

Abstract
Recent epidemiological and clinical data suggest that persons with low folic acid levels and elevated homocysteine levels are at increased risk of Alzheimer's disease (AD), but the underlying mechanism is unknown. We tested the hypothesis that impaired one-carbon metabolism resulting from folic acid deficiency and high homocysteine levels promotes accumulation of DNA damage and sensitizes neurons to amyloid β-peptide (Aβ) toxicity. Incubation of hippocampal cultures in folic acid-deficient medium or in the presence of methotrexate (an inhibitor of folic acid metabolism) or homocysteine induced cell death and rendered neurons vulnerable to death induced by Aβ. Methyl donor deficiency caused uracil misincorporation and DNA damage and greatly potentiated Aβ toxicity as the result of reduced repair of Aβ-induced oxidative modification of DNA bases. When maintained on a folic acid-deficient diet, amyloid precursor protein (APP) mutant transgenic mice, but not wild-type mice, exhibited increased cellular DNA damage and hippocampal neurodegeneration. Levels of Aβ were unchanged in the brains of folate-deficient APP mutant mice. Our data suggest that folic acid deficiency and homocysteine impair DNA repair in neurons, which sensitizes them to oxidative damage induced by Aβ.
Keywords: apoptosis, comet assay, glycosylase, oxidative stress, transgenic, uracil
Folic acid deficiency may facilitate several different age-related diseases, including coronary artery disease (Swain and St. Clair, 1997), stroke (Elkind and Sacco, 1998), and cancers (Duthie, 1999). By promoting uracil misincorporation and hypomethylation of DNA and by impairing DNA repair, folic acid deficiency can induce DNA damage in mitotic cells (Choi and Mason, 2000). Hyperhomocysteinemia is a consequence of folic acid deficiency that contributes to the pathogenesis of cardiovascular disease and stroke (Refsum et al., 1998) and possibly Alzheimer's disease (AD;Clarke et al., 1998; Miller, 1999; Snowdon et al., 2000) and Parkinson's disease (Kuhn et al., 1998). Homocysteine is a metabolite of methionine, an amino acid that plays a key role in the generation of methyl groups required for numerous biochemical reactions; homocysteine either can be remethylated to methionine by enzymes that require folic acid or can be catabolized by cystathionine β-synthase, a vitamin B6-dependent enzyme, to form cysteine (Scott and Weir, 1998). Patients with severe hyperhomocysteinemia exhibit a wide range of clinical manifestations, including profound neurological abnormalities such as mental retardation, cerebral atrophy, and seizures (Watkins and Rosenblatt, 1989; van den Berg et al., 1995). Recent studies have shown that homocysteine can be directly toxic to cultured neurons; the mechanism may involve the activation of NMDA receptors (Lipton et al., 1997) or apoptosis triggered by DNA damage (Kruman et al., 2000).
In AD the death of neurons in brain regions critical for learning and memory is believed to result from increased production and accumulation of insoluble forms of amyloid β-peptide (Aβ), which may endanger and kill neurons by inducing oxidative stress and disrupting cellular ion homeostasis (Yankner, 1996; Mattson, 1997). Analyses of brain tissue from AD patients and of experimental cell culture and animal models of AD have provided evidence for the involvement of Aβ and apoptotic biochemical cascades in the neurodegenerative process (Mattson, 2000). DNA damage, which is a potent trigger of neuronal apoptosis, has been documented in studies of AD patients and in cell culture and animal models of AD (Mullaart et al., 1990; de la Monte et al., 1998; Torp et al., 1998; Adamec et al., 1999; Harada and Sugimoto, 1999; Love et al., 1999; Seidl et al., 1999). In addition, non-neuronal cells from AD patients exhibit a defect in their ability to repair DNA damage (Li and Kaminskas, 1985; Robison et al., 1987; Boerrigter et al., 1991), suggesting a widespread abnormality in DNA repair mechanisms. Cells from Down's syndrome patients (which have AD-like brain pathology) exhibit hypersensitivity to ionizing radiation-induced DNA damage (Otsuka et al., 1985), and environmentally induced DNA damage may contribute to neurofibrillary degeneration in the ALS–Parkinson–dementia complex of Guam (Kisby et al., 1999). In light of the data implicating increased DNA damage in neurons that degenerate in AD and the evidence that folic acid deficiency and homocysteine can impair DNA repair in non-neuronal cells, we used cell culture and mouse models of AD to test the hypothesis that folic acid deficiency and homocysteine sensitize neurons to Aβ-induced death.
MATERIALS AND METHODS
Hippocampal and cortical cell cultures, experimental treatments, and assessment of neuron survival. Primary hippocampal cell cultures were established from embryonic day 18 (E18) rat embryos by methods described previously (Mark et al., 1995). Dissociated cells were seeded onto polyethylenimine-coated plastic dishes or 22 mm2 glass coverslips and maintained in Neurobasal medium containing B-27 supplements, 2 mml-glutamine, 0.001% gentamycin sulfate, and 1 mm HEPES, pH 7.2. The density of the cultures was 100–150 neurons/mm2 of culture surface. At the time of the experiments (7–10 d in culture) 90–95% of the cells were neurons (MAP-2-immunoreactive); neurons in cultures prepared in an identical manner are vulnerable to apoptosis induced by various insults, including Aβ (Mark et al., 1995). Astrocyte cultures were derived from the cerebral cortex of 1-d-old rat pups and cultured in DMEM/F12 with the addition of 10% fetal calf serum. Cultures were composed primarily of large type-1-like astrocytes and were ∼98% pure as indicated by immunostaining for glial fibrillary acidic protein with a polyclonal antibody (Sigma, St. Louis, MO). Astrocytes were incubated for 24 hr in serum-free DMEM/F12 medium, complete (control) or l-methionine and folic acid (M/F)-deficient; apoptosis and cell proliferation rates were determined. Apoptosis was quantified in cultures stained with fluorescent DNA-binding dye Hoechst 33342 as described previously (Kruman et al., 1997). Proliferation of astrocytes was quantified by cell counting.
Methyl donor-deficient medium was identical to the normal medium except that it lacked folic acid and methionine. In all experiments involving methyl donor-deficient medium the control cultures were subjected to the same number of medium changes of control medium. Experimental treatments were added to cultures by dilution into the culture maintenance medium from concentrated stocks. Aβ (Aβ1-42) was purchased from Bachem (Torrance, CA) and prepared as a 1 mmsolution in water 16 hr before its addition to the cell cultures at a final concentration of 5 μm. This method produces stocks that are in an early state of aggregation (small aggregates and few or no fibrils) at the time of their addition to the cultures (Mattson et al., 1993; Fezoui et al., 2000). l-Methionine, homocysteine, thymidine, hypoxanthine, and methotrexate (Sigma) were prepared as concentrated stocks in sterile water, pH 7.2.
Neuron survival was quantified by counting undamaged neurons in premarked microscope fields before, and at indicated time points after, exposure to experimental treatments via methods described previously (Mark et al., 1995). Neurons that died in the intervals between examination points were usually absent, and the viability of the remaining neurons was assessed by morphological criteria. Neurons with intact neurites and soma with a smooth round appearance were considered viable, whereas neurons with fragmented neurites and vacuolated soma were considered nonviable. Analyses were performed without knowledge of the treatment history of the cultures.
Assessments of DNA damage. DNA damage was assessed by the alkaline single-cell gel electrophoresis “comet assay” method as described previously (Morris et al., 1999). The comet assay has been shown to be a sensitive and reliable measure of DNA strand breaks associated with incomplete excision repair sites and alkali-labile sites. After experimental treatment the neurons were scraped, and cell suspensions (typically 10,000 cells) were embedded into 0.5% low-melting agarose on Trevigen (Gaithersburg, MD) slides. After treatment with lysis buffer (1% Triton X-100, 10% DMSO, 2.5m NaCl, 100 mm EDTA, and 10 mmTris, pH 10) the slides were transferred to a horizontal electrophoresis unit, and electrophoresis was performed at 25 V and 300 mA for 30 min. Then the slides were stained with ethidium bromide and analyzed by using an epifluorescence microscope and the comet image analysis software (Komet 4.0; Kinetic Imaging, Bromborough, UK); 50–100 consecutive cells were analyzed from the center of the slide. Nuclei with damaged DNA have the appearance of a comet with a bright head and a tail, whereas nuclei with undamaged DNA appear round with no tail. The parameters that commonly are measured with the comet assay are tail length, percentage of DNA in the tail, and Olive Tail Moment (OTM). In preliminary studies we found that these three parameters are tightly correlated and therefore chose to use the OTM, which represents the product of the amount of DNA in the tail (expressed as a percentage of the total DNA) and the distance between the centers of mass of the head and tail regions as the measure of DNA damage. As a positive control we used lymphocytes exposed to 1 Gy of γ-irradiation (OTM was 7.1 ± 0.8), and we used untreated lymphocytes as a negative control (OTM was 0.9 ± 0.3). Our assay reliably detected DNA damage in lymphocytes exposed to as little as 5 cGy of γ-irradiation. Two Escherichia coli DNA glycosylases, formamidopyrimidine glycosylase (FPG; Trevigen) and uracil DNA glycosylase (UDG; Roche, Palo Alto, CA) were used separately to convert Aβ- or methyl donor deficiency- and homocysteine-induced DNA base modifications into strand breaks. These strand breaks were detected by comet analysis. For this purpose, after cell lysis the slides were washed with FLARE buffer [containing (in mm) 250 KCl, 25 EDTA, 25 HEPES-KOH, pH 7.4], and the agarose gel was covered with 100 μl of either FPG (0.01 U/μl) or UDG (0.001 U/μl) in FLARE buffer. After incubation at 37°C for 1 hr the slides were subjected to comet analysis performed at a pH of 12.1–12.5. Oxidative DNA damage also was determined as described previously (Struthers et al., 1998; Sattler et al., 2000) with the Biotrin OxyDNA Assay (Biotrin, Dublin, Ireland) according to the manufacturer's protocol. Images of fluorescence were acquired with 488 nm excitation and 510 nm emission.
