

Abstract
Background & Aims:
Little is known regarding the risk of hepatic steatosis (HS) among adult children of affected parents. We examined the association between parental and offspring HS in the multigenerational Framingham Heart Study, which characterized HS using computed tomography.
Methods:
We performed multivariable logistic regression models adjusted for age, sex, alcohol use, and body mass index to generate the odds of HS according to parental HS. We determined the proportion of participants with HS according to parental HS and the presence or absence of hypertension, diabetes, or obesity (BMI ≥30 kg/m2). After excluding heavy alcohol use (n = 126) and missing covariates (n = 1), 785 offspring with at least one parent were included.
Results:
Approximately 23% (183/785) had at least one parent with HS and 1.1% had two affected parents (9/785). In adjusted models, participants with at least one parent with HS had a nearly two-fold increased odds of HS compared to participants without a parental history of HS (OR 1.86, 95% confidence interval 1.15–3.03). Among participants without hypertension, diabetes, or obesity, a higher proportion had HS if they had a parental history of HS compared to those without (16.1% vs 5.2%, P < 0.001). However, for participants with cardiometabolic risk factors, we did not observe a difference in HS among those with and without parental HS (30.3% vs 28.5%, P = 0.78).
Conclusions:
Individuals with a parental history of HS are at increased risk for HS. Specifically, a parental history of HS may be an important factor among those that are otherwise metabolically healthy.
Keywords: computed tomography, disease risk, familial, fatty liver disease, heritability
1 |. INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD), the most common cause of chronic liver disease, is characterized by hepatic steatosis in the absence of excess alcohol intake or other causes.1,2 In a minority of individuals, hepatic steatosis can progress to steatohepatitis and fibrosis though it is not fully understood why some people develop liver fibrosis and others have a relatively benign disease course.3 Hepatic steatosis is associated with features of the metabolic syndrome, including obesity, insulin resistance, hypertension, and dyslipidemia; however, these traits alone are not sufficient to produce hepatic steatosis.4 Recent genetic association studies have suggested an association between the patatin-like phospholipase domain-containing 3 (PNPLA3) genotype, among others, and hepatic steatosis.5–11 However, the PNPLA3 genotype explains only 10–12% of the variance in the trait9; therefore, there remains much to be discovered regarding the genetic susceptibility to hepatic steatosis.
To date, there has been limited investigation regarding the risk of hepatic steatosis among children of affected parents. Prior studies utilizing twin cohorts suggest hepatic steatosis is heritable; however, these studies have been limited by relatively small sample sizes or the use of serum aminotransferase levels as a surrogate for hepatic steatosis.12–14
We examined the association between parental hepatic steatosis and risk of hepatic steatosis in offspring by utilizing the multigenerational Framingham Heart Study cohorts which characterized hepatic steatosis using computed tomography. We hypothesized that offspring with one or more parents with hepatic steatosis will have an increased odds of hepatic steatosis. Further, we hypothesized that this relationship would be maintained after adjusting for potential confounding factors including body mass index (BMI).
2 |. MATERIALS AND METHODS
2.1 |. Study sample
Data for the present study were obtained from second-generation and third-generation cohort participants in the multigenerational Framingham Heart Study (FHS) who had previously undergone multidetector computed tomography (MDCT) from 2002 to 2005 as part of a sub-study.15–17 The MDCT sub-study of the FHS has been described in detail.15 Inclusion criteria for participating in the MDCT sub-study were as follows: residing in the greater New England area; an age of ≥40 for women and age ≥35 years for men; and body weight <450 lbs owing to the weight restrictions of the MDCT scanner. Pregnant women were also excluded. Of the 1883 third-generation participants with adequate measurement of hepatic steatosis in the MDCT study, 912 had at least one parent in the second-generation cohort who also participated in the MDCT sub-study. Participants were excluded if they or a parent participant were significant alcohol users defined as >14 drinks per week for women and >21 drinks per week for men1 (n = 126) or if covariates were missing (n = 1) which yielded a final sample of study sample of 785 third-generation participants (Figure 1). The institutional review boards of the Boston University Medical Center and the Massachusetts General Hospital approved the study protocol. Participants provided written informed consent.
FIGURE 1.
