

Abstract
Chronic exposure to opiates eventually leads to drug addiction, which is believed to involve maladaptive changes in brain function, but the underlying neuronal mechanisms remain primarily unknown. Given the known effects of opiates such as morphine and heroin on hippocampal function, we investigated the potential effect of chronic opiate treatment on long-term potentiation (LTP) at CA1 synapses in rat hippocampus, a leading experimental model for studying synaptic plasticity. Our results revealed that chronic exposure of rats to morphine or heroin, which induced severe drug tolerance and dependence, markedly reduced the capacity of hippocampal CA1 LTP during the period of drug withdrawal (from ∼190% in control to ∼120%). More interestingly, the capacity of LTP could be restored to the normal level by re-exposure of the animals to opiates, indicating that the synaptic function was already adapted to opiates. Morris water maze test, which measures behavioral consequences of synaptic plasticity, showed parallel learning deficits after chronic exposure to opiates. Moreover, the opiate-reduced LTP could also be restored by inhibitors of cAMP-dependent protein kinase A (PKA), suggesting that upregulation of cAMP pathway was likely one of the underlying mechanisms of the observed phenomena. These findings demonstrated that chronic opiate treatment can significantly modulate synaptic plasticity in the hippocampus, leading to an opiate dependence of the plasticity.
Keywords: opiate, rat, hippocampus, long-term potentiation, cAMP, addiction
Opiate addiction, which is defined as tolerance to and dependence on the drugs of abuse (O'Brien, 1997), has been increasingly considered as a neuropsychiatric disorder (Leshner, 1997; Robbins and Everitt, 1999). Accumulating evidences have revealed that long-term exposure to opiates, e.g., morphine and heroin, can significantly alter brain function (Nestler and Aghajanian, 1997;Eisch et al., 2000; Kelley et al., 2000), resulting in the development of the tolerance and dependence of opiates. However, the underlying neuropathological mechanisms for opiate addiction are poorly understood, in comparison to our understanding of their analgesic effects.
Synaptic plasticity has been thought to play a critical role in brain function. Long-term potentiation (LTP), a leading experimental model for measuring activity-dependent synaptic plasticity, has received considerable attention as a possible neural mechanism underling learning and memory (Bliss and Collingridge, 1993; Malenka and Nicoll, 1999). Recently, it has been proposed that drug addiction is an aberrant form of learning, mediated by maladaptive recruitment of certain memory systems in the brain (Robbins and Everitt, 1999). Chronic exposure to opiates can result in cognitive deficits, as shown by poor performances on memory task of heroin users, compared with controls (Guerra et al., 1987). Long-term administration of morphine or heroin decrease neurogenesis in the adult rat hippocampus (Eisch et al., 2000). Moreover, studies from many laboratories, including ours, have shown that molecules critical for hippocampal LTP, e.g., NMDA/AMPA subtypes of glutamate receptors and calcium–calmodulin-dependent kinase II (CaMKII) (Malenka and Nicoll, 1999; Woolf and Salter, 2000), are also essential for the development of opiate tolerance and dependence (Trujillo and Akil, 1991; Nestler and Aghajanian, 1997; Fan et al., 1999; Lou et al., 1999). These findings suggest that chronic use of opiates may lead to some maladaptive changes in hippocampal plasticity. In the present study, we demonstrated that chronic exposure to opiates functionally altered the capacity of rat hippocampal LTP and resulted in an opiate dependence of LTP.
MATERIALS AND METHODS
Animals and chronic daily opiate treatment. Male Sprague Dawley (200–250 gm) rats were obtained from the Laboratory Animal Center, Chinese Academy of Sciences (Shanghai, China). Rats were housed in groups and maintained on a 12 hr light/dark cycle with food and water available ad libitum. All animal treatments were strictly in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animals were chronically treated by subcutaneous injection of morphine (10 mg/kg) or heroin (1 mg/kg) twice per day at 12 hr intervals for 10 d as described (Trujillo and Akil, 1991; Fan et al., 1999). Control rats were treated similarly, except that normal saline (NS) was used.
Electrode implantation and electrophysiological recording in vivo. Electrode implantation in male Sprague Dawley rats (200–250 gm) was undertaken as previously described (Doyle et al., 1996; Xu et al., 1997). Recording was performed either under urethane (1.3 gm/kg, i.p.) anesthetization or 2 weeks later in freely moving rats after their recovery from surgery. Three stainless steel screws (1.0 mm diameter) were inserted into the skull through a drill hole without piercing the dura. One served as a ground electrode (7 mm posterior to bregma and 5 mm left of the midline), another acted as an anchor (opposite the ground screw, 7 mm posterior to bregma and 5 mm right of the midline), and the third served as the reference electrode (8 mm anterior to bregma and 1 mm left of the midline). Recording and stimulating electrodes were made by gluing together a pair of twisted Teflon-coated platinum (90%)–iridium (10%) wire (50 μm inner diameter, 75 μm outer diameter). Recordings of field EPSPs (fEPSPs) were made from the CA1 stratum radiatum of the right hippocampal hemisphere in response to stimulation of the Schaffer collateral–commissural pathway. The recording electrode was inserted 3.4 mm posterior to bregma and 2.5 mm right of the midline, and the stimulating electrode was inserted 4.2 mm posterior to bregma and 3.8 mm right of the midline. The dura mater was pierced, and the electrodes were lowered slowly through the cortex and the upper layers of the hippocampus into the CA1 region until the appearance of a negative-deflecting EPSP. For the recording on freely moving rats, the electrodes then were fixed in place with cyanoacrylate glue and acrylic dental cement. The right placement of the electrodes in the stratum radiatum of the CA1 region of the dorsal hippocampus was verified by postmortem examination.
In all experiments, test fEPSPs were evoked by stimulating with a square-wave constant current pulse of 50 μsec duration at a frequency of 0.033 Hz. At the beginning of each experiment, input–output curves were generated to determine the maximal fEPSP slope, and then the intensity of stimulus was set at a level that evoked an fEPSP slope of 55–65% of the maximum. The slope of fEPSP was measured and averaged every 3 min. LTP was induced by high-frequency stimulation using 20 pulses at 200 Hz, repeated three times at a 30 sec interval. All recording and stimulation was performed using an on-line computerized oscilloscope–stimulator and data analysis interface system. Statistical comparisons between baseline and post-stimulation values were made using Student's t test. Values are mean percentage of the baseline fEPSP amplitude ± SEM.
Hippocampal slice preparation. We prepared hippocampal slices as described (Nishiyama et al., 2000). Dissection was performed using ice-cold artificial CSF (ACSF, gassed with 95% O2 and 5% CO2) containing (in mm): 124 NaCl, 1.25 NaH2PO4, 2 KCl, 1.3 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose. A block of hippocampus was removed and sectioned into 400-μm-thick slices using a vibratome. The slices were maintained in an incubation chamber for at least 1 hr at room temperature. For experiments, individual slices were transferred to a submerged recording chamber and perfused continuously with ACSF at a rate of 3–4 ml/min. The temperature in the recording chamber was 31 ± 0.5°C.