Mice, diets, measurement of homocysteine levels, and brain tissue preparation. Experiments were performed in transgenic mice expressing the “Swedish” amyloid precursor protein (APP) mutation under the control of a prion promoter (Borchelt et al., 1996); the original line of mice was bred into a C57BL/6 background through 19 generations. These mice develop age-dependent deposition of Aβ in their brains, which is first evident in the hippocampus and cerebral cortex beginning after 10 months of age. Seven-month-old APP mutant mice and littermate nontransgenic control mice were maintained on either a control diet or a folic acid-deficient diet. The control diet was a standard mouse diet that contained defined choline and folate and lacked d,l-homocysteine (Dyets, Incorporated, Bethlehem, PA; diet 518754). The experimental diet lacked folic acid and contained 4.5 gm/kg d,l-homocysteine (Dyets, Incorporated; diet 518806). All mice were given water and diet ad libitum. After 3 months on the diets, blood was taken for analysis of homocysteine levels, and the mice were killed by anesthesia overdose and were decapitated; the brains were removed. Homocysteine levels in serum samples were quantified with an IMX immunoassay analyzer (Abbott Laboratories, Irving, TX) according to the protocol provided by the manufacturer. Trunk blood was collected from mice at the time of death, serum was isolated from whole blood, and a 50 μl aliquot was used for analysis. One brain hemisphere was homogenized in calcium- and magnesium-free buffer (1 hemisphere/2 ml buffer); a 100 μl aliquot was taken for use in quantification of DNA damage by comet analyses (see above), and the remaining homogenate was used for ELISA analysis of the levels of Aβ. The other hemisphere was immersion fixed in Bouin's fixative, embedded in paraffin, sectioned in the sagittal plane at 30 μm, deparaffinized, and stained with cresyl violet.
Quantification of hippocampal pyramidal neurons.Nissl-positive undamaged neurons were counted in the entire extent of the pyramidal cell layer, including regions CA1 and CA3 (CA2 was included in counts for CA3), using a stereological approach similar to that described previously (Long et al., 1999; West, 1999; Lee et al., 2000). The level of resolution at which the estimates were performed allows for an easy discrimination, based on cell morphology, between the neurons of different subdivisions and between the neurons and glial cells. Pyramidal cells of the CA3 subfield were recognized as having a larger size than those of the CA1. The transition from CA1 to CA2/3 was recognized as a narrow zone containing large, loosely organized pyramidal cells, in contrast to the characteristically tightly packed pyramidal cells of CA1. Glial cells, characterized by their much smaller size compared with neurons, were not included in the estimates. One of the ways to obtain an unbiased estimate of number of neurons is a two-step process that involves estimating both the numerical density of neurons and the reference volume in which they reside (West, 1999). We used a computer-based system, Stereologer (Systems Planning and Analysis, Alexandria, VA), and methods described previously (Long et al., 1999). Estimates of the reference volume of the delineated region were assessed according to the Cavalieri method (Gundersen and Jensen, 1987). For each section the reference space was delineated at low power (5× objective; on-screen magnification, 138×); identification of CA1 and CA3 neurons was accomplished at high power (100× objective; numerical aperture, 1.4; on-screen magnification, 2722×). Numerical density was calculated by dividing the number of neurons counted by the total volume sampled in the reference space.
ELISA and immunohistochemical analysis of Aβ. ELISA for soluble and plaque-associated Aβ1-42 and Aβ1-40 was performed as described previously (Chishti et al., 2001). The brain tissue was homogenized in ice-cold buffer containing 5 m guanidine HCl and 50 mm Tris-HCl, pH 8.0, and incubated at room temperature for 3–4 hr. After dilution (1:50, v/v) with cold Dulbecco's PBS with 5% BSA, 0.03%Tween-20, and a cocktail of proteinase inhibitors (Calbiochem, La Jolla, CA) and then centrifugation (16,000 × g for 20 min at 40°C), the supernatant was used for ELISA analysis. A solid phase sandwich ELISA for Aβ1-40 and Aβ1-42 peptides was performed according to the protocol provided by the manufacturer (BioSource, Camarillo, CA). Values of Aβ1-42 and Aβ1-40 in brain were expressed as nanomoles per gram wet weight of brain. Immunohistochemical analysis of Aβ in 8-μm-thick brain sections from APP mutant and wild-type mice was performed by using a monoclonal mouse antibody against human Aβ (clone 6E10) purchased from Senetec, Napa, CA). The antibody was applied at 1:1600 to deparaffinized and rehydrated sections after treatment with 50% formic acid and immersion in 0.3% H2O2 in methanol, followed by incubation with 5% normal horse serum. After incubation with biotinylated anti-mouse IgG secondary antibody, the sections were incubated in ABC solution and developed in nickel-enhanced diaminobenzamine. Sections were counterstained with cresyl violet.
RESULTS
Methyl donor deficiency and homocysteine promote apoptosis and increase vulnerability of cultured hippocampal neurons to Aβ-induced death
We first determined the effects of methyl donor deficiency on neuronal survival by maintaining cultured hippocampal neurons in medium that was deficient in folic acid and methionine. Progressive death of neurons in cultures maintained in methyl donor-deficient medium occurred, with ∼80% of the neurons dying within 72 hr (Fig.1a,b). In contrast, only ∼10% of the neurons in control cultures died during the 72 hr period (Fig. 1a,b). The extent of neuronal death caused by combined methionine/folic acid deficiency was greater than that caused by either methionine deficiency or folic acid deficiency alone (Fig.1c). Methotrexate, a dihydrofolate reductase inhibitor that causes folic acid deficiency (Golos and Malec, 1989; Huennekens, 1994), induced a level of neuronal death similar to that of combined folic acid/methionine deficiency (Fig. 1c), consistent with the mechanism of folic acid deficiency-induced neuronal death involving impairment of one-carbon metabolism. In contrast to postmitotic neurons, M/F deficiency was not toxic in astrocytes. Levels of apoptosis were low in control (4 ± 2%) and in M/F-deficient (1.4 ± 1.3) culture media. However, M/F deficiency adversely affected the proliferation of astrocytes; the proliferation rate of astrocytes incubated for 24 hr in M/F-deficient medium was reduced to 65 ± 3% of the proliferation rate of control cultures (mean ± SE of determinations made in six cultures; p < 0.01, paired Student's t test).
Fig. 1.
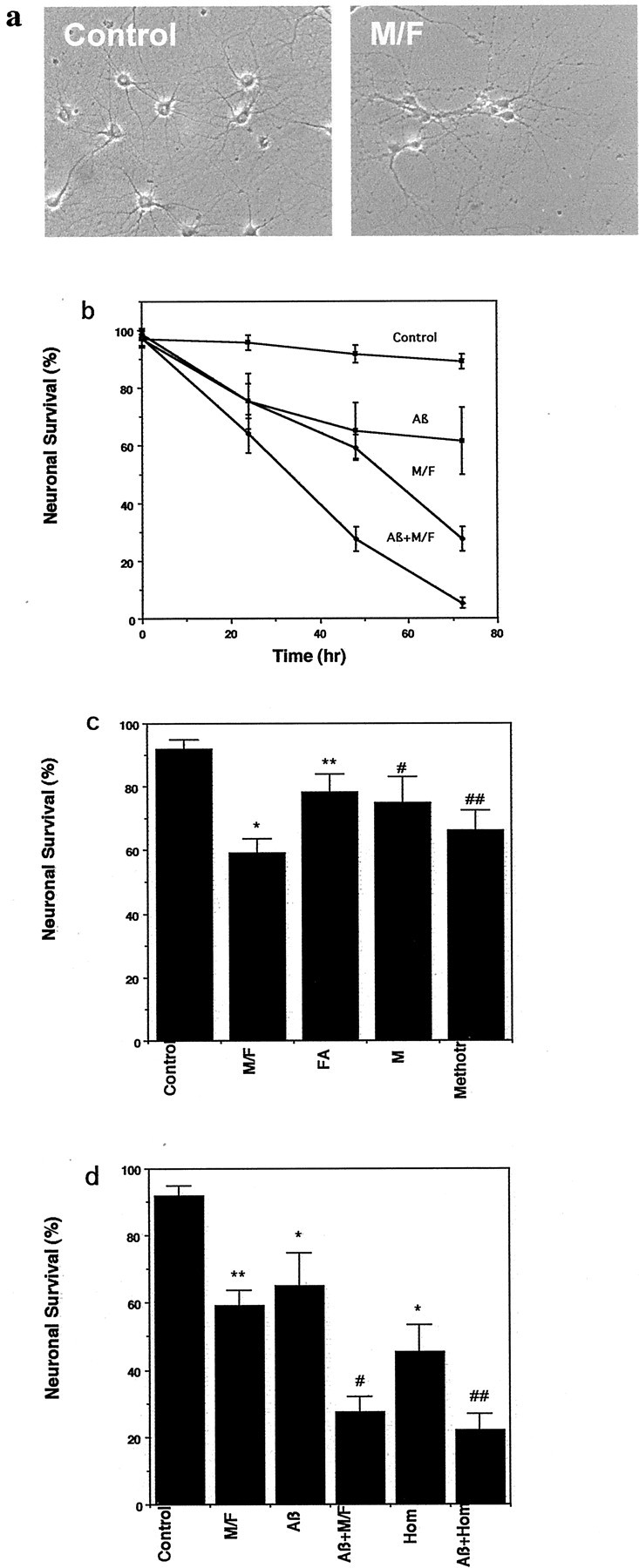
Methyl donor deficiency induces death of hippocampal neurons and potentiates Aβ toxicity. a, Phase-contrast micrographs showing hippocampal neurons in a control culture and a culture that had been maintained for 48 hr in medium lacking methionine and folic acid. b, Cultures were exposed to control medium, medium lacking l-methionine and folic acid (M/F), medium containing 5 μm Aβ1-42 (Aβ), or medium containing a combination of M/F plus Aβ; neuronal survival was quantified at the indicated time points. Neuronal survival is expressed as a percentage of the initial number of neurons present before experimental treatment (see Materials and Methods). Values are the mean and SD of determinations made in six cultures. c, Cultures were incubated for 48 hr in control medium, medium lacking methionine and folic acid (M/F), medium lacking folate (FA), medium lacking methionine but containing folic acid (M), or medium containing 20 μm methotrexate (Methotr). Neuron survival was quantified (mean and SD; n = 6). *p < 0.001 compared with control; **p < 0.01 compared with M/F;#p < 0.05 and##p < 0.01 compared with control (ANOVA with Scheffe's post hoc tests).d, Cultures were exposed for 48 hr to saline (Control), M/F-deficient medium, 5 μm Aβ1-42 (Aβ), a combination of M/F-deficient medium plus 5 μm Aβ1-42 (Aβ+M/F), 250 μm homocysteine (Hom), or a combination of homocysteine plus Aβ (Aβ+Hom). Neuron survival was quantified (mean and SD;n = 6). *p < 0.01, **p < 0.001 compared with control;#p < 0.01 compared with M/F deficiency and with Aβ; ##p < 0.01 compared with homocysteine and with Aβ (ANOVA with Scheffe's post hoc tests).
