aβ has recently gained considerable attention (Roher et al., 1996; Lambert et al., 1998; Hartley et al., 1999; Hsia et al., 1999; Wang et al., 1999; Mucke et al., 2000). Our studies in Tg2576 mice support this concept and provide a potential methodological framework within which to study directly the effects of these small Aβ assemblies on learning and memory. It is important to note that Tg2576 mice, lacking neurofibrillary tangles and significant neuronal loss, are a partial model of AD. Although small Aβ assemblies appear to mediate memory loss in Tg2576 mice in the absence of neuronal destruction, their relative contribution to AD dementia is unknown. The development of treatments for AD may nevertheless benefit from understanding these Aβ assemblies that impair learning and memory.
Footnotes
This work was supported by National Institutes of Health (Grants AG15453 to K.H.A., G.A.C., and S.Y., NS33249 to K.H.A., and MH11834 to M.W.). We thank Stanley B. Prusiner and Mike Scott for the hamster PrP cosmid vector, Michela Gallagher and Paul Chapman for insightful comments, Robert Ehlenfeldt for generating transgenic mice, Teresa Gómez-Isla for valuable help and advice, Liza Moscovice, Stefanie Schrump, Sherry Turner, and Rosa Johannsdottir for technical assistance, Dorothy Aeppli for help with statistical analyses, Hiromi Maeta for animal care, Rob Wolfe from Accutech Plastics for the pool, and Catherine Kraft for secretarial help.
Correspondence should be addressed to Karen H. Ashe, Department of Neurology, Mayo Mail Code 295, 420 Delaware Street Southeast, Minneapolis, MN 55455. E-mail: hsiao005@umn.edu.
REFERENCES
- 1.Arriagada PV, Growdon JH, Hedley-White ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 2.Bach ME, Barad M, Son H, Zhuo M, Lu YF, Shih R, Mansuy I, Hawkins RD, Kandel ER. Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc Natl Acad Sci USA. 1999;96:5280–5285. doi: 10.1073/pnas.96.9.5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartoo GT, Nochlin D, Chang D, Kim Y, Sumi SM. The mean Aβ load in the hippocampus correlates with duration and severity of dementia in subgroups of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:531–540. doi: 10.1097/00005072-199705000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Bellush LL, Wright AM, Walker JP, Kopchick J, Colvin RA. Caloric restriction and spatial learning in old mice. Physiol Behav. 1996;60:541–547. doi: 10.1016/s0031-9384(96)80029-1. [DOI] [PubMed] [Google Scholar]
- 5.Berg L, McKeel DW, Jr, Miller JP, Baty J, Morris JC. Neuropathological indexes of Alzheimer's disease in demented and nondemented persons aged 80 years and older. Arch Neurol. 1993;50:349–358. doi: 10.1001/archneur.1993.00540040011008. [DOI] [PubMed] [Google Scholar]
- 6.Cai XD, Golde TE, Younkin SG. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- 7.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 8.Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RG. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer's disease. Nature. 2000;408:975–979. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- 9.Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zucker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morrisette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of APP695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- 10.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St. George Hyslop P, Selkoe DJ. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 11.Crawley JN. What's wrong with my mouse? Wiley; New York: 2000. [Google Scholar]
- 12.Cummings BJ, Pike CJ, Shankle R, Cotman CW. Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer's disease. Neurobiol Aging. 1996;17:921–933. doi: 10.1016/s0197-4580(96)00170-4. [DOI] [PubMed] [Google Scholar]
- 13.Delaere P, Duyckaerts C, Masters C, Beyreuther K, Piette F, Hauw JJ. Large amounts of neocortical beta A4 deposits without neuritic plaques nor tangles in a psychometrically assessed, non-demented person. Neurosci Lett. 1990;116:87–93. doi: 10.1016/0304-3940(90)90391-l. [DOI] [PubMed] [Google Scholar]
- 14.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13:179–189. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- 15.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 16.Fordyce DE, Wehner JM. Effects of aging on spatial learning and hippocampal protein kinase C in mice. Neurobiol Aging. 1993;14:309–317. doi: 10.1016/0197-4580(93)90116-s. [DOI] [PubMed] [Google Scholar]
- 17.Gallagher M, Burwell R, Burchinal M. Severity of spatial learning impairment in aging: development of a learning index for performance in the Morris water maze. Behav Neurosci. 1993;107:618–626. doi: 10.1037//0735-7044.107.4.618. [DOI] [PubMed] [Google Scholar]