Extracellular recordings on slices. Extracellular recordings on brain slices were performed as described (Silva et al., 1992). A bipolar electrode was used to stimulate Schaffer collateral–commissural afferents. fEPSPs were measured extracellularly with an electrode filled with 150 mm NaCl in CA1 stratum radiatum. Stimulation parameters consisted of a square-wave constant current pulse of 50 μsec duration. Stimulus–response curves were performed at the beginning of each experiment, and pulses at an intensity eliciting ∼50% of a maximum slope was used. The averaged fEPSP slope was calculated and averaged every 3 min. After baseline synaptic responses had been stable for at least 15 min, tetanic stimulation consisted of three trains of 20 pulses at 200 Hz with the intertrain interval of 30 sec was delivered. Data in the graphs are presented as the mean ± SEM.
Whole-cell recordings on slices. Whole-cell perforated-patch recordings were made as described (Selig et al., 1999). Cells were visualized with an upright microscope using infrared illumination. Patch electrodes (1–2 MΩ) were pulled from borosilicate glass (1.6 mm optical density) and filled with intracellular solution containing (in mm): 130 CsMeSO3, 8.0 NaCl, 10 HEPES, and 0.2 EGTA, pH 7.2, with CsOH (290–300 mOsm). Amphotericin B (1.2 mg/ml; Calbiochem, La Jolla, CA) dissolved in DMSO (0.6% final concentration) was added to this solution, triturated, and used to backfill pipettes. EPSCs were recorded in CA1 pyramidal cells while stimulating the Schaffer collateral–commissural afferents. Cells were voltage clamped at −60 mV, and experiments were begun only after the access resistance had stabilized (typically 12–20 MΩ). Slices were stimulated every 30 sec. The amplitude of the EPSCs was taken as the peak of the inward current. Only data with initial EPSC amplitude >70 pA were considered. Data were amplified with an Axopatch 2B amplifier (Axon Instruments, Foster City, CA), filtered at 2 kHz, and digitized at 10 kHz. LTP was induced by pairing 120 stimuli at 1 Hz while voltage clamping the postsynaptic cell at +10 mV. Data in the graphs are presented as the mean ± SEM.
Morris water maze test. Morris water maze test was performed as described (Morris et al., 1986; Zhou et al., 1999). A circular, black painted pool (150 cm diameter, 68 cm height) filled to a depth of 35 cm with water was used. The water was maintained at 20 ± 1°C and made opaque by the addition of 20 ml of Indian ink. The pool was divided into four quadrants with four starting locations called north (N), east (E), south (S), and west (W) at equal distance on the rim. An invisible black platform (10 cm diameter) was submerged 1.5 cm below the water line and placed in the center of the northeast quadrant. Rats were chronically treated with morphine for 10 d as described above, trained, and tested in Morris water maze for subsequent 5 d during which 10 mg/kg morphine was still injected at 12 hr intervals to maintain the chronic drug treatment. The rats were trained, either 2 hr before or 1 hr after the second daily injection of morphine, in the water maze to find and escape onto the hidden platform with a 120 sec cutoff time. Each rat was gently placed into the water, with the nose pointing toward the wall at one of the starting points. The escape latency, the time required for the rats to climb onto the platform, was recorded as the average of four trials. The searching patterns of animals were also recorded when the platform was removed from the pool or visible above water on the day 6. ANOVA was used for statistical comparisons, and p < 0.05 was accepted as significant.
Protein kinase activity assay. PKA activity was determined essentially according to the method described by Lou and Pei (1997). Animals were rapidly decapitated, and hippocampi were dissected rapidly and homogenized on ice in homogenization buffer (25 mm Tris-HCl, 1 mm EDTA, 1 mm DTT, and 100 μmleupeptin). The homogenate was centrifuged at 20,000 ×g for 5 min at 4°C. The resulting supernatant was assayed for PKA activity using PepTag nonradioactive PKA assay kit (Promega, Madison, WI) as described in Promega Technical Bulletin. All reaction components were added on ice in a final volume of 25 μl of the following mixture: 5 μl of PepTag PKA reaction buffer, 5 μl of PepTag A1 Peptide (0.4 μg/μl), 5 μl of cAMP (5 μm), and 5 μl of sample homogenate. The mixture was incubating for 30 min at 30°C. Then, the reaction was stopped by placing the tube in a boiling water bath for 10 min, and the samples were loaded onto the gel for electrophoresis. Before loading samples, 1 μl of 80% glycerol was added to the sample to ensure that it remained in the well. PKA-specific peptide substrate used in this experiment was PepTagA1 Peptide, L-R-R-A-S-L-G (kemptide). The assay was based on the changes in the net charge of the fluorescent PKA substrates before and after phosphorylation. This change allowed the phosphorylated and unphosphorylated versions of the substrate to be rapidly separated on an agarose gel at neutral pH. The phosphorylated species migrated toward the positive electrode, whereas the nonphosphorylated substrate migrated toward the negative electrode. The intensity of fluorescence of phosphorylated peptides, which reflected the activity of PKA, was quantified using a bioimaging system (Syngene, Cambridge, UK). Data in the graphs are presented as the mean ± SEM.
RESULTS
Reduction of LTP after chronic opiate treatments
In the present study, we first examined the effect of chronic treatment of morphine on hippocampal LTP by in vivoextracellular recording at CA1 synapses in freely moving rats. Before morphine treatment, high-frequency stimulation of the Schaffer collateral–commissural inputs to CA1 pyramidal cells resulted in a persistent increase (∼190% of baseline) (Fig.1A,C) in the slope of fEPSP, and the increase lasted for at least 4 hr, which was consistent with a previous report (Xu et al., 1997). For chronic opiate treatment, rats were subcutaneously injected with 10 mg/kg morphine or 1 mg/kg heroin twice per day for 10 d, a procedure known to produce significant tolerance and dependence to the drugs (Trujillo and Akil, 1991; Fan et al., 1999). The capacity of LTP measured 12 hr after the termination of 10 d morphine treatment was greatly reduced (from ∼190% to ∼120% of baseline), compared with that recorded before the treatment for the same group of rats (Fig. 1A-D). Significant reduction of LTP was also observed in the animals after 10 d exposure to heroin (Fig. 1E–H). In contrast, the control rats injected with NS for 10 d showed no reduction of LTP under the same conditions (Fig. 1C,D).
Fig. 1.

Significant reduction of hippocampal LTP by chronic opiate treatment in freely moving rats. LTP was induced before chronic treatment (A, C,E, G) and 12 hr after the termination of chronic treatment (subcutaneously, twice per day for 10 d) with NS, 10 mg/kg morphine (Mor; B, D) or 1 mg/kg heroin (Her; F, H) respectively. A, B, E,F, Results were from a representative animal before and after drug treatment. Insets, Sample fEPSPs (average of 6 consecutive sweeps) at the time indicated by the numbers.C, D, G, H, Results were summarized from all animals (n = 6 in each group). Calibration: 10 msec, 1 mV. Arrows indicate high-frequency stimulation.