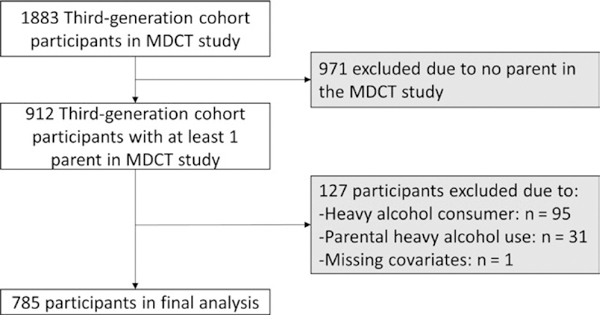
Study sample in the analysis of risk of hepatic steatosis in offspring according to parental history of hepatic steatosis. MDCT, multidetector computed tomography
2.2 |. Measuring hepatic steatosis
Hepatic steatosis was measured from MDCT scans of the abdomen as previously described in detail.4 A total of 25 contiguous images (5 mm slices) of the abdomen were obtained from participants in the supine position (Light Speed Ultra, General Electric, Milwaukee, WI; 120 KVp, 400 mA; gantry rotation time, 500 milliseconds; table feed, 3:1). A radiopaque phantom (Image Analysis, Lexington, KY) was visualized on each image obtained. The mean Hounsfield units (HU) across three separate areas of the liver were averaged to determine the liver HU attenuation from the MDCT scans. We calculated the liver phantom ratio (LPR) as the ratio between the average liver HU and the phantom HU as previously described.18 As the LPR decreases, the fat content of the liver increases. We defined hepatic steatosis by a LPR ≤0.33, which was shown in our prior work to be highly sensitive and specific for detecting liver fat.4
2.3 |. Parental and offspring hepatic steatosis
Our primary exposure of interest was parental hepatic steatosis defined as a LPR ≤0.33 in at least one parent of a participant in the third-generation cohort. Secondary exposures were maternal hepatic steatosis (defined as LPR ≤0.33 in a participant’s mother), paternal hepatic steatosis (defined as LPR ≤0.33 in a participant’s father), and both parents with hepatic steatosis. The primary outcome was the presence of hepatic steatosis defined as a LPR ≤0.33 in the third-generation cohort participants.
2.4 |. Covariates
Covariates were assessed at the first examination (2002–2005) for the FHS third-generation cohort participants at the time of the MDCT scan. Information pertaining to the alcohol use of parent participants in the second-generation FHS study was assessed at the seventh examination (1998–2001). Alcohol use was assessed on the basis of a physician-administrated questionnaire and recorded as drinks per week or drinks per month. Participants were considered current smokers if they had smoked at least one cigarette per day in the year preceding the FHS examination. BMI was computed by participants’ weight in kilograms divided by height in metres squared. Obesity was specified as BMI 30 kg/m2 or higher. Plasma glucose and serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were obtained from fasting morning samples using an automated Roche method (Roche Cobas 501). Elevated ALT or AST was defined as an ALT or AST >19 U/L for women and >30 U/L for men.
Diabetes was defined as fasting plasma glucose ≥126 mg/dL or treatment with a hypoglycaemic agent or insulin. Impaired fasting glucose was defined as fasting glucose ≥100 to < 126 mg/dL in the absence of hypoglycaemic use. The homeostatical model assessment for insulin resistance (HOMA-IR) was calculated using fasting glucose and insulin levels. Dyslipidemia was defined as the use of lipid-lowering therapy. Hypertension was defined as a systolic blood pressure ≥140 mm Hg, a diastolic blood pressure ≥ 90 mm Hg, or the use of antihypertensive medications. Cardiovascular disease was defined as new-onset angina, myocardial infarction, transient ischaemic attack, stroke, heart failure, or intermittent claudication noted at the FHS clinical encounter with a study physician and from available medical records. Cardiovascular disease events were adjudicated by a committee of three FHS investigators.
