

Abstract
Early life, systemic inflammation causes long-lasting changes in behavior. To unmask possible mechanisms associated with this phenomenon, we asked whether the intrinsic membrane properties in hippocampal neurons were altered as a consequence of early life inflammation. C57BL/6 mice were bred in-house and both male and female pups from multiple litters were injected with lipopolysaccharide (LPS; 100 μg/kg, i.p.) or vehicle at postnatal day (P)14, and kept until adolescence (P35–P45) or adulthood (P60–P70), when brain slices were prepared for whole-cell and perforated-patch recordings from CA1 hippocampal pyramidal neurons. In neurons of adult male mice pretreated with LPS, the number of action potentials elicited by depolarizing current pulses was significantly increased compared with control neurons, concomitant with increased input resistance, and a lower action potential threshold. Although these changes were not associated with changes in relevant sodium channel expression or differences in capacitance or dendritic architecture, they were linked to a mechanism involving intracellular chloride overload, revealed through a depolarized GABA reversal potential and increased expression of the chloride transporter, NKCC1. In contrast, no significant changes were observed in neurons of adult female mice pretreated with LPS, nor in adolescent mice of either sex. These data uncover a potential mechanism involving neonatal inflammation-induced plasticity in chloride homeostasis, which may contribute to early life inflammation-induced behavioral alterations.
SIGNIFICANCE STATEMENT Early life inflammation results in long-lasting changes in many aspects of adult physiology. In this paper we reveal that a brief exposure to early life peripheral inflammation with LPS increases excitability in hippocampal neurons in a sex- and age-dependent manner through a chloride homeostasis disruption. As this hyperexcitability was only seen in adult males, and not in adult females or adolescent animals of either sex, it raises the possibility of a hormonal interaction with early life inflammation. Furthermore, as neonatal inflammation is a normal feature of early life in most animals, as well as humans, these findings may be very important for the development of animal models of disease that more appropriately resemble the human condition.
Keywords: GABA reversal potential, hippocampus, inflammation, intrinsic membrane properties, lipopolysaccharide, sex differences
Introduction
Brain maturation is a long process of developmental changes at molecular, cellular, and network levels, which can be affected by processes that are external to the brain. Overwhelming evidence indicates that early life inflammatory and stressful challenges may predispose the brain to several neurological disorders that emerge later in life. For instance, studies in humans have provided substantial findings that an inflammatory response to bacterial or viral infection, or to stressors in early life can lead to an increased risk of subsequent neurodevelopmental dysfunction in children and adults (Hagberg et al., 2012; Danese and Lewis, 2017).
Over the last decade, our research group has extensively characterized the effects of systemic early life inflammation on rodent physiology and behavior in later life. In particular, we have previously shown that a single episode of inflammation evoked by lipopolysaccharide (LPS), a Gram-negative bacteria endotoxin, during a critical period of development, can alter many aspects of adult physiology including neuroimmune function, behavioral responses, and endocrine function (Rana et al., 2012; Doenni et al., 2016, 2017). These central alterations are associated with altered vulnerability of the organism to subsequent pathological challenges, including hypoxic ischemia (Spencer et al., 2006), autoimmune disease (Ellestad et al., 2009), and seizure susceptibility (Heida et al., 2005; Galic et al., 2008; Reid et al., 2012; Lewis et al., 2018). Interestingly, many of the changes observed in adult animals involve the hippocampus; these include changes in spatial memory, and cognitive and learning deficits (Bilbo et al., 2005, 2006; Harré et al., 2008; Wang et al., 2013; Dinel et al., 2014). Additionally, at the molecular level, altered expression of glutamatergic and GABAergic receptor subunits, and chloride cotransporters have been reported in response to early life inflammation (Harré et al., 2008; Reid et al., 2013). However, to date we are unaware of any studies that have addressed the effects of neonatal inflammation on cellular and/or network activities. This prompted us to hypothesize that early life inflammation may also cause long-term alterations in the normal development of neuronal electrical properties. Therefore, we induced early life inflammation with LPS in postnatal day (P)14 mice and examined the intrinsic electrophysiological properties of CA1 hippocampal neurons in adolescence and adulthood. Considering that there have been relatively few studies comparing sex differences with respect to effects of neonatal inflammation, male and female offspring, both prepubertal, as adolescents, and after sexual maturation were evaluated.
Materials and Methods
Neonatal inflammation.
All the experiments were conducted in accordance with the Canadian Council on Animal Care regulations and approval of the University of Calgary Animal Care committee. C57BL/6 mice (Charles River Laboratories) were maintained under standard specific pathogen-free conditions at 20–21°C, on a 12 h light/dark cycle (light onset at 07:00 A.M.) with food and water available ad libitum. Female mice were bred, then pregnant single females (N = 34) were separated and monitored for the offspring's birth day. On P14, offspring of both sexes received pyrogen-free saline (vehicle; 1 ml/kg) or lipopolysaccharide (LPS, Escherichia coli, serotype 026:B6 100 μg/kg, i.p.). LPS is a molecule derived from Gram-negative bacteria that has been shown to induce a transient fever and a mild peripheral and central inflammatory response in the host (Heida et al., 2004; Ellis et al., 2005; Galic et al., 2009). Although it does not mimic all of the effects of a true infection (Schwarz and Bilbo, 2011), it provides a defined stimulus and has been used previously to induce an inflammatory state in nearly all of the experiments that provide the basis for our electrophysiological study. Nonetheless, when we refer to our animals as “neonatally inflamed” it should be understood that this refers only to LPS-induced inflammation. All experiments involved both female and male offspring to reveal any sex-specific interaction and pups were taken from multiple litters over a 3 year period to mitigate possible differences in individual maternal care, litter, and cohort effects. Males and females were chosen at the appropriate ages in a sequential fashion from each litter. Groups were distributed approximately equally for sex and treatment within each litter. Ears were marked for later identification, and then pups were returned to their dams, weaned at P21, and housed three to five per cage by sex and random treatment. The offspring growth was not altered by neonatal inflammation, with no significant effect on body mass (data not shown) as previously described (Spencer et al., 2007).
In vitro hippocampal electrophysiology.
To investigate intrinsic electrophysiological properties in later life, adolescent (P35–P45) and adult mice (P60–P70) of both sexes, treated with either LPS or vehicle (Veh) on P14, were anesthetized with isoflurane and transcardially perfused with ice-cold slicing solution containing the following (in mm): 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 d-glucose, and 75 sucrose. Brains were quickly dissected and transverse slices (300 μm) of the dorsal hippocampus were taken at a 45° angle from the coronal plane using a Vibratome VT1200S (Leica). In this study we used only the dorsal hippocampus because of known differences in pyramidal cell properties and function of the hippocampus along its dorsal and ventral extent (Strange et al., 2014). Slices were immediately transferred to a recovery solution containing warm (32°C) artificial CSF (aCSF) composed of the following (in mm): 126 NaCl, 2.5 KCl, 26 NaHCO3, 2.5 CaCl2, 1.5 MgCl2, 1.25 NaH2PO4, and 10 d-glucose for 60 min, and then maintained at room temperature for at least 30 min before recording. Both slicing and aCSF solutions were continuously bubbled with a gas mixture of 95% O2 and 5% CO2 to maintain a pH at 7.4.
Hippocampal slices were placed in a recording chamber and perfused with aCSF (32°C) at a flow rate of ∼1 ml/min. Pyramidal neurons were then identified under differential contrast/infrared illumination by their location in the cell body layer and by their pyramidal shape, and selected based on their large apical dendrites and multiple basal dendrites. Any cell exhibiting fast-spiking, prominent after hyperpolarization potentials, and a train of nondecrement spikes without frequency adaptation was considered an interneuron (Han, 1994; Zemankovics et al., 2010) and was not studied further. Whole-cell recordings were made sequentially starting lateral (near the CA2/CA1 border) and proceeding medially (toward, but not into the subiculum) at well separated locations. We used borosilicate glass micropipettes (3–6 MΩ) filled with a recording solution containing the following (in mm): 108 K-gluconate, 8 Na-gluconate, 2 MgCl2, 8 KCl, 1 EGTA, 4 K2-ATP, 0.3 Na-GTP, and 10 HEPES, with pH adjusted to 7.2 with KOH and osmolarity to ∼285 mOsm. Signals were amplified and filtered (low pass at 1 kHz) with a MultiClamp 700B amplifier (Molecular Devices), and digitized at 10 kHz using a Digidata 1322A (Molecular Devices). Cell capacitance and access resistance (initial value <20 MΩ) were monitored during experiments, and recordings were accepted for analysis if either variable did not change by >20%. The amplifier was set to zero to null the liquid junction potential between the pipette and the bath solution (Neher, 1992).
To obtain reliable recordings, patched cells were allowed to stabilize for at least 5 min, and resting membrane potential (RMP) was then recorded as the voltage with no injected current. Input resistance (Rin) was determined as the slope of the linear portion of a voltage-current plot constructed from the steady-state voltage response to step current injections from RMP ranging from −100 up to 50 pA in 30 pA increments and 500 ms duration. To investigate neuronal excitability, firing frequency was calculated in response to current steps (80–320 pA/500 ms) starting from RMP. Action potential (AP) threshold and rheobase were then determined as the membrane potential and the minimum injected current needed respectively, at the initiation of the first spike point with dV/dt exceeding 10 mV/ms in responses to up to 8 linear ramps (50 pA increments and 2 s). AP kinetic properties including amplitude, half-width, 10–90% rise time, and 90–10% decay time, were also measured from the first spike elicited, and a linear interpolation between data points was used to enhance the temporal resolution.
To estimate chloride concentration the GABA reversal potential (EGABA) was evaluated. Neurons were held at −70 mV and the membrane potential was stepped to various test potentials. During each membrane potential step, local electrical stimulation via a concentric bipolar electrode (SNEX-100, Kopf Instruments) placed in stratum radiatum ∼100 μm from the recorded neuron was used to activate IPSCs in the presence of 20 μm 6,7-dinitroquinoxaline-2,3-dione and 40 μm dl-2-amino-5-phosphonopentanoic acid. A linear regression of the IPSC amplitude versus membrane potential was calculated, and the intercept of this line with the abscissa was taken as EGABA. The sodium-potassium-chloride cotransporter 1 (NKCC1) blocker bumetanide (10 μm) was added into the bath as needed. In some experiments, gramicidin perforated patch recordings were performed to interfere as little as possible with intracellular anion concentration (Kyrozis and Reichling, 1995). Before each perforated patch experiment, gramicidin (ABCD, Sigma) was dissolved in dimethyl sulfoxide (50 mg/ml) and added to the intracellular solution to obtain a final concentration of 50 μg/ml. Electrodes were always loaded with a small volume of gramicidin-free internal solution then backfilled with the gramicidin solution to facilitate the seal formation (seal resistance, >3 GΩ). Within ∼20–40 min after gigaseal formation, the access resistance dropped and stabilized at ∼25–35 MΩ. Data were collected via a MultiClamp 700B amplifier (Molecular Devices) and were stored on a computer using pClamp 9 software (Molecular Devices). Analysis was performed off-line with Clamp-fit 9 program (Molecular Devices).
All electrophysiological datasets comprised results from animals (reported as N in results, with n = number of cells) obtained from a minimum of three litters. In addition, data were collected over a period of over 3 years from multiple cohorts of animals.
Golgi staining.