In a separate series of experiments, the opiate-treated rats were urethane-anesthetized and inserted with the electrodes to perform fEPSP recording, which would minimize the influence of stress induced by surgery treatment or environmental stimulation. The profound reduction of LTP was also recorded 12 hr after the termination of opiate treatment, as observed in the freely moving rats (from ∼190% to ∼130% of baseline) when compared with the control rats injected with NS (Fig. 2A).
Fig. 2.
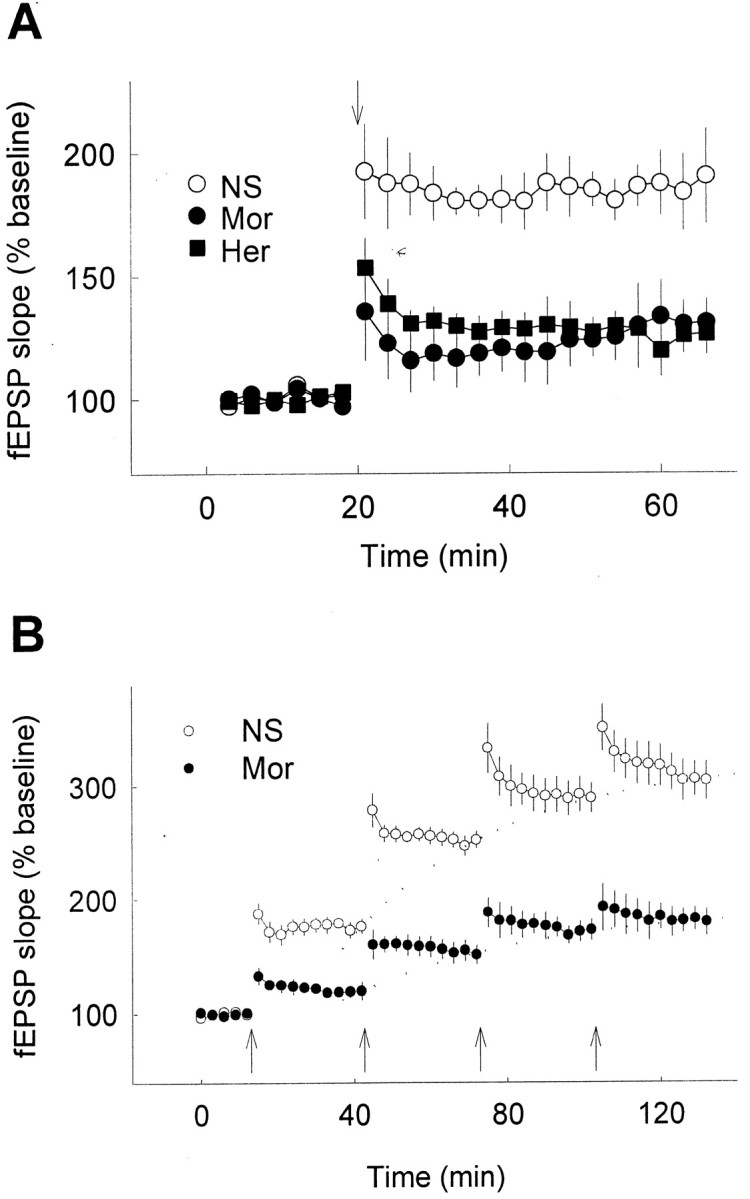
Significant reduction of hippocampal LTP by chronic opiate treatment in anesthetized rats. A, Hippocampal LTP was determined 12 hr after the termination of chronic treatment (subcutaneously, twice per day for 10 d) of normal saline (NS), 10 mg/kg morphine (Mor), or 1 mg/kg heroin (Her) as indicated (n= 7–9 in each group). B, LTP was induced by repeated high-frequency stimulation in rats treated with NS (n = 4) or morphine (n = 5) for 10 d. Arrows indicate high-frequency stimulation.
To further elucidate the cause of the apparent reduction in the capacity of LTP by chronic opiate treatment, we examined the extent of LTP produced by repeated application of tetanic stimulation, as shown in Figure 2B. Repeated tetanic stimulation induced LTP in control animals (receiving 10 d of NS) to a saturated level much higher than that of chronic morphine-treated rats (for 10 d). This indicates that there is indeed a reduction of capacity in chronic drug-treated rats in generating LTP, rather than a reduction of efficiency in the LTP induction.
Since development of opiate tolerance and dependence is well known to depend on repeated exposure to opiates (Nestler and Aghajanian, 1997), whether the reduction of LTP also requires the chronic use of opiates was further tested. As shown in Figure3A, a single injection of morphine or heroin had no significant effect on hippocampal LTP, a result consistent with that of an earlier report (Stringer et al., 1983). In contrast, LTP was significantly reduced in rats after 5 d of daily treatment with opiates, and the reduction seemed to reach a plateau level after 10 d (data for >10 d not shown). The reduction of hippocampal LTP by opiates appeared in a time-dependent manner: the magnitude of induced LTP was significantly reduced 6, 12, and 18 hr after the termination of 10 d opiate treatment, during the period of drug withdrawal. The hippocampal LTP recorded 12 hr after the termination of chronic treatment was apparently reduced to the lowest level, therefore it was chosen as the standard time point in this study. The reduction of LTP by chronic opiate treatment was reversible and was gradually recovered to an apparent normal level ∼24 hr after the termination of chronic drug treatment (Fig.3B).
Fig. 3.
