

SUMMARY
Single-cell transcriptome profiling of heterogeneous tissues can provide high-resolution windows into developmental dynamics and environmental responses, but its application to plants has been limited. Here, we used the high-throughput Drop-seq approach to profile >12,000 cells from Arabidopsis roots. This identified numerous distinct cell types, covering all major root tissues and developmental stages, and illuminated specific marker genes for these populations. In addition, we demonstrate the utility of this approach to study the impact of environmental conditions on developmental processes. Analysis of roots grown with or without sucrose supplementation uncovers changes in the relative frequencies of cell types in response to sucrose. Finally, we characterize the transcriptome changes that occur across endodermis development and identify nearly 800 genes with dynamic expression as this tissue differentiates. Collectively, we demonstrate that single-cell RNA-seq can be used to profile developmental processes in plants and show how they can be altered by external stimuli.
Graphical Abstract
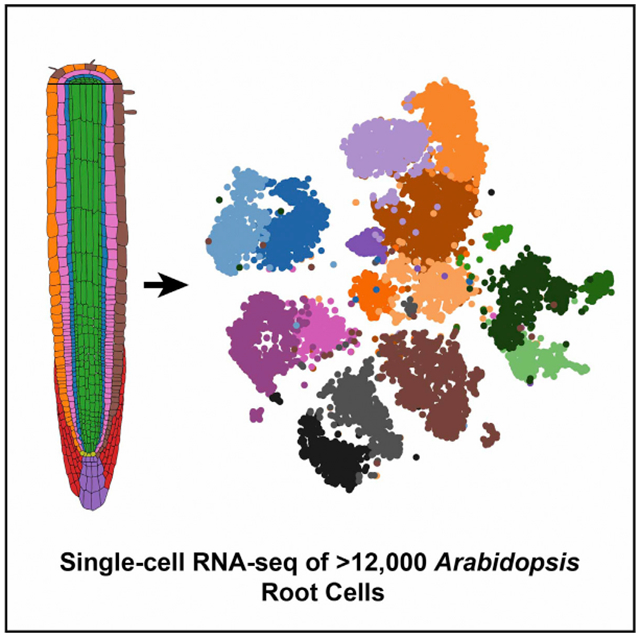
In Brief
The application of single-cell transcriptome profiling to plants has been limited. Shulse et al. performed Drop-seq on Arabidopsis roots, generating a transcriptional resource for >12,000 cells across major populations. This revealed marker genes for distinct cell types, cell frequency changes resulting from sucrose addition, and genes dynamically regulated during development.
INTRODUCTION
Single-cell transcriptomic technologies are revolutionizing molecular studies of heterogeneous tissues and organs, enabling the elucidation of new cell type populations and revealing the cellular underpinnings of key developmental processes (Efroni et al., 2016; Patel et al., 2014; Villani et al., 2017). Recently developed high-throughput single-cell RNA sequencing (scRNA-seq) techniques, such as Drop-seq (Macosko et al., 2015), use a microfluidic device to encapsulate cells in emulsified droplets, allowing for the profiling of hundreds or even thousands of cells in a single experiment. Despite this remarkable advance, the large and non-uniform size of plant cells, as well as the presence of cell walls, has hindered the application of this technology to plant tissues. Applying high-throughput scRNA-seq methods to plants would negate the need for specialized reporter lines that are widely used for the capture of specific cell type populations. Single-cell technologies have the potential to provide a detailed spatiotemporal characterization of distinct cell types present in plants, their developmental trajectories, and their transcriptional regulatory pathways (Efroni and Birnbaum, 2016).
In the present study, we report gene expression profiles for >12,000 single cells isolated from the Arabidopsis root. This compendium includes all common cell types and allowed the identification of highly specific marker genes for each population profiled. We compared cellular profiles of roots grown with or without sucrose, which illuminated differences in cell type frequency and tissue-specific gene expression resulting from this external stimulus. Finally, we used pseudotime analysis to characterize gene expression changes during endodermis development, which highlighted genes that likely direct the differentiation of this tissue. Collectively, these results show Arabidopsis root development at high resolution.
RESULTS
We performed high-throughput, microfluidic-enabled scRNA-seq of plant tissue, following the Drop-seq method and using protoplasts isolated from 5- and 7-day-old Arabidopsis thaliana whole roots (Figure 1; Table S1). We generated 10 libraries: 3 libraries for cells from plants grown with 1% sucrose supplementation and 7 libraries for cells from plants grown without sucrose. Across all replicates, we obtained transcriptomes for 12,198 individual root cells, each with a minimum of 1,000 unique molecular identifier (UMI)-tagged transcripts (Figure S1A; STAR Methods). Protoplasts are delicate and prone to bursting, releasing free-floating mRNA into suspension. To assess the quality of the protoplasts, we spiked cultured human or mouse cells into the plant cell preparations before each run. Plotting the number of control (human or mouse) UMIs versus Arabidopsis UMIs for each cell allowed us to confirm that the cell preparations were of high quality (Figure S1B). In addition, because the process of protoplasting plant roots can lead to changes in gene expression, we confirmed that Drop-seq captured a representative population of cells present in the root, as well as their native gene expression, by combining the transcriptomes of all captured cells into a pseudobulk profile and comparing this profile to a conventional mRNA-seq profile of non-protoplasted 5-day-old Arabidopsis root tissue (Figure S1C). The pseudobulk transcriptome showed high correlation with the bulk root mRNA sequencing (mRNA-seq) profile (Spearman’s rho: 0.79 for all genes, 0.80 when known protoplast response genes [Birnbaum et al., 2003] were excluded) and much lower correlation with previously reported (Zhang et al., 2018) bulk whole-flower mRNA expression (Spearman’s rho: 0.44–0.46) (Figure S1D).
Figure 1. Single-Cell RNA-Seq of 12,198 Arabidopsis Root Cells Captures Diverse Cell Types.

(A) Cartoon representing the cell types that comprise the Arabidopsis root.