As our electrophysiological analyses indicated striking differences between properties of adult male mice treated with LPS at P14, it was important to determine whether the cells were morphologically similar in the LPS and control groups. Golgi silver impregnation was done using a commercially available kit (FD Rapid GolgiStain Kit, FD NeuroTechnologies) as we previously described (Acharjee et al., 2018). Adult male mice treated with either LPS or vehicle at P14 were anesthetized with sodium pentobarbital (80 mg/kg, i.p.) and the brains harvested. Coronal sections (100 μm) were cut on a vibratome (VT 1000S, Leica), mounted on gelatin-coated slides and developed following the protocol in FD Rapid GolgiStain Kit. CA1 pyramidal neurons from the dorsal hippocampus were imaged using reflective confocal microscopy (DM5500 B, Leica) with a 488 nm laser and 40× objective (1.62 NA). Three to six neurons were imaged from at least three slices per animal (N = 3/group, each from a separate litter). Care was taken to ensure (1) that imaged neurons did not extend processes to the very end part of the sections to avoid dendritic stumps, and (2) that processes of the neurons could be clearly identified. Further, neurons chosen were sufficiently distant from their neighbors to minimize the overlap of dendritic trees. Confocal image stacks were imported into Imaris TM software (Bitplane) for analysis. The grayscale images were inverted, and histogram adjusted to optimize visualization. Dendrites were reconstructed in a semiautomated way using Filament Tracer in Imaris TM software. For each reconstructed neuron, total dendritic length was calculated and Sholl analysis was performed to quantify the number of dendritic intersections using concentric spheres from the center of the soma with radii increasing in 10 μm increments. All analyses were completed blindly as to the experimental/control status of the animal
Hippocampal dissection for qPCR and Western blot.
With the purpose of identifying possible mechanisms for the intrinsic electrophysiological property alterations, additional mice were injected using the same neonatal LPS-induced inflammation paradigm and the CA1 hippocampus was assessed using mRNA and protein expression-based molecular approaches. Under deep isoflurane anesthesia, adult female (N = 20 Veh; 20 LPS) and male (N = 16 Veh; 16 LPS) mice were decapitated, and brains were quickly removed. For quantitative PCR (qPCR) and Western blot experiments, we used the dorsal hippocampal CA1 area. Briefly, brains were mounted on a Vibratome VT1200s (Leica Biosystems) in ice-cold slicing solution, 500 μm sections were then taken, and the CA1 area was dissected over ice and under microscopic observation by hand using a microsurgical blade. The tissue samples were snap frozen on dry ice and stored at −80°C until later processing.
RNA isolation and qPCR.
Samples were prepared and stored at −80°C as described above. Tissue samples containing the dorsal CA1 area were disrupted using QIAzol reagent (Qiagen) and homogenized using 5 mm stainless steel beads in a TissueLyser LT (Qiagen) for 5 min at an oscillation frequency of 50 Hz. Cell homogenates were processed according to the QIAzol protocol before mRNA was purified using the RNeasy Mini Kit (Qiagen). cDNA was synthesized by the QuantiTect Reverse Transcription Kit, and qPCR was then performed by the QuantiTect SYBR Green PCR kit (Qiagen), using an Eppendorf ep gradient S Mastercycler. Validated primers for the voltage-gated sodium channel (Nav), type 1.1 and 1.2 (SCN1α and SCN2) were designed by Qiagen. GAPDH (Qiagen) was amplified from the same samples and used as a reference gene for normalization. Cycling conditions included: initial heat activation at 95°C for 15 min, denaturation at 94°C for 15 s (40 rounds), annealing at 55°C for 30 s and extension at 72°C for 30 s. Melt curves were used to confirm reaction specificity. Data analysis was accomplished using the 2−ΔΔCt method (Livak and Schmittgen, 2001; Schmittgen and Livak, 2008).
Protein extraction and immunoblotting.
Hippocampal tissue was homogenized in RIPA buffer using a TissueLyser LT (Qiagen). Insoluble fractions were separated from the homogenate by centrifugation and resuspended in RIPA. Proteins were quantified using a BCA Assay before SDS-PAGE separation of proteins using 10% TGX Stain-Free acrylamide gels (Bio-Rad). Proteins and trihalo compounds were activated in-gel using a ChemiDoc MP (Bio-Rad) before transfer onto to a 0.44 μm Immobilon-P PVDF membrane (MilliporeSigma). Membranes were imaged after transfer to obtain the total protein profile using the ChemiDoc MP then blocked for 1 h in 3% BSA. Membranes were then incubated overnight with anti-Nav1.1 (K74/71, RRID:AB_2238842, UC Davis/NIH NeuroMab Facility), anti-Nav1.2 (K69/3, RRID:AB_2184030, UC Davis/NIH NeuroMab Facility), anti-phosphoNKCC1 (Millipore, ABS1004), and anti-phosphoKCC2 (PhosphoSolutions, RRID:AB_2492213) primary antibodies at 1:1000 in 3% BSA. HRP labeled secondary antibodies, rabbit anti-mouse-HRP 1:10,000 (48-370-H, Antibodies Incorporated) and goat anti-rabbit-HRP 1:10,000 (48-156-H, Antibodies Incorporated), were added to the membrane and incubated for 30 min before being imaged using the ECL Substrate Kit (ThermoFisher Scientific, WP20005) on the ChemiDoc MP. Proteins-of-interest were quantified by normalizing band densities against the corresponding lane containing the total vehicle protein profile from the same membrane using the Image Lab software 6.0 (Bio-Rad). Each membrane analyzed proteins from one vehicle and one LPS-treated mouse of each sex.
Data analysis.
All statistical analyses were done using GraphPad Prism v8. Data were verified for normal distribution with D'Agostino and Pearson test. Intrinsic membrane and synaptic properties, Golgi staining, qPCR and Western blot were analyzed using three-way or two-way ANOVA with Tukey post hoc test, χ2 test, or unpaired Student's t test as appropriate. Results are reported as mean ± SEM. Statistical significance was set a p < 0.05 based on the number of cells for electrophysiological data and on the animal number for molecular biology data. Mean data from each animal are also depicted in graphs for information.
Results
Neonatal inflammation with LPS altered intrinsic electrophysiological properties of CA1 pyramidal neurons in adult but not adolescent mice
We first addressed the effect of neonatal inflammation with LPS (i.p.) at P14 on firing frequency in the CA1 pyramidal neurons of adolescent (P35–P45) and adult (P60–P70) C57BL/6 mice. Figure 1A shows voltage responses and APs in response to the indicated step current injections for representative control (Veh, black traces) and neonatally inflamed (LPS, red traces) CA1 pyramidal neurons from adolescent and adult mice. When firing rate was plotted against injected current steps and analyzed using three-way ANOVA test, we found significant differences among groups (Table 1). We observed an age-dependent (F(1,117) = 7.662, p = 0.0066) and sex-dependent (F(1,116) = 5.518, p = 0.0205) effect of the early life LPS treatment on firing rate in CA1 pyramidal neurons. Multiple comparisons by Tukey post hoc analyses revealed that the statistically significant effect was attributed to a higher firing rate in CA1 pyramidal neurons of adult male mice exposed to LPS treatment compared with controls (for all significant values p < 0.05; Fig. 1B2; Table 1-1). On the other hand, early life inflammation had no significant effects on firing rate of CA1 pyramidal neurons in adult female, or in adolescent female and male mice compared with controls (Fig. 1; Table 1), indicating an increased LPS-induced excitability only in adult male mice.
Figure 1.

Neonatal inflammation alters the firing frequency of hippocampal neurons from adult male mice. A, Voltage response traces from CA1 pyramidal neurons from (A1) adolescent and (A2) adult mice treated with Veh (dark line) or LPS (red line) at postnatal day 14, elicited by 500 ms duration current injections ranging from 80 to 320 pA in 30 pA increments. Calibration: 30 mV/170 pA, 200 ms. B1, Summary data show no differences in the number of APs for Veh and LPS-treated adolescent female and male mice (n = 28–32 cells, 5–6 mice/group). B2, Firing frequency of CA1 pyramidal neurons from LPS-treated adult male mice was significantly higher than Veh control group (n = 29–31 cells, 5–8 mice/group). Number of APs for adult female groups was unchanged (n = 28–32 cells, 6–7 mice/group). Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Table 1.
Three-way ANOVA for comparison of age, sex, and treatment among groups on firing rate
| Group | Variable | p | Treatment | p | Interaction | p | |
|---|---|---|---|---|---|---|---|
| Age | |||||||
| Female, ♀ | Firing rate | F(1,116) = 5.900 | 0.017 | F(1,116) = 0.02043 | 0.8866 | F(1,116) = 1.468 | 0.2282 |
| Male, ♂ | F(1,117) = 9.205 | 0.003 | F(1,117) = 11.91 | 0.0008 | F(1,117) = 7.662 | 0.0066 | |
| Sex | |||||||
| Adolescent, ♀♂ | Firing rate | F(1,117) = 11.50 | 0.0009 | F(1,117) = 0.1715 | 0.6796 | F(1,117) = 0.9688 | 0.327 |
| Adult, ♀♂ | F(1,116) = 3.592 | 0.0606 | F(1,116) = 13.08 | 0.0004 | F(1,116) = 5.518 | 0.0205 | |
| ♀ Vehicle | ♀ LPS | ♂ Vehicle | ♂ LPS | ||||
| Adolescent | n: 32, N: 6 | n: 28, N: 5 | n: 32, N: 5 | n: 29, N: 5 | |||
| Adult | n: 32, N: 6 | n: 28, N: 7 | n: 29, N: 8 | n: 31, N: 5 | |||
Treatment indicates Veh/LPS. Female & male, ♀♂; n, number of cells; N, number of mice.
Post hoc analysis is provided in extended data Table 1-1.
Tukey post hoc for comparison of age, sex and treatment among groups on firing rate. Download Table 1-1, DOCX file (26.2KB, docx)
It has been reported that hippocampal pyramidal cells consist of at least two populations, bursting cells and non-bursters (Jarsky et al., 2008; Graves et al., 2012). We considered the possibility that neonatal inflammation somehow altered the relative proportions of these two cell types in adult males only. We found however, that the majority of our recorded cell population was comprised of non-bursters, and this proportion did not change in any of the groups as a function of either sex or neonatal inflammation (Fig. 2; Table 2).
Figure 2.

Distribution of regular and bursting spiking pattern of hippocampal neurons. A, CA1 pyramidal neurons respond to step current injections by firing APs in two distinct patterns: regular spiking (top) or bursting (bottom). Bursting neurons display a burst of two or more APs with an instantaneous frequency >100 Hz, whereas regular spiking neurons display single spikes. However, when the current injection is increased, bursting neurons typically fire additional bursts before switching to single APs at the end of the step, and regular neurons fire single spikes with decreased interspike intervals. Insets, Magnified view of high-frequency bursts. Calibration: 20 mV/250 pA, 200 ms; inset, 20 mV/10 ms. B, Pie charts show percentages of all regular and bursting CA1 pyramidal cells recorded, and no changes in any of the groups as a function of either sex or neonatal inflammation were found.
Table 2.
χ2 test for comparison of the proportions between regular and bursting cells among groups
| Group | Variable | χ2 (df) | z | p |
|---|---|---|---|---|
| Adolescent | ♀ Veh vs LPS | 0.00924 (1) | 0.096 | 0.923 |
| ♂ Veh vs LPS | 0.00501 (1) | 0.071 | 0.944 | |
| Adult | ♀ Veh vs LPS | 0.00924 (1) | 0.096 | 0.923 |
| ♂ Veh vs LPS | 0.00230 (1) | 0.048 | 0.962 | |
| ♀ Vehicle | ♀ LPS | ♂ Vehicle | ♂ LPS | |
| Adolescent | n: 32, N: 6 | n: 28, N: 5 | n: 32, N: 5 | n: 29, N: 5 |
| Adult | n: 32, N: 6 | n: 28, N: 7 | n: 29, N: 8 | n: 31, N: 5 |
n, Number of cells; N, number of mice.