We next determined whether the vulnerability of hippocampal neurons to Aβ-induced death was affected by methyl donor deficiency. Exposure to Aβ1-42 caused death of ∼30% of the neurons in control culture medium during a 48 hr exposure period. When cultures were exposed to Aβ1-42 in methyl donor-deficient medium, a highly significant enhancement of Aβ-induced neuronal death occurred such that >70% of the neurons were killed (Fig. 1d). Because homocysteine levels are elevated in AD patients and because folic acid deficiency can cause hyperhomocysteinemia, we determined whether homocysteine modifies neuronal vulnerability to Aβ. Neurons incubated in the presence of homocysteine exhibited a significant increase in their sensitivity to being killed by Aβ1-42 (Fig. 1d).
Folic acid deficiency increases homocysteine levels and sensitizes hippocampal neurons to death in APP mutant transgenic mice
Transgenic mice overexpressing the Swedish mutation in APP under the control of a prion promoter exhibit increased levels of soluble Aβ1-42 and progressive age-dependent Aβ deposits in their hippocampus and cerebral cortex (Borchelt et al., 1996). In the transgenic line used in the present study amyloid deposits first become detectable in the brain at ∼10 months of age. We placed 7-month-old APP mutant mice and age-matched wild-type mice on either the normal diet or a diet deficient in folic acid and containing excess homocysteine. After 3 months on the diets, serum levels of homocysteine were elevated by ∼10-fold in both APP mutant and wild-type mice maintained on the experimental diet compared with mice on the control diet (Fig. 2a). Then the mice were killed, and their brains were prepared for histological analysis and assessment of DNA damage. Examination of sections stained with cresyl violet suggested that there was damage to hippocampal CA3 neurons in APP mutant mice that had been maintained on the folic acid-deficient diet compared with mutant mice maintained on the normal diet and that there was damage to wild-type mice maintained on the folic acid-deficient diet (Fig. 2b). We therefore performed stereology-based counts to determine the numerical densities of neurons in regions CA3 and CA1 of APP mutant and wild-type mice that had been maintained on control and folic acid-deficient diets. The analyses in CA3 of hippocampus revealed a highly significant 20% loss of neurons in APP mutant mice on the folic acid-deficient diet compared with mutant mice on the control diet and with wild-type mice maintained on the folic acid-deficient diet (Fig. 2c). The total numbers of neurons within the CA3 reference volume that were measured were 888 ± 61 for nontransgenic mice on the normal diet, 880 ± 52 mm3 for nontransgenic mice on the experimental diet, 896 ± 66 mm3 for APP mutant mice on the normal diet, and 712 ± 32 mm3 for APP mutant mice on the experimental diet (p < 0.01 compared with each of the other values; ANOVA with Scheffe's post hoc tests). There were no differences in the volume densities of neurons in region CA1 among the four different groups of mice (Fig. 2c). The analyzed reference volumes for specific hippocampal regions among the four groups of animals did not differ significantly (p > 0.1); for the CA3 region the values were 0.0028 ± 0.0007 mm3 for nontransgenic mice on the normal diet, 0.0028 ± 0.0008 mm3 for nontransgenic mice on the experimental diet, 0.0026 ± 0.0006 mm3 for APP mutant mice on the normal diet, and 0.0032 ± 0.0009 mm3 for APP mutant mice on the experimental diet. Thus the decrease in CA3 neuronal density is attributable to cell loss and was not the result of a change in the volume of this structure.
Fig. 2.
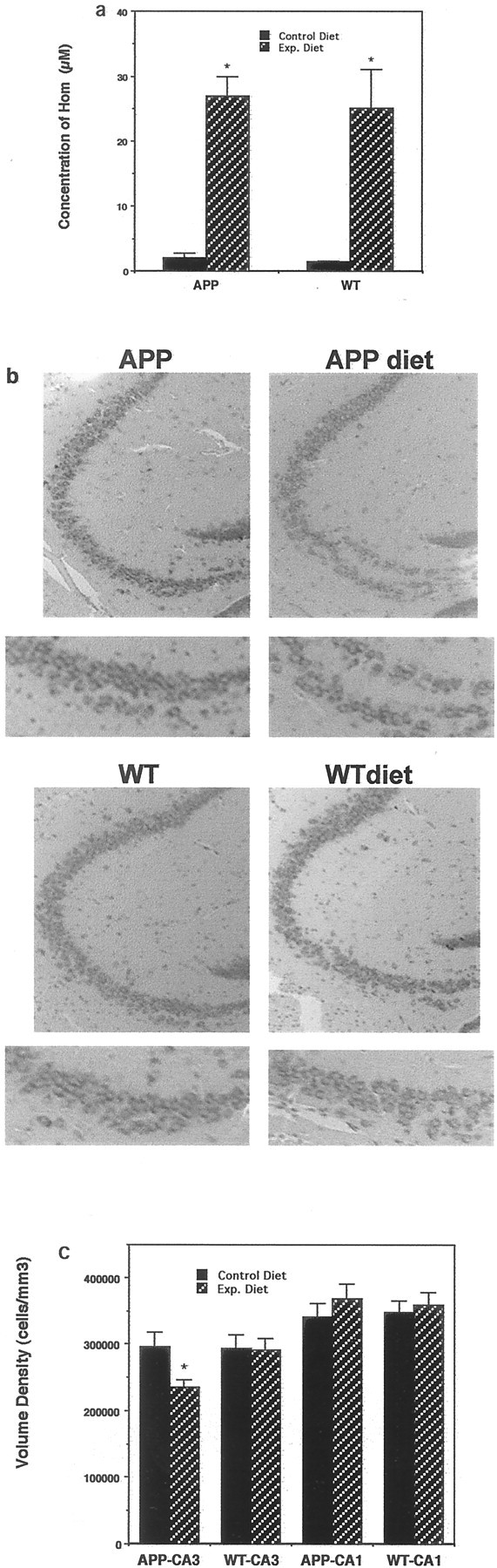
A methyl donor-deficient diet induces hyperhomocysteinemia and promotes neuronal degeneration in APP mutant mice. a, Levels of homocysteine in serum samples from wild-type (WT) and APP mutant mice that had been maintained for 3 months on the normal control diet or the experimental folic acid-deficient diet were quantified. Values are the mean and SD (n = 8). *p < 0.0001 compared with corresponding control value (ANOVA with Scheffe's post hoc tests). b, Micrographs showing cresyl violet-stained sections of hippocampus (region CA3) from wild-type and APP mutant mice maintained for 3 months on either the control diet or the folic acid-deficient diet (APP diet; WT diet). The micrographs at thebottom show high magnification of the lower limb of CA3.c, Numerical densities of neurons in regions CA3 and CA1 of hippocampus were quantified in the brains of wild-type and APP mutant mice maintained for 3 months on either control or folic acid-deficient diets. Values are the mean and SD (n= 8). *p < 0.01 compared with APP mutant or wild-type mice maintained on the control diet and compared with wild-type mice on the experimental diet (ANOVA with Scheffe'spost hoc tests).
