

Abstract
Serotonin-gated ion channels (5-HT3) are members of the ligand-gated channel family, which includes channels that are opened directly by the neurotransmitter acetylcholine, GABA, glycine, or glutamate. Although there is general agreement that the second transmembrane domain (M2) lines the pore, the position of the gate in the M2 is less certain. Here, we used substituted cysteine accessibility method (SCAM) to provide new evidence for a centrally located gate that moves during channel activation. In the closed state, three cysteine substitutions, located on the extracellular side of M2, were modified by methanethiosulfonate (MTS) reagents. In contrast, 13 cysteine substitutions were modified in the open state with MTS reagents. The pattern of inhibition (every three to four substitutions) was consistent with an α helical structure for the middle and cytoplasmic segments of the M2 transmembrane domain. Unexpectedly, open-state modification of two amino acids in the center of M2 with three different MTS reagents prevented channels from fully closing in the absence of neurotransmitter. Our results are consistent with a model in which the central region of the M2 transmembrane domain is inaccessible in the closed state and moves during channel activation.
Keywords: 5-HT3R, substituted cysteine accessibility method, serotonin, gating, ligand-gated ion channel, ion channel structure, ionotropic receptor
5-HT3 channels are nonselective, cationic ion channels that are activated directly by serotonin. They are expressed in the enteric nervous system, the spinal cord, and various regions of the brain, including the hippocampus, striatum, neocortex, amygdala, and raphe nucleus (Tecott et al., 1993). Activation of 5-HT3 receptors leads to membrane depolarization and Ca2+ influx (MacDermott et al., 1999). 5-HT3 channels have been implicated in some forms of long-term potentiation (Alhaider et al., 1991; Maeda et al., 1994; Alkadhi et al., 1996), antinociception (Khasabov et al., 1999), and emesis (Perez, 1999). They exist as a homopentamer of the 5-HT3a subunit or as a heteropentamer of 5-HT3a and 5-HT3b subunits in native tissue and cell lines (Maricq et al., 1991; Hussy et al., 1994; Davies et al., 1999; Dubin et al., 1999).
5-HT3 channels are members of a superfamily of ligand-gated ion channels in which the extracellular N-terminal domain encodes the entire ligand-binding site. The second putative transmembrane domain, M2, lines part of the channel pore and is involved in determining ion selectivity (see Fig. 1) (Corringer et al., 2000). The mechanism by which the binding of neurotransmitter leads to channel opening is not known. One structural component in this transduction process is the gate of the channel, the region that prevents the flow of ions in the absence of neurotransmitter and presumably moves during ligand activation.
Fig. 1.

Properties of wild-type and cysteine-substituted 5-HT3a channels expressed in Xenopusoocytes. A, Alignment of the M2 transmembrane domain sequences from mouse 5HT3a (M74425), mouse 5-HT3b (AF155045), mouse nAChRα7 (P49582), and mouse nAChRα1 (M17640) subunits. “C” indicates cysteine substitutions.Bold delimits two possible positions for the gate in nAChR, −2′G through 2′T (Akabas et al., 1994; Wilson and Karlin, 1998) and 9′L (Unwin, 1995). The proposed membrane topology is shown below.B, Removal of extracellular Ca2+slowed the rate of desensitization with 5-HT (10 μm).Dashed line indicates zero current level. All currents were recorded at −80 mV. C, Exposure of wild-type 5-HT3a channels to 1 mm MTSET in the closed or open (+10 μm 5-HT) state did not irreversibly change 5-HT-induced current. D, Continuous current recording from oocyte expressing V296C shows the direct activation with 1 mm MTSET. Bar graph shows the agonist activity (MTSET-induced current divided by the 5-HT-induced current) of 1 mm MTSET for S290C, V291C, I295C, and V296C (striped bar). The 5-HT3a antagonist MDL-72222 (1 μm; MDL) suppressed the agonist activity of MTSET (black bar) and MTSEA (data not shown). For I295C, MDL-72222 plus MTSET decreased the inward current, giving rise to a negative percentage of 5-HT-induced current.
The location of the gate in the nicotinic acetylcholine-gated receptor (nAChR) has been equivocal. In one set of studies, the location of the gate was inferred using the substituted cysteine accessibility method (SCAM) (Akabas et al., 1994; Wilson and Karlin, 1998; Wilson and Karlin, 2001). In SCAM, each amino acid in a domain (e.g., M2) is changed to cysteine one at a time, and each cysteine mutant is then examined for accessibility to cysteine modifying reagents [methanethiosulfonate (MTS)] using electrophysiological methods. Using this approach, Akabas et al. (1994) proposed that the gate and the narrowest region of the nAChR pore are located on the cytoplasmic end of the M2 transmembrane domain, between the −2′G and 2′T [this nomenclature allows the comparison of homologous amino acids in the M2 transmembrane domain from different types of neurotransmitter-gated ion channels (refer to Fig. 1A)]. Mutagenesis studies of a conserved leucine (9′L), on the other hand, implicated the middle of the M2 transmembrane domain in the gating of nAChR. Mutating the 9′L dramatically changed the EC50 for activation (Filatov and White, 1995; Labarca et al., 1995), increased spontaneous channel activity (Bertrand et al., 1997), and permitted inappropriate activation by antagonists (Palma et al., 1996, 1999). Moreover, structural data derived from high-voltage electron microscopy (4.6–9 Å resolution) of nAChR channels in the closed and open states suggested that the gate in a closed channel is situated in the center of the M2, near the 9′L implicated previously in gating (Unwin, 1995;Miyazawa et al., 1999). To learn more about the secondary structure and gating of neurotransmitter-activated ion channels, we performed a SCAM analysis of the M2 transmembrane domain in 5-HT3achannels.
MATERIALS AND METHODS
Molecular biology. Mouse 5-HT3acDNA (M74425; kindly provided by Dr. Julius, UCSF, San Francisco, CA) was subcloned into pGEMHE for high expression inXenopus oocytes (Maricq et al., 1991). Sequencing of the mouse 5-HT3a revealed three differences from the published sequence (E31 is A31+R32; ΔA304). Cysteine mutants were created by overlap PCR or whole-plasmid PCR. The numbering nomenclature for the mutants refers to the amino acid number in the corrected full-length sequence of 5-HT3a. All PCR-generated products were subjected to DNA sequencing (Salk Sequencing Facility) for potential errors generated by Taq polymerase. In vitro methyl-capped cRNA was made from linearized cDNA and T7 RNA polymerase (Stratagene, La Jolla, CA). The concentration and quality of cRNA were assessed using an ethidium-stained formaldehyde gel and RNA molecular weight marker. Xenopus oocytes were isolated as described previously (Slesinger et al., 1996). Stage V/VI oocytes were injected with a 46 nl solution containing 0.1–10 ng of channel cRNA. Oocytes were incubated in 96 mmNaCl, 2 mm KCl, 1 mmCaCl2, 1 mmMgCl2, and 5 mm HEPES, pH 7.6 with NaOH, for 1–7 d at 16°C.
Electrophysiology. Macroscopic currents were recorded from oocytes with a two-electrode voltage-clamp amplifier (Geneclamp 500;Axon Instruments, Union City, CA), filtered at 0.05–2 kHz, digitized (0.1–2 kHz) with a Digidata 1200 analog-to-digital interface (Axon Instruments), and stored on a laboratory computer. Electrodes were filled with 3 m KCl and had resistances of 0.4–2 MΩ. Oocytes were perfused continuously with a solution containing 86 mm NaCl (Sigma-Aldrich, St. Louis, MO), 2 mm KCl, 4.7 mmMgCl2 (free Mg2+ of 3.3 mm), 0.5 mm EGTA, and 5 mm HEPES, pH 7.5 with ∼5 mm NaOH. For experiments with zero divalents, MgCl2 was omitted. Programmable pinch valves (Warner Instruments, Hamden, CT) were used to change the extracellular solution flowing through a small chamber (0.3175 × 1.524 cm). The extracellular solution was connected to ground via a 3m KCl agarose bridge. Because of instability of MTS (Toronto Research Chemicals, North York, Ontario, Canada), MTSET [(2-(trimethylammonium) ethyl)-methanethiosulfonate], MTSEA (2-aminoethyl-methanethiosulfonate-bromide), and MTSES [sodium (2-sulfonatoethyl)-methanethiosulfonate] stocks were made on the day of the experiment, stored on ice, and diluted immediately before perfusion. For experiments with DTT, 50 mm DTT was prepared in 0.5× of the extracellular solution to minimize osmotic shock. MDL-72222 (Tocris, Ballwin, MO) stock of 10 mm was diluted (1 μm) just before use. For MDL-72222 experiments, we compared the change in 5-HT-induced current after exposure to MDL-72222 (control) with that of MDL-72222 plus MTS, after 10 min of wash to allow for sufficient removal of MDL-72222.