The initial membrane potential may influence neuronal firing rate to current injection, however, statistical analysis shows no significant effect of age, sex or treatment on RMP among groups as displayed in Table 3 (adolescent: female vehicle −66.8 ± 0.54 mV, female LPS −67.1 ± 0.64 mV, male vehicle −65.7 ± 0.70 mV, male LPS −66.6 ± 0.55 mV; adult: female vehicle −67.9 ± 0.53 mV, male LPS −68.0 ± 0.73 mV, male vehicle −66.6 ± 0.69, male LPS −67.6 ± 0.56 mV).
Table 3.
Two-way ANOVA for comparison of age, sex, and treatment among groups on RMP, Rin, and capacitance
| Group | Variable | p | Treatment | p | Interaction | p | |
|---|---|---|---|---|---|---|---|
| Age | |||||||
| Female, ♀ | RMP | F(1,116) = 1.04 | 0.309 | F(1,116) = 0.29 | 0.593 | F(1,116) = 0.005 | 0.944 |
| Rin | F(1,116) = 3.08 | 0.082 | F(1,116) = 0.08 | 0.770 | F(1,116) = 0.01 | 0.917 | |
| Capacitance | F(1,116) = 2.96 | 0.088 | F(1,116) = 0.00002 | 0.996 | F(1,116) = 2.59 | 0.110 | |
| Male, ♂ | RMP | F(1,117) = 0.78 | 0.379 | F(1,117) = 0.83 | 0.363 | F(1,117) = 0.001 | 0.982 |
| Rin | F(1,117) = 15.29 | 0.0002 | F(1,117) = 19.79 | <0.0001 | F(1,117) = 11.11 | 0.001 | |
| Capacitance | F(1,117) = 1.73 | 0.190 | F(1,117) = 2.65 | 0.107 | F(1,117) = 0.091 | 0.764 | |
| Sex | |||||||
| Adolescent, ♀♂ | RMP | F(1,117) = 2.94 | 0.089 | F(1,117) = 0.48 | 0.491 | F(1,117) = 0.05 | 0.830 |
| Rin | F(1,117) = 2.55 | 0.113 | F(1,117) = 0.19 | 0.660 | F(1,117) = 0.38 | 0.536 | |
| Capacitance | F(1,117) = 1.16 | 0.283 | F(1,117) = 2.29 | 0.133 | F(1,117) = 0.03 | 0.859 | |
| Adult, ♀♂ | RMP | F(1,116) = 3.20 | 0.076 | F(1,116) = 0.57 | 0.451 | F(1,116) = 0.026 | 0.873 |
| Rin | F(1,116) = 17.86 | <0.0001 | F(1,116) = 15.35 | 0.0002 | F(1,116) = 18.82 | <0.0001 | |
| Capacitance | F(1,116) = 3.77 | 0.054 | F(1,116) = 0.015 | 0.902 | F(1,116) = 2.95 | 0.089 | |
| ♀ Vehicle | ♀ LPS | ♂ Vehicle | ♂ LPS | ||||
| Adolescent | n: 32, N: 6 | n: 28, N: 5 | n: 32, N: 5 | n: 29, N: 5 | |||
| Adult | n: 32, N: 6 | n: 28, N: 7 | n: 29, N: 8 | n: 31, N: 5 | |||
Treatment indicates Veh/LPS. Female & male, ♀♂; n, number of cells; N, number of mice.
Post hoc analysis is provided in extended data Table 3-1.
Tukey post hoc for comparison of age, sex and treatment among groups on Rin. Download Table 3-1, DOCX file (16.4KB, docx)
Given that resting membrane potential was unaltered, other factors that could contribute to the firing frequency alteration are the Rin and the threshold voltage for AP generation. Therefore, the effect of neonatal inflammation on these intrinsic membrane properties in the hippocampal neurons was also investigated. Figure 3 shows current–voltage curves (Fig. 3B) constructed by measuring the voltage response of CA1 pyramidal neurons elicited by step current injections from RMP (Fig. 3A). When comparing effects of age and early life LPS treatment on Rin, significant differences were observed (Table 3). We found an age-dependent effect of the LPS treatment on Rin in CA1 pyramidal neurons of male mice, as the interaction between both variables was highly significant (F(1,117) = 11.11, p = 0.0012). Multiple comparisons revealed that the interaction was attributed to a ∼34% increase in Rin from CA1 pyramidal neurons of adult (vehicle 103.5 ± 3.46 vs LPS 138.7 ± 5.42 ΜΩ, p < 0.0001; Fig. 3B2; Table 3-1), but not adolescent male mice (vehicle 100.8 ± 4.37 vs LPS 105.9 ± 4.41 ΜΩ, p = 0.8579; Fig. 3B1; Table 3-1), exposed to early life inflammation compared with controls. Also, no significant effects of the early life LPS inflammation on Rin in CA1 pyramidal neurons of adolescent (vehicle 108.5 ± 4.38 vs LPS 110.6 ± 5.56 ΜΩ) and adult (vehicle 104.0 ± 3.65 vs LPS 102.2 ± 4.10 ΜΩ) female mice were found (F(1,116) = 0.01, p = 0.917; Table 3). Consistently, a sex-dependent effect of the early LPS treatment was observed in adult mice (F(1,116) = 18.82, p < 0.0001; Table 3), where comparisons confirmed the significant increase in Rin only in male mice (p < 0.0001; Table 3-1). To better address whether the differences in Rin seen in male mice could depend on the size of the neurons, we compared the effects of age, sex, and early life LPS treatment on specific capacitance for each cell (Table 3). We found that the capacitances were similar in control and neonatally inflamed CA1 pyramidal neurons from adolescent (female vehicle 121.4 ± 6.62 pF, female LPS 110.6 ± 5.57 pF; male vehicle 127.2 ± 6.67 pF, male LPS 118.6 ± 6.52 pF, F(1,117) = 0.03, p = 0.859) and adult mice (female vehicle 122.2 ± 6.34 pF, female LPS 133 ± 8.16 pF; male vehicle 121.3 ± 5.9 pF, male LPS 109.7 ± 6.1 pF, F(1,116) = 2.95, p = 0.089). This was further corroborated by Golgi staining. We performed a detailed analysis of the dendritic architecture, and assessed the length of the apical dendrites. Sholl analysis of CA1 pyramidal neurons from adult male mice exposed to LPS at P14, shows no significant differences in dendritic intersections with all Sholl spheres analyzed (multiple Student's t test, for all values p > 0.05; Fig. 4B), or in total dendritic length compared with controls (vehicle 1896 ± 150 μm vs LPS 2055 ± 177 μm, unpaired Student's t test, t(23) = 0.681, p = 0.50; Fig. 4C; Table 4). Thus, a possible contribution of the dendritic tree size or extent was unlikely to have contributed to the Rin differences.
Figure 3.

Neonatal inflammation increases the input resistance of hippocampal neurons from adult male mice. A, Sample traces of voltage responses to hyperpolarizing and depolarizing current steps from −100 to 50 pA in 30 pA increments (500 ms duration) of CA1 pyramidal neurons from (A1) adolescent and (A2) adult mice treated with Veh (dark line) or LPS (red line) at P14. Calibration: 10 mV/120 pA, 100 ms. B, Current–voltage (I–V) plots for (B1) adolescent and (B2) adult mice showing mean ± SEM voltage responses to current injections as depicted in A. Significant differences between I–V curves were only observed for Veh and LPS-treated adult male mice (B2, right; p < 0.0001, n = 29–31 cells, 5–8 mice/group). B1, B2, Insets, Plots depicting Rin of individual CA1 pyramidal neurons (▿) and average values per mouse (females, ♀; males, ♂) from respective groups calculated from the slope of the current voltage plots. ****p < 0.0001.
Figure 4.
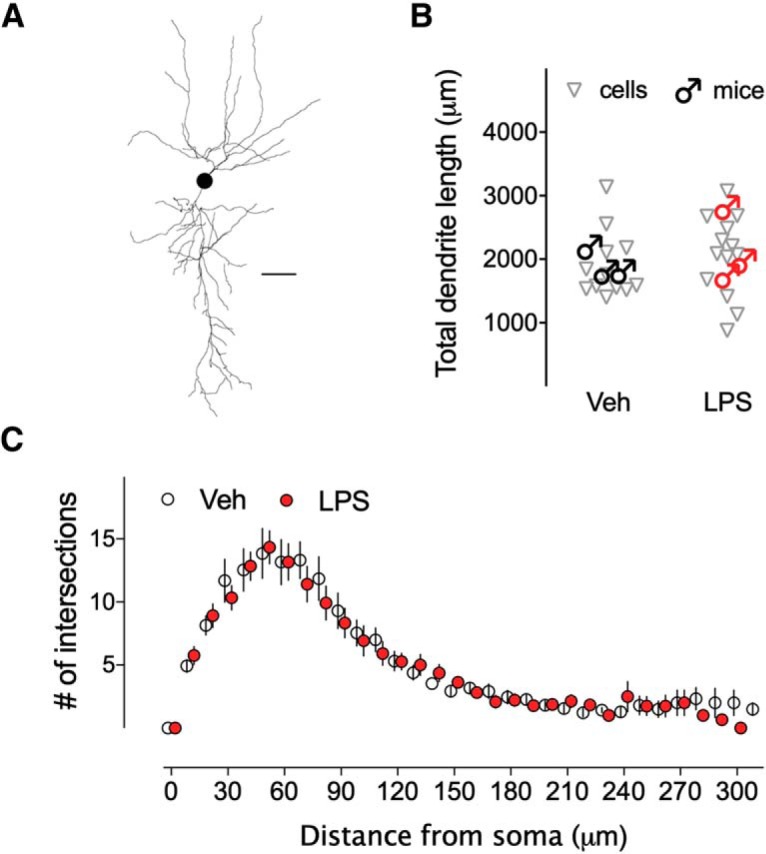
General morphology of CA1 pyramidal neurons in adult male mice is unaltered by neonatal LPS. A, Three-dimensional reconstruction of a representative pyramidal neuron showing the dendritic architecture. Calibration, 30 μm. B, Scatter graph shows the total dendritic length of individual cells and average per mouse (n = 12–13 cells, 3 mice/group). C, Sholl analysis of apical dendrites of CA1 pyramidal neurons depict the number of intersections for each concentric circle, starting from the point in the center of the cell body, and analyzed per 10 μm concentric circle. Error bars indicate SEM.
Table 4.
Unpaired Student's t test for comparison the total dendritic length among vehicle and LPS groups of adult male mice
| Group | Variable | t | df | p |
|---|---|---|---|---|
| Adult | ♂ Veh & LPS | 0.68 | 23 | 0.5025 |
| Veh n: 12, N: 3 | LPS n: 13, N: 3 |
n, Number of cells, N; number of mice.