Amyloid β-peptide levels are unchanged in APP mutant mice maintained on a folic acid-deficient diet
APP mutant mice exhibit increased production of Aβ1-42 and age-dependent deposition of extracellular Aβ in the brain, particularly in the hippocampus and cerebral cortex (Games et al., 1995; Borchelt et al., 1996; Hsiao et al., 1996). We therefore quantified levels of Aβ1-40 and Aβ1-42 in brain tissues from APP mutant and nontransgenic mice that had been maintained on control or folate-deficient diets. As expected, levels of each Aβ species were below the limit of detection in nontransgenic mice (data not shown). Levels of Aβ1-40 and Aβ1-42 were ∼4 and 2 nmol/gm wet brain weight, respectively, in APP mutant mice that had been maintained on either the control or folate-deficient diets, with no significant differences in levels of either Aβ species between mice on control or folate-deficient diets (Fig.3a). The ratio Aβ1-42/Aβ1-40 also was not changed by the folate-deficient diet (Fig. 3b). Examination of brain sections immunostained with an antibody against Aβ revealed no evidence of extracellular amyloid deposition in APP mutant mice that had been maintained on either diet (Fig. 3c). Collectively, the data suggest that folate deficiency renders hippocampal CA3 neurons in APP mutant mice vulnerable to death by a mechanism that does not involve increased Aβ production or deposition.
Fig. 3.
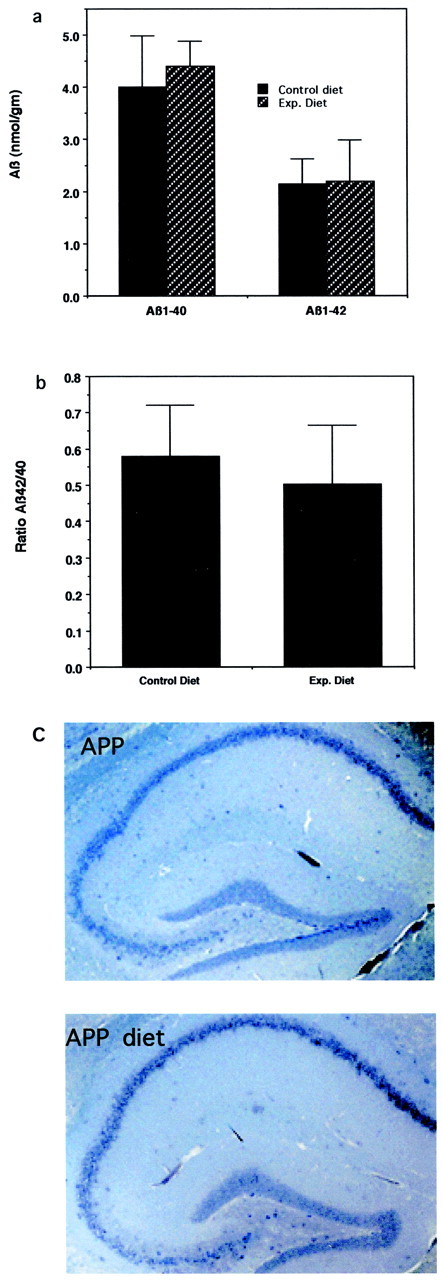
Dietary folic acid deficiency does not affect levels of Aβ in the brains of APP mutant mice. a, Levels of Aβ1-40 and Aβ1-42 were quantified by ELISA analysis in cerebral cortex of APP mutant mice that had been maintained on either a control diet or a folate-deficient diet (Exp. Diet). Values are the mean and SE of determinations (6 mice per group).b, Levels of Aβ, expressed as a ratio of Aβ1-42/Aβ1-40 in brain tissue from APP mutant mice that had been maintained for 3 months on folic acid-deficient or control diets. Values are the mean and SE of measurements made in samples from six mice on each diet. c, Micrographs showing Aβ immunoreactivity (brown) in sections of hippocampus from APP mutant transgenic mice that had been maintained for 3 months on a folic acid-deficient diet (APP diet) or a control diet. Note the absence of plaques in mice on either diet.
Folic acid deficiency and homocysteine enhance Aβ-induced DNA damage in cultured hippocampal neurons and APP mutant mice
DNA damage has been documented in association with neuronal degeneration in brain tissues of AD patients (Torp et al., 1998; Adamec et al., 1999). We therefore determined whether methyl donor deficiency was sufficient to promote DNA damage in neurons and whether Aβ might exacerbate such DNA damage. Hippocampal cultures were incubated in medium that was deficient in folic acid and methionine or in the normal culture medium; DNA damage was measured via the comet assay, a sensitive technique that has proved useful in measuring the DNA damage and repair capacity in cells subjected to chemical- or radiation-induced DNA lesions (Fairbairn et al., 1995). DNA damage was increased significantly in neurons within 8 hr of incubation in the methyl donor-deficient medium (Fig.4a,b). Although Aβ1-42 alone did not induce DNA damage during a 5 hr exposure (this time of treatment was based on the dynamics of DNA damage induced by folic acid deficiency; Fig. 4b), accumulation of DNA damage in neurons maintained in folic acid-deficient medium was enhanced significantly in the presence of Aβ1-42 (Fig. 4c). A significant increase in DNA damage also occurred in neurons exposed to Aβ1-42 in the presence of homocysteine, but the magnitude of the increase was not greater than that seen with either treatment alone. To determine whether the folic acid-deficient diet promoted DNA damage to hippocampal neurons in vivo, we performed comet analysis to detect cells with DNA strand breaks in hippocampal tissue samples from APP mutant and wild-type mice maintained on either the normal diet or the folic acid-deficient diet. DNA damage was increased significantly in APP mutant mice maintained on the folic acid-deficient diet compared with mutant mice on the control diet and with wild-type mice maintained on either control or folic acid-deficient diets (Fig.4d).
Fig. 4.

Amyloid β-peptide enhances DNA damage in neurons under conditions of methyl donor deficiency. a, Comet assay analysis of nuclear DNA in neurons in a control culture and a culture that had been exposed for 8 hr in methionine/folic acid-deficient medium. b, DNA damage was quantified by comet analysis in hippocampal neurons after the indicated time periods of incubation in methionine/folic acid-deficient medium. Values are the mean and SD of determinations made in six cultures; *p < 0.01. c, DNA damage was quantified in neurons that had been exposed for 4 hr to the indicated treatments (5 μm Aβ1-42; 250 μm homocysteine). Values are the mean and SD (n = 6). **p < 0.01 compared with control. #p < 0.05,##p < 0.01 compared with Aβ alone (ANOVA with Scheffe's post hoc test). d, DNA damage was quantified by comet assay in hippocampal tissue samples from wild-type and APP mutant mice that had been maintained for 3 months on either the control diet or the methyl donor-deficient experimental diet. Values are the mean and SD (n = 8). *p < 0.01 compared with the value for APP mutant mice on the control diet and with WT mice on either diet.
Folic acid deficiency promotes uracil misincorporation and impairs DNA repair under conditions of exposure to Aβ
The increased accumulation of damaged DNA in neurons subjected to folic acid deficiency or exposed to homocysteine might result from either an increase in damage or a decrease in DNA repair. Folic acid/methionine deficiency or Aβ alone did not produce significant DNA damage within 5 hr (Fig. 4c). Cultured hippocampal neurons were maintained for 5 hr in the presence of homocysteine alone, in the presence of homocysteine plus Aβ1-42, or in medium deficient in folic acid and methionine in the presence of Aβ1-42; then the neurons were incubated for an additional 12 hr in the normal culture medium containing folic acid and methionine. The level of DNA damage was increased in neurons incubated in the presence of homocysteine alone and was restored to basal levels after homocysteine was removed (Fig. 5a), indicating that the DNA damage was reversible. In contrast, the DNA damage was not restored to basal levels when neurons were incubated in the presence of Aβ1-42 together with either homocysteine or folic acid-deficient medium (Fig.5a). Thus, Aβ impairs the ability of neurons to repair DNA damage under conditions of methyl donor deficiency.
Fig. 5.

Impaired DNA repair and increased accumulation of oxidatively modified bases in neurons exposed to Aβ under methyl donor-deficient conditions. a, Cultured neurons were exposed for 5 hr to the indicated treatments (5 μm Aβ1-42; 250 μm homocysteine). Then the cultures either were processed for analysis of DNA damage (Treatment) or were incubated for an additional 12 hr in the absence of treatments (Recovery) and then processed for analysis of DNA damage. Values are the mean and SD (n = 6). *p < 0.01 compared with the corresponding treatment value. b, Neurons were exposed for 5 hr to the indicated treatments and then subjected to comet analysis of DNA damage, using FPG treatment as described in Materials and Methods. Values are the mean and SD (n = 6). *p < 0.01, **p < 0.001 compared with the corresponding buffer value. c, Cultured neurons were exposed for 5 hr to the indicated treatments (5 μm Aβ1-42). Cultures either were processed for analysis of DNA damage (Treatment) or were incubated for an additional 12 hr in the absence of treatments (Recovery) and subjected to comet analysis of DNA damage by FPG treatment. Values are the mean and SD (n = 6). *p < 0.01 compared with the corresponding treatment value. d, Micrographs showing 8-oxoguanine immunoreactivity in cultured hippocampal neurons subjected to the indicated treatments. Cultures were exposed to the indicated treatments for 5 hr and either were analyzed at that time (treatment) or were allowed to recover for 12 hr in the absence of treatment (recovery). Control, Exposure of 5 hr to buffer; Aβ, 5 μmAβ1-42; M/F def., methionine- and folic acid-deficient medium.