The majority of cysteine-substituted mutants expressed in oocytes yielded small (<300 nA) agonist-independent currents and 5-HT-induced currents between 0.5 and 15 μA. L286C, G288C, Y289C, and F292C were coinjected with wild-type 5-HT3a cRNA (e.g., F292Cwt) in ∼1:1 ratio because injection of the mutant cRNA alone resulted in little or no 5-HT-induced currents. Injection of the cRNA for I295C sometimes gave rise to larger agonist-independent currents and/or unstable agonist-independent currents. Only recordings that were stable and displayed reproducible 5-HT-induced currents were included in the analyses.
Analysis. The dose–response curve for 5-HT was determined by recording the current (voltage clamped at −80 mV) elicited by different concentrations of 5-HT. The 5-HT-induced current for each concentration was divided by the maximal 5-HT-induced current (I/Io) and plotted as a function of [5-HT]. The induced current, I, was the peak current measured with 5-HT current minus the initial agonist-independent current. The data collected from each oocyte was fit with the Hill equationI/Io = 1/(1 + (EC50/[X])∧h), where the EC50 is the concentration at which there is half-activation, and h is the Hill coefficient. The deactivation time (T50) was determined by measuring the time taken for the current to decrease to 50% of its level before removing the 5-HT.
The irreversible effect of MTS on 5-HT-induced currents was examined using one of two protocols. Protocol 1 consisted of two 5–10 sec pulses of 10 μm 5-HT (I1,I2), 2–3 min of wash, 1 min of MTS (1 mm) reagent in the absence or presence of 10 μm 5-HT, 3.5 min of wash, and two 5–10 sec pulses of 5-HT (I3,I4). The percentage inhibition–potentiation was calculated using the equation (I4/I2− 1) * 100. For measuring the Mg2+inhibition, an additional 15–20 sec 5-HT pulse in 0 mm Mg2+ was applied between the two 5–10 sec 5-HT pulses. Mg2+-dependent inhibition was calculated using the equation (1 −I1/IMg(0)) * 100. Protocol 2 was used to measure the rate of modification: a 5–10 sec pulse of 10 μm 5-HT (I0), 1–2 min of wash, a 5–10 sec pulse of MTS (0.04–2 mm) reagent in the absence or presence of 10 μm 5-HT, and a 1–2 min wash (times varied for different mutants to allow recovery from desensitization). The cycle was repeated until complete modification occurred (I1 −In). The 5-HT-induced current (I0 −In) was plotted as a function of cumulative exposure time to MTS. To account for the decrease in the current attributable to desensitization for V291C, several pulses of 10 μm 5-HT were delivered first before starting the rate experiment. The rate of modification for G288Cwt was not measured. The data from each oocyte were fit with a single exponential: y =y0 + ae−t/τ for potentiation or y = y0 + a(1 − e−t/τ) for inhibition. The bimolecular rate constant equals 1/(τ * [MTS]). The change in agonist-independent current was quantified by taking the difference between the basal current just beforeI3 (after the 3.5 min wash) and the initial basal current (after I2) and then dividing by I3 (this represents the total amount of current that can pass through modified channels in the open state).
All values were reported as mean ± SEM. Data were analyzed for statistical significance (SigmaStat 2.0; SPSS, Chicago, IL) using one-way ANOVA, followed by Bonferroni's post hoc test with wild-type as control or using a paired t test for Mg2+-dependent inhibition. Values ofp < 0.05 were considered significant.
RESULTS
Effect of cysteine substitutions on 5-HT3a
Neurotransmitter stimulation of ligand-gated channels produces a mixed population of closed, open, and desensitized states. For example, in the presence of a saturating dose of 5-HT (10 μm), 5-HT3a channels opened rapidly and then quickly entered a nonconducting, desensitized state (Fig.1B). Wilson et al. (2001) demonstrated recently that the pattern of MTS reactivity in desensitized nAChR channels differs from that of channels in the open or closed state. To minimize the contribution of the desensitized state, we recorded 5-HT-induced currents in the absence of extracellular Ca2+ (Fig.1B), which dramatically slowed the rate of desensitization (Yakel et al., 1993).
To localize the position of the gate in 5-HT3a, we examined the accessibility of cysteine substitutions in the M2 domain of open and closed mutant channels. If treatment with MTS leads to an irreversible reduction in the amplitude of agonist-induced current, then the cysteine side chain is likely positioned in or near the pore of the channel. If a cysteine-substituted channel is modified equally well in the open and closed states with extracellularly applied MTS reagents, then the channel gate is located on the cytoplasmic side of the engineered cysteine.
We first examined the effect of 1 mm MTSET (extracellular) on the 5-HT-induced current through wild-type channels (Fig.1C). Wild-type 5-HT3a channels exposed to MTSET in either the closed or open state showed no decrement in 5-HT-induced current. We therefore used the wild-type 5-HT3a channel for the introduction of new cysteine substitutions and assumed that the eight native cysteines remained silent. Twenty-three amino acids in the M2 transmembrane domain (D274–V296) were systematically replaced with a cysteine (one at a time) (Fig. 1A). Nineteen 5-HT3a cysteine mutants gave rise to measurable 5-HT-induced currents after injection of the cRNA intoXenopus oocytes. In contrast to our results with S290C,Reeves et al. (2001) reported that 5-HT3achannels containing the S290C substitution failed to express inXenopus oocytes. It is possible that differences in the noncoding region or the use of a splice variant (short vs long 5-HT3a) allowed S290C to express in our study. Four mutants that gave little or no 5-HT-induced currents were studied by coinjecting the mutant cRNA with wild-type cRNA in ∼1:1 ratio (L286Cwt, G288Cwt, Y289Cwt, and F292Cwt). Although it is probable that a small percentage of current came from wild-type channels, currents recorded from coinjected eggs had properties (e.g., Mg2+-dependent inhibition and deactivation) unique from those of wild-type, indicating coassembly of both wild-type and mutant subunits (Table1).
Table 1.
