

Abstract
Introgression among parasite species has the potential to transfer traits of biomedical importance across species boundaries. The parasitic blood fluke Schistosoma haematobium causes urogenital schistosomiasis in humans across sub-Saharan Africa. Hybridization with other schistosome species is assumed to occur commonly, because genetic crosses between S. haematobium and livestock schistosomes, including S. bovis, can be staged in the laboratory, and sequencing of mtDNA and rDNA amplified from microscopic miracidia larvae frequently reveals markers from different species. However, the frequency, direction, age, and genomic consequences of hybridization are unknown. We hatched miracidia from eggs and sequenced the exomes from 96 individual S. haematobium miracidia from infected patients from Niger and the Zanzibar archipelago. These data revealed no evidence for contemporary hybridization between S. bovis and S. haematobium in our samples. However, all Nigerien S. haematobium genomes sampled show hybrid ancestry, with 3.3–8.2% of their nuclear genomes derived from S. bovis, providing evidence of an ancient introgression event that occurred at least 108–613 generations ago. Some S. bovis-derived alleles have spread to high frequency or reached fixation and show strong signatures of directional selection; the strongest signal spans a single gene in the invadolysin gene family (Chr. 4). Our results suggest that S. bovis/S. haematobium hybridization occurs rarely but demonstrate profound consequences of ancient introgression from a livestock parasite into the genome of S. haematobium, the most prevalent schistosome species infecting humans.
Keywords: Schistosoma haematobium, Schistosoma bovis, parasite, adaptation, M8 metalloprotease
Introduction
Introgressive hybridization occurs when hybrid offspring repeatedly backcross with one or both parental types, acting as a conduit for genetic exchange between species. Once present in the “new” genetic background, introgressed loci are broken up through recombination and exposed to selection. Deleterious alleles are purged while advantageous alleles may increase in frequency if they escape loss by drift. For example, genes impacting skin pigmentation (Vernot and Akey 2014) and immune response (Gittelman et al. 2016) were transferred between Neanderthals and humans during the out-of-Africa migration(s). The gene underlying seasonal coat color changes in snowshoe hares are introgressed from jackrabbits (Jones et al. 2018). Resistance to warfarin-containing-pesticides was transferred to mice from closely related wild species (Song et al. 2011), whereas resistance to insecticide-treated bed nets was transferred between Anopheles gambiae into A. coluzzii (Norris et al. 2015). Introgression allows for complex phenotypic traits to be quickly introduced into a population over the course of a few generations allowing for rapid adaptation to new environments that may not be possible when relying on mutation or standing variation alone (Hedrick 2013).
Gene transfer occurs frequently between bacterial species through plasmid transfer or scavenging of environmental DNA, with profound ecological and biomedical consequences. Among parasitic organisms, hybridization and introgression is a concern since this provides a potential path for genes encoding biomedically important traits including virulence, host specificity or drug resistance to be transferred between species (King et al. 2015). Schistosomes are dioecious trematodes that cause the debilitating disease schistosomiasis which infects more than 220 million people in 78 developing countries (World Health Organization 2019). Schistosoma haematobium, the focus of this article, infects 112 million people (World Health Organization 2019) and is responsible for extensive morbidity including bladder cancer, genital schistosomiasis, and other pathologies associated with the urogenital tract. Schistosomiasis is associated with increased susceptibility to HIV and progression to AIDS (Colley et al. 2014). Although primarily confined to Africa and adjacent regions recent schistosomiasis outbreaks in Corsica caused by hybrids between S. haematobium and a closely related livestock species S. bovis have raised concern that this hybridization may have promoted range expansion and transmission potential (Boissier et al. 2016; Kincaid-Smith et al. 2019).