2.5 |. Statistical analysis
Generalized estimating equation (GEE) models were used to account for familial correlation. We applied GEE using multivariable logistic regression to generate the odds of hepatic steatosis (LPR ≤0.33) in individuals according to the exposure category. The primary exposure category was at least one parent with hepatic steatosis compared to those without a parental history of hepatic steatosis (referent group). Secondary exposures included: maternal hepatic steatosis, paternal hepatic steatosis, and both parents with hepatic steatosis. Models were adjusted for age, sex, and alcohol use (base model). A multivariable model additionally adjusted for BMI. We performed two sensitivity analyses. We had available HOMA-IR and lipid-lowering treatment on 679 participants and added adjustment for log-transformed HOMA-IR and lipid-lowering treatment to the regression models. Additionally, we repeated the multivariable regression analyses without excluding heavy alcohol consumers, which increased the sample size to n = 816.
Given that a few genetic determinants of hepatic fat content have recently been identified in genome-wide association studies (GWAS), we performed an exploratory analysis to additionally adjust by the genetic risk for NAFLD. We have genetic data available on 741/785 (94.4%) of Third Generation participants. We have previously created a weighted genetic risk score (GRS)19 for NAFLD based on a published GWAS in adults of European ancestry that had replication or functional studies available for validation.8 Five SNPs (rs738409, rs2228603, rs12137855, rs780094, rs4240624) were selected. The weighted GRS was created by summing together the product of the number of NAFLD-associated risk alleles and the corresponding regression coefficient derived from the GWAS.19 For the exploratory analysis, we additionally adjusted for the GRS in the regression models.
In a secondary subgroup analysis, we determined the proportion of offspring with hepatic steatosis according to parental hepatic steatosis status. We additionally stratified according to the presence or absence of cardiometabolic diseases including hypertension, diabetes, or obesity (BMI ≥30 kg/m2), which have previously been associated with hepatic steatosis. Participants without hypertension, diabetes, or obesity were considered to have an “optimal” risk factor profile while those with at least one of these conditions were considered to be at elevated cardiometabolic risk. A chi-square test was performed to determine the difference in proportions between groups.
All analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC) software. All p-values were two-sided and a P < 0.05 was considered significant.
3 |. RESULTS
3.1 |. Study sample characteristics
Approximately 23% of our sample (183/785) had at least one parent with hepatic steatosis (maternal hepatic steatosis (n = 95), paternal hepatic steatosis (n = 79), both parents affected (n = 9)). Participants with a parent history of hepatic steatosis were slightly younger (42.5 ± 5.1 years vs 43.9 ± 5.6 years; P=0.002) and slightly more likely to smoke (18% vs 12%; P=0.048) compared to those without a parental history of hepatic steatosis; however, there were no significant differences between cardiometabolic traits (Table 1).
TABLE 1.
Characteristics of third-generation cohort participants (n = 785) by presence of parental hepatic steatosis
| At least one parent with hepatic steatosis |
No parental hepatic steatosis |
P-value | |
|---|---|---|---|
| n (%) | 183 (23.3%) | 602 (76.7%) | |
| Age, years | 42.5 ± 5.1 | 43.9 ± 5.6 | 0.004 |
| Women (%) | 74 (40.4%) | 282 (46.8%) | 0.13 |
| White (%) | 183 (100%) | 602 (100%) | - |
| Alcohol intake, drinks per week | 4.6 ± 5.4 | 4.1 ± 4.8 | 0.27 |
| Current smoking (%) | 32 (18.2%) | 72 (12.3%) | 0.05 |
| BMI, kg/m2 | 27.4 ± 5.1 | 26.8 ± 5.1 | 0.13 |
| Systolic blood pressure, mm Hg | 118 ± 13 | 117 ± 134 | 0.54 |
| Total cholesterol, mg/dL | 192 ± 35 | 190 ± 33 | 0.40 |
| HDL cholesterol, mg/dL | 52 ± 15 | 53 ± 16 | 0.32 |
| Triglycerides, mg/dL | 121 ± 86 | 117 ± 91 | 0.59 |
| LDL cholesterol, mg/dL | 116 ± 32 | 113 ± 30 | 0.25 |
| Use of lipid treatment (%) | 10 (5.7%) | 49 (8.4%) | 0.24 |
| Diabetes (%) | 6 (3%) | 15 (3%) | 0.60 |
| Impaired fasting glucose (%) | 35 (21%) | 123 (22%) | 0.80 |
| HOMA-IR (median, IQR) | 2.7 ± 1.0 | 2.5 ± 1.1 | 0.19 |
| Hypertension (%) | 29 (16%) | 101 (17%) | 0.79 |
| Cardiovascular disease (%) | 3 (2%) | 6 (1%) | 0.44 |
| ALT, U/L | 29.1 ± 19.1 | 26.5 ± 16.6 | 0.10 |
| AST, U/L | 24.8 ± 10.2 | 23.7 ± 8.8 | 0.17 |
| Elevated ALT (%)a | 76 (42%) | 236 (39%) | 0.57 |
| Elevated AST (%)a | 18 (10%) | 37 (6%) | 0.09 |
| Hepatic steatosis (%)b | 39 (21%) | 74 (12%) | 0.002 |
| Genetic risk score | 5.7 ± 1.3 | 5.5 ± 1.3 | 0.10 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; HOMA-IR, homeostatical model assessment for insulin resistance.