Properties of cysteine-substituted 5-HT3achannels
| EC50 | Hill | Mg inhibition | Deactivation | |||||
|---|---|---|---|---|---|---|---|---|
| Before MTSET | After MTSET | T50 ratio | ||||||
| μm SEM | h SEM | n | % SEM | % SEM | n | Ratio SEM | n | |
| WT | 1.67 ± 0.09 | 2.0 ± 0.1 | 8 | −41 ± 3 | −39 ± 3 | 5 | 0.95 ± 0.03 | 5 |
| V296C | *0.22 ± 0.01 | 1.8 ± 0.1 | 5 | −26 ± 3 | −29 ± 3 | 7 | 1.03 ± 0.05 | 7 |
| I295C | *0.27 ± 0.02 | 1.9 ± 0.2 | 4 | 1-160−37 ± 3 | −29 ± 2 | 10 | *2.68 ± 0.27 | 10 |
| I294C | *0.94 ± 0.06 | 2.6 ± 0.1 | 7 | 1-160−71 ± 2 | −59 ± 2 | 7 | 1.46 ± 0.11 | 7 |
| L293C | 1.37 ± 0.12 | 1.6 ± 0.1 | 6 | −42 ± 3 | −38 ± 3 | 7 | 0.74 ± 0.02 | 4 |
| F292Cwt | nt | nt | −41 ± 3 | −39 ± 2 | 6 | *2.09 ± 0.16 | 5 | |
| V291C | *0.35 ± 0.02 | 3.0 ± 0.2 | 4 | −53 ± 5 | −61 ± 4 | 9 | 1.00 ± 0.07 | 9 |
| S290C | *0.92 ± 0.11 | 2.3 ± 0.3 | 5 | 1-160−15 ± 2 | −28 ± 3 | 7 | 0.91 ± 0.06 | 7 |
| Y289Cwt | nt | nt | −14 ± 3 | −14 ± 3 | 4 | 1.07 ± 0.04 | 4 | |
| G288Cwt | nt | nt | −75 ± 3 | −63 ± 3 | 11 | *2.21 ± 0.13 | 6 | |
| L287C | *0.30 ± 0.01 | *3.0 ± 0.4 | 9 | 1-160−56 ± 1 | −40 ± 1 | 5 | 1.54 ± 0.16 | 9 |
| L286Cwt | nt | nt | −41 ± 7 | −35 ± 6 | 4 | 1.01 ± 0.05 | 4 | |
| L285Cwt | *2.32 ± 0.06 | 2.8 ± 0.1 | 4 | −40 ± 1 | −40 ± 2 | 5 | 0.88 ± 0.05 | 10 |
| T284C | *1.15 ± 0.11 | 3.0 ± 0.1 | 5 | 1-160−37 ± 2 | −47 ± 2 | 5 | 0.87 ± 0.03 | 15 |
| I283C | *1.00 ± 0.02 | *3.2 ± 0.1 | 5 | −18 ± 2 | −18 ± 2 | 4 | 0.97 ± 0.04 | 13 |
| K282C | 1.46 ± 0.12 | 2.2 ± 0.2 | 8 | −32 ± 5 | −29 ± 5 | 5 | 1.02 ± 0.04 | 14 |
| F281C | *0.83 ± 0.02 | 2.0 ± 0.1 | 6 | −14 ± 2 | −14 ± 1 | 7 | 1.40 ± 0.07 | 21 |
| S280C | *2.21 ± 0.03 | *3.3 ± 0.1 | 10 | 1-160−26 ± 2 | −36 ± 2 | 5 | 1.09 ± 0.07 | 8 |
| V279C | 1.87 ± 0.15 | 2.7 ± 0.3 | 6 | −29 ± 2 | −28 ± 1 | 5 | 1.03 ± 0.03 | 15 |
| R278C | 0.78 ± 0.16 | 1.2 ± 0.2 | 12 | 1-160−52 ± 4 | −16 ± 2 | 10 | 1.37 ± 0.07 | 12 |
| E277C | 1.36 ± 0.07 | 2.0 ± 0.2 | 7 | 1-160−13 ± 1 | −7 ± 1 | 7 | 1.01 ± 0.04 | 15 |
| G276C | *1.18 ± 0.01 | 2.7 ± 0.2 | 5 | −67 ± 2 | −68 ± 2 | 5 | 1.06 ± 0.03 | 6 |
| S275C | 1.62 ± 0.07 | *3.2 ± 0.2 | 4 | 1-160−40 ± 3 | −15 ± 2 | 5 | *3.77 ± 0.79 | 7 |
| D274C | 1.27 ± 0.12 | 2.3 ± 0.2 | 6 | −18 ± 2 | −20 ± 2 | 11 | *1.97 ± 0.13 | 18 |
The EC50 and Hill coefficient were determined for all mutants that expressed as homomultimers. L286Cwt, G288Cwt, Y289Cwt, and F292Cwt, which were coexpressed (1:1) with wild-type 5-HT3a, were not tested (nt). The percentage of Mg2+ inhibition was measured before and then after 1 min exposure to MTSET (1 mm). The T50 ratio was measured with MTSET (1 mm) for the majority of cysteine mutants and MTSEA (1 mm) for F292Cwt and L293C.
p < 0.05 indicates statistical difference from wild-type using one-way ANOVA.
F1-160: p < 0.05 indicates statistical difference between modified and unmodified levels using a Student's t test. All values are mean ± SEM with the indicated n value.
To assess whether the cysteine mutation grossly altered the channel structure, we compared the dose–response curves (EC50) for serotonin activation of the cysteine-substituted mutants that formed homomultimers. The EC50 changed less than twofold for 13 cysteine substitutions and twofold to eightfold for the remaining six cysteine substitutions. The largest shifts in EC50occurred with cysteine substitutions near the extracellular half of the M2 transmembrane domain. These changes in EC50are relatively small considering that each homopentamer contained five cysteine mutations. For comparison, some single cysteine mutations in two of five nAChR subunits produced 50-fold changes in the EC50 (Akabas et al., 1994). The Hill coefficient for 5-HT3 mutants varied from 1.2 to 3.3 compared with ∼2 for wild-type channels (Table 1). The change in Hill coefficient suggests that the cooperativity between agonist binding and channel activation changed with the mutation, although this was not investigated further. Some mutations were also directly activated by MTS reagents (Fig. 1D). A shift in the EC50 and/or inappropriate activation by a ligand (e.g., MTSET and MTSEA) are not unexpected outcomes for point mutations in an allosteric protein. Mutations in nAChR have been reported to convert antagonists to agonists and shift the EC50 for ACh activation (Labarca et al., 1995;Palma et al., 1996, 1999). Although mutations in the channel protein can alter its function, the general mechanism governing channel activation appears to be preserved in the majority of these mutants. Overall, the small shift in EC50 for the majority of 5-HT3a mutant channels suggests that large structural changes were minimal in 5-HT3a mutant channels (Table 1).
State-dependent MTS modification of 5-HT3a mutants
We first examined the MTS sensitivity of all 5-HT3a cysteine mutant channels with MTSET in either the absence (closed state) or presence (open state) of 10 μm 5-HT (this dose was 4.5–45 times the EC50). We chose MTSET because it has a permanent positive charge and is not membrane permeant (Holmgren et al., 1996). In the closed state, MTSET (1 mm) treatment for 1 min produced no irreversible change in the 5-HT-induced current for 19 cysteine mutants (Fig.2B). Four mutants, S290C, V291C, I295C, and V296C, however, appeared to be directly opened by MTSET in the absence of 5-HT (Fig. 1D). To study these channels in their closed state, we coapplied the 5-HT3 antagonist MDL-72222 (Maricq et al., 1991), which was found to inhibit the MTSET-activated current (Fig.1D). In the presence of MDL-72222 (1 μm; ∼300 times the IC50), MTSET treatment did not irreversibly alter the amplitude of the 5-HT-induced current for S290C, V291C, and V296C mutants. For I295C, however, MTSET treatment in the presence of MDL-72222 irreversibly potentiated the 5-HT-induced current, although there was no detectable direct activation with MTSET. Thus, only I295C appears to be modified with MTSET in the closed state.
Fig. 2.

Reactivity of cysteine-substituted 5-HT3a channels in open and closed states with MTSET.A, Continuous current recorded from oocyte injected with cRNA for wild-type, I295C, S290C, S280C, or R278C. Solid bar indicates 10 μm 5-HT, hatched bar indicates 1 mm MTSET, and + and − refers to extracellular solution with (+) or without (−) Mg2+. MTSET exposure irreversibly inhibited the 5-HT-induced current for S290C and S280C. The 5-HT-induced current for R278C increased in the +Mg2+ but not the −Mg2+ solution after MTSET treatment. I295C appeared to have slower deactivation kinetics after MTSET treatment (see Table 1). B, Summary of the percentage inhibition or potentiation after 1 min exposure to MTSET in either the closed (open bars) or open (solid bars) states (n = 4–17). L286Cwt, G288Cwt, Y289Cwt, and F292Cwt designate coexpression with wild-type 5-HT3a. MTSET was coapplied with 1 μmMDL-72222 to study closed-state inhibition for S290C, V291C, I295C, and V296C (MDL). The 5-HT-induced current was adjusted for desensitization for S290C (∼12%), V291C (∼16%), I294C (∼8%), and I295C (∼6%) that occurred after 1 min of 5-HT stimulation alone. *p < 0.05 indicates statistical difference from wild-type channels using one-way ANOVA.
The accessibility of cysteine mutants in the open state, on the other hand, was strikingly different from that of closed channels. MTSET irreversibly reduced the 5-HT-induced current by 40–80% of control for six cysteine 5-HT3a mutants and potentiated the 5-HT-induced current for four mutants (Fig.2A,B). Although G276C and V296C fell out of statistical significance using one-way ANOVA, they both appeared to be inhibited after MTSET treatment in the open state. Our results with MTSET are similar but not identical to those of Reeves et al. (2001), who recently used SCAM to study 5-HT3a in the open state. The use of a splice variant in their study might explain some of the differences. In addition, we accounted for a reduction in current caused by desensitization and an apparent potentiation caused by a change in Mg2+-dependent inhibition. Our results show that, of the mutants modified in the open state with MTSET, only I295C was modified in both closed and open states. These results suggest that 5-HT3a channels possess a gate on the extracellular side of the M2 transmembrane domain. Alternatively, the extracellular portion of 5-HT3a might be too narrow for MTSET to modify exposed cysteines in the closed state.