A second factor that could influence the firing frequency is the threshold for AP generation. Figure 5A–D shows representative voltage responses to ramp current injections from RMP for control and neonatally inflamed CA1 pyramidal neurons of adolescent and adult mice. Interestingly, statistical analyses uncovered an age-dependent (F(1,109) = 16.5, p < 0.0001) and sex-dependent (F(1,110) = 22.26, p < 0.0001) effect of the LPS treatment on AP threshold in CA1 pyramidal neurons (Table 5). This was attributed solely to a more hyperpolarized AP threshold in adult male mice exposed to early life inflammation compared with controls (vehicle −41.33 ± 0.6 vs LPS −46.49 ± 0.5 mV, p < 0.0001; Fig. 5D; Table 5-1). On the other hand, neonatal inflammation had no effect on the AP threshold in adult female (vehicle −41.96 ± 0.48 vs LPS −42.22 ± 0.47, p = 0.985; Fig. 5C), or adolescent female (vehicle −42.9 ± 0.5 vs LPS −42.4 ± 0.7 mV, p = 0.944; Fig. 5A) and male mice (vehicle −40.5 ± 0.6 vs LPS −41.2 ± 0.6 mV, p = 0.937; Fig. 5B). Moreover, the hyperpolarized AP threshold in CA1 pyramidal neurons from adult male offspring exposed to LPS, was paralleled by a ∼25% reduction in rheobase (vehicle 162.2 ± 9.7 vs LPS 122.4 ± 7.4 pA, p = 0.0055; Fig. 5D; Table 5-1). Together, these data suggest that the Rin and AP threshold changes seen in CA1 pyramidal neurons of male adult offspring exposed to LPS as neonates are possible explanations for the increased firing rate seen in response to depolarizing current.
Figure 5.

Neonatal inflammation alters the firing threshold of hippocampal neurons from adult male but not female mice. A–D, Representative traces of voltage responses to the indicated ramp current injections (2 s duration) of CA1 pyramidal neurons of adolescent and adult mice treated with Veh (dark line) or LPS (red line) at P14. Calibration: 20 mV/200 pA, 500 ms. Scatter graphs on left show AP thresholds (APTs) of individual cells and average values per mouse (n = 26–31 cells, 5–8 mice/group). The fractional distribution of cells firing first AP relative to rheobase (depicted as vertical bars and smoothed line on right) are overlaid by their respective cumulative probabilities. Insets, Rheobase plotted for individual cells and average values per mouse (n = 26–31 cells, 5–8 mice/group). **p < 0.01, ****p < 0.0001.
Table 5.
Two-way ANOVA for comparison of age, sex, and treatment among groups on action potential threshold and rheobase
| Group | Variable | p | Treatment | p | Interaction | p | |
|---|---|---|---|---|---|---|---|
| Age | |||||||
| Female, ♀ | Threshold | F(1,110) = 0.90 | 0.344 | F(1,110) = 0.023 | 0.880 | F(1,110) = 0.413 | 0.522 |
| Rheobase | F(1,110) = 0.097 | 0.756 | F(1,110) = 0.034 | 0.855 | F(1,110) = 0.137 | 0.712 | |
| Male, ♂ | Threshold | F(1,109) = 25.27 | <0.0001 | F(1,109) = 23.81 | <0.0001 | F(1,109) = 16.5 | <0.0001 |
| Rheobase | F(1,109) = 7.189 | 0.009 | F(1,109) = 4.159 | 0.044 | F(1,109) = 5.152 | 0.025 | |
| Sex | |||||||
| Adolescent, ♀♂ | Threshold | F(1,109) = 7.227 | 0.008 | F(1,109) = 0.002 | 0.965 | F(1,109) = 0.579 | 0.448 |
| Rheobase | F(1,109) = 1.882 | 0.173 | F(1,109) = 0.128 | 0.721 | F(1,109) = 0.021 | 0.885 | |
| Adult, ♀♂ | Threshold | F(1,110) = 12.4 | 0.001 | F(1,110) = 27.23 | <0.0001 | F(1,110) = 22.26 | <0.0001 |
| Rheobase | F(1,110) = 0.988 | 0.322 | F(1,110) = 6.302 | 0.014 | F(1,110) = 5.315 | 0.023 | |
| ♀ Vehicle | ♀ LPS | ♂ Vehicle | ♂ LPS | ||||
| Adolescent | n: 30, N: 6 | n: 26, N: 5 | n: 30, N: 5 | n: 27, N: 5 | |||
| Adult | n: 31, N: 6 | n: 27, N: 7 | n: 27, N: 8 | n: 29, N: 5 | |||
Treatment indicates Veh/LPS. Female & male, ♀♂; n, number of cells; N, number of mice.
Post hoc analysis is provided in extended data Table 5-1.
Tukey post hoc for comparison of age, sex and treatment among groups on action potential threshold and rheobase. Download Table 5-1, DOCX file (19.9KB, docx)
Because changes in AP threshold may reflect alterations in sodium channels, and different sodium currents may affect other AP properties (Platkiewicz and Brette, 2010), we examined AP waveform. Comparison among groups displayed a significant age-dependent (F(1,109) = 7.834, p = 0.0061) and sex-dependent (F(1,110) = 9.262, p = 0.0029) effect of early life LPS inflammation on AP amplitude only in adult male mice (Table 6). Figure 6 shows that in offspring exposed to LPS, AP amplitude was significantly larger by ∼4% in adult male mice (vehicle 87.5 ± 0.41 vs LPS 90.6 ± 0.53 mV, Tukey post hoc analysis, p = 0.0011; Table 6-1); however, this is most likely a reflection of the hyperpolarized AP threshold, as there was no change in the maximum depolarization attained by the spike. Unlike amplitude, AP kinetics including half-width, 10–90% rise time, and 90–10% decay time, were not modified by neonatal inflammation across all conditions, suggesting similar regenerative sodium and potassium conductances.
Table 6.
Two-way ANOVA for comparison of age, sex, and treatment among groups on AP kinetics
| Group | Variable | p | Treatment | p | Interaction | p | |
|---|---|---|---|---|---|---|---|
| Age | |||||||
| Female, ♀ | Amplitude | F(1,110) = 1.728 | 0.1914 | F(1,110) = 1.981 | 0.1621 | F(1,110) = 0.761 | 0.3849 |
| Half-width | F(1,110) = 7.634 | 0.0067 | F(1,110) = 2.738 | 0.1009 | F(1,110) = 3.369 | 0.0692 | |
| Rise time | F(1,110) = 14.41 | 0.0002 | F(1,110) = 3.036 | 0.0842 | F(1,110) = 6.892 | 0.0099 | |
| Decay time | F(1,110) = 1.873 | 0.1739 | F(1,110) = 1.703 | 0.1946 | F(1,110) = 2.294 | 0.1327 | |
| Male, ♂ | Amplitude | F(1,109) = 1.183 | 0.2792 | F(1,109) = 6.706 | 0.0109 | F(1,109) = 7.834 | 0.0061 |
| Half-width | F(1,109) = 31 | <0.0001 | F(1,109) = 0.079 | 0.7787 | F(1,109) = 2.093 | 0.1509 | |
| Rise time | F(1,109) = 36.97 | <0.0001 | F(1,109) = 0.108 | 0.7426 | F(1,109) = 2.353 | 0.1280 | |
| Decay time | F(1,109) = 31.02 | <0.0001 | F(1,109) = 0.268 | 0.6060 | F(1,109) = 2.544 | 0.1136 | |
| Sex | |||||||
| Adolescent, ♀♂ | Amplitude | F(1,109) = 0.023 | 0.8804 | F(1,109) = 1.475 | 0.2272 | F(1,109) = 1.044 | 0.3091 |
| Half-width | F(1,109) = 2.67 | 0.1052 | F(1,109) = 4.153 | 0.0440 | F(1,109) = 0.252 | 0.6165 | |
| Rise time | F(1,109) = 1.09 | 0.2987 | F(1,109) = 6.663 | 0.0112 | F(1,109) = 0.907 | 0.3428 | |
| Decay time | F(1,109) = 7.207 | 0.0084 | F(1,109) = 4.36 | 0.0391 | F(1,109) = 0.004 | 0.9510 | |
| Adult, ♀♂ | Amplitude | F(1,110) = 5.638 | 0.0193 | F(1,110) = 6.056 | 0.0154 | F(1,110) = 9.262 | 0.0029 |
| Half-width | F(1,110) = 5.238 | 0.0240 | F(1,110) = 1.052 | 0.3074 | F(1,110) = 0.631 | 0.4288 | |
| Rise time | F(1,110) = 3.396 | 0.0681 | F(1,110) = 1.845 | 0.1772 | F(1,110) = 0.068 | 0.7952 | |
| Decay time | F(1,110) = 5.18 | 0.0248 | F(1,110) = 0.707 | 0.4022 | F(1,110) = 0.379 | 0.5394 | |
| ♀ Vehicle | ♀ LPS | ♂ Vehicle | ♂ LPS | ||||
| Adolescent | n: 30, N: 6 | n: 26, N: 5 | n: 30, N: 5 | n: 27, N: 5 | |||
| Adult | n: 31, N: 6 | n: 27, N: 7 | n: 27, N: 8 | n: 29, N: 5 | |||
Treatment indicates Veh/LPS. Female & male, ♀♂; n, number of cells; N, number of mice.
Post hoc analysis is provided in extended data Table 6-1.
Figure 6.

Neonatal inflammation does not affect AP kinetics of CA1 hippocampal neurons from adolescent or adult mice. A, AP waveforms for CA1 pyramidal neurons from (A1) adolescent and (A2) adult mice, are depicted as average voltage response of the first AP elicited by ramp current injections from the total traces per each group (Veh, dark line; LPS, red line). Calibration, 2 ms. B1, B2, Summary scatter graphs plotting individual cells and average values per mouse (n = 26–31 cells, 5–8 mice/group) for AP amplitude, half-width, 10–90% rise time, and 90–10% decay time. Only AP amplitude for adult male LPS pretreated mice was changed. **p < 0.01.
Tukey post hoc for comparison of age, sex and treatment among groups on AP amplitude. Download Table 6-1, DOCX file (16.3KB, docx)
Neonatal inflammation did not change Nav channel expression
As changes in AP properties may result from changes in the expression level of Nav channels (Platkiewicz and Brette, 2010), we analyzed the mRNA and protein expression pattern of the Nav1.1 and 1.2, which have been shown to be important in setting the AP threshold. qPCR data however, revealed that mRNA expression of both genes analyzed was similar in the hippocampus of adult, neonatally inflamed and control mice as displayed in Table 7 (SCN1α, females: vehicle 1.04 ± 0.09, LPS 1.08 ± 0.17; males: vehicle 1.08 ± 0.19, LPS 0.80 ± 0.12, F(1,28) = 1.032, p = 0.319; SCN2, females: vehicle 1.09 ± 0.13, LPS 1.09 ± 0.17; males: vehicle 1.02 ± 0.09, LPS 0.85 ± 0.03; F(1,28) = 0.338, p = 0.565; Figure 7A). Consistent with this, immunoblotting also revealed no significant differences in hippocampal Nav1.1 and 1.2 protein expression between adult inflamed and control mice (values normalized to vehicle controls; Nav1.1, females: 0.93 ± 0.04, unpaired Student's t test, t = −1.79, p = 0.12; males: 0.99 ± 0.065, t = −0.165, p = 0.87; Nav1.2, females: 1.12 ± 0.07, t = 1.68, p = 0.14; males: 1.07 ± 0.06, t = 1.30, p = 0.242; Fig. 7B; Table 7). Thus, the absence of changes in AP dynamics, along with no evident changes in Nav1.1 or Nav1.2 expression suggests that other mechanisms may account for the differences in excitability that we observed.
Table 7.