(B) t-Distributed Stochastic Neighbor Embedding (t-SNE) dimensional reduction of 12,198 single Arabidopsis root cells that were profiled using Drop-seq. Cells were clustered into 17 populations using Seurat (Butler et al., 2018). Points indicate individual cells and are colored by assigned cell type and cluster according to the legend.
(C) Same as (B), except colored according to the top index of cell identity (ICI) classification for each cell regardless of statistical significance. ICI assignments passing statistical significance are shown in Figures S3B and S3C.
See also Figures S1–S4 and Table S1.
To identify distinct cell type populations and to directly compare cell type identity in the presence and absence of sucrose, we performed unsupervised clustering analysis of all 12,198 single cells with the canonical correlation analysis function of Seurat (Butler et al., 2018; Satija et al., 2015) (Figure 1B; Figure S1E). All 17 cell type clusters identified contained cells from each replicate, and replicates within the same growth conditions had similar proportions of each cell type (Figures S1F and S1G). This supports the high reproducibility of the approach and an absence of substantial batch effects. To assign individual clusters to known major root cell types, we (1) examined the expression of commonly used marker genes (Brady et al., 2007), (2) compared our single-cell profiles to reported whole-transcriptome profiles of root cell types isolated from reporter lines (Brady et al., 2007; Li et al., 2016), and (3) computed an index of cell identity (ICI) score (Efroni et al., 2015) for each cell (Figures 1B and 1C; Figures S2 and S3; see also STAR Methods). Cell clusters were primarily assigned an overall cell type identity based on a majority of statistically significant ICI calls (p < 0.05) (Figures S3B and S3C). Nearly all major cell or tissue types present in the root were captured by at least one cluster, including stele (four clusters), endodermis (two clusters), cortex (two clusters), hair cells (one cluster), non-hair cells (four clusters), and columella (two clusters). In addition, we identified two clusters in which the cells have an apparently mixed identity, with transcriptomes similar to both cortex and hair cells (Figures 1B and 1C; Figures S2A, S2B and S3C). We could not assign a distinct cell cluster to quiescent center (QC) cells, likely due to their rarity (Cartwright et al., 2009), but cells with a suggestive quiescent center identity (by ICI score) are found in clusters 2, 4, and 5 (non-hair cell epidermis) (Figure S3A). Consistent with this, clusters 2, 4, and 5 have a less mature (i.e., meristematic) developmental identity, and their expression profiles correlate well with cells expressing WOL and SCR (Figure S2). We also could not assign a specific cluster to lateral root cap cells, which are not included in the ICI model and are not well defined by known marker genes. To better locate lateral root cap (LRC) cells within our captured cell populations, we examined the expression of suggested LRC-associated genes, including PASPA3, BFN1, SMB, and RCP1 (Fendrych et al., 2014; Olvera-Carrillo et al., 2015; Tsugeki and Fedoroff, 1999). These genes were found to be predominantly expressed by cells within the non-hair cell and columella clusters (Figure S4). Furthermore, cells isolated from the LRC enhancer trap reporter line (Birnbaum et al., 2003) show strong transcriptional similarity to several non-hair cell clusters identified by Drop-seq (Figure S2A). Collectively, these observations suggest that LRC cells are clustering with other epidermal cell types rather than forming a distinct cluster.
We next compared the proportions of each cell type captured by Drop-seq to published microscopy-based surveys of root cell types (Cartwright et al., 2009), which highlighted a relative depletion of stele cells in our data (Figure S3D). This is at least partially explained by the higher proportion of mature tissue in our samples (whole roots) versus the microscopy-based study (root tips only to the first lateral root primordia), because mature xylem cells undergo programmed cell death (Fukuda, 1996) and phloem cells undergo nuclear degradation (Furuta et al., 2014). Despite this possible depletion, we captured several known subpopulations of stele cells, including protoxylem (cluster 13), phloem-like (cluster 14), meristematic xylem (cluster 15), and pericycle (cluster 16) (Figure 2). Aside from the stele tissue, other cell types were well captured by Drop-seq, including epidermal, cortical, and endodermal cells. Overall, these results demonstrate that Drop-seq accurately captures the complexity of cell types present in the plant root and illuminates subpopulations for many tissues.
Figure 2. Stele Cell Subpopulations.

Left: t-SNE representation, as in Figure 1B, of all captured single cells, showing the stele clusters (13–16) colored in shades of green. Right: t-SNE showing just these stele clusters, with each cell colored by the stele subpopulation identity assigned by ICI. Gray cells had no stele cell type identity that reached statistical significance (p < 0.05).
Expression analysis of marker genes commonly used to label specific cell populations revealed that many exhibit limited specificity or very low expression levels (Figure 3A). Therefore, we used the scRNA-seq data to identify a new set of robust marker genes, quantitatively optimized for specificity (see STAR Methods). The top candidate marker gene for each cluster is shown in Figure 3B, with the top 50 candidate marker genes for each cluster listed in Table S2. This unbiased approach identified genes with previously established roles in the biology of some cell populations. For example, all five genes encoding components of the Casparian strip (CASP1 through CASP5) are among the top 20 marker genes for one of the endodermis clusters (cluster 12), and the root hair-specific gene ADF8 (Ruzicka et al., 2007) is a top candidate marker gene for hair cells (cluster 17). However, genes with well-established roles in root development represent a relatively small subset of the highly specific marker genes we identified, and many candidate marker genes have little to no functional annotation data available. As evidence of this, 58% of the top 50 marker genes, including 7 of the 17 top (rank 1) marker genes across all clusters, have no gene symbol beyond an Arabidopsis Genome Initiative (AGI) gene identifier (Figure 3B; Table S2). We expect that these marker genes may have important and previously unrecognized roles in the development of these cells types and are thus candidates for investigation through traditional reverse genetic studies. While most new marker genes are specific to the cluster from which they derive, a large proportion of markers identified for meristematic clusters are shared (Table S2, clusters 2, 4, 5, and 15). This observation may explain the lack of a quiescent center-specific cluster identified from our data, because the transcriptional profiles from quiescent center cells may be similar to other meristematic cell types. Altogether, these marker genes provide several avenues to explore the transcriptional processes that underlie root cell type specification and function.