DNA damage is associated with a variety of modifications of DNA bases, among which the formation of 8-oxyguanine has been reported widely; FPG excises oxidized nucleotides, including 8-oxyguanine (Laval et al., 1998). The relative amount of such oxidatively modified bases incorporated into nuclear DNA can be determined by performing comet analysis after incubation of the nuclei in the presence of recombinant FPG, which removes the oxidatively modified bases, resulting in an increase in DNA strand breaks. Cultured hippocampal neurons were exposed for 8 hr to Aβ1-42 alone, methionine/folic acid-deficient medium alone, or the combination of methionine/folic acid-deficient medium and Aβ1-42. Slides containing cells were treated with FPG or buffer, and comet analysis was performed. Aβ1-42 induced an increase in oxidatively modified FPG-sensitive sites, whereas methionine/folic acid deficiency alone did not (Fig. 5b). However, the extent of oxidative DNA damage induced by Aβ was enhanced significantly in neurons subjected to methionine/folic acid-deficient medium. To determine whether methyl donor deficiency enhances oxidative modification of bases in neuronal DNA or impairs DNA repair, we exposed neurons to Aβ alone or in combination with folic acid-deficient medium for 5 hr, washed out the treatments, and then assessed DNA damage 12 hr later. Aβ1-42 induced oxidative DNA damage that recovered to basal levels in neurons maintained in normal medium containing folic acid and methionine (Fig. 5c). In contrast, neurons maintained in folic acid-deficient medium were unable to repair their DNA after exposure to Aβ1-42. To confirm these results, we used the Biotrin OxyDNA Assay to detect oxidized nucleotides (see Materials and Methods). Levels of 8-oxoguanine were very low in control cultures and increased within 8 hr of exposure to Aβ (Fig. 5d). When the Aβ was removed from the cultures, levels of 8-oxyguanine immunoreactivity returned to a low level within 12 hr. Aβ caused an increase in 8-oxyguanine in cultures maintained in folic-deficient medium, and the neurons were unable to repair the DNA damage after washout of the Aβ (Fig. 5d).
Because methyl donor deficiency can cause misincorporation of uracil into DNA of proliferating cells caused by impairment of deoxynucleoside triphosphate pools (Pogribny et al., 1997), we determined the effects of folic acid deficiency on uracil misincorporation in neurons by incubating cells in the presence of UDG, an enzyme that removes misincorporated uracil (Duthie and McMillan, 1997; Mol et al., 1999). Recognition of the misincorporated uracil by UDG and subsequent exposure to alkaline conditions result in DNA strand breaks that can be detected by comet assay. There was a highly significant increase in uracil misincorporation in neurons when they were incubated in folic acid/methionine-deficient medium (Fig.6a). Aβ1-42 alone did not cause uracil misincorporation (Fig. 6a). The misincorporation of uracil appears to be a key event in the neurotoxic effects of folic acid deficiency and homocysteine, because pretreatment with thymidine and hypoxanthine (a precursor of purines) to the culture medium reduced neuronal cell death induced by methyl donor deficiency and homocysteine (Fig. 6b). We have shown previously that DNA damage and poly (ADP-ribose) polymerase (PARP) activation occur before evidence of mitochondrial alterations or caspase activation after exposure of neurons to homocysteine and that a PARP inhibitor and a caspase inhibitor prevented homocysteine-induced cell death (Kruman et al., 2000). In light of the latter data and because elevated homocysteine levels mediate the cytotoxic actions of folate deficiency, it appears that the early DNA damage after folic acid deprivation is a trigger of apoptosis. To confirm this, we performed an additional experiment to determine whether the inhibition of PARP can protect neurons against death induced by folic acid deficiency. Cultures were incubated for 24 hr in folic acid-deficient medium in the absence or presence of the PARP inhibitor 3-aminobenzamide (5 mm), and neuronal survival was quantified. Values for neuronal survival (mean ± SE; n = 6 cultures) included the following: control (folate-containing medium), 91 ± 3.1%; 3-aminobenzamide in folate-containing medium, 89 ± 2.1%; folate-deficient medium, 59 ± 4.5%; 3-aminobenzimide in folate-deficient medium, 82 ± 5.9% (p < 0.01, ANOVA with Scheffe's post hoc test). Collectively, these data suggest that folic acid deficiency and homocysteine can promote neuronal death and sensitize neurons to Aβ toxicity by impairing DNA repair.
Fig. 6.
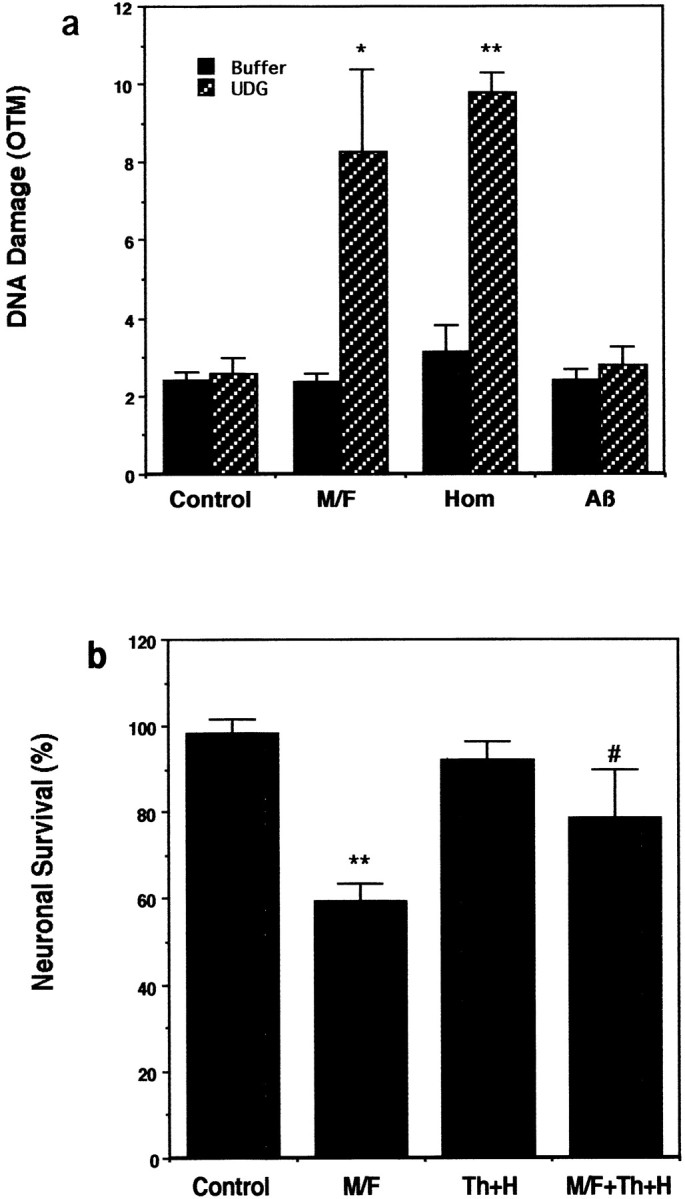
Methyl donor deficiency-induced neuronal death involves misincorporation of uracil into DNA. a, Neurons were exposed for 5 hr to saline (Control), methionine/folic acid-deficient medium (M/F), or 250 μm homocysteine (Hom) and then were subjected to comet analysis of DNA damage via UDG treatment. Next the nuclei were subjected to comet analysis of DNA damage. Values are the mean and SD (n = 6). *p < 0.01, **p < 0.001 compared with control value and with the corresponding buffer value. b, Cultures were exposed for 48 hr to saline (Control), methionine/folic acid-deficient medium (M/F), 15 μm thymidine plus 30 μm hypoxanthine (Th+H), or the combination of methionine/folic acid-deficient medium plus Th+H. Neuron survival was quantified; the values are the mean and SD (n = 6). **p < 0.01 compared with control;#p < 0.05 compared with the methionine/folic acid-deficient value.
DISCUSSION
The ability of folic acid deficiency and elevated homocysteine levels to increase the vulnerability of cultured hippocampal neurons to Aβ-induced death and to promote neuronal degeneration in APP mutant transgenic mice suggests a mechanism whereby individuals with low folic acid intake and a resulting elevation of homocysteine levels might be at increased risk of AD. Aβ can induce oxidative stress and DNA damage in cultured neurons (Loo et al., 1993; Mark et al., 1995, 1997), and both oxidative stress and DNA damage have been documented in neurons associated with Aβ-containing plaques in the brains of AD patients (for review, see Mattson, 1997, 2000). Our analyses of DNA damage and repair suggest that folic acid deficiency and homocysteine promote the accumulation of DNA damage in neurons by impairing DNA repair. Thus, neuronal death promoted by methyl donor deficiency was attenuated by purine and thymidine supplementation, suggesting that limitation of the availability of purines and thymidine for DNA repair plays a key role in the endangering actions of methyl donor deficiency. The difference in dynamics of DNA damage induced by the combination of folic acid deficiency and Aβ, compared with the combination of homocysteine and Aβ, may result from a more rapid direct effect of homocysteine and a more delayed effect of folic acid deficiency requiring the production of endogenous homocysteine. Because cellular RNA also can contain oxidizable bases, it could be a target for Aβ-induced oxidative modification. However, the FPG recombinant enzyme that we used is specific for 8-oxodeoxyguanosine, and the conditions of the alkaline comet assay will not cause RNA to be degraded. The immunostaining protocol we used is also specific for DNA base modifications, and the extranuclear localization of immunoreactivity shown in Figure 5 could reflect oxidative modifications of mitochondrial DNA.