Statistical comparisons of sex and treatment among groups on Nav channel expression
| Two-way ANOVA |
Sex | p | Treatment | p | Interaction | p | |
|---|---|---|---|---|---|---|---|
| Group | Variable | ||||||
| Adult, ♀♂ | SCN1α | F(1,28) = 0.559 | 0.461 | F(1,28) = 0.564 | 0.459 | F(1,28) = 1.032 | 0.319 |
| SCN2 | F(1,28) = 1.085 | 0.307 | F(1,28) = 0.330 | 0.570 | F(1,28) = 0.338 | 0.565 | |
| Unpaired Student's t test |
♀ | ♂ | |||
|---|---|---|---|---|---|
| Group | Variable | t | p | t | p |
| Adult, ♀♂ | Nav 1.1 | −1.79 | 0.12 | −0.165 | 0.87 |
| Nav 1.2 | 1.68 | 0.14 | 1.30 | 0.242 | |
| Group | Variable | ♀ Vehicle | ♀ LPS | ♂ Vehicle | ♂ LPS |
| qPCR | SCN | N: 10 | N: 10 | N: 6 | N: 6 |
| Western | Nav | N: 4 | N: 4 | N: 4 | N: 4 |
Treatment indicates Veh/LPS. Female & male, ♀♂; Female, ♀, Male, ♂; N, number of mice.
Figure 7.

Neonatal inflammation did not modify the mRNA and protein expression of voltage gated sodium channel subunits in the hippocampus of adult mice. A, Summary scatter graph of mean (horizontal line) and individual animal (6–10 mice/group) relative expression of SCN1α and SCN2 (normalized to vehicle, 2−ΔΔCt) in adult male and female mice treated with LPS or vehicle on P14. B, Top, Immunoblots of Nav1.1 and 1.2 (∼240 kDa) from hippocampal membranes in control and neonatally inflamed adult mice. Quantification of Nav1.1 and 1.2 band-density measurements were normalized to the corresponding vehicle whole-lane total protein of the same membrane (lower gel, Total protein) using Image Lab 6.0 software. No significant change in Nav1.1 and 1.2 protein levels was detected as a result of the LPS treatment (N = 4 mice/group; each symbol represents 1 animal and the horizontal lines are the mean).
Chloride overload influence on AP threshold and Rin of CA1 neurons
We further tested for a possible mechanism that would explain lower AP threshold and higher Rin following early life inflammation. It has been known for several years that chloride gradients modify the properties of the axon initial segment where APs are generated (Khirug et al., 2008). Recent observations demonstrate that AP threshold and Rin can be substantially modified by changes in transmembrane chloride gradients in hippocampal pyramidal neurons (Rinke et al., 2010; Sørensen et al., 2018). The gradient for this ion is determined, in part, by chloride influx through NKCC1 and efflux through K+-Cl− cotransporter 2 (KCC2; Farrant and Kaila, 2007), and a shift in the expression and activity of these transporters alters chloride homeostasis. Interestingly, a recent study, performed only in male rats, from our laboratory demonstrated that early life inflammation causes an increase in NKCC1 expression in the hippocampus (Reid et al., 2013). This enabled us to hypothesize that the alterations in AP threshold and in Rin induced in adults by neonatal inflammation may be due to changes in chloride homeostasis.
The GABA reversal potential (EGABA), is dependent largely upon chloride ion concentration. Thus, to directly test for changes in chloride homeostasis following neonatal inflammation, we measured EGABA in slices obtained from adult mice treated with LPS at P14. We examined the effect of postsynaptic membrane voltage on evoked inhibitory postsynaptic GABA currents in CA1 pyramidal neurons (Fig. 8A). Statistical analysis showed a sex-dependent effect of the early life LPS treatment on EGABA (F(1,63) = 14.69, p = 0.0003; Table 8). This significant effect was attributed to adult male mice, where whole-cell recordings showed that neonatal inflammation caused a significant depolarizing shift in EGABA (vehicle −57.71 ± 0.94 vs LPS −49.13 ± 1.33 mV, p < 0.0001; Fig. 8A1; Table 8-1). However, no effect of neonatal inflammation was seen in adult females (vehicle −57.30 ± 1.06 vs LPS −59.09 ± 0.79 mV, p = 0.999; Fig. 8A1). Our observations indicate that neonatal inflammation causes a depolarizing shift in EGABA in adult males, suggesting a high intracellular chloride concentration; thus, this finding parallels the observations of altered excitability, Rin, and AP threshold only in neonatally inflamed adult male mice.
Figure 8.

Whole-cell and gramicidin perforated-patch recordings show that EGABA is depolarized in hippocampal CA1 pyramidal neurons of neonatally inflamed male mice. A1, Left, Top diagram illustrating chloride homeostasis in a vehicle-treated adult pyramidal cell; low relative activity of NKCC1 and high activity of KCC2 maintains low intracellular chloride (blue fill). Bottom diagram illustrates chloride homeostasis in an adult neonatally inflamed pyramidal cell with a more active (larger blue barrel) NKCC1 and normal activity KCC2 (red barrel) maintaining a higher intracellular chloride level (blue fill). Right, whole-cell, representative evoked IPSC recordings of CA1 pyramidal neurons from adult male control (dark lines) and neonatally inflamed (red lines) mice. Calibration: 100 pA, 20 ms. Whole-cell, Current–voltage (I–V) curve of evoked IPSCs recorded at different holding potentials from −80 to −30 mV in 10 mV increments of 15–19 hippocampal neurons from control (black) or inflamed (red) mice. Inset, Plot shows individual cell EGABA values and average values per mouse obtained from all I–V curves indicating a depolarizing shift in inflamed male, but not female mice (n = 15–19 cells, 4 mice/group). A2, Top, Immunoblots of p-NKCC1 (∼140 kDa) and p-KCC2 (∼140 and ∼270 kDa) from CA1 hippocampal tissue in control and neonatally inflamed adult mice. Quantification of p-NKCC1 and p-KCC2 band density measurements were normalized to the corresponding vehicle whole-lane total protein of the same membrane (lower gel, Total protein) using Image Lab 6.0 software. Significant increase in p-NKCC1 protein level was found only in adult male mice previously exposed to LPS at P14 (N = 6 mice/group; each symbol represents one animal and the horizontal lines are the mean). Whole-cell (B1) and gramicidin (B2) recordings show that acutely applied bumetanide (10 μm) is without effect on EGABA in vehicle-treated animals (control, black; bumetanide, gray). Insets, Representative IPSC recordings and EGABA of individual cells and average values per mouse in the two groups. Interestingly, acutely applied bumetanide (10 μm; LPS, red; bumetanide, pink) reversed the neonatal inflammation-induced depolarizing shift in EGABA; insets show representative IPSCs and individual EGABA of individual cells and average values per mouse in the two groups (n = 12–20 cells, 4 mice/group). Error bars indicate SEM. *p < 0.05, ***p = 0.0001, ****p < 0.0001.
Table 8.
Statistical comparisons of sex and treatment among groups on EGABA, and NKCC1 and KCC2 expression levels
| Two-way ANOVA |
Sex | p | Treatment | p | Interaction | p | |
|---|---|---|---|---|---|---|---|
| Group | Variable | ||||||
| Adult | |||||||
| ♀♂1 | EGABA | F(1,63) = 11.99 | 0.001 | F(1,63) = 16.17 | 0.0002 | F(1,63) = 14.69 | 0.0003 |
| Treatment |
Drug |
||||||
| ♂2 | EGABA | F(1,46) = 9.839 | 0.003 | F(1,46) = 8.768 | 0.0048 | F(1,46) = 12.35 | 0.001 |
| ♂3 | EGABA | F(1,72) = 49.14 | <0.0001 | F(1,72) = 20.06 | <0.0001 | F(1,72) = 4.193 | 0.0443 |
| Unpaired Student's t test |
♀ |
♂ |
|||
|---|---|---|---|---|---|
| Group | Variable | t | p | t | p |
| Adult, ♀♂ | NKKC1 | 1.71 | 0.15 | 2.87 | 0.03 |
| KCC2 | 0.70 | 0.52 | 1.03 | 0.33 | |
| Group | ♀ Vehicle | ♀ LPS | ♂ Vehicle | ♂ LPS | |
| Whole-cell1 | EGABA | n: 16, N: 4 | n: 15, N: 4 | n: 17, N: 4 | n: 19, N: 4 |
| Whole-cell2 | n: 12, N: 4 | n: 13, N: 4 | |||
| Perforated3 | n: 18, N: 4 | n: 20, N: 4 | |||
| Western | NKCC/KCC | N: 6 | N: 6 | N: 6 | N: 6 |
Treatment indicates Veh/LPS. Drug, Bumetanide (−/+); female & male, ♀♂; female, ♀, male, ♂; n, number of cells; N, number of mice.
Post hoc analysis is provided in extended data Table 8-1.
Tukey post hoc for comparison of age, sex and treatment among groups on EGABA. Download Table 8-1, DOCX file (16.1KB, docx)
Changes in Cl− homeostasis can be the result of either changes in the transporter expression and/or activity of NKCC1 and KCC2. To explore differences in the functional expression of chloride cotransporters that could account for the differences in EGABA, we analyzed phosphorylated protein levels of NKCC1 and KCC2 in membrane extracts from dissected dorsal CA1 hippocampal tissue. Immunoblots from both control and neonatally inflamed adult mice, showed two prominent bands for NKCC1 at ∼140 kDa, consistent with the previously reported presence of multiple sites where phosphate binds to the NKCC1 protein (Darman and Forbush, 2002); and two bands for KCC2 at ∼140 kDa and ∼270 kDa, which correspond to a monomeric and dimeric form (Blaesse et al., 2006; Hewitt et al., 2009). Densitometric analysis revealed that in offspring exposed to LPS, NKCC1 protein expression was significantly higher in CA1 hippocampal tissue from adult male mice (values normalized to vehicle controls; NKCC1 1.519 ± 0.08, unpaired Student's t test, t = 2.87, p = 0.03; Fig. 8A2; Table 8), with no changes in KCC2 expression levels (KCC2 1.231 ± 0.22, t = 1.03, p = 0.33; Fig. 8A2; Table 8). Also, no effect of neonatal inflammation on NKCC1 and KCC2 protein expression was seen in adult female mice (NKCC1 0.91 ± 0.05, t = 1.71, p = 0.15, KCC2 1.04 ± 0.09, t = 0.70, p = 0.52; Fig. 8A2; Table 8). Together, these results strongly imply increased active transport via NKCC1 in CA1 pyramidal neurons of adult male mice exposed to LPS at P14, which would result in high intracellular chloride concentration that in turn could contribute to aberrant excitability in these pyramidal neurons. For the following experiments, only males were used because females did not display altered chloride homeostasis and intrinsic electrophysiological properties.
To determine whether the high NKCC1 expression level is responsible for the neonatal inflammation-induced shift in EGABA, we examined the effects of bumetanide (10 μm), a pharmacological inhibitor of NKCC1, on slices obtained from neonatally inflamed animals. In these conditions, bumetanide reversed the shift in EGABA (LPS −47.3 ± 1.69; plus bumetanide −56.08 ± 1.16 mV, p = 0.0001; Fig. 8B1; Table 8-1); in contrast, bumetanide caused no changes in EGABA in hippocampal neurons in the control group. A depolarizing shift in EGABA was also observed in CA1 pyramidal neurons of adult male mice neonatally exposed to LPS when gramicidin perforated patch recordings were performed (vehicle −68.8 ± 0.8 vs LPS −60.9 ± 0.9 mV, p < 0.0001; Fig. 8B2). Furthermore, bumetanide again reversed the shift in EGABA in these conditions (LPS −60.9 ± 0.9 mV; plus bumetanide −66.6 ± 0.8 mV, p < 0.0001; Fig. 8B2). These data demonstrate that NKCC1 blockade, by reducing the intracellular chloride concentration only in neonatally inflamed, adult male CA1 pyramidal cells, re-establishes an intracellular chloride concentration approximating that seen in control adult males.