Figure 3. Identification of Highly Specific Cell Type Marker Genes.

(A) Violin plots showing the expression of 10 commonly used cell type marker genes across all clusters. Labels to the left of the gene names indicate the cell population each gene is used to designate.
(B) Violin plots showing the cluster-specific expression of the top-ranking novel candidate marker gene for each cell population identified by this study, color-coded as in Figure 1B.
See also Table S2.
Sucrose Results in Altered Cell-Type Frequencies and Gene Expression
Next, we assessed differences in cell type populations and gene expression between plants grown with or without sucrose. Sucrose is known to result in changes to cell signaling (Tognetti et al., 2013), and its presence or absence is a common distinction between experimental growth protocols. While several reports have documented the impact of sucrose on root system architecture (Macgregor et al., 2008), hormone signaling (León and Sheen, 2003; Tanimoto et al., 1995), and response to abiotic stress (Jain et al., 2007), its direct role on root cell type differentiation has not been well studied (Tognetti et al., 2013). Half of the 12,198 cells profiled were from each growth condition (6,102 sucrose+, 6,096 sucrose−). All 17 cell type clusters included cells from both sucrose+ and sucrose− plants (Figure 4A), which suggests that sucrose does not result in fundamentally different cell type identities. However, there are notable differences between sucrose+ and sucrose− in the proportion of cells from each cell type cluster (Figure 4B; Figure S1F). For example, cells grown with sucrose were strongly enriched among hair cells (cluster 17), consistent with previous morphological observations (Jain et al., 2007). Cells grown without sucrose were found to be enriched in the four meristematic cell clusters (clusters 2, 4, 5, and 15). These results suggest that availability of sucrose results in overall higher proportions of developmentally mature cells.
Figure 4. Sucrose Alters Root Cell Type Proportions and Tissue-Specific Gene Expression.

(A) t-SNE representation as in Figure 1, colored by growth condition (sucrose±).
(B) Proportion of cells in each of the 17 clusters by growth condition (sucrose±), colored as in (A). Across all experiments, 50% of cells profiled were from each growth condition (red line). Clusters with significant differences (p < 0.05 by χ2 test) between the percentages of sucrose+ and sucrose− cells are denoted by red asterisks above the column.
(C) Bar plot characterizing genes that are differentially expressed between sucrose+ and sucrose− growth conditions. Genes were classified by the condition in which they had higher expression (sucrose−, no shading; sucrose+, gray shading) and how many tissue or cell types exhibited differential expression of that gene. Nearly half of all differentially expressed genes have altered expression in only one tissue or cell type.
(D and E) Examples of genes with tissue-specific (D) or universal (E) differential expression between sucrose+ conditions (filled violins) or sucrose− conditions (open violins). Violin plots show normalized gene expression by cell or tissue type and growth condition. Red asterisks indicate statistically significant differences (see Table S3 for exact p values).
We next assessed gene expression differences between sucrose+ and sucrose− conditions. In total, 1,838 genes were differentially expressed between conditions, with 974 genes upregulated in sucrose+, 838 genes upregulated in sucrose−, and 26 genes upregulated in both but in different cell or tissue types (Figures 4C–4E; Table S3). Gene expression differences were largely tissue or cell type specific, with 49% of the differentially expressed genes altered in a single tissue or cell type and only 1% altered throughout the root (Figure 4C). These results indicate that the addition of sucrose as a carbon source results in altered proportions of cell populations, along with gene expression changes that are often highly specific to distinct tissues or cell populations.
Gene Expression Changes across Endodermis Development
Finally, a particular strength of the application of scRNA-seq to developing tissues is the ability to infer developmental trajectories of individual cell populations through pseudotime analysis (Trapnell et al., 2014). Individual root cell types form an apical-basal anatomical gradient of temporal differentiation states, with undifferentiated (i.e., meristematic) cells near the root tip and the most mature cells located nearer the root-shoot junction. Therefore, we hypothesized that the various subpopulations identified for some cell types may reflect these trajectories. We carried out a pseudotime analysis using Monocle (Trapnell et al., 2014) and focusing on endodermal cells, which have a known trajectory from undifferentiated, to state I (defined by the formation the Casparian strip), to state II (defined by the formation of a secondary cell wall made of suberin) (Andersen et al., 2015) (Figure 5A). Pseudotime analysis performed separately on endodermis cells from sucrose+ or sucrose− growth conditions showed similar trajectories, so all 1,660 endodermis cells were combined for a single analysis (Figure 5B). To properly orient the pseudotime axis relative to known endodermis differentiation, we examined the profiles of genes known to be dynamically expressed during endodermal development (Figure 5C). This resulted in a bifurcating developmental trajectory, with an immature population splitting into two developmentally mature branches that had significant but overall subtle gene expression differences between them (Figure S5A; Table S4). While it is unlikely that protoplasting yields endodermis cells that are heavily suberized, due to the recalcitrance of these cells’ walls to digestion, we speculate that the differences in expression levels between the two mature branches may represent the patchy nature of initial suberization at these early stages of differentiation. We examined the expression of genes implicated in suberization or co-expressed with GPAT5, considered the rate-limiting step in endodermal suberization (Beisson et al., 2007). GPAT5 expression was not robustly captured in any cell, possibly due to it being expressed later in the endodermis developmental trajectory or at a level that is below the sensitivity of Drop-seq (Figure S5B). However, we identified numerous cells expressing GPAT7 and/or ABCG16, both implicated in suberization (Yadav et al., 2014; Yang et al., 2012), and these cells are co-localized predominantly along the major developmental axis (Figure S5B).
Figure 5. Transcriptional Dynamics of Endodermis Development.

(A) Left: schematic representation of the trajectory of endodermis development. Right: t-SNE representation, as in Figure 1B, with only the endodermis cells colored blue.
(B) Pseudotime trajectory of all 1,660 endodermis cells, generated using Monocle (Trapnell et al., 2014) (see STAR Methods). Each point indicates a unique cell, color coded by the original endodermis cluster as in (A). “Major trajectory” indicates the pseudotime trajectory with most cells.