To test this idea, we studied the effect of MTSEA, which is smaller than MTSET [3.6 vs 5.8 Å for the head group (see Fig. 5)], on cysteine substitutions L286C through V296C. In the closed state, V296C and L293C were both modified by 1 mm MTSEA (Fig.3A,C). Interestingly, MTSEA treatment increased the 5-HT-induced current by 350% for L293C in the closed state, whereas MTSET had little effect (Fig. 3A,C). Although L287C exhibited some potentiation of 5-HT-induced current with MTSEA treatment in the closed state, this increase was not statistically significant. In the open state, MTSEA treatment modified six cysteine substitutions [G288Cwt, S290C, V291C, F292Cwt, L293C, and V296C (Fig. 3C)]. Of these, F292Cwtand L293C were not significantly modified with MTSET, suggesting that they lie within a region of the channel that is too small for MTSET to access but large enough for MTSEA to enter. Additionally, MTSET modification of residues S290C and V291C inhibited 5-HT-induced current, whereas modification with MTSEA did not. One possibility is that the pore is relatively wide at this level in the channel, and attachment of a smaller MTS moiety, such as MTSEA, may have negligible effects on current flow. Combining the results from experiments with MTSEA and MTSET, we conclude that amino acids L293 through V296 are accessible in the closed and open state, suggesting that the gate is on the cytoplasmic side of L293.
Fig. 5.

MTS modification in the open state produces agonist-independent currents for cysteine substitutions in the middle of the M2 transmembrane domain. Continuous current recorded from oocyte injected with the cRNA for V291C (A), S290C (B), or L287C (C). One minute exposure to MTS reagent (1 mm) in the presence of 5-HT (10 μm) produced an agonist-independent current for L287C, S290C, V291C, and I295C (data not shown) with MTSET and for S290C and V291C with MTSEA and MTSES. The structure of MTS moiety that would covalently attach to the cysteine sulfhydryl is shown above.D, Continuous current recorded from oocyte injected with cRNA for V291C. In the absence of 5-HT, 50 mm DTT decreased the agonist-independent current to the level before MTSET modification. Note that 5-HT (10 μm) fully activated the V291C channel after DTT treatment.
Fig. 3.

Reactivity of cysteine-substituted 5-HT3a channels in open and closed states with MTSEA.A, B, Continuous current recorded from oocyte injected with the cRNA for L293C or G288Cwt. The 5-HT-induced current through L293C was potentiated with MTSEA (1 mm) in the closed and open states. G288Cwt was modified only in the open state.C, Summary of the percentage of inhibition or potentiation of current for mutants L286Cwt through V296C after 1 min exposure to MTSEA (1 mm) in the closed (open bars) or open (solid bars) states (n = 4–10). V296C and L293C were modified in both the closed and open states. The inhibition or potentiation for S290C, V291C, I294C, I295C, and V296C were adjusted as described in Figure 2. *p < 0.05 indicates statistical difference from wild-type channels using one-way ANOVA.
To quantitate the difference in accessibility of cysteines in the open and closed states, we measured the bimolecular rate constant for MTS modification (Fig. 4). We used a protocol in which the MTS reagent was applied repeatedly in the absence or presence of 5-HT for 5–10 sec (see legend of Fig. 4). The rates of closed-state modification were not measured for E277C, R278C, S280C, T284C, G288Cwt, F292Cwt, and I294C because 1 mm MTS treatment did not significantly affect the 5-HT-induced current (Fig. 2B). The rate of modification for S290C, V291C, L293C, I295C, and V296C in the closed state was <50 m/sec. However, the rates for S290C, V291C, I295C, and V296C are overestimates because these mutants were directly opened by MTSET (Figs. 1D,4C, a) (MDL-72222 washed out too slowly for using in rate measurements). Because there was no significant modification of S290C and V291C when MTS was coapplied with MDL-72222 (Fig.2B), the actual rate of modification in the closed state was likely <10 m/sec (Fig. 4C,dotted lines). In contrast to the closed state, the rate of modification for open channels varied from ∼500 to ∼5000m/sec, with a majority of the faster rates occurring on the cytoplasmic side of M2 transmembrane domain. For comparison, these rate constants are comparable with those reported for modification of nAChR channels (Pascual and Karlin, 1998). Together, the rate of modification increased 100- to 1000-fold during activation for cysteine substitutions on the cytoplasmic side of F292C and increased only onefold to 10-fold for cysteine substitutions on the extracellular side of F292C. This difference in state dependence of modification is consistent with data collected by steady-state modification (Figs. 2, 3), supporting the conclusion that the amino acids on the cytoplasmic side of L293C are primarily inaccessible in the closed state and become readily available for modification in the open state (see Fig. 7B).
Fig. 4.

Rate constants for gated access to cysteine substitutions in the M2 transmembrane domain of 5-HT3a.A–C, Determination of the rate constant for modification with MTSET or MTSEA. A, Continuous current recorded from oocyte injected with the cRNA for E277C. Alternating pulses of 5-HT alone and 5-HT with MTSET (0.1 mm) were delivered. B, The amplitude of 5-HT-induced current was plotted as a function of cumulative exposure time to MTSET and fit with a single exponential having a time constant of 3.8 sec.C, Average rate constants for modification with MTSET (whitecircles, blackcircles) or MTSEA (circleswithhorizontal stripes, circles with diagonal stripes) in the closed and open states, respectively (n = 4–6). Circles with crosses indicate that closed-channel modification was not statistically different from wild type and was likely ≤10m/sec (see Fig. 2B). Note that closed rates are overestimates for those mutants in which MTSET opened the channel directly, denoted by a. Dashed line for S290C and V291C indicates there was no detectable effect of MTSET or MTSEA when coapplied with MDL-72222.
Fig. 7.
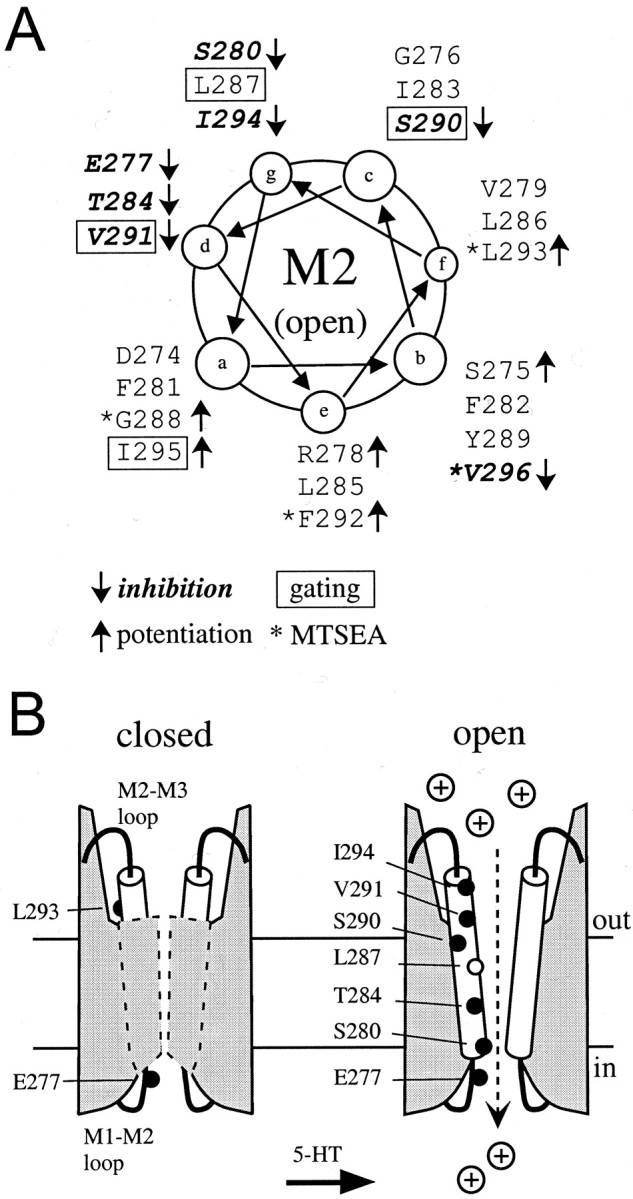
A, Helical wheel representation of the MTS-dependent changes in current. Note the majority of cysteine substitutions that are inhibited with MTS reagents (boldand italicized, ↓) fall along the same side of the helix, as do those that are potentiated (↑). Mutants that are locked open are boxed; mutants modified by MTSEA have anasterisk. B, Schematic model shows the extent of the gate in 5-HT3a, which is intracellular to L293. For clarity, only two of five subunits are shown with the M2 transmembrane domain. The M2 is modeled as a straight α helix in an open channel (the structure of M2 in the closed state is unknown). To explain the MTS accessibility of four cysteine substitutions located on the extracellular side of F292, the α helix is postulated to be water accessible on more than one face of the helix. The dashed area indicates that these cysteine substitutions (see Figs.2B, 3C) are accessible in the open but not closed state, suggesting that the gate extends from F292 to a more intracellular residue whose position can be determined by application of intracellular MTS. In the open state, the modification of cysteine substitutions leading to an inhibition of current are shown (black circles). L287C (9′) (white circle) was implicated previously in gating of nAChR. The narrowest region of the pore in nAChR is between E277 and S280 (Corringer et al., 2000).