Finally, to investigate whether the lower AP threshold and higher Rin following neonatal inflammation are linked to a chloride overload, we examined the effects of bumetanide on intrinsic electrophysiological membrane alterations induced by neonatal inflammation (Fig. 9; Table 9). NKCC1 blockade with bumetanide had no effect on AP threshold (p = 0.5944; Fig. 9A; Table 9-1), Rin (p = 0.999; Fig. 9B), or excitability (for all values p > 0.05; Fig. 9C) in animals that were treated with vehicle as neonates. However, we found that by acutely blocking NKCC1 in neonatally inflamed adult male CA1 pyramidal neurons, the atypical hyperpolarized AP threshold induced by neonatal inflammation was brought back close to control values (LPS −47.21 ± 0.62; plus bumetanide −44.46 ± 0.88 mV, p = 0.0003; Fig. 9A; Table 9-1). Furthermore, the increase in Rin evoked in hippocampal neurons by neonatal inflammation was also lowered close to control levels by NKCC1 blockade (LPS 144.4 ± 5.7 plus bumetanide 127.6 ± 5.3 ΜΩ, p = 0.011; Fig. 9B), as was also seen for the firing rate (for all significant values p < 0.05; Fig. 9C). These data suggest that the mechanisms underlying the alterations in AP threshold, Rin and excitability of adult CA1 pyramidal neurons caused by neonatal inflammation may involve an intracellular chloride overload.
Figure 9.

Inhibition of chloride overload with acutely applied bumetanide resets the AP threshold and Rin, and reverses the hyperexcitability in adult neonatally inflamed CA1 pyramidal cells. A, Representative traces of voltage responses to the indicated ramp current injections before (top traces) and after (bottom traces) bath application of bumetanide (10 μm) from CA1 pyramidal neurons of adult male mice treated with Veh (black/gray) or LPS (red/pink) at P14. Calibration: 20 mV/200 pA, 500 ms. Bottom summary scatter graphs of individual cells and average per mouse show that bumetanide restored the atypical hyperpolarized AP threshold induced by neonatal inflammation without any change in controls. B, Bumetanide also reversed the neonatal inflammation-induced increased in Rin (colors as in A). Top, Representative voltage responses to current injections. Calibration: 10 mV/100 pA, 100 ms. Lower individual cellular Rin and average per mouse before and after bumetanide in Veh and LPS pretreated groups. C, Bumetanide reduced firing frequency (top, representative traces; calibration: 20 mV/170 pA, 200 ms; bottom, plots of firing frequency in response to current injections; colors as in A), in neonatally inflamed (LPS) cells, without any significant effect in hippocampal neurons in control group. n = 15–17 cells, 5 mice/group for each dataset. *p < 0.05, ***p < 0.001, ****p < 0.0001.
Table 9.
Statistical comparisons of treatment among groups on intrinsic membrane properties
| Group | Variable | Treatment | p | Drug | p | Interaction | p |
|---|---|---|---|---|---|---|---|
| Two-way ANOVA | |||||||
| Adult, ♂ | AP Threshold | F(1,60) = 44.11 | <0.0001 | F(1,60) = 15.36 | 0.0002 | F(1,60) = 4.364 | 0.0410 |
| Rin | F(1,60) = 48.55 | <0.0001 | F(1,60) = 4.782 | 0.0327 | F(1,60) = 4.769 | 0.0329 | |
| Three-way ANOVA | |||||||
| Adult, ♂ | Firing rate | F(1,30) = 5.088 | 0.0315 | F(1,30) = 46.44 | <0.0001 | F(1,30) = 20.61 | <0.0001 |
Treatment indicates vehicle (n:15, N:5)/LPS (n:17, N:5). Drug, Bumetanide (−/+); male, ♂; n, number of cells; N, number of mice
Post hoc analysis is provided in extended data Table 9-1.
Tukey post hoc for comparison of treatment among groups on intrinsic membrane properties. Download Table 9-1, DOCX file (22KB, docx)
Discussion
This study provides new evidence that early life inflammation with LPS alters the intrinsic electrophysiological properties in CA1 pyramidal neurons, in a manner that varied by age and sex. These changes included increases in Rin and a more hyperpolarized AP threshold, alterations that may underlie increased excitability seen in response to depolarizing current. There was also a depolarizing shift in EGABA that was reversed by acute application of the NKCC1 blocker, bumetanide. Interestingly, the intrinsic changes in Rin, AP threshold and excitability seen in neurons after early life inflammation were also reversed by acute application of bumetanide. A second remarkable finding here is that all of these changes were only seen in adult males, and not in adult females or adolescent animals of either sex. Together, these observations demonstrate that a single, transient episode of neonatal inflammation caused CA1 pyramidal neurons of adult male mice to be intrinsically more excitable than controls. This could be a contributing factor to altered network excitability during development and raises the possibility that such changes may underlie some of the many changes in behavior seen in adult animals after early life inflammation.
The increased excitability seen in response to depolarizing current pulses could result from either the increased Rin (∼34%), or the shift of AP threshold to more hyperpolarized potentials (∼5 mV). Although we do not know at this time what accounts for the alteration in Rin the fact that resting membrane potential was unaltered argues against a change in a potassium-mediated leak current. A possibility is the hyperpolarization-activated current, which is a voltage-dependent and cAMP-mediated cationic inward current known to regulate Rin (Shah, 2014), and which is normally somewhat active also at resting potential. Likewise the M-current also contributes to resting membrane conductance and acts to clamp the membrane potential at rest (Harnett et al., 2015). Interestingly, both channels are known to be affected by inflammation (Djouhri et al., 2015; Tzour et al., 2017). Also, the voltage- and chloride-sensitive channels (ClC-2), which are expressed in CA1 pyramidal neurons, have been implicated in regulating the Rin and AP threshold (Rinke et al., 2010). Obviously, the potential roles of alteration in membrane ion channels in neonatally inflamed animals will be fruitful areas for future investigation.
We believe it unlikely that early life inflammation with LPS results in either a seizure-induced or a toxic effect to cause cell death of specific cell populations in adult male mice. There has never been any evidence that acute LPS alone causes a seizure in P14 rodents unless they are seizure-prone, although there is ample evidence that neuroinflammation contributes to seizures in an epilepsy prone animal (Vezzani et al., 2013). Even if LPS did cause a seizure, it has long been known that, with the possible exception of lithium-pilocarpine-induced status epilepticus (Torolira et al., 2016), even tonic-clonic seizures of 1–2 h duration in young animals (e.g., <18 d) causes no histopathological damage, unlike what is seen in animals older than P30 (Ben-Ari et al., 1984; Nitecka et al., 1984; Sperber et al., 1991). With respect to LPS alone (i.e., no seizure) causing injury, neither in adult (Mouihate and Pittman, 1998) nor in neonatal rodents (Lee et al., 2000; Heida et al., 2004, 2005) has any evidence been found for neuronal damage in CA1 (or elsewhere) after acute doses of LPS in the amounts we use. This is corroborated by our analysis that revealed a similar distribution of bursting versus nonbursting CA1 pyramidal cell types in the LPS and vehicle pretreated adult males, the identical cell capacitances and our Golgi analysis that indicated similar dendritic trees in the two populations.
The hyperpolarizing shift in AP threshold associated with a 25% decrease of rheobase suggests a change in the properties of the underlying voltage-dependent sodium, potassium, or calcium ion channels (Platkiewicz and Brette, 2010). The inherent space clamp problems associated with recording from pyramidal cells with their large dendrites precludes voltage-clamp experiments to identify activation or inactivation thresholds. However, it is noteworthy, and unexpected, that when we examined in current-clamp aspects like rise time, AP width, and height, the only change was in amplitude and this most likely arises from the hyperpolarized initiation of the AP. Taking into account that the major sodium channel subtype expressed in the hippocampus is Nav1.2 (Westenbroek et al., 1989), and that Nav1.1 has been shown to play a critical role in hyperexcitability conditions (Escayg and Goldin, 2010), the expression of their mRNA transcript and protein was measured. However, in keeping with the identical AP waveform, our data from qPCR and Western blot did not reveal any significant differences in voltage-gated sodium channel expression. An important caveat here is that our approach in which we measured mRNA expression as well as protein levels in CA1 hippocampus, does not rule out possible change in intracellular versus membrane distribution, or redistribution to different parts of the somatodendritic membrane (Trimmer and Rhodes, 2004), nor would it have the necessary fidelity to identify significant changes only in the pyramidal cell in ruling out their contribution to modifying the AP threshold in inflamed CA1 pyramidal neurons.
AP threshold and Rin have recently been shown to be substantially modified by changes in transmembrane chloride gradients in hippocampal pyramidal neurons (Rinke et al., 2010; Sørensen et al., 2018), changes that may underlie some of the increased excitability associated with raised intracellular chloride (Alfonsa et al., 2015). Neuronal chloride homeostasis is maintained primarily by chloride influx through NKCC1 and efflux through KCC2 (Kaila et al., 2014). Under physiological conditions in adult brain, intracellular chloride is kept at a low concentration, normally driving its flow into the neurons (Misgeld et al., 1986; Farrant and Kaila, 2007; Blaesse et al., 2009). High intracellular chloride levels however, are found in immature neurons (Ben-Ari et al., 1989, 1997; Owens et al., 1996; Sulis Sato et al., 2017) and in epileptic brain (Okabe et al., 2002; Ben-Ari, 2006; Barmashenko et al., 2011; Raimondo et al., 2015), where chloride flows in the opposite direction and depolarizes neurons. Here, we provide strong evidence indicating that early life inflammation induced by LPS at P14 increases intracellular chloride concentration in CA1 pyramidal neurons from adult male, but not female mice; this is revealed as a depolarizing shift in the EGABA, suggesting that these changes may contribute to the hyperexcitability found within these neurons. Previous findings from our research group reported that early life inflammation induced increased NKCC1 expression in the hippocampus of male adult rats (Reid et al., 2013). Here we report additional data indicating that adult male, but not female mice exposed to neonatal inflammation, displayed higher NKCC1 protein expression in CA1 hippocampal tissue compared with controls. Hence, the (presumably) high intracellular chloride levels found here may be caused by a change in NKCC1 expression in CA1 neurons toward levels found in immature neurons. Supporting this hypothesis, we also found that inhibiting chloride influx through NKCC1 with the selective inhibitor, bumetanide, reversed the shift in EGABA. Of course, as intracellular chloride levels are regulated by the relative activity of both NKCC1 and KCC2, we cannot differentiate whether the bumetanide, through inhibition of NKCC1, is effective in restoring the depolarized reversal potential to control levels by inhibiting excessive NKCC1 activity, or is merely reducing its (normal) activity to enable a compromised KCC2 to function more effectively. Our biochemical and molecular data would suggest the former explanation. It is interesting that levels of NKCC1 normally begin to decline between postnatal weeks 1 and 2 (Ben-Ari et al., 2012), around the time when neonatal LPS is administered. This timing may suggest that inflammation somehow prolongs its expression/activity into adulthood, similar to how neonatal seizures induced a progressive NKCC1-dependent intracellular chloride accumulation in the hippocampus that is prolonged into adulthood (Dzhala et al., 2010). On the other hand, it has been shown that the expression level of KCC2 in male rodents at P14 is lower compared with females (Galanopoulou et al., 2003; Murguía-Castillo et al., 2013), therefore we cannot overlook the possibility that a sex-specific KCC2 expression at the time of LPS exposure may also contribute to male vulnerability when LPS is injected. Whichever transporter is involved, our results further corroborate previous findings showing alteration of GABA function, and in particular a positive shift in EGABA associated with increased intrinsic excitability in hippocampal neurons in several pathological conditions (Barmashenko et al., 2011; Deidda et al., 2015; MacKenzie and Maguire, 2015; Ben-Ari, 2017), where either under expression/activity of KCC2 or over expression/activity of NKCC1 has been shown to play an essential role.