(C) Expression profiles along pseudotime for genes known to have dynamic expression during endodermal development. Each point indicates the log-normalized expression of the indicated gene in a single cell.
(D) Right: heatmap showing the scaled and centered gene expression (Z scores) of 798 genes that show dynamic expression over the major endodermis developmental trajectory by scRNA-seq (see STAR Methods). For this plot, all endodermis cells were divided into 20 bins, distributed uniformly across pseudotime. Canonical endodermal development genes are noted in black along the left axis, with red indicating genes whose expression was validated in this study. Genes were grouped by expression pattern, from top to bottom: early, middle, and late expression. Left: top 5 gene ontology (GO) terms in each group by fold enrichment (all significantly enriched; see Table S5). Numbers in parentheses indicate the number of genes in each category.
(E) AT4CL1 promoter-driven GFP is highest in mature endodermis, consistent with the AT4CL1 expression pattern predicted from the pseudotime analysis in (D). Scale bar: 200 μm.
Once we established the correctly oriented pseudotime trajectory, we used it to ascertain additional genes that are dynamically regulated during endodermis development, focusing on the primary developmental trajectory that included most endodermis cells (red arrow in Figure 5B; see also STAR Methods). This identified 798 total dynamically expressed genes, which fell into three patterns: early, middle, and late expression (Figure 5D; Table S5). The early expression gene group includes MYB36, which is known to regulate the initial stages of endodermis differentiation (Liberman et al., 2015), and this group is enriched for gene ontology (GO) terms associated with water or fluid transport (Figure 5D; Table S5). Genes whose expression peaks in the middle of the pseudotime axis are enriched for GO terms associated with response to desiccation and toxin metabolism. Genes whose expression peaks late include canonical Casparian strip genes (e.g., CASP1), and this category is enriched in GO terms associated with cell junction assembly and lignin metabolism. From these expression groups, we identified promoter-driven GFP transgene lines for two of the genes with distinct expression patterns over endodermis pseudotime, AT4CL1 and MSL4 (Figure S5C). These reporter lines confirmed the strong expression pattern of AT4CL1 in late endodermis (Figure 5E) and the endodermis-specific expression of MSL4 (Figure S5D). The specific transcriptional pathways that directly regulate state II differentiation are not well defined (Andersen et al., 2015). Thus, late-expressing genes are candidates for the cellular components that direct mature endodermis differentiation. Transcription factor genes found in this late-expressing group include B70, HAT22, BZIP65, and TLP11 (Table S5). Collectively, these results highlight the power of scRNA-seq for generating hypotheses that may lead to a deeper understanding of cellular development in plant tissues.
DISCUSSION
This study demonstrates that Drop-seq is a powerful method to quickly profile the diverse cell types that comprise heterogenous plant tissue. We profiled >12,000 single cells and captured subpopulations of many major cell types. By comparing cells from roots grown on sucrose+ or sucrose− media, we discovered that sucrose does not substantially alter cell type identity but does lead to changes in the relative proportions of cell types and tissue-specific gene expression. Overall, sucrose has a surprisingly subtle effect on root cell populations given its relationship to abscisic acid (ABA) and ethylene signaling (León and Sheen, 2003), its known influence on root system architecture (Macgregor et al., 2008), and its effect on responses to abiotic stress (Jain et al., 2007). Going forward, we anticipate Drop-seq will be broadly used to characterize the effects of many environmental conditions, such as drought, microbial infection, marginal soils, salt, and pH levels.
Overall, our results suggest that Drop-seq will be applicable to any tissue or plant species for which protoplasts can be obtained. Efficient protoplasting techniques and scRNA-seq optimization will be essential for isolating all cell types uniformly. The rare quiescent center population has proven particularly challenging to capture using microfluidic-based scRNA-seq methods (Ryu et al., 2019), and more robust characterization of these cells may require the profiling of larger numbers of cells or specialized enrichment strategies. Despite this, current methods can be used to study many aspects of plant development and environmental responses (this study; Ryu et al., 2019; Denyer et al., 2019). We anticipate that the dataset described herein will be a valuable resource to other investigators for the study of root cell type differentiation and that Drop-seq will open exciting inroads of exploration into the identities and functions of plant cell types.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Diane E. Dickel (dedickel@lbl.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Plant material and growth conditions
Arabidopsis (Arabidopsis thaliana) ecotype Columbia (Col-0) seeds were surface sterilized in 70% ethanol for 2 minutes, followed by 50% bleach and 0.1% Triton X-100 for 5 - 10 minutes. Sterilized seeds were washed 4 - 6 times with sterile water and imbibed in the dark for 3 days at 4°C. Seeds were sown at a density of approximately 125 seeds per row on sterile nylon mesh filters placed on top of plant growth media consisting of 1X Murashige and Skoog Basal Medium (MilliporeSigma, St. Louis, MO) supplemented with 1% agar (MilliporeSigma) and 2.6 mM 2-(N-Morpholino)ethanesulfonic acid (MES; MilliporeSigma), and adjusted to pH 5.7. Medium for plants grown in the presence of sucrose was supplemented with 1% sucrose (MilliporeSigma). Petri dishes were positioned vertically in an incubator (Percival Scientific, Perry, IA) under a long-day photoperiod (16 h of light) at 22°C.