Data are presented as mean ± SD or n (%), unless otherwise noted.
Elevated ALT or AST was defined as an ALT or AST > 19 U/L for women and > 30 U/L for men.
Hepatic steatosis was defined by a liver phantom ratio of ≤ 0.33.
3.2 |. Odds of hepatic steatosis in offspring according to parent hepatic steatosis status
Participants with at least one parent with hepatic steatosis had a two-fold increased odds of hepatic steatosis compared to participants without a parental history of hepatic steatosis (OR 2.0, 95% confidence interval (CI) 1.29, 3.11). This association was slightly attenuated but remained statistically significant in multivariable models adjusted for age, sex, alcohol use, and BMI (OR 1.86, CI 1.15–3.03) (Table 2). Specifically, maternal hepatic steatosis was associated with increased odds of hepatic steatosis in offspring in age-, sex-, and alcohol-adjusted models, (OR 1.86, CI 1.08–3.21), though the association was no longer significant after the model was additionally adjusted for baseline BMI. The magnitude of the OR was similar for paternal hepatic steatosis as for maternal hepatic steatosis; however, the models were not statistically significant (Table S1). Though only a small number of participants had 2 parents with hepatic steatosis, we observed a significant increased odds of hepatic steatosis in offspring after adjusting for age, sex, and alcohol use (OR 6.79, CI 1.79, 25.81); however, after BMI was added to the multivariable model, the association was attenuated and no longer statistically significant (OR 3.31, OR 0.34, 32.29) (Table 2). Additional adjustment for log-transformed HOMA-IR and lipid-lowering treatment did not significantly change the results (Table 2). Expanding the sample size to include heavy alcohol consumers (n = 126) also did not significantly change the results (Table S2).
TABLE 2.
Unadjusted and multivariable-adjusted odds of hepatic steatosis in Third Generation Cohort participants grouped by parental hepatic steatosis status
| Odds of hepatic steatosis compared to those without parent hepatic steatosis (reference) |
||
|---|---|---|
| OR (95% CI) | P-value | |
| At least one parent with hepatic steatosis | ||
| n | 183 | |
| Unadjusted | 2.00 (1.29, 3.11) | 0.002 |
| Age, sex, alcohol adjusted | 1.95 (1.25, 3.05) | 0.003 |
| Age, sex, alcohol, BMI adjusted | 1.86 (1.15, 3.03) | 0.01 |
| Age, sex, alcohol, HOMA-IRa, lipid-lowering treatment adjusted | 1.95 (1.15, 3.31) | 0.01 |
| Age, sex, alcohol, HOMA-IRa, lipid-lowering treatment, BMI adjusted | 1.92 (1.13, 3.27) | 0.02 |
| Both parents with hepatic steatosis | ||
| n | 9 | |
| Unadjusted | 6.43 (1.46, 28.32) | 0.01 |
| Age, sex, alcohol adjusted | 6.79 (1.79, 25.81) | 0.005 |
| Age, sex, alcohol, BMI adjusted | 3.31 (0.34, 32.29) | 0.30 |
| Age, sex, alcohol, HOMA-IRa, lipid-lowering treatment adjusted | 4.89 (0.99, 24.20) | 0.05 |
| Age, sex, alcohol, HOMA-IRa, lipid-lowering treatment, BMI adjusted | 3.66 (0.52, 25.78) | 0.19 |
BMI, body mass index; GRS, genetic risk score; HOMA-IR, homeostatical model assessment for insulin resistance.