One of the most exciting findings is that the mechanism behind the increased Rin and the hyperpolarized AP threshold induced by early life inflammation is probably linked to a high intracellular chloride concentration. This is supported by our results showing a reversal of the Rin changes, and the atypical hyperpolarized AP threshold and altered rheobase by bumetanide, ultimately controlling the aberrant excitation in inflamed CA1 neurons. A caveat here is that there may be off-target effects of bumetanide of which we are unaware. However, a mechanism that involves a chloride overload in hippocampal pyramidal neurons induced by optogenetic activation has also been shown to significantly lower AP threshold and led to enhanced excitatory activity in these neurons (Sørensen et al., 2018). Further, ClC-2 channels have been proposed to help regulate intracellular chloride concentration. The direction of chloride flow through ClC-2 channels has been debated because of the rectification properties of the channel, but because chloride flux is dictated by the electrochemical chloride gradient, ClC-2 channels normally let chloride flow into the neuron (Ratté and Prescott, 2011). Only in conditions where the electrochemical gradient for chloride is reversed, ClC-2 serves to extrude chloride (Staley, 1994; Rinke et al., 2010). Indeed, a role of CIC-2 in regulating neuronal excitability through changes in AP threshold and Rin was found (Staley et al., 1996; Rinke et al., 2010). Interestingly, it has been shown that CIC-2 activates at membrane potential more negative that the EGABA (Madison et al., 1986; Staley et al., 1996). As EGABA was substantially depolarized in the neonatally inflamed CA1 pyramidal cells from adult male mice, it is unlikely that ClC-2 is less active in these neurons. Thus, reduced ClC-2 is unlikely to be responsible for the increased input resistance we see. Additional studies however, are required to further elucidate the exact mechanisms by which high intracellular chloride affects AP threshold, Rin and firing rate.
Finally, we report sex-specific differences in response to neonatal inflammation in adult mice that only appear post puberty. Although beyond the scope of the current paper, future studies must be directed to determine the mechanism(s) for this. Whether estrogens suppress the early life effects of LPS, or whether androgens are permissive, is not currently known and will entail extensive experimentation involving manipulation of sex hormones. However, it would appear that the resistance to early life LPS induced changes in the three groups that lack testosterone (prepubertal males and females and postpubertal females) suggests a permissive action of the male hormones. Another possibility is that the differential steroidogenesis seen in male and female neonatal brain (Konkle and McCarthy, 2011), or the different developmental trajectory of the male and female brain in early life (Schwarz et al., 2012) may come into play are only some of the interesting questions raised by this study.
Footnotes
This work was supported by the Natural Sciences and Engineering Research Council of Canada and the Canadian Institutes of Health Research (Q.J.P.) and CONACyT- the Mexican Council of Science and Technology (C.D.G.). We thank Drs. Mio Tsutsui, Kiarash Riazi, and Adrienne Benediktsson for their advice and technical support, and Drs. Kai Kaila and Mel Woodin for valuable advice on this research.
The authors declare no competing financial interests.
References
- Acharjee S, Verbeek M, Gomez CD, Bisht K, Lee B, Benoit L, Sharkey KA, Benediktsson A, Tremblay ME, Pittman QJ (2018) Reduced microglial activity and enhanced glutamate transmission in the basolateral amygdala in early CNS autoimmunity. J Neurosci 38:9019–9033. 10.1523/JNEUROSCI.0398-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonsa H, Merricks EM, Codadu NK, Cunningham MO, Deisseroth K, Racca C, Trevelyan AJ (2015) The contribution of raised intraneuronal chloride to epileptic network activity. J Neurosci 35:7715–7726. 10.1523/JNEUROSCI.4105-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmashenko G, Hefft S, Aertsen A, Kirschstein T, Köhling R (2011) Positive shifts of the GABAA receptor reversal potential due to altered chloride homeostasis is widespread after status epilepticus. Epilepsia 52:1570–1578. 10.1111/j.1528-1167.2011.03247.x [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. (2006) Seizures beget seizures: the quest for GABA as a key player. Crit Rev Neurobiol 18:135–144. 10.1615/CritRevNeurobiol.v18.i1-2.140 [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. (2017) NKCC1 chloride importer antagonists attenuate many neurological and psychiatric disorders. Trends Neurosci 40:536–554. 10.1016/j.tins.2017.07.001 [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Tremblay E, Berger M, Nitecka L (1984) Kainic acid seizure syndrome and binding sites in developing rats. Brain Res 316:284–288. 10.1016/0165-3806(84)90314-6 [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL (1989) Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol 416:303–325. 10.1113/jphysiol.1989.sp017762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Khazipov R, Leinekugel X, Caillard O, Gaiarsa JL (1997) GABAA, NMDA and AMPA receptors: a developmentally regulated 'menage a trois'. Trends Neurosci 20:523–529. 10.1016/S0166-2236(97)01147-8 [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Khalilov I, Kahle KT, Cherubini E (2012) The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 18:467–486. 10.1177/1073858412438697 [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Biedenkapp JC, Der-Avakian A, Watkins LR, Rudy JW, Maier SF (2005) Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. J Neurosci 25:8000–8009. 10.1523/JNEUROSCI.1748-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Rudy JW, Watkins LR, Maier SF (2006) A behavioural characterization of neonatal infection-facilitated memory impairment in adult rats. Behav Brain Res 169:39–47. 10.1016/j.bbr.2005.12.002 [DOI] [PubMed] [Google Scholar]
- Blaesse P, Guillemin I, Schindler J, Schweizer M, Delpire E, Khiroug L, Friauf E, Nothwang HG (2006) Oligomerization of KCC2 correlates with development of inhibitory neurotransmission. J Neurosci 26:10407–10419. 10.1523/JNEUROSCI.3257-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K (2009) Cation-chloride cotransporters and neuronal function. Neuron 61:820–838. 10.1016/j.neuron.2009.03.003 [DOI] [PubMed] [Google Scholar]
- Danese A, Lewis SJ (2017) Psychoneuroimmunology of early-life stress: the hidden wounds of childhood trauma? Neuropsychopharmacology 42:99–114. 10.1038/npp.2016.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Forbush B (2002) A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J Biol Chem 277:37542–37550. 10.1074/jbc.M206293200 [DOI] [PubMed] [Google Scholar]
- Deidda G, Parrini M, Naskar S, Bozarth IF, Contestabile A, Cancedda L (2015) Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of down syndrome. Nat Med 21:318–326. 10.1038/nm.3827 [DOI] [PubMed] [Google Scholar]
- Dinel AL, Joffre C, Trifilieff P, Aubert A, Foury A, Le Ruyet P, Layé S (2014) Inflammation early in life is a vulnerability factor for emotional behavior at adolescence and for lipopolysaccharide-induced spatial memory and neurogenesis alteration at adulthood. J Neuroinflammation 11:155. 10.1186/s12974-014-0155-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouhri L, Al Otaibi M, Kahlat K, Smith T, Sathish J, Weng X (2015) Persistent hindlimb inflammation induces changes in activation properties of hyperpolarization-activated current (Ih) in rat C-fiber nociceptors in vivo. Neuroscience 301:121–133. 10.1016/j.neuroscience.2015.05.074 [DOI] [PubMed] [Google Scholar]
- Doenni VM, Gray JM, Song CM, Patel S, Hill MN, Pittman QJ (2016) Deficient adolescent social behavior following early-life inflammation is ameliorated by augmentation of anandamide signaling. Brain Behav Immun 58:237–247. 10.1016/j.bbi.2016.07.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doenni VM, Song CM, Hill MN, Pittman QJ (2017) Early-life inflammation with LPS delays fear extinction in adult rodents. Brain Behav Immun 63:176–185. 10.1016/j.bbi.2016.11.022 [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Kuchibhotla KV, Glykys JC, Kahle KT, Swiercz WB, Feng G, Kuner T, Augustine GJ, Bacskai BJ, Staley KJ (2010) Progressive NKCC1-dependent neuronal chloride accumulation during neonatal seizures. J Neurosci 30:11745–11761. 10.1523/JNEUROSCI.1769-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellestad KK, Tsutsui S, Noorbakhsh F, Warren KG, Yong VW, Pittman QJ, Power C (2009) Early life exposure to lipopolysaccharide suppresses experimental autoimmune encephalomyelitis by promoting tolerogenic dendritic cells and regulatory T cells. J Immunol 183:298–309. 10.4049/jimmunol.0803576 [DOI] [PubMed] [Google Scholar]
- Ellis S, Mouihate A, Pittman QJ (2005) Early life immune challenge alters innate immune responses to lipopolysaccharide: implications for host defense as adults. FASEB J 19:1519–1521. 10.1096/fj.04-3569fje [DOI] [PubMed] [Google Scholar]
- Escayg A, Goldin AL (2010) Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51:1650–1658. 10.1111/j.1528-1167.2010.02640.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Kaila K (2007) The cellular, molecular and ionic basis of GABAA receptor signalling. Prog Brain Res 160:59–87. 10.1016/S0079-6123(06)60005-8 [DOI] [PubMed] [Google Scholar]
- Galanopoulou AS, Kyrozis A, Claudio OI, Stanton PK, Moshé SL (2003) Sex-specific KCC2 expression and GABAA receptor function in rat substantia nigra. Exp Neurol 183:628–637. 10.1016/S0014-4886(03)00213-9 [DOI] [PubMed] [Google Scholar]
- Galic MA, Riazi K, Heida JG, Mouihate A, Fournier NM, Spencer SJ, Kalynchuk LE, Teskey GC, Pittman QJ (2008) Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci 28:6904–6913. 10.1523/JNEUROSCI.1901-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galic MA, Spencer SJ, Mouihate A, Pittman QJ (2009) Postnatal programming of the innate immune response. Integr Comp Biol 49:237–245. 10.1093/icb/icp025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves AR, Moore SJ, Bloss EB, Mensh BD, Kath WL, Spruston N (2012) Hippocampal pyramidal neurons comprise two distinct cell types that are countermodulated by metabotropic receptors. Neuron 76:776–789. 10.1016/j.neuron.2012.09.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H, Gressens P, Mallard C (2012) Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol 71:444–457. 10.1002/ana.22620 [DOI] [PubMed] [Google Scholar]
- Han ZS. (1994) Electrophysiological and morphological differentiation of chandelier and basket cells in the rat hippocampal formation: a study combining intracellular recording and intracellular staining with biocytin. Neurosci Res 19:101–110. 10.1016/0168-0102(94)90013-2 [DOI] [PubMed] [Google Scholar]