Interspecific laboratory crosses between members of the S. haematobium species group, which contains nine closely related species, have produced viable hybrid offspring (e.g., see Rollinson et al. 1990; Bremond et al. 1993; Tchuenté et al. 1997; Southgate et al. 1998; Webster et al. 2013) through to the F7 generation (Mutani et al. 1985). In some cases, laboratory-reared, hybrid offspring have displayed hybrid vigor in the form of increased egg production (Mutani et al. 1985), increased infectivity to intermediate and definitive hosts (Wright and Ross 1980), and expansion of suitable intermediate and definitive hosts (Huyse et al. 2013). In natural populations, hybridization is diagnosed via a combination of pathology, egg morphology, and genetic identification via mitochondrial and nuclear discordance from single gene sequencing (reviewed in Leger and Webster 2017). In West Africa, parasites carrying ITS-rDNA from S. haematobium, but mtDNA from S. bovis are frequently observed and occasional parasites with ITS-rDNA copies from both species have been documented (table 1). These results suggest hybridization, but the limited genotyping cannot fully distinguish whether this results from recent hybridization (F1 or F2 parasites), ancient hybridization and introgression, or incomplete sorting of ancestral lineages.
Table 1.
Summary of Studies Examining Hybridization between S. bovis and S. haematobium.
| Study | Molecular Markers | Country | Num. Samples | Hybrid Profile Types |
||
|---|---|---|---|---|---|---|
| Nonhybrid Origin | Mitonuclear Disordance | Heterozygous Nuclear loci | ||||
| Huyse et al. (2009) | cox1/ITS-rDNA | Senegal | 158 | 126 | 30 | 2 |
| Webster et al. (2013) a | cox1/ITS-rDNA | Senegal | 681 | 598 | 35 | 48 |
| Boissier et al. (2016) | cox1/ITS-rDNA | France (Corsica) | 73 | 43 | 30 | 0b |
| Moné et al. (2015) | cox1/ITS-rDNA | France (Corsica) | 4 | 0 | 3 | 1 |
| Leger et al. (2016) | cox1/ITS-rDNA | Niger | 42 | 24 | 13 | 5c |
| Soentjens et al. (2016) | cox1/ITS-rDNA | Mali | 2 | 0 | 2 | 0 |
| Boon et al. (2017) | cox1/ITS-rDNA | Senegal | 66 | 47 | 19 | 0 |
| Catalano et al. (2018) | cox1/ITS-rDNA | Senegal | 6 | 5 | 1 | 0 |
| Boon et al. (2018) | cox1 | Senegal | 1,132 | 907 | 225 | NA |
| Boon et al. (2018) | ITS-rDNA | Senegal | 193 | 190 | NA | 3 |
| Kincaid-Smith et al. (2019) | Whole genome | France (Corsica) | 1 | 0 | 1d | |
| Sene-Wade et al. (2018) | cox1/ITS-rDNA | Senegal | 81 | 51 | 23 | 7 |
| Djuikwo-Teukeng et al. (2019) | cox1/ITS-rDNA, microsats | Cameroon | 218 | 218 | 0 | 0 |
| Oey et al. (2019) | Whole genome | Egypt | 1 | 0 | 0 | 1 |
| Oey et al. (2019) | Exome enrichmente | Zanzibar | 8 | 8 | 0 | 0 |
Also identified 31 and 28 possible S. haematobium and curassoni hybrids from mitonuclear discordance and heterozygous ITS-rDNA markers, respectively.
Includes one nonhybrid S. bovis.
Five samples contained an S. bovis cox1 and a heterozygous S. curassoni/haematobium ITS-rDNA profile.
Genome data were generated from pooled samples of lab-passaged parasites. The sample(s) contained the S. bovis mitochondrial genome and 77% and 23% of the nuclear genome reads derived from S. haematobium and S. bovis, respectively.
Data from Le Clec’h et al. (2018).
Schistosomiasis ranks second only to malaria as a parasitic disease impacting human health. Yet unlike malaria for which thousands of parasite genomes have been sequenced (Pearson et al. 2016; Amato et al. 2018), population genomic analysis of schistosomes is in its infancy, mainly because adult worms live in the blood vessels of human hosts and cannot be easily sampled. In this study, we examine hybridization and introgression in S. haematobium, leveraging a method that allows whole-genome amplification followed by exome capture or whole-genome sequencing from single miracidia larvae (Le Clec’h et al. 2018). We sequenced exomes from parasite populations from both East Africa (Zanzibar) and West Africa (Niger). We used these data to examine the distribution of haplotype blocks containing S. bovis alleles within the S. haematobium autosomal genome to determine 1) whether hybridization is ancient or ongoing, 2) to evaluate evidence for introgression, and 3) to determine whether introgressed alleles show signatures of selection.