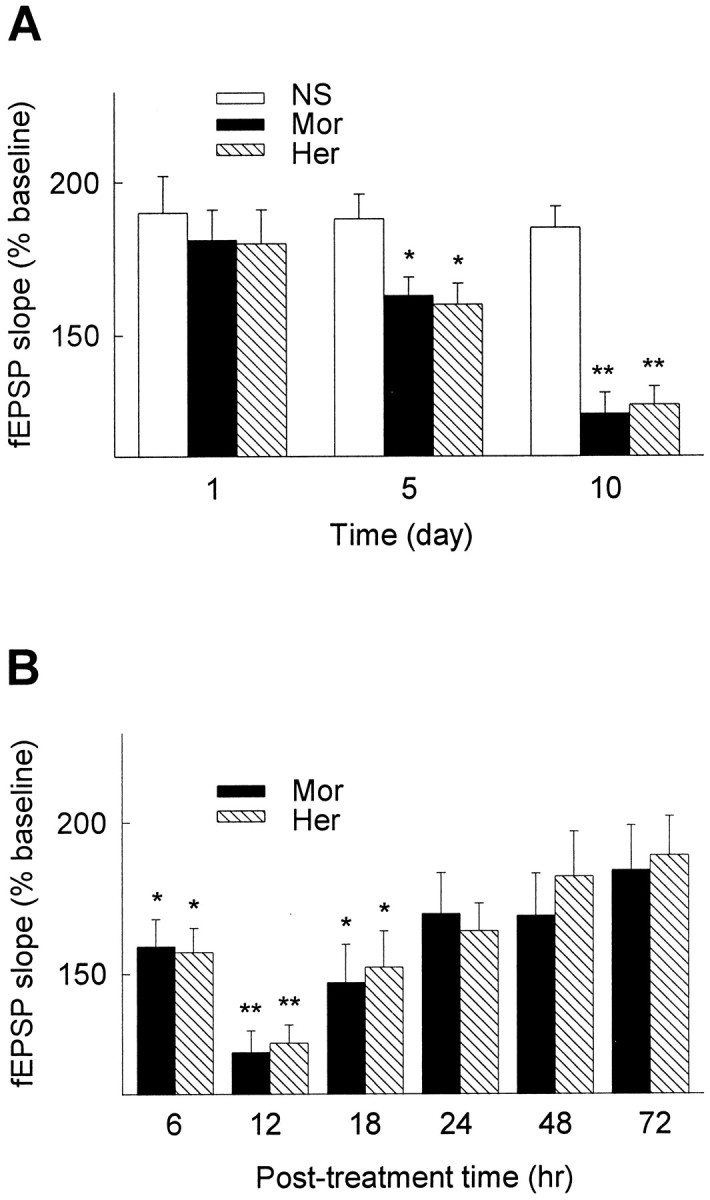
Development and recovery of opiate-reduced LTP in hippocampus. A, Hippocampal LTP was recorded 12 hr after the termination of opiate treatment for 1, 5, and 10 d (n = 6 in each group). B, Hippocampal LTP was recorded 6, 12, 24, 48, and 72 hr after the termination of 10 d chronic opiate treatment (n = 6–10 in each group). LTP was recorded using rats under anesthetization. The mean fEPSP amplitude at 30 min after LTP induction is shown. *p < 0.05; **p < 0.01 compared with NS control.
Restoration of hippocampal LTP by re-exposure to opiates
When opiate dependence is developed, the neural systems adapt to the repeated drug exposure and only function normally in the presence of the drug (Koob and Moal, 1997). Therefore, we further examined whether the capacity of LTP can be influenced by opiate re-exposure. It was found that a single injection (10 mg/kg, s.c.; 30 min before LTP induction) of morphine or heroin at 12 hr after the termination of chronic opiate treatment could indeed restore the capacity of LTP to the normal level in either freely moving (Fig.4A,B) or anesthetized rats (Fig. 4C,D). The restoration of LTP by morphine could be blocked by NMDA receptor antagonist MK-801 (Fig. 4C), suggesting that the rescued LTP in chronic drug exposure animals shares the same underlying mechanism of NMDA receptor dependence as the LTP at normal conditions in control animals. Similar results of opiate-facilitated recovery of LTP were also observed when morphine or heroin was administrated intracerebroventricularly (data not shown).
Fig. 4.

Restoration of the reduced LTP by re-exposure of the chronic drug-treated animals to opiates. Twelve hours after the termination of chronic opiate treatment, rats were reinjected with opiates or NS (subcutaneously, as indicated by thetriangle), respectively, 30 min before LTP induction.A, B, In freely moving animals, reinjection of 10 mg/kg morphine (Mor;A) or 1 mg/kg heroin (Her;B) 30 min before high-frequency stimulation correspondingly restored the reduced LTP to the normal level compared with the NS group. C,D, The reduced LTP after chronic opiate treatment as above was also restored in anesthetized rats by subcutaneous administration of 10 mg/kg morphine (Mor;C) or 1 mg/kg heroin (Her;D) 30 min before LTP induction, respectively, compared with the NS control (n = 6–8 in each group). NMDA receptor antagonist MK-801 (3 mg/kg, i.p.), could block the restoration of LTP by morphine (C). Arrowsindicate high-frequency stimulation; triangles indicate drug injection.
The reduction of hippocampal LTP by chronic opiate treatment and subsequent restoration of the capacity of LTP by re-exposure to opiates were also confirmed using extracellular recording in rat hippocampal slices (Fig. 5). Although the restoration effect of morphine on the reduced capacity of LTP in vitrowas not as robust as that found in vivo, it demonstrated that the restoration by morphine was attributable at least in part to its direct effect on the hippocampus. We also tested the effect of bicuculline (10 μm), a GABAA receptor antagonist, on the reduced LTP in brain slices from chronic morphine-treated animals, and no significant difference in LTP was observed in the presence of bicuculline as compared with the control group (Fig. 5). This result indicated that changes of GABAergic activity appear not be involved in the opiate-induced reduction of LTP.
Fig. 5.
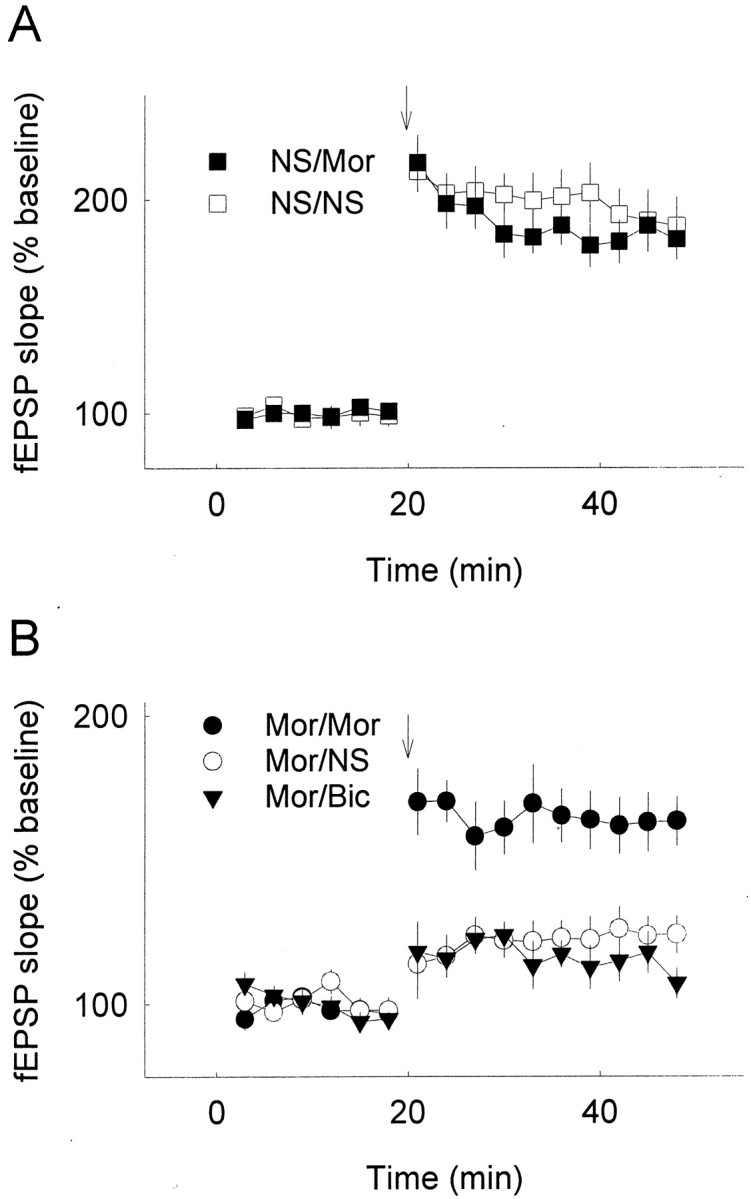
Reduction and restoration of LTP in slices of chronic morphine-treated animals from extracellular recording. Rats were chronically subcutaneously treated with normal saline (NS) or morphine (Mor) for 10 d as above. Slices were prepared 9 hr after the last opiate injection and incubated in ACSF with vehicle (NS), morphine (Mor; 5 μm), or bicuculline (Bic; 10 μm). fEPSP slopes were measured and averaged. LTP was induced by high-frequency stimulation;n = 6–8 in each group. Labelsindicate chronic treatment–incubation. A, No significant difference was observed in LTP between slices from 10 d NS-treated animals incubated in normal ACSF (NS/NS) and ACSF with morphine (NS/Mor). B, LTP of slices from chronic opiate-treated rats (Mor/NS) was reduced compared with the slices from normal saline control (NS/NS) or from acute opiate incubation group (NS/Mor). Opiate incubation (Mor/Mor, morphine was added 30 min before tetanic stimulation and kept throughout the recording period) could partially restore the reduced capacity of LTP, whereas no change was seen after bicuculline incubation (Mor/Bic). Arrows indicate high-frequency stimulation.