HOMA-IR was log-transformed.
In an exploratory analysis, after accounting for a NAFLD GRS in the multivariable models, the association with parental hepatic steatosis and risk of hepatic steatosis in the offspring remained essentially unchanged (Table S3).
3.3 |. Proportion with hepatic steatosis by parental hepatic steatosis status
Among participants with a parental history of hepatic steatosis, 21.3% also had hepatic steatosis. In contrast, a significantly lower proportion of participants without a parental history of hepatic steatosis had hepatic steatosis (21.3% vs 12.6%, P = 0.004) (Figure 2).
FIGURE 2.
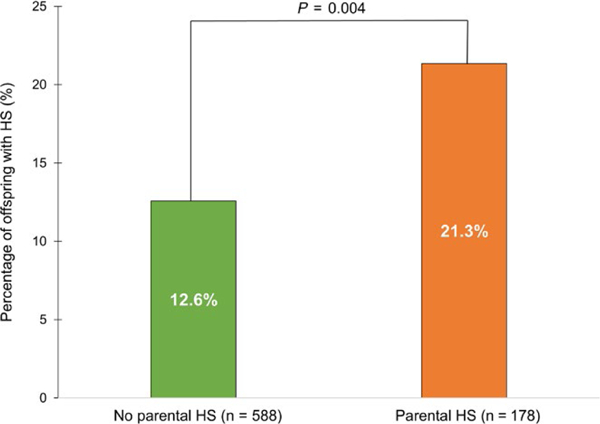
Proportion of third-generation cohort participants with hepatic steatosis stratified by parental hepatic steatosis status. In our cohort, 21.3% of participants with hepatic steatosis had a history of parental hepatic steatosis. In contrast, among participants without parental hepatic steatosis, only 12.6% had hepatic steatosis (21.3% vs 12.6%, P = 0.004)
In a subgroup analysis, 39 participants with an optimal risk factor profile (no hypertension, diabetes, or obesity (n = 514)) had hepatic steatosis on MDCT scan. A higher proportion of those with optimal risk factors and hepatic steatosis had a history of parental hepatic steatosis compared to those with optimal risk factors and no parental hepatic steatosis (16.1% vs 5.2%, P < 0.001) (Figure S1). For participants with elevated cardiometabolic risk (defined by the presence of hypertension, diabetes, or obesity (n = 252)), 73 had hepatic steatosis on MDCT scan. Among participants with elevated cardiometabolic risk and hepatic steatosis, there was no difference in the proportion of those with a parental history of hepatic steatosis compared to without a history of parental hepatic steatosis (30.3% vs 28.5%, P = 0.78) (Figure S1).
4 |. DISCUSSION
In a community-based sample, parental hepatic steatosis predicted an increased risk of hepatic steatosis among offspring after accounting for alcohol use20 and BMI,21 which are known to have genetic components. Our findings suggest that shared genetic and environmental mechanisms contribute to the pathogenesis of NAFLD even among unselected, community-based individuals. Our models were not significantly changed after accounting for known genetic risk factors for NAFLD, which suggests additional genetic risk factors or shared environmental factors may account for the increased risk observed. A higher proportion of participants with a parental history of hepatic steatosis themselves had hepatic steatosis. In particular, participants who were relatively healthy, but with a family history of hepatic steatosis, were more likely to have hepatic steatosis compared to those with optimal risk factors but without a parental history of hepatic steatosis. However, parental hepatic steatosis was not a significant risk factor for hepatic steatosis among those with elevated cardiometabolic risk.