METHOD DETAILS
Drop-seq of Arabidopsis root protoplasts
Protoplast suspensions were prepared from 5- and 7-day-old Arabidopsis seedlings using previously described methods (Birnbaum et al., 2005). Briefly, whole Arabidopsis roots were finely chopped with a Feather Disposable Scalpel #10 (Andwin Scientific, Schaumburg, IL) and distributed among 70-μm cell strainers (each containing 1,250-1,500 roots). Blades were changed every 5 to 6 plates to prevent dulling of the blade and consequent bruising of the tissue. Each strainer was then immersed in a solution of cellulysin (MilliporeSigma) and pectolyase (MilliporeSigma) for 60 minutes with agitation at 85 rpm on an orbital shaker at room temperature. After cell wall digestion, each strainer, containing remnants of roots that were not protoplasted, was removed and discarded. Protoplasts were then pelleted, resuspended, and filtered through a 40-μm cell strainer to eliminate any clumped cells. The cells were then kept either at their starting concentration or diluted in Solution A (Birnbaum et al., 2005) (10 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 0.1% bovine serum albumin, 2 mM MES, 600 mM mannitol). Cultured human cells (HEK293T/17 from ATCC) or E14Tg2a.4 mouse embryonic stem cells (Skarnes, 2000) were spiked in at a concentration of 2.5% or 10% to estimate capture purity. Drop-seq was performed as described previously (Macosko et al., 2015) and in Drop-seq Laboratory Protocol version 3.1 (http://mccarrolllab.org/dropseq/). Drop-seq beads (ChemGenes) used were from lots 011416B and 072817. The libraries were prepared and uniquely indexed using the Nextera XT DNA Library Preparation Kit (Illumina) and sequenced on a HiSeq 4000, HiSeq 2500, or NextSeq (Illumina). See Table S1 for the details of each library, including plant age, growth conditions, cell concentrations, and replicate relationships.
Read alignment and generation of digital gene expression data
A digital expression matrix of Single-cell Transcriptomes Attached to MicroParticles (STAMPs) was created from the raw sequencing data as described in the Drop-seq Core Computational Protocol version 1.0.1 (http://mccarrolllab.org/dropseq/). Sequence data were aligned to a combined Arabidopsis-human (Libraries A-C) or Arabidopsis-mouse (Libraries D-J) mega-reference using STAR v2.5.2b (Dobin et al., 2013) with the default settings. The following genome versions were used to generate the mega-references: TAIR 10 (Arabidopsis thaliana), GRCh38/hg38 (human, full dataset), mm10 (mouse, full dataset). Uniquely mapped reads were grouped by cell barcode. To identify true STAMPs, we discarded any cell barcodes with fewer than 1,000 unique transcripts (i.e., UMIs) and expression of fewer than 200 genes. A STAMP was then considered as derived from Arabidopsis (rather than human, mouse, or mixed) if 98% or more of the mapped reads mapped to Arabidopsis. For constructing barnyard plots, we mapped the Drop-seq data to reduced mega-reference genomes, excluding alternative or unknown chromosome sequences from the human and mouse genomes.
Data alignment and clustering analysis
Data from the ten independent Drop-seq runs were combined to create two individual single cell datasets with the Seurat R package (version 2.3.4) (Butler et al., 2018) after removing genes known to be induced during protoplasting (Birnbaum et al., 2003). The first dataset contained 6,102 single cell transcriptomes isolated from roots grown on media containing sucrose, and the second contained 6,096 single cell transcriptomes isolated from roots grown on media without sucrose. Seurat was then used to 1) normalize and scale these two datasets (regressing out variance due to the number of UMI counts per cell, as well as the percent of mitochondria- or chloroplast-derived transcripts using the ScaleData function), and 2) identify highly variable genes (1,058 and 1,021 genes for the sucrose-minus and -plus datasets, respectively) among the single cell expression profiles. Seurat was then used to combine the two media-specific datasets into a single dataset using Canonical Correlation Analysis (Butler et al., 2018) with the RunCCA function and the intersection of the two variable gene sets (1549 genes). The top 35 canonical components were used to align the two datasets with the AlignSubspace function. The canonical correlation vectors and the RunTSNE function in Seurat were then used to compute a t-SNE (van der Maaten and Hinton, 2008) dimensional reduction and to group cells into distinct clusters. Clustering was performed with the FindClusters function in the Seurat package, using a resolution of 0.6 and default parameters.
Assignment of clusters to known cell types
To align cell population clusters from the unsupervised scRNA-seq to known cell types, we assessed 1) expression of known cell type-specific marker genes identified from the literature, 2) expression profiles of cell populations isolated from reporter lines (Brady et al., 2007; Li et al., 2016), and 3) Index of Cell Identity (ICI) scores (Efroni et al., 2015). For marker gene expression-guided assignments, a set of known or inferred marker genes was identified from the literature (e.g., Brady et al., 2007). The average normalized expression for these genes for each cell was computed and used to guide assignments of cell clusters to putative cell types. Additionally, for each cluster, the average expression for all genes was computed and compared to data from two previous studies: Brady et al. (2007) and Li et al. (2016). For comparison to the Brady et al. study, a Spearman’s correlation coefficient was calculated by comparing the average gene expression values for each cluster to the expression values of the 7,769 genes representing the top 50% most variable genes across each reporter line. For comparison to the Li et al. study, isoform FPKM values were summed for each gene to determine gene-level expression values for each of the datasets described. Variance was calculated for each gene across all Li et al. datasets, and genes with mean expression less than 1 FPKM or a variance of zero were filtered out. The top 50% of these genes (9,513 by decreasing variance) were used to calculate a Spearman’s correlation coefficient with the average normalized gene expression values for each cluster. Finally, we used the ICI algorithm, which assesses the expression of hundreds of genes to calculate a probability that a single cell belongs to a specific Arabidopsis root cell type and returns the best cell type match. Probabilities associated with the cell type assignments were estimated using a bootstrapped permutation method (1000 iterations) and adjusted using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995). Entire clusters of cells within the t-SNE graph were assigned an overall cell type based on the preponderance of statistically significant ICI calls (p < 0.05, Figures S3B and S3C), taking mixed cell type identities into account (i.e., if a STAMP had a significant ICI score for more than one cell type). Once assignments were made for each cluster, the numerical cluster identity from Seurat (i.e., 0 through 16) was re-ordered (to 1 through 17) alphabetically based on the cell type assignment.
Marker gene identification
For each cluster, the FindAllMarkers function in Seurat was used to identify marker genes. Following this analysis, the marker gene table was filtered for genes having a significant adjusted p value (< 0.01), an average natural log-fold change greater than 0.5, and then further filtered for genes expressed in more than 75% of the cells within that cluster. This list of genes is provided as Table S2. For identification of genes differentially expressed between sucrose growth conditions, clusters were first combined into broader tissue or cell type categories based on shared type assignment (e.g., grouping together cells within clusters 1-4, all of which have a non-hair cell identity). Gene expression was then compared within each cell type category for cells from the two growth conditions, using the FindMarkers function in Seurat. The resulting gene set was filtered for genes with a significant adjusted p value (< 0.01), and an average natural log-fold change greater than 0.5.