We next searched for patterns of inhibition or potentiation with MTS reagents in the open state. In an α helix, amino acids that align along one face of the helix are spaced three to four amino acids apart. This pattern is readily seen in a helical wheel plot (see Fig.7A). Except for V296C, all of the cysteine substitutions that were inhibited by MTS reagents cluster on one side of the helix (see Fig. 7A, ↓). Interestingly, those cysteine substitutions that were potentiated by MTS cluster on the opposite side of the helix (see Fig. 7A, ↑). If we define pore-facing amino acids as those that were irreversibly inhibited with MTS reagents, then the M2 transmembrane domain appears to be α helical, with hydrophilic amino acids on the cytoplasmic side and hydrophobic residues on the extracellular side of the M2 transmembrane domain.
Could the introduction of the cysteine in the mutant increase the MTS reactivity of one or more of the native cysteines? To test this idea, the S280C mutation was generated in the background of a 5HT3a subunit in which two native cysteines positioned close to the M2 transmembrane domain, C270 (in M1) and C316 (in M3), were mutated to alanine. In this triple mutant, MTSET irreversibly reduced the 5-HT-induced current by ∼70%, similar to the reduction observed with S280C alone. Removing all of the native cysteines in 5-HT3a is impractical, because a canonical pair of cysteines is located in the ligand-binding domain of most ligand-gated channels and is essential for function (Corringer et al., 2000). Although we have not studied all mutants in the C270A/C316A background, it appears unlikely that native cysteines contributed to the MTS effects reported here.
MTS-dependent potentiation of 5-HT-induced current
The potentiation of 5-HT-induced current with MTSET or MTSEA for six cysteine substitutions (S275C, R278C, G288Cwt, F292Cwt, L293C, and I295C) was unexpected. We anticipated that covalent attachment of a positively charged moiety to a cysteine in the channel would reduce the 5-HT-induced current, because 5-HT3a is a cationic channel. One possible explanation for potentiation of current is that the MTS treatment shifted the EC50 to lower concentrations of 5-HT. This possibility was unlikely because a maximal dose of 5-HT (10 μm) was used to activate wild-type and mutant channels. Alternatively, MTSET modification might have altered divalent inhibition; 5-HT3 channels are inhibited by extracellular divalent cations (Lovinger, 1991; Maricq et al., 1991). To explore this possibility, we quantified the inhibition produced by extracellular Mg2+ before and after exposure to MTSET. For S275C and R278C, the Mg2+-dependent inhibition decreased after modification with MTSET, which led to an apparent potentiation of 5-HT-induced current when recorded in the Mg2+-containing solution. In contrast, the current flowing through these channels while in the Mg2+-free solution was not changed after modification (Fig. 2A, R278C; Table1).
A change in Mg2+-dependent inhibition, on the other hand, did not appear to explain the potentiation of 5-HT-induced current for G288Cwt, F292Cwt, L293C, and I295C mutants (Table1). An alternative explanation is that MTS modification increased the single-channel conductance or open-channel probability, which would produce a potentiation of 5-HT-induced current. The small conductance of wild-type 5-HT3a channels (<1 pS), however, precluded measuring the mean open-channel lifetime or conductance directly (Hussy et al., 1994). We measured the deactivation time (T50) of the channel, which is the time taken for channels to close after removing 5-HT. TheT50 depends on the single-channel open and closed lifetime, as well as the off rate of 5-HT from the ligand-binding site (Wang et al., 1999). The deactivation times for G288Cwt, F292Cwt, and I295C mutants were approximately two to three times slower after modification with MTSET (Table 1). For comparison, MTSET treatment of wild-type, E277C, S280C, or V291C channels produced no significant change in the deactivation rate (Table 1). The deactivation time for L293C, however, was slightly faster after MTSEA treatment, suggesting that the single-channel conductance increased after modification of this cysteine mutant. Although we do not fully understand the underlying mechanism of potentiation, the results demonstrate that cysteines substitutions at S275, R278, G288, F292, L293, and I295 are accessible to MTS reagents in the open state. Interestingly, cysteine mutants that give rise to potentiation during MTS modification cluster on one side of the helix, whereas those that are inhibited cluster on the opposite side, thought to face the pore (see Fig. 7A).
Middle of M2 transmembrane domain implicated in conformational change
Surprisingly, MTSET treatment of four cysteine mutants (L287C, S290C, V291C, and I295C) in the open state increased the basal current, which persisted in the absence of 5-HT (“agonist-independent current”) (Figs. 2A,5). The amplitude of this agonist-independent current increased to 15–50% of the modified 5-HT-induced current, which reflects the total amount of current that can flow through the modified channels (Fig.6). For comparison, MTS modification of a cysteine-substituted voltage-gated K+channel also produced constitutively open channels (Liu et al., 1997). We tested whether this change depended on the type of MTS reagent. MTSEA (1 mm) and MTSES (1 mm) did not induce an agonist-independent current for I295C and L287C. In contrast, both MTSEA and MTSES increased the agonist-independent current for S290C and V291C in the open state (Figs. 5, 6). For S290C and V291C, MTSES administered in the closed state did not give rise to an agonist-independent current (data not shown), consistent with our previous conclusion that these two sites are inaccessible in the closed state. Thus, regardless of the charge or size of the MTS reagent, MTS treatment in only the open state produced a large, agonist-independent current for S290C and V291C.
Fig. 6.

Summary of the change in agonist-independent current produced by MTSET, MTSEA, and MTSES. Bar graphs show the average amplitude of agonist-independent current after MTS treatment, expressed as a percentage of the 5-HT-induced current after MTS modification (see Materials and Methods) (n = 3–13). $ indicates that the pre-modified 5-HT-induced current was used to calculate the percentage change in agonist-independent current because MTSEA treatment eliminated >95% of the 5-HT-induced current. * indicates statistical difference from wild type using one-way ANOVA. nt, Not tested.
One explanation for the agonist-independent current is that modified channels become locked open after removal of 5-HT (see Fig. 8). To test this idea, we examined the effect of DTT (50 mm) in the absence of 5-HT. If MTSET-modified channels were locked open, then DTT would be expected to have access to the pore and, during reduction of the newly formed disulfide bond, enable the mutant channel to close normally. Indeed, the agonist-independent current declined to within ∼5% of the premodified level when V291C was exposed to DTT in the absence of 5-HT (Fig. 5E). DTT treatment also restored the agonist-independent current to within 5% of the premodified level for L287C, S290C, and I295C channels modified with MTSET. In addition to the recovery of agonist-independent current, the amplitude of 5-HT-induced current returned to 80–130% of the original level after DTT treatment.
Fig. 8.