Apoptosis of cultured neurons has been observed in response to a variety of DNA-damaging agents, including UV irradiation, cytosine arabinoside, and the topoisomerase I inhibitor camptothecin (Park et al., 1998). Postmitotic cells have been shown to be more vulnerable to DNA damage than mitotic cells, probably because of the lack of efficacy of DNA repair. It was shown that neurons (Morris and Geller, 1996;Gobbel et al., 1998) and neuroblastoma cells become extremely UV-sensitive after terminal differentiation (McCombe et al., 1976). Patients with hereditary disorders such as Cockayne's syndrome and xeroderma pigmentosum, deficient in DNA repair on a par with increased predisposition to cancer, have severe neurological abnormalities (Allen et al, 2001). Currently, little is known about the repair capabilities of neurons. Nevertheless, although DNA repair ability may decrease during brain maturation, neurons in the adult brain can repair damaged DNA effectively (Korr and Schultze, 1989). This implies that impairment of DNA repair ability in neurons could be an important factor in the accumulation of DNA damage and neuronal death in neurodegenerative disorders. The ability of folic acid deficiency to potentiate Aβ toxicity was associated with oxidative DNA modification caused by decreased DNA repair, consistent with previous studies in non-neuronal cells showing that folate deficiency increases genetic damage caused by alkylating agents and γ-irradiation (Branda and Blickensderfer, 1993). Interestingly, we found that postmitotic neurons are more vulnerable to being killed by homocysteine and folic acid deficiency than are mitotic astrocytes. In dividing cells DNA damage can inhibit cell proliferation and DNA repair and lead to mutagenesis and malignant transformation (Blount et al., 1997; Green and Miller, 1999). Thus, folic acid deficiency and elevated homocysteine also may have an adverse effect on mitotic cells in the nervous system, including glia and neural progenitor cells.
Whereas the APP mutant transgenic mice used in the present study exhibited no evidence of neuronal degeneration when maintained on the usual rodent diet, a significant loss of neurons occurred in these mice when they were maintained for 3 months on a folic acid-deficient diet that caused a >10-fold elevation of circulating levels of homocysteine. In contrast, no significant neuronal loss occurred in wild-type mice maintained on the folic acid-deficient diet despite a similar elevation of circulating homocysteine levels. The major consequence of overexpression of mutant APP in mice is to increase the production of Aβ1-42, which then forms plaque-like deposits in the hippocampus and cerebral cortex (Games et al., 1995; Borchelt et al., 1996; Hsiao et al., 1996). As expected, we found that levels of soluble Aβ1-42/Aβ1-40 were increased in the brains of APP mutant mice, as were levels of intracellular Aβ immunoreactivity in hippocampal neurons of the mutant mice. However, levels of Aβ appeared unchanged in APP mutant mice maintained on the folic acid-deficient diet. When taken together with our cell culture data showing that methyl donor deficiency and homocysteine sensitize hippocampal neurons to Aβ toxicity, our findings in the APP mutant mice suggest that the increased production of Aβ1-42 in the brains of these mice renders their neurons vulnerable to homocysteine. In this scenario, neuronal death would be triggered when the extent of DNA damage reaches a critical threshold level, which is lowered by a folic acid-deficient diet and age-related increases in the accumulation of Aβ. The selective loss of CA3, as opposed to CA1, neurons in brains of APP mutant mice after a folate-deficient diet regimen might result from the selective vulnerability of CA3 pyramidal neurons of the hippocampus to DNA damage and/or excitotoxicity. Previous studies have shown that CA3 neurons are vulnerable to excitotoxicity (Nadler et al., 1980) and that Aβ increases neuronal vulnerability to excitotoxicity (Mattson et al., 1992). Consistent with this possibility are data showing that oxidative stress, DNA damage, and activation of p53 mediate excitotoxicity (Copani et al., 2001).
Accumulating evidence suggests that cell cycle-related proteins such as cyclins or cyclin-dependent kinases are reexpressed in neurons committed to death in response to a variety of insults, including Aβ (Copani et al., 2001). It even has been reported that DNA replication can be triggered in postmitotic neurons (Smith et al., 2000; Yang et al., 2001). In proliferating cells specific proteins detect DNA damage (e.g., PARP and p53) and can trigger cell cycle arrest and promote DNA repair. If the damage is too extensive to be repaired, the same factors trigger apoptosis. Such a mechanism appears to occur in differentiated postmitotic cells such as neurons. Aβ-induced cell death may be mediated by p53 (Culmsee et al., 2001), which responds to DNA damage and activates a transcriptional gene program that induces the expression of proapoptotic genes such as Bax. We have shown that homocysteine induces DNA damage, p53 activation, and neuronal cell death (Kruman et al., 2000). Folate deficiency also induces DNA damage, followed by cell death. The capability of postmitotic neurons to replicate their DNA during conditions of DNA precursor deficiency induced by homocysteine or folate deficiency would explain our observation of a relatively rapid effect of these treatments on uracil misincorporation and cell death.
There is increasing evidence supporting a role for DNA damage and apoptosis in the pathogenesis of several neurodegenerative disorders, including AD. Neurons in vulnerable brain regions of AD patients exhibit several alterations suggestive of apoptosis, including caspase activation (Masliah et al., 1998; Chan et al., 1999), increased levels of the proapoptotic protein Par-4 (Guo et al., 1998), and increased expression of Bax (Su et al., 1997). Exposure of cultured neurons to Aβ induces caspase activation (Mattson et al., 1998; Chan et al., 1999) and increased production of Par-4 (Guo et al., 1998) and Bax (Paradis et al., 1996), each of which appears to play an important role in the cell death process. Apoptosis triggered by DNA damage typically involves activation of PARP and induction and activation of the tumor suppressor protein p53 (Smulson et al., 2000). Increased PARP activity and p53 levels have been documented in association with degenerating neurons in AD patients (de la Monte et al., 1998; Love et al., 1999) and in cultured neurons exposed to Aβ (Culmsee et al., 2001). Moreover, a chemical inhibitor of p53 can protect neurons against Aβ toxicity (Culmsee et al., 2001), suggesting a key role for this DNA damage-responsive cell death pathway in the pathogenesis of AD. Our data suggest that, by impairing the DNA repair capacity in neurons, folic acid deficiency and elevated homocysteine levels may lower the threshold level of DNA damage that is required to trigger neuronal death. In this view, folic acid deficiency and elevated homocysteine levels accelerate the accumulation of DNA damage that is promoted by age-related increases in oxidative stress and by accumulation of Aβ. Neurons are more vulnerable to DNA damage than non-neuronal cells (McCombe et al., 1976; Gueneau et al., 1979), suggesting that the brain may be particularly sensitive to diets deficient in folic acid and other nutritional and genetic factors associated with one-carbon metabolism.
In humans the normal range of homocysteine concentrations in plasma is 5–15 μm, and levels of homocysteine in CSF and brain tissue are reported to range from 0.5 to 10 μm(Welch and Loscalzo, 1998). The levels of homocysteine in the blood of wild-type mice and APP mutant mice in the present study were not different, being the range of 1–3 μm when fed the usual diet. However, homocysteine levels increased to 20–30 μmin wild-type and APP mutant mice maintained on the methyl donor-deficient diet. Homocysteine can be taken up rapidly by neurons via a specific membrane transporter (Grieve et al., 1992), and, based on previous studies of non-neuronal cells (Blount et al., 1997) and the present study of neurons, homocysteine can induce DNA strand breaks by inducing thymidine insufficiency and promoting uracil misincorporation. Plasma folic acid levels decrease and homocysteine levels increase with age (Andersson et al., 1992; Brattstrom et al., 1994) and to an even greater extent in patients with AD and PD (Clarke et al., 1998; Kuhn et al., 1998; Snowdon et al., 2000), consistent with a possible contribution of disturbed de novo synthesis of purines and thymidine and increased accumulation of damaged DNA to the pathogenesis of several different age-related neurodegenerative disorders. If our findings in the present studies of cultured neurons exposed to Aβ and APP mutant mice reflect the pathogenic process in humans, then dietary supplementation with folic acid would be expected to reduce risk of sporadic forms of AD and also might suppress the neurodegenerative process in familial AD cases.
Footnotes
We thank D. Borchelt for providing initial breeding pairs of mice, J. Lee for technical assistance, and D. Ingram for valuable discussions.
Correspondence should be addressed to Mark P. Mattson, Laboratory of Neurosciences, National Institute on Aging Gerontology Research Center, 5600 Nathan Shock Drive, Baltimore, MD 21224. E-mail:mattsonm@grc.nia.nih.gov.