Pseudotime analysis of endodermis clusters
The branched developmental trajectory (pseudotime) for the two endodermal cell clusters was computed using the Monocle 2 R package (version 2.8.0) (Trapnell et al., 2014). As with analyses described above, known protoplast-response genes were removed. Briefly, raw single cell transcriptomes from cells belonging to clusters 11 or 12 were used to construct a CellDataSet object within Monocle, which was then normalized using the BiocGenerics (Huber et al., 2015) (version 0.26.0) functions, estimateSizeFactors and estimateGeneDispersions. Genes to be used for dimension reduction and ordering of these cells were then determined using the differentialGeneTest function in Monocle, assaying for genes that differ between clusters 11 and 12 from the Seurat analysis. These genes were then filtered for the top 1000 genes that are expressed in at least 10% of cells, with a q-value < 0.01, and then sorted by q-value. The normalized cells and ordering gene set was then used to compute a pseudotime graph using the reduceDimension function (using the DDRTree method), followed by the orderCells function. The root state for the resulting pseudotime graph was assigned as the state with the lowest mean expression of CASP1, a known marker gene of mature endodermis. The main branch point was, therefore, the first branch point from the root state. The major branch was defined as the branch following from the first branch point with the highest number of cells, including any sub-branches. The minor branch was defined as the branch with the fewest number of cells following from the main branch point, including any sub-branches. For all genes, mean normalized expression was calculated for a set of 20 equally spaced (in pseudotime) bins (binned expression in pseudotime) independently for the major and minor branches. Genes that are differentially expressed between branches were determined using the FindMarkers function in Seurat, comparing gene expression from cells belonging only to the main branch to those cells belonging only to the minor branch, and restricting the resulting gene list to genes having an average absolute natural log-fold change greater than 0.5 and an adjusted p value below 0.01. Genes that significantly vary along the main branch were determined using the differentialGeneTest function in Monocle using pseudotime as a predictor, on a subset of the cellDataSet object containing only genes expressed in > 10% of cells, and cells along the main branch. The genes that vary along the main branch were further filtered to include only those genes with a significant Monocle-defined q-value (< 0.01 for whether expression is variable along pseudotime), and those genes whose variance of binned expression in pseudotime was greater than 0.125. The 1000 genes that vary between branches, and the 798 genes identified as variable along the main pseudotime branch, were separately clustered using the plot_genes_ heatmap_branched, and plot_pseudotime_heatmap functions (respectively) in Monocle, using the “complete” hierarchical clustering method. Three main clades of genes with differential expression over the main endodermis developmental trajectory were identified using the cutree method in R. These three clades were reordered by gene expression timing (i.e., early, middle, late) for better visualization in Figure 5D.
Analysis of enriched GO terms and co-expression analysis
Gene Ontology (GO) analysis was performed using a PANTHER overrepresentation test (Mi et al., 2013) against the Arabidopsis thaliana genome (GO Onotology database, released 2018-10-08). Genes co-expressed with GPAT5 were identified using ATTED-II (Obayashi et al., 2018), with the top 17 candidates (by Arabidopsis co-expression score) used for analysis of pseudotime-based expression.
Bulk root tissue mRNA-seq and pseudo-bulk analysis
To compare Drop-seq expression data to standard mRNA-seq data, we isolated total RNA from flash frozen 5-day-old Arabidopsis root material using the QIAGEN RNeasy kit (Carlsbad, CA). The RNA was used in the TruSeq Stranded mRNA Library Prep Kit (Illumina, RS-122-2101) according to the manufacturer’s Low Sample (LS) Protocol. The library was uniquely indexed and then sequenced on a single lane of a HiSeq 4000. Transcript counts for each gene were then tabulated. In parallel, a pseudo-bulk dataset was generated from the Digital Gene Expression matrix from scRNA-seq of the root protoplasts. A separate transcript count profile from Arabidopsis floral bud tissue was also downloaded from the NCBI GEO database (GSM2616967). These three expression profiles (filtered to contain only genes with FPKM > 1) were then normalized together using the DESeq2 package (Love et al., 2014). The Spearman correlation coefficients between the pseudo-bulk and whole-root and whole-flower datasets were computed in R before or after filtering out known protoplast-inducible genes.
Confocal imaging of gene reporter lines
The At4CL1p:GFP (Taylor-Teeples et al., 2015) and MSL4p:GFP-GUS (gift of Elizabeth Haswell) lines were grown as described above using 1% sucrose. Plants were harvested after 5 days and imaged using a Zeiss LSM 700 Confocal. Images were captured at 20X magnification. In order to capture the length of the whole root at this magnification, multiple images were obtained at exactly the same gain settings and stitched together in Affinity Design into a single image. In order to improve visibility of GFP reporter abundance, brightness was adjusted for the entire stitched images.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise indicated, statistical analysis and plot generation were performed using R (https://www.r-project.org/). Technical and biological replicate Drop-seq experiments were performed. Sets A and B represent technical replicates from the same protoplast preparation. Sets F and G represent biological replicates, independently sampled from the same (larger) cohort of seedlings. Sets I and J are also biological replicates from the same (larger) cohort of seedlings. All other sets represent experimental replicates, with seedlings and protoplasts derived from independent experimental workflows (see Method Details section Drop-seq of Arabidopsis root protoplasts and Table S1).