Our study is in line with prior studies which have identified family clustering in individuals with more advanced liver disease.14,22,23 A study of 90 patients with non-alcoholic steatohepatitis found that 18% had a first-degree relative with advanced liver disease.23 Additionally, another small study found higher hepatic fat content in parents and siblings of obese children with NAFLD compared to obese children without NAFLD.14 Importantly, this association remained after accounting for age, sex, ethnicity, and BMI, indicating a possible genetic component to hepatic steatosis separate from general adiposity. In a Finnish twin study, hepatic steatosis as measured by magnetic imaging spectroscopy in a small sub-set of monozygotic twins was highly correlated between twin pairs, after accounting for BMI.24 A twin study derived from a Southern California cohort showed that both hepatic steatosis and hepatic fibrosis are heritable traits13 with significant shared gene effects.25 A prospective study of probands with cirrhosis from non-alcoholic steatohepatitis showed that both the odds of NAFLD and advanced fibrosis were significantly increased among the first-degree relatives of patients with NASH cirrhosis compared to healthy controls.26 These data implicate the role of familial factors in increasing susceptibility towards NAFLD. Our study adds to the current literature by describing the impact of parental hepatic steatosis on the risk of hepatic steatosis in offspring without apparent liver disease.
In an exploratory analysis, the risk of hepatic steatosis among offspring of affected parents remained after accounting for multiple genes associated with NAFLD (PNPLA3, NCAN, LYPLAL1, GCKR, and PP1R3B) in a GRS. However, the GRS did not fully explain the heritability of NAFLD and additional genes may also contribute to NAFLD genetic risk. A variant in the locus for the membrane bound O-acyltransferase domain-containing 7 gene (MBOAT7 or LPIAT1) was recently associated with increased liver fat in alcoholic and non-alcoholic fatty liver disease,27 though its role in NAFLD is not yet completely understood.28 The transmembrane 6 superfamily 2 (TM6SF2) gene has also been associated with NAFLD in multiple studies29–31 though it did not reach the genome-wide significant threshold in the GWAS study used to create the GRS.8 Additional genes, including rare variants, which have yet to be identified, may add to our understanding of NAFLD genetics. Inherited epigenetic changes, such as DNA methylation, histone modifications, and non-coding RNA mediated gene silencing, may also contribute to the heritability of NAFLD, though more research is needed.32,33
In our study, participants with optimal cardiometabolic disease defined as lack of hypertension, diabetes, or obesity were more likely to have hepatic steatosis if they had a parental history of hepatic steatosis compared to those without; however, this difference was not apparent among participants with cardiometabolic disease risk factors. Prior studies of adults demonstrate a strong association between NAFLD and metabolic disease.2,4,34 It is possible that once individuals develop metabolic disease, the contribution of genetic risk factors for hepatic steatosis becomes less important. It is not known if individuals with NAFLD but without metabolic disease will later develop liver or cardiovascular consequences of NAFLD or will continue to have a benign course. One prior study noted that volunteers with NAFLD because of a genotype associated with hepatic steatosis, PNPLA3, did not have potentially harmful adipose tissue inflammation, which was observed in those with obesity and NAFLD.35 However, other studies have demonstrated an association between the PNPLA3 genotype and the severity of liver disease.36–38 Additional genetic studies in younger aged-NAFLD cohorts or in cohorts of individuals without metabolic disease are needed to further explore the genetics of NAFLD when gene-gene and gene-environment interactions may be minimized.39
The strengths of our study include the definition of hepatic steatosis based on MDCT imaging in both the offspring and parents which reduced the potential for misclassification had we relied on non-imaging based surrogate markers of liver fat such as serum aminotransferase levels. Additionally, our study included unselected, community-based participants which reduces the likelihood that our sample had unique mechanisms underlying hepatic steatosis or that our sample included families with rare genetic conditions.
Limitations of our study include the few cases (n = 9) with two affected parents and a large number of participants with only one parent with available data (n = 643) which potentially led to differential misclassification and biased our results towards the null. In the subgroup analysis of the separate contribution of maternal or paternal hepatic steatosis to hepatic steatosis in offspring, our sample size and power were low and; therefore, our type II error rate was high. Moreover, our sample was 100% White of European ancestry and our results may not be generalizable to individuals of different race or ethnicity. Additionally, we cannot exclude early family environmental influences which may account for the observed familial association; however, this is less likely given the mean age of the offspring was 43 years. In our subgroup analysis, we define those without obesity, diabetes, or hypertension as having “optimal metabolic risk”; however, many of the participants in this category with hepatic steatosis also were overweight with central obesity and atherogenic dyslipidemia so cannot be considered metabolically healthy.