REFERENCES
- 1.Adamec E, Vonsattel JP, Nixon RA. DNA strand breaks in Alzheimer's disease. Brain Res. 1999;849:67–77. doi: 10.1016/s0006-8993(99)02004-1. [DOI] [PubMed] [Google Scholar]
- 2.Allen DM, van Praag H, Ray J, Weaver Z, Winrow CJ, Carter TA, Braquet R, Harrington E, Ried T, Brown KD, Gage FH, Barlow C. Ataxia telangiectasia mutated is essential during adult neurogenesis. Genes Dev. 2001;15:554–566. doi: 10.1101/gad.869001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersson A, Brattstrom L, Israelsson B, Isaksson A, Hamfelt A, Hultberg B. Plasma homocysteine before and after methionine loading with regard to age, gender, and menopausal status. Eur J Clin Invest. 1992;22:79–87. doi: 10.1111/j.1365-2362.1992.tb01940.x. [DOI] [PubMed] [Google Scholar]
- 4.Blount BC, Mack MM, Wehr CM, MacGregor JT, Hiatt RA, Wang G, Wickramasinghe SN, Everson RB, Ames BN. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implication for cancer and neuronal damage. Proc Natl Acad Sci USA. 1997;94:3290–3295. doi: 10.1073/pnas.94.7.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boerrigter ME, van Duijn CM, Mullaart E, Eikelenboom P, van der Togt CM, Knook DL, Hofman A, Vijg J. Decreased DNA repair capacity in familial, but not in sporadic, Alzheimer's disease. Neurobiol Aging. 1991;12:367–370. doi: 10.1016/0197-4580(91)90024-e. [DOI] [PubMed] [Google Scholar]
- 6.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 7.Branda RF, Blickensderfer DB. Folate deficiency increases genetic damage caused by alkylating agents and gamma-irradiation in Chinese hamster ovary cells. Cancer Res. 1993;53:5401–5408. [PubMed] [Google Scholar]
- 8.Brattstrom L, Lindgren A, Israelsson B, Andersson A, Hultberg B. Homocysteine and cysteine: determinants of plasma levels in middle aged and elderly subjects. J Intern Med. 1994;236:633–641. doi: 10.1111/j.1365-2796.1994.tb00856.x. [DOI] [PubMed] [Google Scholar]
- 9.Chan SL, Griffin WS, Mattson MP. Evidence for caspase-mediated cleavage of AMPA receptor subunits in neuronal apoptosis and Alzheimer's disease. J Neurosci Res. 1999;57:315–323. doi: 10.1002/(SICI)1097-4547(19990801)57:3<315::AID-JNR3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 10.Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- 11.Choi SW, Mason JB. Folate and carcinogenesis: an integrated scheme. J Nutr. 2000;130:129–132. doi: 10.1093/jn/130.2.129. [DOI] [PubMed] [Google Scholar]
- 12.Clarke R, Smith AD, Jobst KA, Refsum H, Sutton L, Ueland PM. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch Neurol. 1998;55:1449–1455. doi: 10.1001/archneur.55.11.1449. [DOI] [PubMed] [Google Scholar]
- 13.Copani A, Uberti D, Sortino AM, Bruno V, Nicoletti F, Memo M. Activation of cell cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci. 2001;24:25–31. doi: 10.1016/s0166-2236(00)01663-5. [DOI] [PubMed] [Google Scholar]
- 14.Culmsee C, Zhu Z, Yu QS, Chan SL, Camandola S, Guo Z, Greig N, Mattson MP. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid β-peptide. J Neurochem. 2001;77:220–228. doi: 10.1046/j.1471-4159.2001.t01-1-00220.x. [DOI] [PubMed] [Google Scholar]
- 15.de la Monte SM, Sohn YK, Ganju N, Wands JR. p53- and CD95-associated apoptosis in neurodegenerative diseases. Lab Invest. 1998;78:401–411. [PubMed] [Google Scholar]
- 16.Duthie SJ. Folic acid deficiency and cancer: mechanisms of DNA instability. Br Med Bull. 1999;55:578–592. doi: 10.1258/0007142991902646. [DOI] [PubMed] [Google Scholar]
- 17.Duthie SJ, McMillan P. Uracil misincorporation in human DNA detected using single cell gel electrophoresis. Carcinogenesis. 1997;18:1709–1714. doi: 10.1093/carcin/18.9.1709. [DOI] [PubMed] [Google Scholar]
- 18.Elkind MS, Sacco RL. Stroke risk factors and stroke prevention. Semin Neurol. 1998;18:429–440. doi: 10.1055/s-2008-1040896. [DOI] [PubMed] [Google Scholar]
- 19.Fairbairn DW, Olive PL, O'Neill KL. The comet assay: a comprehensive review. Mutat Res. 1995;339:37–59. doi: 10.1016/0165-1110(94)00013-3. [DOI] [PubMed] [Google Scholar]
- 20.Fezoui Y, Hartley DM, Harper JD, Khurana R, Walsh DM, Condron MM, Selkoe DJ, Lansbury PT, Jr, Fink AL, Teplow DB. An improved method of preparing the amyloid β-protein for fibrillogenesis and neurotoxicity experiments. Amyloid. 2000;7:166–178. doi: 10.3109/13506120009146831. [DOI] [PubMed] [Google Scholar]
- 21.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Lieberburg I, Schenk D. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 22.Gobbel GT, Bellinzona M, Vogt AR, Gupta N, Fike JR, Chan PH. Response of postmitotic neurons to X-irradiation: implications for the role of DNA damage in neuronal apoptosis. J Neurosci. 1998;18:147–155. doi: 10.1523/JNEUROSCI.18-01-00147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golos B, Malec J. Enhancement of methotrexate-induced growth inhibition, cell killing, and DNA lesions in cultured L5178Y cells by the reduction of DNA repair efficiency. Biochem Pharmacol. 1989;38:1743–1748. doi: 10.1016/0006-2952(89)90407-3. [DOI] [PubMed] [Google Scholar]
- 24.Green R, Miller JW. Folate deficiency beyond megaloblastic anemia: hyperhomocysteinemia and other manifestations of dysfunctional folate status. Semin Hematol. 1999;36:47–64. [PubMed] [Google Scholar]
- 25.Grieve A, Butcher SP, Griffiths R. Synaptosomal plasma membrane transport of excitatory sulphur amino acid transmitter candidates: kinetic characterization and analysis of carrier specificity. J Neurosci Res. 1992;32:60–68. doi: 10.1002/jnr.490320108. [DOI] [PubMed] [Google Scholar]
- 26.Gueneau G, Drouet J, Privat A, Court L. Differential radiosensitivity of neurons and neuroglia of the hippocampus in the adult rabbit. Acta Neuropathol (Berl) 1979;48:199–209. doi: 10.1007/BF00690520. [DOI] [PubMed] [Google Scholar]
- 27.Gundersen HJ, Jensen EB. The efficacy of systematic sampling in stereology and its prediction. J Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- 28.Guo Q, Fu W, Xie J, Luo H, Sells SF, Geddes JW, Bondada V, Rangnekar VM, Mattson MP. Par-4 is a mediator of neuronal degeneration associated with the pathogenesis of Alzheimer disease. Nat Med. 1998;4:957–962. doi: 10.1038/nm0898-957. [DOI] [PubMed] [Google Scholar]
- 29.Harada J, Sugimoto M. Activation of caspase-3 in β-amyloid-induced apoptosis of cultured rat cortical neurons. Brain Res. 1999;842:311–323. doi: 10.1016/s0006-8993(99)01808-9. [DOI] [PubMed] [Google Scholar]
- 30.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 31.Huennekens FM. The methotrexate story: a paradigm for development of cancer chemotherapeutic agents. Adv Enzyme Regul. 1994;34:397–419. doi: 10.1016/0065-2571(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 32.Kisby GE, Kabel H, Hugon J, Spencer P. Damage and repair of nerve cell DNA in toxic stress. Drug Metab Rev. 1999;31:589–618. doi: 10.1081/dmr-100101937. [DOI] [PubMed] [Google Scholar]
- 33.Korr H, Schultze B. Unscheduled DNA synthesis in various types of cells of the mouse brain in vivo. Exp Brain Res. 1989;74:573–578. doi: 10.1007/BF00247359. [DOI] [PubMed] [Google Scholar]
- 34.Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kruman II, Culmsee C, Chan SL, Kruman Y, Guo Z, Penix L, Mattson MP. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci. 2000;20:6920–6926. doi: 10.1523/JNEUROSCI.20-18-06920.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuhn W, Roebroek R, Blom H, van Oppenraaij D, Przuntek H, Kretschmer A, Buttner T, Woitalla D, Muller T. Elevated plasma levels of homocysteine in Parkinson's disease. Eur Neurol. 1998;40:225–227. doi: 10.1159/000007984. [DOI] [PubMed] [Google Scholar]
- 37.Laval J, Jurado J, Saparbaev M, Sidorkina O. Anti-mutagenic role of base-excision repair enzymes upon free radical-induced DNA damage. Mutat Res. 1998;402:93–102. doi: 10.1016/s0027-5107(97)00286-8. [DOI] [PubMed] [Google Scholar]
- 38.Lee J, Duan W, Long JM, Ingram KD, Mattson MP. Dietary restriction increases survival of newly generated neural cells and induces BDNF expression in the dentate gyrus of rats. J Mol Neurosci. 2000;15:99–108. doi: 10.1385/JMN:15:2:99. [DOI] [PubMed] [Google Scholar]
- 39.Li JC, Kaminskas E. Deficient repair of DNA lesions in Alzheimer's disease fibroblasts. Biochem Biophys Res Commun. 1985;129:733–738. doi: 10.1016/0006-291x(85)91953-9. [DOI] [PubMed] [Google Scholar]