To identify and quantify high-quality individual cell transcriptomes within the data for each replicate set, we used the Drop-seq Core Computational Protocol version 1.0.1, using default parameters (http://mccarrolllab.org/dropseq/). The 12,198 cell gene expression profiles with greater than 1000 UMIs, with more than 98% of UMIs mapping to the Arabidopsis TAIR10 genome, were retained for further analysis (see Method Details section Data alignment and clustering analysis). A detailed breakdown of the number of cells from each replicate set is provided in Table S1. An ICI score was determined for each cell for all 15 possible cell identities, with empirical, adjusted p values generated from 1,000 random permutations of the specificity score matrix (see Method Details section Assignment of clusters to known cell types). Dimensional reduction and clustering were performed using the Seurat (version 2.3.4) R package (see Method Details section Data alignment and clustering analysis). Canonical coordinates for dimension reduction and clustering were selected using the MetageneBicorPlot function in Seurat, with 35 CCs selected as a number where the shared correlation strength stops strongly decreasing. Marker genes for each cluster and genes differentially regulated by sucrose were identified using the FindMarkers function within the Seurat R package (see Method Details section Marker gene identification; exact p values for this analysis are reported in Tables S2 and S3, respectively). Lastly, Monocle 2 was used to generate a pseudotime trajectory and identify developmentally variable genes for endodermis cells (clusters 11 and 12, n = 1,660 cells), using cluster information previously identified using Seurat (see Method Details section Pseudotime analysis of endodermis clusters). Exact p values for genes showing differential expression between the major and minor endodermis branches are reported in Table S4. Exact p values for ontology terms enriched among genes expressed in early, middle, or late endodermis development are reported in Table S5.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE122687. No software was generated for this project.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cellulysin | MilliporeSigma | 219466 |
| Pectolyase | MilliporeSigma | P3026 |
| Mannitol | MilliporeSigma | M4125; CAS 69-65-8 |
| MES hydrate | MilliporeSigma | M2933; CAS 1266615-59-1 |
| Murashige and Skoog Basal Medium | MilliporeSigma | M5519 |
| Critical Commercial Assays | ||
| Nextera XT DNA Library Preparation Kit | Illumina | FC-131-1024 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | 20020594 |
| Barcoded dT Beads for Drop-Seq | Macosko et al., 2015; ChemGenes | Lot# 011416B and 072817 |
| Deposited Data | ||
| Raw and analyzed Drop-seq and bulk tissue RNA-seq data from Arabidopsis root | This paper | GEO: GSE122687 |
| Cell type-specific microarray data of Arabidopsis root | Brady et al., 2007 | N/A (Table S12 of referenced paper) |
| Cell type-specific RNA-seq data of Arabidopsis root - raw sequence reads | Li et al., 2016 | SRA: BioProject PRJNA323955 |
| RNA-seq data of Arabidopsis unopened floral bud tissue | Zhang et al., 2018 | GEO: GSM2616967 |
| Experimental Models: Cell Lines | ||
| Human HEK293T/17 cells | ATCC | CRL-11268 |
| Mouse embryonic stem cells | Skarnes, 2000 | E14Tg2a.4 |
| Experimental Models: Organisms/Strains | ||
| Arabidopsis: Col-0 | ABRC | CS70000 |
| Arabidopsis: At4CL1p:GFP | Taylor-Teeples et al., 2015 | N/A |
| Arabidopsis: MSL4p:GFP-GUS | Elizabeth Haswell | N/A |
| Software and Algorithms | ||
| Drop-seq_tools v1.12 | Macosko et al., 2015 | https://github.com/broadinstitute/Drop-seq/releases |
| Seurat R package (version 2.3.4) | Butler et al., 2018 | https://satijalab.org/seurat |
| Monocle 2 R package (version 2.8.0) | Trapnell et al., 2014 | https://bioconductor.org/packages/release/bioc/html/monocle.html |
| STAR Aligner v2.5.2b | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Index of Cell Identity (ICI) algorithm | Efroni et al., 2015 | N/A |
| Other | ||
| Resource website and protocol for Drop-seq | Macosko et al., 2015 | http://mccarrolllab.org/dropseq |
Highlights.
RNA expression profiling of >12,000 single Arabidopsis root cells by Drop-seq
Recovered major tissue types and cell stages and identified specific marker genes
Sucrose alters cell type proportions but does not profoundly change cell identities
Identified ~800 genes dynamically expressed across endodermis development
ACKNOWLEDGMENTS
This work was supported by a grant to D.E.D. from the Laboratory Directed Research and Development Program of Lawrence Berkeley National Laboratory (LBNL). Work was performed at LBNL and the US DOE Joint Genome Institute under U.S. Department of Energy contract DE-AC02-05CH11231, University of California. G.M.T., M.G., and S.M.B. were funded by a Howard Hughes Medical Institute Faculty Scholar Fellowship. G.M.T. was also funded by an NSF predoctoral fellowship and the AAUW Dissertation Completion Fellowship. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH Instrumentation Grant S10OD018174. We thank V. Singan for generating library counts from raw mRNA-seq dataand I. Barozzi for computational advice. The authors acknowledge E. Macosko, M. Goldman, J. Nemesh, and S. McCarroll for providing detailed Drop-seq protocols. In addition, the authors thank T. Grube-Andersen and N. Geldner for discussing endodermis pseudotime ordering, E. Haswell for providing the MSL4p:GFP-GUS reporter line, and A. Visel and J. Vogel for helpful feedback on the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.04.054.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Andersen TG, Barberon M, and Geldner N (2015). Suberization-the second life of an endodermal cell. Curr. Opin. Plant Biol. 28, 9–15. [DOI] [PubMed] [Google Scholar]
- Beisson F, Li Y, Bonaventure G, Pollard M, and Ohlrogge JB (2007). The acyltransferase GPAT5 is required for the synthesis of suberin in seed coat and root of Arabidopsis. Plant Cell 19, 351–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B 57, 289–300. [Google Scholar]
- Birnbaum K, Shasha DE, Wang JY, Jung JW, Lambert GM, Galbraith DW, and Benfey PN (2003). A gene expression map of the Arabidopsis root. Science 302, 1956–1960. [DOI] [PubMed] [Google Scholar]
- Birnbaum K, Jung JW, Wang JY, Lambert GM, Hirst JA, Galbraith DW, and Benfey PN (2005). Cell type-specific expression profiling in plants via cell sorting of protoplasts from fluorescent reporter lines. Nat. Methods 2, 615–619. [DOI] [PubMed] [Google Scholar]