In conclusion, individuals with a parental history of hepatic steatosis are at increased risk for hepatic steatosis, even after accounting for BMI. Specifically, a parental history of hepatic steatosis may be an important factor in NAFLD among those that are otherwise relatively metabolically healthy. Additional genetic studies are needed to identify the genes and mechanisms underlying the familial relationship.
Supplementary Material
Key points.
Multiple risk factors for hepatic steatosis have been previously identified, including hypertension, diabetes, and obesity; however, these conditions do not completely explain the risk for non-alcoholic fatty liver disease.
We demonstrate that participants in the community-based Framingham Heart Study had a nearly two-fold increased odds of hepatic steatosis if they had a family history of imaging-defined hepatic steatosis.
In particular, a parental history of hepatic steatosis may be a more important risk factor for hepatic steatosis among those without other cardiometabolic risk factors.
A family history of hepatic steatosis should be considered when determining an individual’s risk for non-alcoholic fatty liver disease.
Acknowledgments
Funding information
JM is an employee of the National Heart, Lung and Blood Institute and his research is supported by the Division of Intermural Research. MTL is supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases K23 DK113252, the Boston University School of Medicine Department of Medicine Career Investment Award and the Boston University Clinical Translational Science Institute UL1 TR001430. EJB is supported in part by R01 HL128914 and R01 HL092577. RL is supported in part by R01 DK106419.
Abbreviations:
- ALT
Alanine aminotransferase
- AST
Aspartate aminotransferase
- BMI
Body mass index
- CI
Confidence interval
- FHS
Framingham Heart Study
- GEE
Generalized estimating equations
- HU
Hounsfield units
- LPR
Liver phantom ratio
- MDCT
multidetector computed tomography
- NAFLD
Non-alcoholic fatty liver disease
- PNPLA3
patatin-like phospholipase domain-containing 3
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
CONFLICT OF INTEREST
The authors do not have any conflict of interests to report.
REFERENCES
- 1.Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005–2023. [DOI] [PubMed] [Google Scholar]
- 2.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–2273. [DOI] [PubMed] [Google Scholar]
- 3.Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol. 2015;13:643–54.e1–9; quiz e39–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Speliotes EK, Massaro JM, Hoffmann U, et al. Fatty liver is associated with dyslipidemia and dysglycemia independent of visceral fat: the Framingham Heart Study. Hepatology. 2010;51:1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anstee QM, Day CP. The genetics of nonalcoholic fatty liver disease: spotlight on PNPLA3 and TM6SF2. Semin Liver Dis. 2015;35:270–290. [DOI] [PubMed] [Google Scholar]
- 6.Hernaez R, McLean J, Lazo M, et al. Association between variants in or near PNPLA3, GCKR, and PPP1R3B with ultrasound-defined steatosis based on data from the third National Health and Nutrition Examination Survey. Clin Gastroenterol Hepatol. 2013;11:1183–90 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen J, Wong GL, Chan HL, et al. PNPLA3 gene polymorphism accounts for fatty liver in community subjects without metabolic syndrome. Aliment Pharmacol Ther. 2014;39:532–539. [DOI] [PubMed] [Google Scholar]
- 8.Speliotes EK, Yerges-Armstrong LM, Wu J, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rotman Y, Koh C, Zmuda JM, Kleiner DE, Liang TJ, Nash CRN. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology. 2010;52:894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Speliotes EK, Butler JL, Palmer CD, et al. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology. 2010;52:904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loomba R, Hwang SJ, O’Donnell CJ, et al. Parental obesity and offspring serum alanine and aspartate aminotransferase levels: the Framingham heart study. Gastroenterology. 2008;134:953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loomba R, Schork N, Chen CH, et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology. 2015;149:1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwimmer JB, Celedon MA, Lavine JE, et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology. 2009;136:1585–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox CS, Massaro JM, Hoffmann U, et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116:39–48. [DOI] [PubMed] [Google Scholar]