- 40.Lipton SA, Kim WK, Choi YB, Kumar S, D'Emilia DM, Rayudu PV, Arnelle DR, Stamler JS. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-d-aspartate receptor. Proc Natl Acad Sci USA. 1997;94:5923–5928. doi: 10.1073/pnas.94.11.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Long JM, Mouton PR, Jucker M, Ingram DK. What counts in brain aging? Design-based stereological analysis of cell number. J Gerontol A Biol Sci Med Sci. 1999;54:B407–B417. doi: 10.1093/gerona/54.10.b407. [DOI] [PubMed] [Google Scholar]
- 42.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Love S, Barber R, Wilcock GK. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer's disease. Brain. 1999;122:247–253. doi: 10.1093/brain/122.2.247. [DOI] [PubMed] [Google Scholar]
- 44.Mark RJ, Hensley K, Butterfield DA, Mattson MP. Amyloid β-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci. 1995;15:6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP. Amyloid β-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci. 1997;17:1046–1054. doi: 10.1523/JNEUROSCI.17-03-01046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masliah E, Mallory M, Alford M, Tanaka S, Hansen LA. Caspase-dependent DNA fragmentation might be associated with excitotoxicity in Alzheimer disease. J Neuropathol Exp Neurol. 1998;57:1041–1052. doi: 10.1097/00005072-199811000-00007. [DOI] [PubMed] [Google Scholar]
- 47.Mattson MP. Cellular actions of β-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 48.Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 49.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mattson MP, Tomaselli K, Rydel RE. Calcium-destabilizing and neurodegenerative effects of aggregated β-amyloid peptide are attenuated by basic FGF. Brain Res. 1993;621:35–49. doi: 10.1016/0006-8993(93)90295-x. [DOI] [PubMed] [Google Scholar]
- 51.Mattson MP, Keller JN, Begley JG. Evidence for synaptic apoptosis. Exp Neurol. 1998;153:35–48. doi: 10.1006/exnr.1998.6863. [DOI] [PubMed] [Google Scholar]
- 52.McCombe P, Lavin M, Kidson C. Control of DNA repair linked to neuroblastoma differentiation. Int J Radiat Biol Relat Stud Phys Chem Med. 1976;29:523–531. doi: 10.1080/09553007614550621. [DOI] [PubMed] [Google Scholar]
- 53.Miller JW. Homocysteine and Alzheimer's disease. Nutr Rev. 1999;57:126–129. doi: 10.1111/j.1753-4887.1999.tb06936.x. [DOI] [PubMed] [Google Scholar]
- 54.Mol CD, Parikh SS, Putnam CD, Lo TP, Tainer JA. DNA repair mechanisms for the recognition and removal of damaged DNA bases. Annu Rev Biophys Biomol Struct. 1999;28:101–128. doi: 10.1146/annurev.biophys.28.1.101. [DOI] [PubMed] [Google Scholar]
- 55.Morris EJ, Geller HM. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell cycle-independent toxicity. J Cell Biol. 1996;134:757–770. doi: 10.1083/jcb.134.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morris EJ, Dreixler JC, Cheng KY, Wilson PM, Gin RM, Geller HM. Optimization of single-cell gel electrophoresis (SCGE) for quantitative analysis of neuronal DNA damage. Biotechniques. 1999;26:282–289. doi: 10.2144/99262st02. [DOI] [PubMed] [Google Scholar]
- 57.Mullaart E, Boerrigter ME, Ravid R, Swaab DF, Vijg J. Increased levels of DNA breaks in cerebral cortex of Alzheimer's disease patients. Neurobiol Aging. 1990;11:169–173. doi: 10.1016/0197-4580(90)90542-8. [DOI] [PubMed] [Google Scholar]
- 58.Nadler JV, Perry BW, Gentry C, Cotman CW. Degeneration of hippocampal CA3 pyramidal cells induced by intraventricular kainic acid. J Comp Neurol. 1980;192:333–359. doi: 10.1002/cne.901920209. [DOI] [PubMed] [Google Scholar]
- 59.Otsuka F, Tarone RE, Seguin LR, Robbins JH. Hypersensitivity to ionizing radiation in cultured cells from Down syndrome patients. J Neurol Sci. 1985;69:103–112. doi: 10.1016/0022-510x(85)90011-5. [DOI] [PubMed] [Google Scholar]
- 60.Paradis E, Douillard H, Koutroumanis M, Goodyer C, LeBlanc A. Amyloid β-peptide of Alzheimer's disease downregulates Bcl-2 and upregulates Bax expression in human neurons. J Neurosci. 1996;16:7533–7539. doi: 10.1523/JNEUROSCI.16-23-07533.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park DS, Morris EJ, Stefanis L, Troy CM, Shelanski ML, Geller HM, Greene LA. Multiple pathways of neuronal death induced by DNA-damaging agents, NGF deprivation, and oxidative stress. J Neurosci. 1998;18:830–840. doi: 10.1523/JNEUROSCI.18-03-00830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pogribny IP, Muskhelishvili L, Miller BJ, James SJ. Presence and consequence of uracil in preneoplastic DNA from folate/methyl-deficient rats. Carcinogenesis. 1997;18:2071–2076. doi: 10.1093/carcin/18.11.2071. [DOI] [PubMed] [Google Scholar]
- 63.Refsum H, Ueland PM, Nygard O, Vollset SE. Homocysteine and cardiovascular disease. Annu Rev Med. 1998;49:31–62. doi: 10.1146/annurev.med.49.1.31. [DOI] [PubMed] [Google Scholar]
- 64.Robison SH, Munzer JS, Tandan R, Bradley WG. Alzheimer's disease cells exhibit defective repair of alkylating agent-induced DNA damage. Ann Neurol. 1987;21:250–258. doi: 10.1002/ana.410210306. [DOI] [PubMed] [Google Scholar]
- 65.Sattler U, Calsou P, Boiteux S, Salles B. Detection of oxidative base DNA damage by a new biochemical assay. Arch Biochem Biophys. 2000;376:26–33. doi: 10.1006/abbi.2000.1701. [DOI] [PubMed] [Google Scholar]
- 66.Scott JM, Weir DG. Folic acid, homocysteine, and one-carbon metabolism: a review of the essential biochemistry. J Cardiovasc Risk. 1998;5:223–227. [PubMed] [Google Scholar]
- 67.Seidl R, Fang-Kircher S, Bidmon B, Cairns N, Lubec G. Apoptosis-associated proteins p53 and APO-1/Fas (CD95) in brains of adult patients with Down syndrome. Neurosci Lett. 1999;260:9–12. doi: 10.1016/s0304-3940(98)00945-8. [DOI] [PubMed] [Google Scholar]
- 68.Smith D, Leone G, DeGregori J, Ahmed MN, Qumsiyeh MB, Nevins JR. Induction of DNA replication in adult rat neurons by deregulation of the retinoblastoma/E2F G1 cell cycle pathway. Cell Growth Differ. 2000;11:625–633. [PubMed] [Google Scholar]
- 69.Smulson ME, Simbulan-Rosenthal CM, Boulares AH, Yakovlev A, Stoica B, Iyer S, Luo R, Haddad B, Wang ZQ, Pang T, Jung M, Dritschilo A, Rosenthal DS. Roles of poly(ADP-ribosyl)ation and PARP in apoptosis, DNA repair, genomic stability, and functions of p53 and E2F-1. Adv Enzyme Regul. 2000;40:183–215. doi: 10.1016/s0065-2571(99)00024-2. [DOI] [PubMed] [Google Scholar]
- 70.Snowdon DA, Tully CL, Smith CD, Riley KP, Markesbery WR. Serum folate and the severity of atrophy of the neocortex in Alzheimer disease: findings from the Nun study. Am J Clin Nutr. 2000;71:993–998. doi: 10.1093/ajcn/71.4.993. [DOI] [PubMed] [Google Scholar]
- 71.Struthers L, Patel R, Clark J, Thomas S. Direct detection of 8-oxodeoxyguanosine and 8-oxoguanine by avidin and its analogs. Anal Biochem. 1998;255:20–31. doi: 10.1006/abio.1997.2354. [DOI] [PubMed] [Google Scholar]
- 72.Su JH, Deng G, Cotman CW. Bax protein expression is increased in Alzheimer's brain: correlations with DNA damage, Bcl-2 expression, and brain pathology. J Neuropathol Exp Neurol. 1997;56:86–93. doi: 10.1097/00005072-199701000-00009. [DOI] [PubMed] [Google Scholar]
- 73.Swain RA, St. Clair L. The role of folic acid in deficiency states and prevention of disease. J Fam Pract. 1997;44:138–144. [PubMed] [Google Scholar]
- 74.Torp R, Su JH, Deng G, Cotman CW. GADD45 is induced in Alzheimer's disease and protects against apoptosis in vitro. Neurobiol Dis. 1998;5:245–252. doi: 10.1006/nbdi.1998.0201. [DOI] [PubMed] [Google Scholar]
- 75.van den Berg M, van der Knaap MS, Boers GH, Stehouwer CD, Rauwerda JA, Valk J. Hyperhomocysteinaemia; with reference to its neuroradiological aspects. Neuroradiology. 1995;37:403–411. doi: 10.1007/BF00588024. [DOI] [PubMed] [Google Scholar]
- 76.Watkins D, Rosenblatt DS. Functional methionine synthase deficiency (cblE and cblG): clinical and biochemical heterogeneity. Am J Med Genet. 1989;34:427–434. doi: 10.1002/ajmg.1320340320. [DOI] [PubMed] [Google Scholar]
- 77.Welch GN, Loscalzo J. Homocysteine and atherothrombosis. N Engl J Med. 1998;338:1042–1058. doi: 10.1056/NEJM199804093381507. [DOI] [PubMed] [Google Scholar]
- 78.West MJ. Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci. 1999;22:51–61. doi: 10.1016/s0166-2236(98)01362-9. [DOI] [PubMed] [Google Scholar]
- 79.Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci. 2001;21:2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yankner BA. Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]