- Brady SM, Orlando DA, Lee JY, Wang JY, Koch J, Dinneny JR, Mace D, Ohler U, and Benfey PN (2007). A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 318, 801–806. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright DA, Brady SM, Orlando DA, Sturmfels B, and Benfey PN (2009). Reconstructing spatiotemporal gene expression data from partial observations. Bioinformatics 25, 2581–2587. [DOI] [PubMed] [Google Scholar]
- Denyer T, Ma X, Klesen S, Scacchi E, Nieselt K, and Timmermans MCP (2019). Spatiotemporal developmental trajectories in the Arabidopsis root revealed using high-throughput single-cell RNA sequencing. Dev. Cell 48, 840–852.e5. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni I, and Birnbaum KD (2016). The potential of single-cell profiling in plants. Genome Biol. 17, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni I, Ip PL, Nawy T, Mello A, and Birnbaum KD (2015). Quantification of cell identity from single-cell gene expression profiles. Genome Biol. 16, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni I, Mello A, Nawy T, Ip PL, Rahni R, DelRose N, Powers A, Satija R, and Birnbaum KD (2016). Root regeneration triggers an embryo-like sequence guided by hormonal interactions. Cell 165, 1721–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendrych M, Van Hautegem T, Van Durme M, Olvera-Carrillo Y, Huysmans M, Karimi M, Lippens S, Guérin CJ, Krebs M, Schumacher K, and Nowack MK (2014). Programmed cell death controlled by ANAC033/SOMBRERO determines root cap organ size in Arabidopsis. Curr. Biol 24, 931–940. [DOI] [PubMed] [Google Scholar]
- Fukuda H (1996). Xylogenesis: Initiation, progression, and cell death. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47, 299–325. [DOI] [PubMed] [Google Scholar]
- Furuta KM, Yadav SR, Lehesranta S, Belevich I, Miyashima S, Heo JO, Vatén A, Lindgren O, De Rybel B, Van Isterdael G, et al. (2014). Plant development. Arabidopsis NAC45/86 direct sieve element morphogenesis culminating in enucleation. Science 345, 933–937. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating highthroughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Poling MD, Karthikeyan AS, Blakeslee JJ, Peer WA, Titapiwatanakun B, Murphy AS, and Raghothama KG (2007). Differential effects of sucrose and auxin on localized phosphate deficiency-induced modulation of different traits of root system architecture in Arabidopsis. Plant Physiol. 144, 232–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- León P, and Sheen J (2003). Sugar and hormone connections. Trends Plant Sci. 8, 110–116. [DOI] [PubMed] [Google Scholar]
- Li S, Yamada M, Han X, Ohler U, and Benfey PN (2016). High-resolution expression map of the Arabidopsis root reveals alternative splicing and lincRNA regulation. Dev. Cell 39, 508–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman LM, Sparks EE, Moreno-Risueno MA, Petricka JJ, and Benfrey PN (2015). MYB36 regulates the transition from proliferation to differentiation in the Arabidopsis root. Proc. Natl. Acad. Sci 112, 12099–12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macgregor DR, Deak KI, Ingram PA, and Malamy JE (2008). Root system architecture in Arabidopsis grown in culture is regulated by sucrose uptake in the aerial tissues. Plant Cell 20, 2643–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT, and Thomas PD (2013). Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc 8, 1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi T, Aoki Y, Tadaka S, Kagaya Y, and Kinoshita K (2018). ATTED-II in 2018: A plant coexpression database based on investigation of the statistical property of the mutual rank index. Plant Cell Physiol. 59, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olvera-Carrillo Y, Van Bel M, Van Hautegem T, Fendrych M, Huysmans M, Simaskova M, van Durme M, Buscaill P, Rivas S, Coll NS, et al. (2015). A conserved core of programmed cell death indicator genes discriminates developmentally and environmentally induced programmed cell death in plants. Plant Physiol. 169, 2684–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka DR, Kandasamy MK, McKinney EC, Burgos-Rivera B, and Meagher RB (2007). The ancient subclasses of Arabidopsis Actin Depolymerizing Factor genes exhibit novel and differential expression. Plant J. 52, 460–472. [DOI] [PubMed] [Google Scholar]
- Ryu KH, Huang L, Kang HM, and Schiefelbein J (2019). Single-cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiol 179, 1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC (2000). Gene trapping methods for the identification and functional analysis of cell surface proteins in mice. Methods Enzymol. 328, 592–615. [DOI] [PubMed] [Google Scholar]
- Tanimoto M, Roberts K, and Dolan L (1995). Ethylene is a positive regulator of root hair development in Arabidopsis thaliana. Plant J. 8, 943–948. [DOI] [PubMed] [Google Scholar]
- Taylor-Teeples M, Lin L, de Lucas M, Turco G, Toal TW, Gaudinier A, Young NF, Trabucco GM, Veling MT, Lamothe R, et al. (2015). An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature 517, 571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognetti JA, Pontis HG, and Martínez-Noël GMA (2013). Sucrose signaling in plants: a world yet to be explored. Plant Signal. Behav. 8, e23316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugeki R, and Fedoroff NV (1999). Genetic ablation of root cap cells in Arabidopsis. Proc. Natl. Acad. Sci. USA 96, 12941–12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L, and Hinton G (2008). Visualizing Data using t-SNE. J. Mach. Learn. Res 9, 2579–2605. [Google Scholar]
- Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, et al. (2017). Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav V, Molina I, Ranathunge K, Castillo IQ, Rothstein SJ, and Reed JW (2014). ABCG transporters are required for suberin and pollen wall extracellular barriers in Arabidopsis. Plant Cell 26, 3569–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Simpson JP, Li-Beisson Y, Beisson F, Pollard M, and Ohlrogge JB (2012). A land-plant-specific glycerol-3-phosphate acyltransferase family in Arabidopsis: substrate specificity, sn-2 preference, and evolution. Plant Physiol. 160, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Harris CJ, Liu Q, Liu W, Ausin I, Long Y, Xiao L, Feng L, Chen X, Xie Y, et al. (2018). Large-scale comparative epigenomics reveals hierarchical regulation of non-CG methylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 115, E1069–E1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data reported in this paper is GEO: GSE122687. No software was generated for this project.