Extracellular fEPSP recording reveals synaptic properties of large populations of neurons. We next investigated the capacity of LTP at the single cell level by pairing depolarization of a single pyramidal neuron with low-frequency presynaptic stimulation, which produces robust LTP in this system. As shown in Figure6, the reduction of LTP after 10 d opiate treatment was further confirmed, and the capacity of LTP could also be restored by acute exposure of the chronic drug-treated animals to morphine (10 mg/kg; 30 min before killing). These results may avoid the complexity of the postsynaptic processes during tetanus-induced LTP (Chen et al., 1999), and further confirmed that LTP was indeed modified in the animals after chronic drug treatment.
Fig. 6.

Reduction and restoration of LTP in slices of chronic morphine-treated animals from whole-cell perforated patch recording. Rats were subcutaneously injected with normal saline (NS) or morphine (Mor) for 10 d as above. Slices were prepared 9 hr after the last opiate injection, and EPSCs were recorded in CA1 pyramidal cells of hippocampal slices. LTP was induced by a pairing protocol (120 stimuli at 1 Hz with voltage clamping the postsynaptic cell at +10 mV). Labelsindicate chronic treatment–acute exposure. LTP was decreased in the slices from chronic morphine-treated rats (Mor/NS) compared with that from control animals (NS/NS). The reduced capacity of LTP was restored by acute injection of morphine (Mor/Mor; 10 mg/kg, s.c.; 30 min before killing), but not by that of saline (Mor/NS); n = 3–5 in each group. Insets, Representative EPSCs of NS/NS (left), Mor/NS (middle), and Mor/Mor (right) before and after pairing protocol at the time indicated by the numbers. Calibration: 10 msec, 100 pA.Arrows indicate pairing protocol.
Behavioral tests of spatial learning
It has been shown that blockade or saturation of hippocampal LTP in animals can modulate the synaptic plasticity, as measured by Morris water maze test (Morris et al., 1986; Moser et al., 1998; Riedel et al., 1999). The functional consequences of LTP reduction by chronic opiate treatment (10 d morphine) were thus tested in Morris water maze in the rats 2 hr before (group 1) or 1 hr after (group 2) re-exposure to morphine. Results in the test to find the hidden platform on the fifth training day (Fig. 7A) revealed that the rats in group 1 whose hippocampal LTP was severely reduced exhibited poorer performances compared with the rats in control group (group 3, treated with NS throughout). However, the rats in group 2, whose capacity of LTP had been restored to a normal level by re-exposure to morphine, showed nearly normal performances compared with rats in group 3 (Fig. 7A). In the test in which the platform was removed and the time for the rats to swim in the platform quadrant was recorded, rats in group 1 showed impaired, whereas group 2 showed normal performances compared with the control group (Fig.7B,C). However, all three groups of rats performed similarly when the platform was visible (Fig. 7D), indicating that their swimming ability and visual discrimination for performing the task were not affected by the opiate treatment.
Fig. 7.
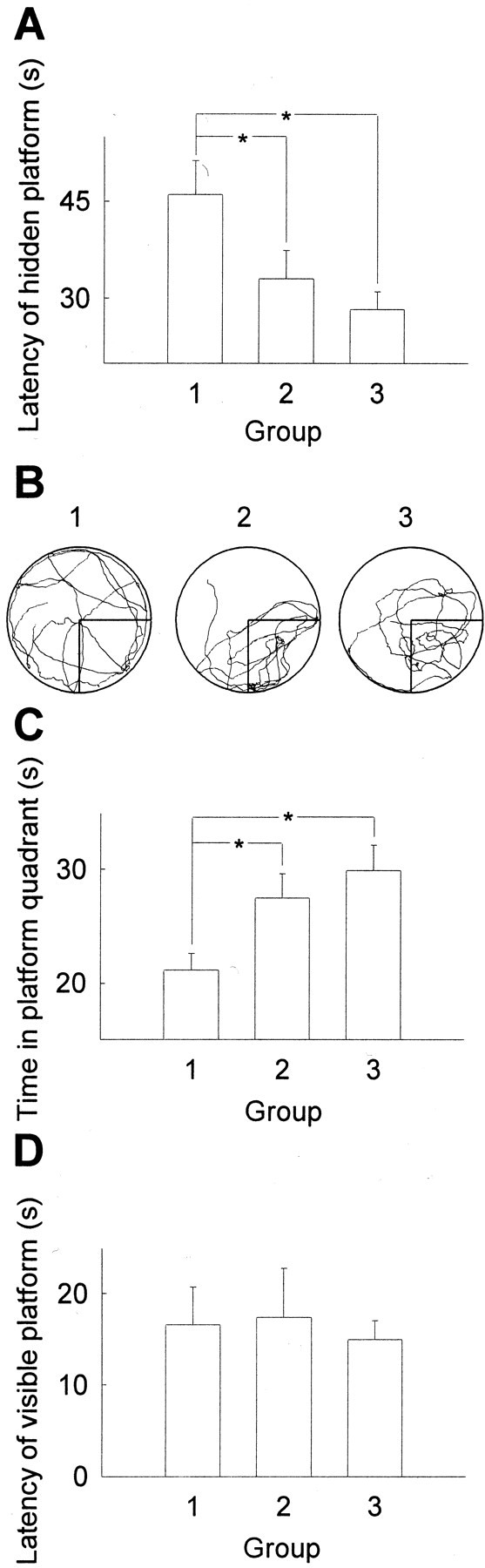
Performance of the opiate-treated rats in the water maze task. Rats were treated with morphine twice per day (10 mg/kg, s.c.) for 15 d. For the last 5 d rats were trained and tested in Morris water maze 2 hr before (group 1) or 1 hr after (group 2) the second daily injection, whereas the control rats were treated with NS (group 3) for 15 d. A, Latency to find the hidden platform on the last day. B, C, A representative searching pattern from each group (B) and time spent in the platform quadrant (C) when the platform was removed.D, Time to find the visible platform. A,C, D,n = 6–9 in each group. *p < 0.05.