- 18.Games D, Adams D, Alessandrini R, Barbour R, Berthelotte P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagoplan S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburh I, Little S, Masliah E, McConlogue L. Alzheimer-type neuropathology in transgenic mice overexpressing V717F b-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 19.Golde TE, Eckman CB, Younkin SG. Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer's disease. Biochim Biophys Acta. 2000;1502:172–187. doi: 10.1016/s0925-4439(00)00043-0. [DOI] [PubMed] [Google Scholar]
- 20.Gordon MN, King DL, Diamond DM, Jantzen PT, Boyett KV, Hope CE, Hatcher JM, DiCarlo G, Gottschall WP, Morgan D, Arendash GW. Correlation between cognitive deficits and Abeta deposits in transgenic APP+PS1 mice. Neurobiol Aging. 2001;22:377–385. doi: 10.1016/s0197-4580(00)00249-9. [DOI] [PubMed] [Google Scholar]
- 21.Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Younkin LH, Suzuki N, Younkin SG. Amyloid beta protein (A beta) in Alzheimer's disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). J Biol Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 22.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 23.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 25.Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29:177–185. doi: 10.1023/a:1021691918517. [DOI] [PubMed] [Google Scholar]
- 26.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 28.Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, Iadecola C, Clark HB, Carlson G. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- 29.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katzman R, Terry R, DeTeresa R, Brown T, Davies P, Fuld P, Renbing S, Peck A. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol. 1988;23:138–144. doi: 10.1002/ana.410230206. [DOI] [PubMed] [Google Scholar]
- 32.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King DL, Arendash GW, Crawford F, Sterk T, Menendez J, Mullan MJ. Progressive and gender-dependent cognitive impairment in the APP(SW) transgenic mouse model for Alzheimer's disease. Behav Brain Res. 1999;103:145–162. doi: 10.1016/s0166-4328(99)00037-6. [DOI] [PubMed] [Google Scholar]
- 34.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meziane H, Dodart JC, Mathis C, Little S, Clemens J, Paul SM, Ungerer A. Memory-enhancing effects of secreted forms of the beta-amyloid precursor protein in normal and amnestic mice. Proc Natl Acad Sci USA. 1998;95:12683–12688. doi: 10.1073/pnas.95.21.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 37.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 38.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 40.Pompl PN, Mullan MJ, Bjugstad K, Arendash GW. Adaptation of the circular platform spatial memory task for mice: use in detecting cognitive impairment in the APP(SW) transgenic mouse model for Alzheimer's disease. J Neurosci Methods. 1999;87:87–95. doi: 10.1016/s0165-0270(98)00169-1. [DOI] [PubMed] [Google Scholar]
- 41.Quon D, Wang Y, Catalano R, Scardina JM, Murakami K, Cordell B. Formation of beta-amyloid protein deposits in brains of transgenic mice. Nature. 1991;352:239–241. doi: 10.1038/352239a0. [DOI] [PubMed] [Google Scholar]
- 42.Roch JM, Masliah E, Roch-Levecq AC, Sundsmo MP, Otero DA, Veinbergs I, Saitoh T. Increase of synaptic density and memory retention by a peptide representing the trophic domain of the amyloid beta/A4 protein precursor. Proc Natl Acad Sci USA. 1994;91:7450–7454. doi: 10.1073/pnas.91.16.7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roher AE, Chaney MO, Kuo YM, Webster SD, Stine WB, Haverkamp LJ, Woods AS, Cotter RJ, Tuohy JM, Krafft GA, Bonnell BS, Emmerling MR. Morphology and toxicity of Abeta-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer's disease. J Biol Chem. 1996;271:20631–20635. doi: 10.1074/jbc.271.34.20631. [DOI] [PubMed] [Google Scholar]
- 44.Scott MR, Kohler R, Foster D, Prusiner SB. Chimeric prion protein expression in cultured cells and transgenic mice. Protein Sci. 1992;1:986–997. doi: 10.1002/pro.5560010804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 46.Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L, Jr, Eckman C, Golde TE, Younkin SG. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 48.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 49.Vassar R, Bennett B, Babu-Khan S, Kahn S, Mendiaz E, Denis P, Teplow D, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski M, Biere AL, Curran E, Burgess T, Louis J-C. β-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]