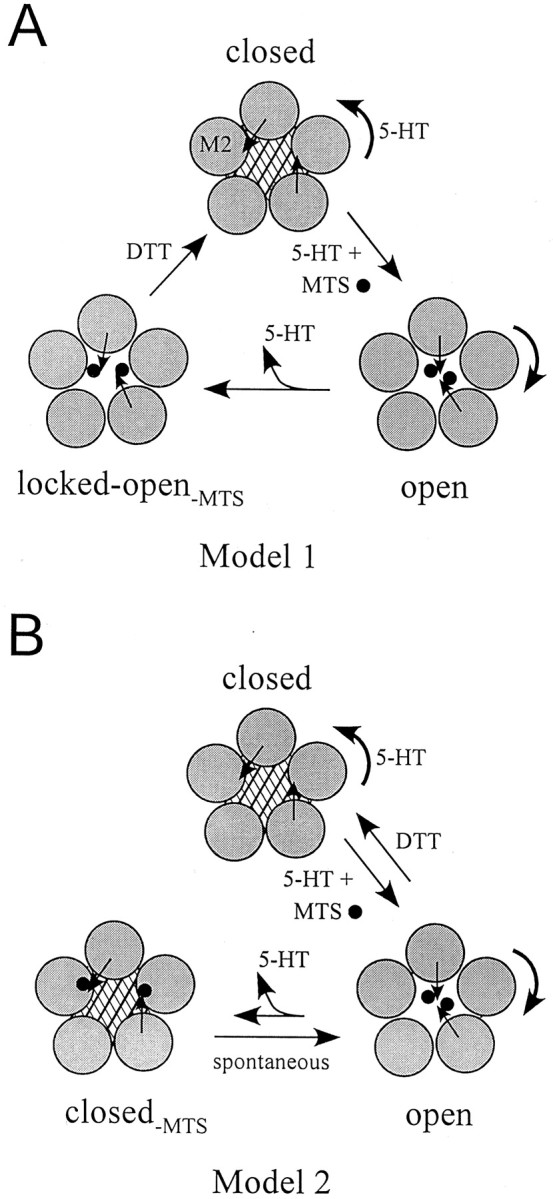
Two working models that explain the agonist-independent current. In both models, 5-HT produces a rotation of M2 transmembrane domain, thereby exposing an ionized sulfhydryl on the cysteine (black arrow), such as at position V291 (only two cysteines are shown for clarity). MTS (black sphere) covalently attaches to the sulfhydryl in the open state (open). A, After removal of 5-HT, the channel fails to close completely in Model 1 because of steric hindrance (locked open-MTS). B, In Model 2, modified channel closes fully in the absence of 5-HT. This closed-MTS state is unstable, however, allowing the channel to spontaneously open. In both models, DTT can reduce the disulfide to restore the channel to its original unmodified closed state.
DISCUSSION
Neurotransmitter-gated ion channels are important for mediating the response of “fast” neurotransmitters and have evolved a transduction mechanism that tightly couples the binding of neurotransmitter to a conformational change in the channel pore. Our analysis of 5-HT3a channels has revealed a cluster of amino acids in the middle of the M2 transmembrane domain that appears to move during channel activation. In addition, our results suggest that the middle and cytoplasmic segments of the M2 transmembrane domain are inaccessible to MTS reagents in the closed state and, during activation with 5-HT, become readily accessible to MTS reagents. Finally, the pattern of inhibition is consistent with an α helical structure for the M2 domain.
SCAM of the M2 in 5-HT3a and nAChR
5-HT3a and nAChR share 29% homology in the M1–M3 transmembrane domains and likely conserve the molecular mechanism underlying channel activation. Consistent with this idea, replacing the N-terminal domain of 5-HT3a with the homologous sequence from α7-nAChR produced a chimeric channel that was activated with ACh but had pore properties of 5-HT3a (Eiselé et al., 1993). We therefore expected the results of SCAM with 5-HT3a channels to be similar to those with muscle nAChR.
The number of cysteine substitutions that were modified in the open state with MTS reagents for both 5-HT3a and nAChR channels is remarkably large. In our study, seven cysteine substitutions modified in 5-HT3a overlap with those modified in cysteine-substituted muscle nAChR channels (Akabas et al., 1994; Wilson and Karlin, 1998). The narrowest region of the 5-HT3a pore, however, appears slightly larger than that in nAChR, because the amino acid at the −1′ position (E277C) was inhibited in 5-HT3a with extracellular MTSET but was inaccessible in nAChR. The inhibition of cysteine substitutions spaced every three to four amino acids apart in 5-HT3a is consistent with an α helical structure for the middle and cytoplasmic segments of the M2 transmembrane domain (Fig.7A). Our finding that M2 is mostly α helical in the open state is consistent with a recent SCAM study using a splice variant of 5HT3a (Reeves et al., 2001). For the nAChR, a similar pattern of MTS inhibition was found for the M2 segment (Akabas et al., 1994; Pascual and Karlin, 1998; Wilson and Karlin, 1998). Moreover, ultrastructural (Unwin, 1995;Miyazawa et al., 1999) and nuclear magnetic resonance studies (Opella et al., 1999) also suggested that the M2 is α helical. Whether the middle of the helix is kinked (Unwin, 1995) or extended (Akabas et al., 1994) in the open state is unclear from our studies. On the extracellular side of M2 (S290-V296), however, we found several cysteine substitutions in a row that were modified in the open state, suggesting that either the helix unwinds at this point or it is positioned in a crevice, in which multiple sides of the helix are accessible (Fig. 7B). Recently, this type of water-filled crevice surrounding a transmembrane helix was reported for a voltage-gated K+ channel (Starace and Bezanilla, 2001).
In contrast to the open state, the accessibility of cysteine substitutions in the closed state appeared to be very different between nAChR and 5-HT3a. With cysteine substitutions S275C through F292Cwt, MTS exposure in the closed state produced no irreversible change in 5-HT-induced current. In contrast, MTS treatment of six cysteine-substituted nAChR channels in the closed state inhibited ∼40–70% of the acetylcholine-induced current (Akabas et al., 1994). In particular, 2′T in nAChR, which is located on the cytoplasmic side of M2 transmembrane domain, was inhibited by ∼70% with extracellular MTSET (Akabas et al., 1994). The homologous amino acid in 5-HT3a, S280C, was clearly inaccessible to MTSET in the closed state, even at high MTSET concentrations (5 mm). The dissimilar pattern of accessibility in the closed state could indicate a slightly different structure for the closed-channel pore in 5-HT3aand nAChR. For example, the extracellular region of the 5-HT3a pore may be smaller or display different electrostatics. Consistent with this idea, 5-HT3aand nAChR differ in their sensitivity to extracellular Ca2+, single-channel conductance, and inhibition by charged local anesthetics (Maricq et al., 1991; Corringer et al., 2000). One experimental difference between SCAM of 5HT3a and nAChR worth noting is that 5-HT3a is a homopentamer, whereas nAChR is a heteropentamer. Thus, 5-HT3a had five cysteine substitutions and nAChR had only two cysteine substitutions (in the α1 subunit). The larger number of cysteine substitutions in 5-HT3a, however, would be expected to increase the probability of closed-state modification.
Working model for the gate in 5-HT3a channel
We define the “gate” as the region of the channel that prevents the inappropriate flow of ions through the channel pore. The activator (i.e., voltage, neurotransmitter, and pH) leads to a conformational change in the protein that moves the gate and allows ions to permeate the open pore. The gate therefore serves two functions: a barrier to ion permeation and a link to the transduction machinery.
There are two important findings with SCAM of 5-HT3a that help pinpoint the location of the gate. First, the middle and cytoplasmic segments of the M2 transmembrane domain (S275 through F292) were accessible in open but not closed states. In contrast, three amino acids on the extracellular side of F292 (L293C, I295C, and V296C) were accessible in both open and closed states. Surprisingly, I294C was only modified in the open state, whereas the more cytoplasmic L293C was modified in both open and closed states. Similarly, residues above the putative gate were also found to exhibit state-dependent modification in the nAChR (Akabas et al., 1994). One possible explanation is that I294C is buried within a region of the protein or “masked” by a residue in the closed state, making it inaccessible to MTS reagents. Nonetheless, the observation that L293C is the last residue to be modified equally well in the open and closed states suggests that the gate is intracellular with respect to L293. It is possible that MTS reagents are sterically or electrostatically restricted from further entry beyond this residue. Additional experiments are necessary to determine whether smaller cations can gain access to sites deeper within the 5-HT3a pore, as was found with cyclic nucleotide-gated channels (Flynn and Zagotta, 2001).