The role of PKA pathway
Stimulation of opioid receptors, which are coupled to inhibitory G-proteins, is known to inhibit both the cAMP formation and the PKA activation (Childers 1991; Chalecka-Franaszek et al., 2000). After chronic suppression by opiate stimulation, the cAMP system significantly rebounds (Koob et al., 1998; Bohn et al., 2000). This upregulation of cAMP pathway in the rewarding system, e.g., nucleus accumbens, ventral tegmental area, and periaqueductal gray is a well known biochemical adaptation that correlates with chronic opiate exposure (Nestler and Aghajanian, 1997). However, whether PKA activity is modulated in hippocampus after chronic opiate treatment remains unknown. In the present study, we found that the PKA activity in rat hippocampus was significantly increased after 10 d morphine treatment as compared with saline control group (Fig.8A). Furthermore, re-exposure to opiate reduced the upregulated PKA activity to the normal level in saline control group (Fig. 8A). Finally, application of H89 or H7, inhibitors of PKA, was found to restore the capacity of LTP, mimicking the effect of morphine readministration (Fig. 8B,C). Similar to morphine, the LTP rescued by the specific PKA inhibitor H89 was blocked by NMDA receptor antagonist MK-801 (Fig. 8B). In contrast to PKA inhibitors, a specific inhibitor of protein kinase C, Gö6976 could not restore the capacity of LTP under the same conditions (Fig.8C). Thus, our results suggested that upregulation of the cAMP pathway in hippocampus might be a possible molecular mechanism underlying the suppression of LTP by chronic opiate treatment.
Fig. 8.
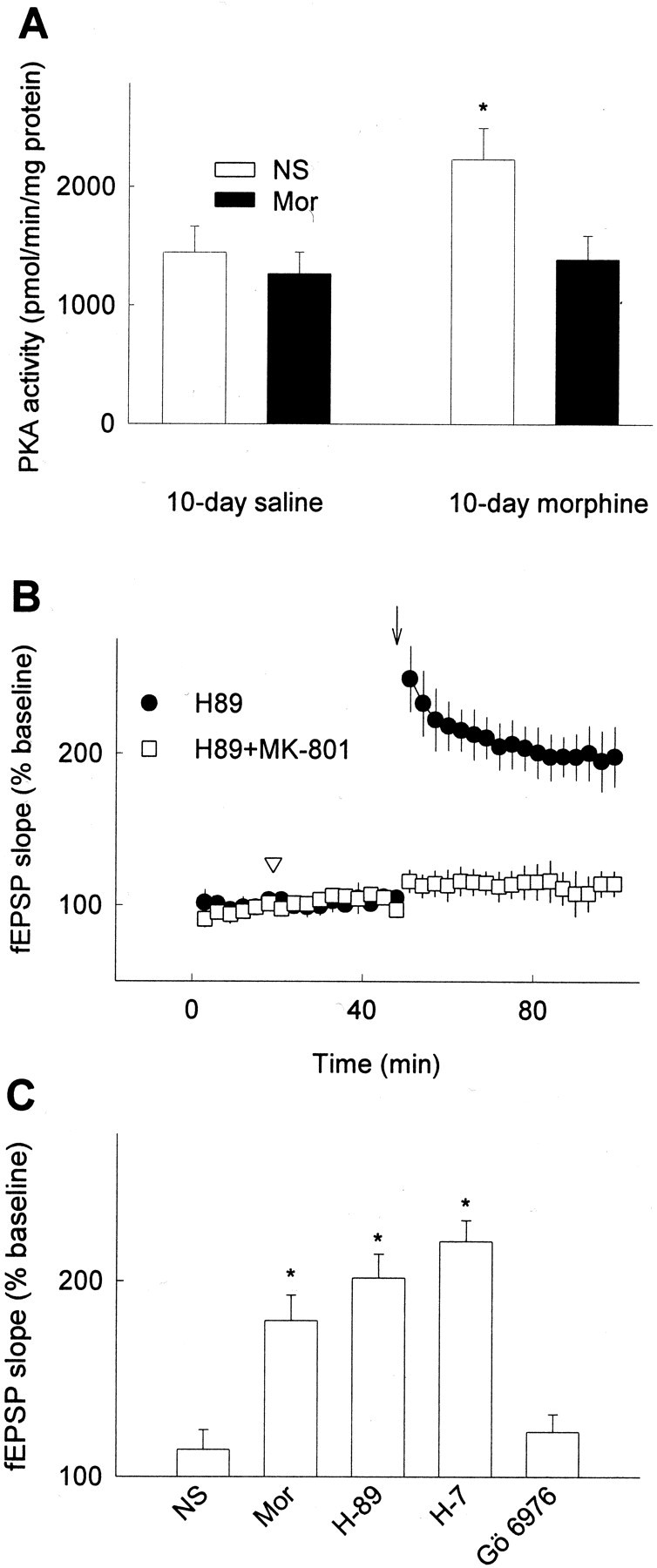
Upregulation of PKA activity and restoration of reduced LTP in chronic morphine-treated rats by PKA inhibitors.A, Hippocampal PKA activity was tested in chronic saline or morphine (10 mg/kg)-treated rats (subcutaneously for 10 d). Twelve hours after the termination of chronic drug treatment, rats were injected with normal saline (NS, white bars) or 10 mg/kg morphine (Mor, black bars), then hippocampal region was obtained 30 min after injection. Data represent the mean ± SEM of three independent experiments. B, Administration of PKA inhibitor H-89 (0.5 mm; 2 μl, i.c.v) restored the capacity of LTP in chronic morphine-treated rats (10 mg/kg, s.c. for 10 d), which mimicked the effect of opiate readministration. NMDA receptor antagonist MK-801 (3 mg/kg, i.p.) blocked the restoration of LTP by H-89. C, Morphine (Mor, 1 mm; 2 μl, i.c.v,) and PKA inhibitors (2 μl, i.c.v) H-89 (0.5 mm) or H-7 (10 mm) but not PKC inhibitor Gö 6976 (0.1 mm; 2 μl, i.c.v) restored the reduced LTP in chronic morphine-treated rats. In vivo recording was performed on anesthetized rats. The mean fEPSP amplitude at 30 min after LTP induction is shown. *p < 0.05 compared with NS control (2 μl, i.c.v); n = 4–6 in each group.