- 16.Dawber TR, Meadors GF, Moore FE Jr. Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nation’s Health. 1951;41:279–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Splansky GL, Corey D, Yang Q, et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol. 2007;165:1328–1335. [DOI] [PubMed] [Google Scholar]
- 18.Speliotes EK, Massaro JM, Hoffmann U, et al. Liver fat is reproducibly measured using computed tomography in the Framingham Heart Study. J Gastroenterol Hepatol. 2008;23:894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma J, Hennein R, Liu C, et al. Improved diet quality associates with reduction in liver fat, particularly in individuals with high genetic risk scores for nonalcoholic fatty liver disease. Gastroenterology. 2018;155:107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verhulst B, Neale MC, Kendler KS. The heritability of alcohol use disorders: a meta-analysis of twin and adoption studies. Psychol Med. 2015;45:1061–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27:325–351. [DOI] [PubMed] [Google Scholar]
- 22.Struben VM, Hespenheide EE, Caldwell SH. Nonalcoholic steatohepatitis and cryptogenic cirrhosis within kindreds. Am J Med. 2000;108:9–13. [DOI] [PubMed] [Google Scholar]
- 23.Willner IR, Waters B, Patil SR, Reuben A, Morelli J, Riely CA. Ninety patients with nonalcoholic steatohepatitis: insulin resistance, familial tendency, and severity of disease. Am J Gastroenterol. 2001;96:2957–2961. [DOI] [PubMed] [Google Scholar]
- 24.Makkonen J, Pietilainen KH, Rissanen A, Kaprio J, Yki-Jarvinen H. Genetic factors contribute to variation in serum alanine amino-transferase activity independent of obesity and alcohol: a study in monozygotic and dizygotic twins. J Hepatol. 2009;50:1035–1042. [DOI] [PubMed] [Google Scholar]
- 25.Cui J, Chen CH, Lo MT, et al. Shared genetic effects between hepatic steatosis and fibrosis: a prospective twin study. Hepatology. 2016;64:1547–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caussy C, Soni M, Cui J, et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J Clin Investig. 2017;127:2697–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mancina RM, Dongiovanni P, Petta S, et al. The MBOAT7-TMC4 Variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology. 2016;150:1219–30.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott E, Anstee QM. Genetics of alcoholic liver disease and non-alcoholic steatohepatitis. Clin Med. 2018;18(Suppl 2):s54–s59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dongiovanni P, Petta S, Maglio C, et al. Transmembrane 6 super-family member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology. 2015;61:506–514. [DOI] [PubMed] [Google Scholar]
- 30.Goffredo M, Caprio S, Feldstein AE, et al. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: a multiethnic study. Hepatology. 2016;63:117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu YL, Reeves HL, Burt AD, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. 2014;5:4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moran-Salvador E, Mann J. Epigenetics and liver fibrosis. Cell Mol Gastroenterol Hepatol. 2017;4:125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Page A, Paoli P, Moransalvador E, White S, French J, Mann J. Hepatic stellate cell transdifferentiation involves genome-wide remodeling of the DNA methylation landscape. J Hepatol. 2016;64:661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lonardo A, Sookoian S, Pirola CJ, Targher G. Non-alcoholic fatty liver disease and risk of cardiovascular disease. Metabolism. 2015;65:1136–1150. [DOI] [PubMed] [Google Scholar]
- 35.Lallukka S, Sevastianova K, Perttila J, et al. Adipose tissue is inflamed in NAFLD due to obesity but not in NAFLD due to genetic variation in PNPLA3. Diabetologia. 2013;56:886–892. [DOI] [PubMed] [Google Scholar]
- 36.Romeo S, Sentinelli F, Cambuli VM, et al. The 148M allele of the PNPLA3 gene is associated with indices of liver damage early in life. J Hepatol. 2010;53:335–338. [DOI] [PubMed] [Google Scholar]
- 37.Sookoian S, Castano GO, Burgueno AL, Gianotti TF, Rosselli MS, Pirola CJ. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res. 2009;50:2111–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valenti L, Alisi A, Galmozzi E, et al. I148M patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology. 2010;52:1274–1280. [DOI] [PubMed] [Google Scholar]
- 39.Blackett PR, Sanghera DK. Genetic determinants of cardiometabolic risk: a proposed model for phenotype association and interaction. J Clin Lipidol. 2013;7:65–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.