- Harnett MT, Magee JC, Williams SR (2015) Distribution and function of HCN channels in the apical dendritic tuft of neocortical pyramidal neurons. J Neurosci 35:1024–1037. 10.1523/JNEUROSCI.2813-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harré EM, Galic MA, Mouihate A, Noorbakhsh F, Pittman QJ (2008) Neonatal inflammation produces selective behavioural deficits and alters N-methyl-d-aspartate receptor subunit mRNA in the adult rat brain. Eur J Neurosci 27:644–653. 10.1111/j.1460-9568.2008.06031.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heida JG, Boissé L, Pittman QJ (2004) Lipopolysaccharide-induced febrile convulsions in the rat: short-term sequelae. Epilepsia 45:1317–1329. 10.1111/j.0013-9580.2004.13704.x [DOI] [PubMed] [Google Scholar]
- Heida JG, Teskey GC, Pittman QJ (2005) Febrile convulsions induced by the combination of lipopolysaccharide and low-dose kainic acid enhance seizure susceptibility, not epileptogenesis, in rats. Epilepsia 46:1898–1905. 10.1111/j.1528-1167.2005.00286.x [DOI] [PubMed] [Google Scholar]
- Hewitt SA, Wamsteeker JI, Kurz EU, Bains JS (2009) Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat Neurosci 12:438–443. 10.1038/nn.2274 [DOI] [PubMed] [Google Scholar]
- Jarsky T, Mady R, Kennedy B, Spruston N (2008) Distribution of bursting neurons in the CA1 region and the subiculum of the rat hippocampus. J Comp Neurol 506:535–547. 10.1002/cne.21564 [DOI] [PubMed] [Google Scholar]
- Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J (2014) Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci 15:637–654. 10.1038/nrn3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Yamada J, Afzalov R, Voipio J, Khiroug L, Kaila K (2008) GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J Neurosci 28:4635–4639. 10.1523/JNEUROSCI.0908-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkle AT, McCarthy MM (2011) Developmental time course of estradiol, testosterone, and dihydrotestosterone levels in discrete regions of male and female rat brain. Endocrinology 152:223–235. 10.1210/en.2010-0607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrozis A, Reichling DB (1995) Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J Neurosci Methods 57:27–35. 10.1016/0165-0270(94)00116-X [DOI] [PubMed] [Google Scholar]
- Lee SH, Han SH, Lee KW (2000) Kainic acid-induced seizures cause neuronal death in infant rats pretreated with lipopolysaccharide. Neuroreport 11:507–510. 10.1097/00001756-200002280-00016 [DOI] [PubMed] [Google Scholar]
- Lewis ML, Kesler M, Candy SA, Rho JM, Pittman QJ (2018) Comorbid epilepsy in autism spectrum disorder: implications of postnatal inflammation for brain excitability. Epilepsia 59:1316–1326. 10.1111/epi.14440 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- MacKenzie G, Maguire J (2015) Chronic stress shifts the GABA reversal potential in the hippocampus and increases seizure susceptibility. Epilepsy Res 109:13–27. 10.1016/j.eplepsyres.2014.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Malenka RC, Nicoll RA (1986) Phorbol esters block a voltage-sensitive chloride current in hippocampal pyramidal cells. Nature 321:695–697. 10.1038/321695a0 [DOI] [PubMed] [Google Scholar]
- Misgeld U, Deisz RA, Dodt HU, Lux HD (1986) The role of chloride transport in postsynaptic inhibition of hippocampal neurons. Science 232:1413–1415. 10.1126/science.2424084 [DOI] [PubMed] [Google Scholar]
- Mouihate A, Pittman QJ (1998) Lipopolysaccharide-induced fever is dissociated from apoptotic cell death in the rat brain. Brain Res 805:95–103. 10.1016/S0006-8993(98)00675-1 [DOI] [PubMed] [Google Scholar]
- Murguía-Castillo J, Beas-Zárate C, Rivera-Cervantes MC, Feria-Velasco AI, Ureña-Guerrero ME (2013) NKCC1 and KCC2 protein expression is sexually dimorphic in the hippocampus and entorhinal cortex of neonatal rats. Neurosci Lett 552:52–57. 10.1016/j.neulet.2013.07.038 [DOI] [PubMed] [Google Scholar]
- Neher E. (1992) Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol 207:123–131. 10.1016/0076-6879(92)07008-C [DOI] [PubMed] [Google Scholar]
- Nitecka L, Tremblay E, Charton G, Bouillot JP, Berger ML, Ben-Ari Y (1984) Maturation of kainic acid seizure-brain damage syndrome in the rat: II. Histopathological sequelae. Neuroscience 13:1073–1094. 10.1016/0306-4522(84)90289-6 [DOI] [PubMed] [Google Scholar]
- Okabe A, Ohno K, Toyoda H, Yokokura M, Sato K, Fukuda A (2002) Amygdala kindling induces upregulation of mRNA for NKCC1, a Na+, K+-2Cl− cotransporter, in the rat piriform cortex. Neurosci Res 44:225–229. 10.1016/S0168-0102(02)00093-7 [DOI] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MB, Kriegstein AR (1996) Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci 16:6414–6423. 10.1523/JNEUROSCI.16-20-06414.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platkiewicz J, Brette R (2010) A threshold equation for action potential initiation. PLoS Comput Biol 6:e1000850. 10.1371/journal.pcbi.1000850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondo JV, Burman RJ, Katz AA, Akerman CJ (2015) Ion dynamics during seizures. Front Cell Neurosci 9:419. 10.3389/fncel.2015.00419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana SA, Aavani T, Pittman QJ (2012) Sex effects on neurodevelopmental outcomes of innate immune activation during prenatal and neonatal life. Horm Behav 62:228–236. 10.1016/j.yhbeh.2012.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratté S, Prescott SA (2011) ClC-2 channels regulate neuronal excitability, not intracellular chloride levels. J Neurosci 31:15838–15843. 10.1523/JNEUROSCI.2748-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid AY, Pittman QJ, Teskey GC (2012) A prolonged experimental febrile seizure results in motor map reorganization in adulthood. Neurobiol Dis 45:692–700. 10.1016/j.nbd.2011.10.013 [DOI] [PubMed] [Google Scholar]
- Reid AY, Riazi K, Campbell Teskey G, Pittman QJ (2013) Increased excitability and molecular changes in adult rats after a febrile seizure. Epilepsia 54:e45–48. 10.1111/epi.12061 [DOI] [PubMed] [Google Scholar]
- Rinke I, Artmann J, Stein V (2010) ClC-2 voltage-gated channels constitute part of the background conductance and assist chloride extrusion. J Neurosci 30:4776–4786. 10.1523/JNEUROSCI.6299-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Bilbo SD (2011) LPS elicits a much larger and broader inflammatory response than Escherichia coli infection within the hippocampus of neonatal rats. Neurosci Lett 497:110–115. 10.1016/j.neulet.2011.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Sholar PW, Bilbo SD (2012) Sex differences in microglial colonization of the developing rat brain. J Neurochem 120:948–963. 10.1111/j.1471-4159.2011.07630.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM. (2014) Cortical HCN channels: function, trafficking and plasticity. J Physiol 592:2711–2719. 10.1113/jphysiol.2013.270058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen AT, Ledri M, Melis M, Nikitidou Ledri L, Andersson M, Kokaia M (2018) Altered chloride homeostasis decreases the action potential threshold and increases hyperexcitability in hippocampal neurons. eNeuro 4:ENEURO.0172–17.2017. 10.1523/ENEURO.0172-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SJ, Auer RN, Pittman QJ (2006) Rat neonatal immune challenge alters adult responses to cerebral ischaemia. J Cereb Blood Flow Metab 26:456–467. 10.1038/sj.jcbfm.9600206 [DOI] [PubMed] [Google Scholar]
- Spencer SJ, Mouihate A, Galic MA, Ellis SL, Pittman QJ (2007) Neonatal immune challenge does not affect body weight regulation in rats. Am J Physiol Regul Integr Comp Physiol 293:R581–R589. 10.1152/ajpregu.00262.2007 [DOI] [PubMed] [Google Scholar]
- Sperber EF, Haas KZ, Stanton PK, Moshé SL (1991) Resistance of the immature hippocampus to seizure-induced synaptic reorganization. Brain Res Dev Brain Res 60:88–93. 10.1016/0165-3806(91)90158-F [DOI] [PubMed] [Google Scholar]
- Staley K. (1994) The role of an inwardly rectifying chloride conductance in postsynaptic inhibition. J Neurophysiol 72:273–284. 10.1152/jn.1994.72.1.273 [DOI] [PubMed] [Google Scholar]
- Staley K, Smith R, Schaack J, Wilcox C, Jentsch TJ (1996) Alteration of GABAA receptor function following gene transfer of the CLC-2 chloride channel. Neuron 17:543–551. 10.1016/S0896-6273(00)80186-5 [DOI] [PubMed] [Google Scholar]
- Strange BA, Witter MP, Lein ES, Moser EI (2014) Functional organization of the hippocampal longitudinal axis. Nat Rev Neurosci 15:655–669. 10.1038/nrn3785 [DOI] [PubMed] [Google Scholar]
- Sulis Sato S, Artoni P, Landi S, Cozzolino O, Parra R, Pracucci E, Trovato F, Szczurkowska J, Luin S, Arosio D, Beltram F, Cancedda L, Kaila K, Ratto GM (2017) Simultaneous two-photon imaging of intracellular chloride concentration and pH in mouse pyramidal neurons in vivo. Proc Natl Acad Sci U S A 114:E8770–E8779. 10.1073/pnas.1702861114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torolira D, Suchomelova L, Wasterlain CG, Niquet J (2016) Widespread neuronal injury in a model of cholinergic status epilepticus in postnatal day 7 rat pups. Epilepsy Res 120:47–54. 10.1016/j.eplepsyres.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS, Rhodes KJ (2004) Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol 66:477–519. 10.1146/annurev.physiol.66.032102.113328 [DOI] [PubMed] [Google Scholar]
- Tzour A, Leibovich H, Barkai O, Biala Y, Lev S, Yaari Y, Binshtok AM (2017) KV 7/M channels as targets for lipopolysaccharide-induced inflammatory neuronal hyperexcitability. J Physiol 595:713–738. 10.1113/JP272547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Aronica E, Mazarati A, Pittman QJ (2013) Epilepsy and brain inflammation. Exp Neurol 244:11–21. 10.1016/j.expneurol.2011.09.033 [DOI] [PubMed] [Google Scholar]
- Wang KC, Fan LW, Kaizaki A, Pang Y, Cai Z, Tien LT (2013) Neonatal lipopolysaccharide exposure induces long-lasting learning impairment, less anxiety-like response and hippocampal injury in adult rats. Neuroscience 234:146–157. 10.1016/j.neuroscience.2012.12.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Merrick DK, Catterall WA (1989) Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron 3:695–704. 10.1016/0896-6273(89)90238-9 [DOI] [PubMed] [Google Scholar]
- Zemankovics R, Káli S, Paulsen O, Freund TF, Hájos N (2010) Differences in subthreshold resonance of hippocampal pyramidal cells and interneurons: the role of h-current and passive membrane characteristics. J Physiol 588:2109–2132. 10.1113/jphysiol.2009.185975 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tukey post hoc for comparison of age, sex and treatment among groups on firing rate. Download Table 1-1, DOCX file (26.2KB, docx)
Tukey post hoc for comparison of age, sex and treatment among groups on Rin. Download Table 3-1, DOCX file (16.4KB, docx)
Tukey post hoc for comparison of age, sex and treatment among groups on action potential threshold and rheobase. Download Table 5-1, DOCX file (19.9KB, docx)
Tukey post hoc for comparison of age, sex and treatment among groups on AP amplitude. Download Table 6-1, DOCX file (16.3KB, docx)
Tukey post hoc for comparison of age, sex and treatment among groups on EGABA. Download Table 8-1, DOCX file (16.1KB, docx)
Tukey post hoc for comparison of treatment among groups on intrinsic membrane properties. Download Table 9-1, DOCX file (22KB, docx)