DISCUSSION
Studies on opiate addiction have been focused on some brain regions such as prefrontal cortex, nucleus accumbens, ventral tegmental area, and striatum, which are generally thought to be major components of the rewarding system (Punch et al., 1997; Robbins and Everitt, 1999). Hippocampus plays a key role in information encoding and retrieving in the CNS. It expresses opioid receptors widely (Tempel and Zukin, 1987; Mansour et al., 1995; Madamba et al., 1999;Svoboda et al., 1999), but the potential effects of chronic use of opiates on the function of hippocampus are poorly understood. Recent reports suggest that hippocampus is critical in the rewarding response (Nestler, 2001) and drug-seeking behavior (Vorel et al., 2001). The current study further demonstrates that LTP of hippocampus is significantly altered by the long-term administration of morphine or heroin, indicating alteration in the function of hippocampus after chronic opiate exposure.
As a kind of brain disease, drug addiction has been considered as a neuronal adaptation with the altered functions of neuronal circuits, including changes in neuronal plasticity (Nestler and Aghajanian, 1997;Zhang et al., 1998; Robbins and Everitt, 1999). Here we provide the direct experimental evidence that after chronic opiate treatment the capacity of LTP in hippocampus was significantly reduced during drug withdrawal, indicating that opiates indeed induce changes in neuronal plasticity. More interestingly, the reduced capacity of LTP could be restored by re-exposure of the animals to opiates, revealing that the hippocampal plasticity becomes opiate-dependent after chronic opiate treatment. In other words, the hippocampal function has been adapted to the presence of opiates. Furthermore, such opiate-induced conditional dependence of neuronal plasticity is likely to take place in other brain regions in view of the wide occurrence of LTP (Malenka and Nicoll, 1999) and the extensive expression of opioid receptors throughout the mammalian brain (Mansour et al., 1995).
Morris water maze task has been established as a useful behavioral test for the spatial learning associated with the hippocampal function. Previous reports have shown that blockade of LTP or saturation of LTP in hippocampus of rats can impair their performances in Morris water maze test (Morris et al., 1986; Moser et al., 1998; Riedel et al., 1999), whereas enhancement of the capacity of hippocampal LTP increases their performances (Tang et al., 1999). Our results demonstrated that the chronic use of opiates led to impairment of their performances in Morris water maze test, in parallel with a reduction of hippocampal LTP, whereas re-exposure of animals to opiates restored both of the capacity of LTP and the performances in the water maze test. These results thus suggest that opiate-induced modulation of LTP may be functionally relevant to some of the abnormal behaviors in chronic opiate-treated animals.
PKA has been shown to be a critical element in hippocampal CA1 LTP (Blitzer et al., 1995) by modulating activity of AMPA receptors and CaMKII (Roche et al., 1996; Blitzer et al., 1998), the key components of the molecular machinery of CA1 LTP (Silva et al., 1992; Malenka and Nicoll, 1999; Lee et al., 2000). Recently, there is further evidence showing that upregulation of PKA activity by application of forskolin, an adenylate cyclase activator, induces a long-lasting synaptic potentiation and completely blocks the induction of LTP in amygdala (Huang and Kandel, 1998). This is reminiscent of the finding that expression of a constitutively active form of CaMKII, which increases postsynaptic activity of CaMKII, can effectively block the LTP (Pettit et al., 1994). It has been widely reported that chronic opiate exposure leads to upregulation of cAMP pathway in brain regions such as locus coeruleus, nucleus accumbens, and ventral tegmental area (Terwilliger et al., 1991; Bonci and Williams, 1997; Punch et al., 1997). In the present study, we demonstrated that the chronic morphine treatment could lead to the upregulation of PKA activity also in rat hippocampus, which was consistent with the previous studies. Furthermore, our study showed that the blockade of the upregulated PKA activity is sufficient to mimic the effect of morphine in restoring the capacity of hippocampal LTP in chronic morphine-treated rats. Taken together, it seems that the activity of PKA may play a dual role in modulation of the hippocampal LTP: at the normal level to maintain LTP, and at the overactivated level to saturate LTP via phosphorylation of AMPA receptors or upregulation of CaMKII activity, thus blocking further induction of LTP. That is, in chronic opiate-treated animals, upregulation of cAMP pathway would become fully functional after removal of the opiate (Nestler and Aghajanian, 1997), and the overactivated PKA thus plays a role of inhibiting the capacity of LTP, whereas, the capacity of LTP may then be restored when the overactivity of PKA in CA1 pyramidal neurons is suppressed by re-exposure to opiates or by application of its inhibitors. Therefore, upregulation of cAMP pathway appears to be responsible for the reduction of hippocampal LTP after the chronic opiate exposure, and suppression of high PKA activity by opiate may account for the restoration of LTP.
L.P. and G.-B.B. contributed equally to this work.
This work was supported by grants from Ministry of Science and Technology (G1999053907 and G1999054003), Chinese Academy of Sciences (KSCX2–2-02), the National Natural Science Foundation of China (39825110 and 30024003), and the German Max-Planck Society. We greatly thank Dr. M.-m. Poo for his critical comments on this manuscript and Drs. S.-m. Duan, B.-m. Li, and P. Xia for their technical help.
Correspondence should be addressed to Dr. Gang Pei, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, People's Republic of China. E-mail: gpei@sibs.ac.cn.