The second finding localizing the gate, however, is that exposure of two cysteine mutants in the middle of M2, S290C and V291C, to all three MTS reagents generated agonist-independent currents. Notably, this change in current occurred only when MTS was applied in the open state. Similarly, Reeves et al. (2001) reported that MTSES modification of V291C produced agonist-independent currents. How does MTS modification lead to agonist-independent currents? Two models can explain this change in gating. First, MTS attachment could sterically hinder channel closure, thereby locking the channel in an unliganded open state (Fig.8A, Model 1). This unliganded open state is not fully open, however, because reapplication of 5-HT elicits more inward current. Alternatively, covalent attachment of the bulky reaction group to the cysteine could increase the probability of spontaneously opening, producing an agonist-independent current (Fig. 8B, Model 2). If the MTS moiety is accessible to DTT in either the locked–open-MTS state (Model 1) or the modified open state (Model 2), then reduction of the disulfide bond and the removal of MTS from the channel pore could allow for the passage of current before the closing of the channel. The sudden increase in amplitude of current during the initial response to DTT in the absence of 5-HT is consistent with this conclusion (Fig.5E). Such a response is expected for a channel in which the rate of removing the inhibitor is faster than the rate of channel closure. Single-channel recordings of MTS-modified channels will be necessary to distinguish between these two models. For either model, however, S290C and V291C appear to be important in the activation of 5-HT3a.
Together, our data are consistent with a model in which the extracellular barrier to ion permeation exists in the middle of the M2 domain of 5-HT3a (Fig. 7B). During ligand binding, the M2 helix likely rotates (Figs. 7, 8), as has been proposed for the nAChR (Unwin, 1995). The intracellular side of the gate of 5-HT3a is not known, however, but can be addressed in the future by examining the state dependence of intracellularly applied MTS reagents. With this method, Wilson et al. (1998) determined that the intracellular region of the nAChR gate was located near the −1′ position (E277C in 5-HT3a) (Fig. 7B). We speculate that the M2 helices are tilted in the open state so that the narrowest region of the pore is located on the cytoplasmic side of the channel (Corringer et al., 2000).
Studies on other ligand-gated channels also support a model for a centrally located gate that includes amino acids at the 12′ and 13′ positions, which are homologous to S290 and V291. First, amino acids in the 12′ and 13′ positions (and 9′) are extremely well conserved (85%–100%) in cationic ligand-gated channels (Le Novere and Changeux, 1999). Second, some mutations in the gate would be expected to open channels in the absence of neurotransmitter (unliganded open state). Indeed, mutations of the 9′L, 12′S/T, or 13′V in the M2 transmembrane domain increased the frequency of spontaneous openings or increased the mean open time (Bertrand et al., 1997; Pan et al., 1997;Chang and Weiss, 1998; Chen and Auerbach, 1998; Dalziel et al., 2000;Dang et al., 2000). In another study, MTS modification of a cysteine substitution of the 13′V in nAChR also increased the mean open lifetimes (Zhang and Karlin, 1998). Finally, England et al. (1999)showed that mutations of amino acids in the 13′, 16′, and 19′ positions in nAChR produced larger shifts in the EC50 than did mutations in the 9′ position.
Our conclusions concerning the structure of the M2 are inferred after a systematic analysis of cysteine-substituted channels exposed to extracellular MTS reagents in closed and open states. Is there good concordance between the structure of a protein determined by SCAM and that by x-ray crystallography? In the handful of cases in which there is a three-dimensional structure, there appears to be good agreement. For example, the narrowest region of the α-hemolysin pore identified with SCAM closely matches the region visualized in the three-dimensional structure (Song et al., 1996; Movileanu et al., 2001). For G-protein-coupled neurotransmitter receptors, the location of the ligand-binding pocket inferred with SCAM overlaps with that observed in the x-ray crystallographic structure of rhodopsin (Ballesteros et al., 2001).
S.P. and H.C. contributed equally to this work.
This work was supported in part by the Sloan Foundation, the McKnight Endowment for Neuroscience, and the Fritz-Burns Foundation. S.P. is a Merck Fellow in the University of California, San Diego Neurosciences Graduate Program. P.A.S. is a McKnight Scholar and an Alfred P. Sloan Research Fellow. We thank D. Julius for providing the 5-HT3a cDNA, Drs. D. Berg, S. Heinemann, N. Unwin, and members of the Slesinger lab for providing discussion and comments on this and previous versions of this manuscript.
Correspondence should be addressed to Paul A. Slesinger, Peptide Biology Laboratory, The Salk Institute, 10010 N. Torrey Pines Road, La Jolla, CA 92037. E-mail: slesinger@salk.edu.
H. Cruz's present address: University of Geneva, Department of Biochemistry, 30, Quai Ernest-Ansermet, 1211 Geneva 4, Switzerland.
REFERENCES
- 1.Akabas MH, Kaufmann C, Archdeacon P, Karlin A. Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the α subunit. Neuron. 1994;13:919–927. doi: 10.1016/0896-6273(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 2.Alhaider AA, Lei SZ, Wilcox GL. Spinal 5-HT3 receptor-mediated antinociception: possible release of GABA. J Neurosci. 1991;11:1881–1888. doi: 10.1523/JNEUROSCI.11-07-01881.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alkadhi KA, Salgado-Commissariat D, Hogan YH, Akpaudo SB. Induction and maintenance of ganglionic long-term potentiation require activation of 5-hydroxytryptamine (5-HT3) receptors. J Physiol (Lond) 1996;496:479–489. doi: 10.1113/jphysiol.1996.sp021700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballesteros JA, Shi L, Javitch JA. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol. 2001;60:1–19. [PubMed] [Google Scholar]
- 5.Bertrand S, Devillers-Thiery A, Palma E, Buisson B, Edelstein SJ, Corringer P-J, Changeux J-P, Bertrand D. Paradoxical allosteric effects of competitive inhibitors on neuronal α7 nicotinic receptor mutants. Mol Neurosci. 1997;8:3591–3596. doi: 10.1097/00001756-199711100-00034. [DOI] [PubMed] [Google Scholar]
- 6.Chang Y, Weiss D. Substitutions of the highly conserved M2 leucine create spontaneously opening ρ1 γ-aminobutyric acid receptors. Mol Pharmacol. 1998;53:511–523. doi: 10.1124/mol.53.3.511. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Auerbach A. A distinct contribution of the δ subunit to acetylcholine receptor channel activation revealed by mutations of the M2 segment. Biophys J. 1998;75:218–225. doi: 10.1016/S0006-3495(98)77508-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corringer P-J, Le Novère N, Changeux J-P. Nicotinic receptors at the amino acid level. Annu Rev Pharmacol Toxicol. 2000;40:431–458. doi: 10.1146/annurev.pharmtox.40.1.431. [DOI] [PubMed] [Google Scholar]
- 9.Dalziel J, Cox G, Gage P, Birnir B. Mutating the highly conserved second membrane-spanning region 9′ leucine residue in the α1 or β1 subunit produces subunit-specific changes in the function of human α1β1 γ-aminobutyric acid-A receptors. Mol Pharmacol. 2000;57:875–882. [PubMed] [Google Scholar]
- 10.Dang H, England PM, Farivar SS, Dougherty DA, Lester HA. Probing the role of a conserved M1 proline residue in 5-hydroxytryptamine(3) receptor gating. Mol Pharmacol. 2000;57:1114–1122. [PubMed] [Google Scholar]
- 11.Davies PA, Pistis M, Hanna MC, Peters JA, Lambert JJ, Hales TG, Kirkness EF. The 5-HT3B subunit is a major determinant of serotonin-receptor function. Nature. 1999;397:359–363. doi: 10.1038/16941. [DOI] [PubMed] [Google Scholar]