REFERENCES
- 1.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 2.Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 3.Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1943. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- 4.Bohn LM, Gainetdinov RR, Lin F-T, Lefkowitz RJ, Caron MG. μ-opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 5.Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chalecka-Franaszek E, Weems HB, Crowder AT, Cox BM, Côté TE. Immunoprecipitation of high-affinity, guanine nucleotide-sensitive, solubilized μ-opioid receptors from rat brain: coimmunoprecipitation of the G proteins Gαo, Gαi1, Gαi3. J Neurochem. 2000;74:1068–1078. doi: 10.1046/j.1471-4159.2000.0741068.x. [DOI] [PubMed] [Google Scholar]
- 7.Chen H-X, Otmakhov N, Lisman J. Requirements for LTP induction by pairing in hippocampal CA1 pyramidal cells. J Neurophysiol. 1999;82:526–532. doi: 10.1152/jn.1999.82.2.526. [DOI] [PubMed] [Google Scholar]
- 8.Childers SR. Opioid receptor-coupled second messenger systems. Life Sci. 1991;48:1991–2003. doi: 10.1016/0024-3205(91)90154-4. [DOI] [PubMed] [Google Scholar]
- 9.Doyle C, Hölscher C, Rowan MJ, Anwyl R. The selective neuronal NO synthase inhibitor 7-nitor-indazole blocks both long-term potentiation and depotentiation of field EPSPs in rat hippocampal CA1 in vivo. J Neurosci. 1996;16:418–424. doi: 10.1523/JNEUROSCI.16-01-00418.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisch AJ, Barrot M, Schad CA, Self DW, Nestler EJ. Opiates inhibit neurogenesis in the adult rat hippocampus. Proc Natl Acad Sci USA. 2000;97:7579–7584. doi: 10.1073/pnas.120552597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan G-H, Wang L-Z, Qiu H-C, Ma L, Pei G. Inhibition of calcium/calmodulin-dependent protein kinase II in rat hippocampus attenuates morphine tolerance and dependence. Mol Pharmacol. 1999;56:39–45. doi: 10.1124/mol.56.1.39. [DOI] [PubMed] [Google Scholar]
- 12.Guerra D, Sole A, Cami J, Tobena A. Neuropsychological performance in opiate addicts after rapid detoxification. Drug Alcohol Depend. 1987;20:261–270. doi: 10.1016/0376-8716(87)90036-6. [DOI] [PubMed] [Google Scholar]
- 13.Huang YY, Kandel ER. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron. 1998;21:169–178. doi: 10.1016/s0896-6273(00)80524-3. [DOI] [PubMed] [Google Scholar]
- 14.Kelley AE, Bakshi VP, Fleming S, Holahan MR. A pharmacological analysis of the substrates underlying conditioned feeding induced by repeated opioid stimulation of the nucleus accumbens. Neuropsychopharmacology. 2000;23:455–467. doi: 10.1016/S0893-133X(00)00117-2. [DOI] [PubMed] [Google Scholar]
- 15.Koob GF, Moal ML. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- 16.Koob GF, Sanna PP, Bloom FE. Neuroscience of addiction. Neuron. 1998;21:467–476. doi: 10.1016/s0896-6273(00)80557-7. [DOI] [PubMed] [Google Scholar]
- 17.Lee H-K, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 18.Leshner AI. Addiction is a brain disease, and it matters. Science. 1997;278:45–47. doi: 10.1126/science.278.5335.45. [DOI] [PubMed] [Google Scholar]
- 19.Lou LG, Pei G. Modulation of protein kinase C and cAMP-dependent protein kinase by δ-opioid. Biochem Biophys Res Commun. 1997;236:626–629. doi: 10.1006/bbrc.1997.7027. [DOI] [PubMed] [Google Scholar]
- 20.Lou L, Zhou T, Wang P, Pei G. Modulation of Ca2+/calmodulin-dependent protein kinase II activity by acute and chronic morphine administration in rat hippocampus: differential regulation of α and β isoforms. Mol Pharmacol. 1999;55:557–563. [PubMed] [Google Scholar]
- 21.Madamba SG, Schweitzer P, Siggins GR. Dynorphin selectively augments the M-current in hippocampal CA1 neurons by an opiate receptor mechanism. J Neurophysiol. 1999;82:1768–1775. doi: 10.1152/jn.1999.82.4.1768. [DOI] [PubMed] [Google Scholar]
- 22.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 23.Mansour A, Fox CA, Akil H, Watson SJ. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 1995;18:22–29. doi: 10.1016/0166-2236(95)93946-u. [DOI] [PubMed] [Google Scholar]
- 24.Morris RGM, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- 25.Moser EI, Krobert KA, Moser MB, Morris RGM. Impaired spatial learning after saturation of long-term potentiation. Science. 1998;281:2038–2042. doi: 10.1126/science.281.5385.2038. [DOI] [PubMed] [Google Scholar]
- 26.Nestler EJ. Total recall—the memory of addiction. Science. 2001;292:2266–2267. doi: 10.1126/science.1063024. [DOI] [PubMed] [Google Scholar]
- 27.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 28.Nishiyama M, Hong K, Mikoshiba K, Poo M-m, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- 29.O'Brien CP. A range of research-based pharmacotherapies for addiction. Science. 1997;278:66–70. doi: 10.1126/science.278.5335.66. [DOI] [PubMed] [Google Scholar]
- 30.Pettit DL, Perlman S, Malinow R. Potentiated transmission and prevention of further LTP by increased CaMKII activity in postsynaptic hippocampal slice neurons. Science. 1994;266:1881–1885. doi: 10.1126/science.7997883. [DOI] [PubMed] [Google Scholar]
- 31.Punch LJ, Self DW, Nestler EJ, Taylor JR. Opposite modulation of opiate withdrawal behaviors on microinfusion of a protein kinase A inhibitor versus activator into the locus coeruleus or periaqueductal gray. J Neurosci. 1997;17:8520–8527. doi: 10.1523/JNEUROSCI.17-21-08520.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riedel G, Micheau J, Lam AGM, Roloff E, Martin SJ, Bridge H, Hoz L, Poeschel B, McCulloch J, Morris RGM. Reversible neural inactivation reveals hippocampal participation in several memory processes. Nat Neurosci. 1999;2:898–905. doi: 10.1038/13202. [DOI] [PubMed] [Google Scholar]
- 33.Robbins TW, Everitt BJ. Drug addiction: bad habits add up. Nature. 1999;398:567–570. doi: 10.1038/19208. [DOI] [PubMed] [Google Scholar]
- 34.Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 35.Selig DK, Nicoll RA, Malenka RC. Hippocampal long-term potentiation preserves the fidelity of postsynaptic responses to presynaptic bursts. J Neurosci. 1999;19:1236–1246. doi: 10.1523/JNEUROSCI.19-04-01236.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in α-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- 37.Stringer JL, Greenfield LJ, Hackett JT, Guyenet PG. Blockade of long-term potentiation by phencyclidine and ς opiates in the hippocampus in vivo and in vitro. Brain Res. 1983;280:127–138. doi: 10.1016/0006-8993(83)91180-0. [DOI] [PubMed] [Google Scholar]
- 38.Svoboda KR, Adams CE, Lupica CR. Opioid receptor subtype expression defines morphologically distinct classes of hippocampal interneurons. J Neurosci. 1999;19:85–95. doi: 10.1523/JNEUROSCI.19-01-00085.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- 40.Tempel A, Zukin RS. Neuroanatomical patterns of the μ, δ, and κ opioid receptors of rat brain as determined by quantitative in vitro autoradiography. Proc Natl Acad Sci USA. 1987;84:4308–4312. doi: 10.1073/pnas.84.12.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Terwilliger RZ, Beitner-Johnson D, Sevarino KA, Crain SM, Nestler EJ. A general role for adaptations in G-proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function. Brain Res. 1991;548:100–110. doi: 10.1016/0006-8993(91)91111-d. [DOI] [PubMed] [Google Scholar]
- 42.Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- 43.Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL. Relapse to cocaine-seeking after hippocampal theta burst stimulation. Science. 2001;292:1175–1178. doi: 10.1126/science.1058043. [DOI] [PubMed] [Google Scholar]
- 44.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1768. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 45.Xu L, Anwyl R, Rowan MJ. Behavioural stress facilitates the induction of long-term depression in the hippocampus. Nature. 1997;387:497–500. doi: 10.1038/387497a0. [DOI] [PubMed] [Google Scholar]
- 46.Zhang X, Hu X, White FJ. Whole-cell plasticity in cocaine withdrawal: reduced sodium currents in nucleus accumbens neurons. J Neurosci. 1998;18:488–498. doi: 10.1523/JNEUROSCI.18-01-00488.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou H, Ge X, Wang L-Z, Ma L, Pei G. Attenuation of morphine tolerance and dependence in scopolamine-treated rats. NeuroReport. 1999;10:2007–2010. doi: 10.1097/00001756-199907130-00003. [DOI] [PubMed] [Google Scholar]