- 12.Dubin AE, Huvar R, D'Andrea MR, Pyati J, Zhu JY, Joy KC, Wilson SJ, Galindo JE, Glass CA, Luo L, Jackson MR, Lovenberg TW, Erlander MG. The pharmacological and functional characteristics of the serotonin 5-HT3A receptor are specifically modified by a 5-HT3B receptor subunit. J Biol Chem. 1999;274:30799–30810. doi: 10.1074/jbc.274.43.30799. [DOI] [PubMed] [Google Scholar]
- 13.Eiselé J-L, Bertrand S, Galzi J-L, Devillers-Thiéry A, Changeux J-P, Bertrand D. Chimaeric nicotinic-serotonergic receptor combines distinct ligand binding and channel specificities. Nature. 1993;366:479–483. doi: 10.1038/366479a0. [DOI] [PubMed] [Google Scholar]
- 14.England PM, Zhang Y, Dougherty DA, Lester HA. Backbone mutations in transmembrane domains of a ligand-gated ion channel: implications for the mechanism of gating. Cell. 1999;96:89–98. doi: 10.1016/s0092-8674(00)80962-9. [DOI] [PubMed] [Google Scholar]
- 15.Filatov G, White M. The role of conserved leucines in the M2 domain of the acetylcholine receptor in channel gating. Mol Pharmacol. 1995;48:379–384. [PubMed] [Google Scholar]
- 16.Flynn GE, Zagotta WN. Conformational changes in S6 coupled to the opening of cyclic nucleotide-gated channels. Neuron. 2001;30:689–698. doi: 10.1016/s0896-6273(01)00324-5. [DOI] [PubMed] [Google Scholar]
- 17.Holmgren M, Liu Y, Xu Y, Yellen G. On the use of thiol-modifying agents to determine channel topology. Neuropharmacology. 1996;35:797–804. doi: 10.1016/0028-3908(96)00129-3. [DOI] [PubMed] [Google Scholar]
- 18.Hussy N, Lukas W, Jones KA. Functional properties of a cloned 5-hydroxytryptamine ionotropic receptor subunit: comparison with native mouse receptors. J Physiol (Lond) 1994;481.2:311–323. doi: 10.1113/jphysiol.1994.sp020441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khasabov SG, Lopez-Garcia JA, Asghar AUR, King AE. Modulation of afferent-evoked neurotransmission by 5-HT3 receptors in young rat dorsal horn neurones in vitro: a putative mechanism of 5-HT3 induced anti-nociception. Br J Pharmacol. 1999;127:843–852. doi: 10.1038/sj.bjp.0702592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Labarca C, Nowak MW, Zhang H, Tang L, Deshpande P, Lester HA. Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature. 1995;376:514–516. doi: 10.1038/376514a0. [DOI] [PubMed] [Google Scholar]
- 21.Le Novere N, Changeux JP. The ligand-gated ion channel database. Nucleic Acids Res. 1999;27:340–342. doi: 10.1093/nar/27.1.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Holmgren M, Jurman ME, Yellen G. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 1997;19:175–184. doi: 10.1016/s0896-6273(00)80357-8. [DOI] [PubMed] [Google Scholar]
- 23.Lovinger DM. Inhibition of 5-HT3 receptor-mediated ion current by divalent metal cations in NCB-20 neuroblastoma cells. J Neurophysiol. 1991;66:1329–1337. doi: 10.1152/jn.1991.66.4.1329. [DOI] [PubMed] [Google Scholar]
- 24.MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- 25.Maeda T, Kaneko S, Satoh M. Inhibitory influence via 5-HT3 receptors on the induction of LTP in mossy fiber-CA3 system of guinea-pig hippocampal slices. Neurosci Res. 1994;18:277–282. doi: 10.1016/0168-0102(94)90163-5. [DOI] [PubMed] [Google Scholar]
- 26.Maricq AV, Peterson AS, Brake AJ, Myers RM, Julius D. Primary structure and functional expression of the 5HT3 receptor, a serotonin-gated ion channel. Science. 1991;254:432–437. doi: 10.1126/science.1718042. [DOI] [PubMed] [Google Scholar]
- 27.Miyazawa A, Fujiyoshi Y, Stowell M, Unwin N. Nicotinic acetylcholine receptor at 4.6 Å resolution: transverse tunnels in the channel wall. J Mol Biol. 1999;288:765–786. doi: 10.1006/jmbi.1999.2721. [DOI] [PubMed] [Google Scholar]
- 28.Movileanu L, Cheley S, Howorka S, Braha O, Bayley H. Location of a constriction in the lumen of a transmembrane pore by targeted covalent attachment of polymer molecules. J Gen Physiol. 2001;117:239–252. doi: 10.1085/jgp.117.3.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Opella SJ, Marassi FM, Gesell JJ, Valente AP, Kim Y, Oblatt-Montal M, Montal M. Structures of the M2 channel-lining segments from nicotinic acetylcholine and NMDA receptors by NMR spectroscopy. Nat Struct Biol. 1999;6:374–379. doi: 10.1038/7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palma E, Mileo AM, Eusebi F, Miledi R. Threonine-for-leucine mutation within domain M2 of the neuronal α7 nicotinic receptor converts 5-hydroxytryptamine from antagonist to agonist. Proc Natl Acad Sci USA. 1996;93:11231–11235. doi: 10.1073/pnas.93.20.11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palma E, Fucile S, Barabino B, Miledi R, Eusebi F. Strychnine activates neuronal α7 nicotinic receptors after mutations in the leucine ring and transmitter binding site domains. Proc Natl Acad Sci USA. 1999;96:13421–13426. doi: 10.1073/pnas.96.23.13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan Z-H, Zhang D, Zhang X, Lipton S. Agonist-induced closure of constitutively open γ-aminobutyric acid channels with mutated M2 domains. Proc Natl Acad Sci USA. 1997;94:6490–6495. doi: 10.1073/pnas.94.12.6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pascual JM, Karlin A. State-dependent accessibility and electrostatic potential in the channel of the acetylcholine receptor. J Gen Physiol. 1998;111:717–739. doi: 10.1085/jgp.111.6.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez EA. 5-HT3 antiemetic therapy for patients with breast cancer. Breast Cancer Res Treat. 1999;57:207–214. doi: 10.1023/a:1006233802863. [DOI] [PubMed] [Google Scholar]
- 35.Reeves DC, Goren EN, Akabas MH, Lummis SCR. Structural and electrostatic properties of the 5-HT3 receptor pore revealed by substituted cysteine accessibility mutagenesis. J Biol Chem. 2001;276:42035–42042. doi: 10.1074/jbc.M106066200. [DOI] [PubMed] [Google Scholar]
- 36.Slesinger PA, Patil N, Liao YJ, Jan YN, Jan LY, Cox DR. Functional effects of the mouse weaver mutation on G protein-gated inwardly rectifying K+ channels. Neuron. 1996;16:321–331. doi: 10.1016/s0896-6273(00)80050-1. [DOI] [PubMed] [Google Scholar]
- 37.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 38.Starace DM, Bezanilla F. Histidine scanning mutagenesis of basic residues of the S4 segment of the shaker K+ channel. J Gen Physiol. 2001;117:469–490. doi: 10.1085/jgp.117.5.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tecott LH, Maricq AV, Julius D. Nervous system distribution of the serotonin 5-HT3 receptor mRNA. Proc Natl Acad Sci USA. 1993;90:1430–1434. doi: 10.1073/pnas.90.4.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- 41.Wang H-L, Milone M, Ohno K, Shen X-M, Tsujino A, Batocchi AP, Tonali P, Brengman J, Engel A, Sine S. Acetylcholine receptor M3 domain: stereochemical and volume contributions to channel gating. Nat Neurosci. 1999;2:226–233. doi: 10.1038/6326. [DOI] [PubMed] [Google Scholar]
- 42.Wilson G, Karlin A. Acetylcholine receptor channel structure in the resting, open, and desensitized states probed with the substituted-cysteine-accessibility method. Proc Natl Acad Sci USA. 2001;98:1241–1248. doi: 10.1073/pnas.031567798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson GG, Karlin A. The location of the gate in the acetylcholine receptor channel. Neuron. 1998;20:1269–1281. doi: 10.1016/s0896-6273(00)80506-1. [DOI] [PubMed] [Google Scholar]
- 44.Yakel JL, Lagrutta A, Adelman JP, North RA. Single amino acid substitution affects desensitization of the 5-hydroxytryptamine type 3 receptor expressed in Xenopus oocytes. Proc Natl Acad Sci USA. 1993;90:5030–5033. doi: 10.1073/pnas.90.11.5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Karlin A. Contribution of the beta subunit M2 segment to the ion-conducting pathway of the acetylcholine receptor. Biochemistry. 1998;37:7952–7964. doi: 10.1021/bi980143m. [DOI] [PubMed] [Google Scholar]