Results
Parasite Samples, Exome Capture, and Sequencing
We used individual, archived S. haematobium miracidia larvae from Niger and Zanzibar (Tanzania) fixed on Whatman FTA cards from the Schistosome Collection at the Natural History Museum (SCAN; Emery et al. 2012). To minimize any impact of within host population structure (Steinauer et al. 2013), we used a single miracidium from each of 96 different S. haematobium infected people (nZanzibar = 48, nNiger = 48) for this study (supplementary table S1, Supplementary Material online). The Zanzibar samples were collected in 2011 and came from 26 locations on Unguja and Pemba islands spaced up to 160.9 km apart (fig. 1). The Niger samples were collected in 2013 from school-aged children from ten locations along the Niger River located up to 125 km apart. We sequenced exomes from 88 miracidia from Niger (n = 48) and Zanzibar (n = 40) using whole-genome amplification and exome capture (Le Clec’h et al. 2018).
Fig. 1.

Collection localities. Schistosoma haematobium samples were collected in Niger and on the Zanzibar Archipelago off the coast of Tanzania.
The exome capture probe set was designed using the S. haematobium reference sequence and contains 156,071 RNA baits targeting 94% of the exome length (15,002,706 bp/15,895,612 bp) in the nuclear genome as well as 67 RNA baits targeting the mitochondrial genome. We combined our data with eight exome sequences from Zanzibar that have been reported previously for a total of 47 from Zanzibar (Le Clec’h et al. 2018) and 48 from Niger. Probes were sequenced to an average depth of 40.75× (range: 5.58–98.27; supplementary table S1, Supplementary Material online) across the 15 Mb, targeted region after excluding a single outlier sample that did not generate much sequence data. Due to their overlapping nature, the 156,071 RNA probes targeted 52,242 regions of the genome. We looked to see if any of these regions were systematically underrepresented across all samples, potentially representing failure in the probe design. We found 51,873 of the 52,242 targeted sites were covered at ≥95% of bases in 48 or more samples, meaning that 99.3% of targeted regions were fully captured in the majority of samples.
Mitochondrial Variation
Our initial attempts to genotype mitochondrial loci contained within the exome probe set failed in 31 of the Nigerien S. haematobium samples due to poor representation of mitochondrial reads in the sequence data, despite high rates of capture from nuclear loci (fig. 2). We therefore sequenced whole genomes from a subset (n = 12) of parasites to an average coverage of 17.8× and de novo assembled the mitochondrial genomes for each individual, while we genotyped cox1 in other samples by Sanger sequencing. We found that the samples that failed mitochondrial probe capture contained a S. bovis-derived mitochondrial haplotype that was 15–18.05% divergent from that of S. haematobium and therefore poorly captured by our exome capture baits, designed from the S. haematobium reference sequence (Bi et al. 2012). In all, 65% (n = 31) of the Nigerien S. haematobium miracidia examined had a S. bovis mtDNA, similar to other West African S. haematobium populations (Huyse et al. 2009; Webster et al. 2013). In contrast, S. bovis mtDNA was not present in the Zanzibari S. haematobium population.
Fig. 2.

Mitochondrial sampling and phylogeny. (A) The mitochondrial genome was sequenced to high coverage despite large variations in the number of reads generated for most samples. For 31 Nigerien samples, mitochondrial reads were at very low frequency regardless of sequencing depth. This was attributed to the inefficient capture of the highly diverged mitochondria in these 31 Nigerien samples. (B) Maximum likelihood phylogeny of 12 mitochondrial genomes from Schistosoma haematobium and S. bovis and haematobium reference sequences. Three Nigerien S. haematobium samples are related to S. bovis indicating mitochondrial introgression from S. bovis into the Nigerien S. haematobium population.
Exome Variation
The presence of S. bovis mtDNA in Nigerien S. haematobium could result from contemporary hybridization or past introgression events. To differentiate between these two scenarios, we examined autosomal, exonic single nucleotide polymorphisms (SNPs) recovered by the exome capture probes (Chevalier et al. 2014; Le Clec’h et al. 2018). In addition to our 96 S. haematobium samples, we used sequence data from previous studies (Young et al. 2012; Coghlan et al. 2019) of six closely related species in the S. haematobium species group (S. bovis, S. curassoni, S. intercalatum, S. guineensis, S. mattheei, and S. margrebowiei). The genome assembly of S. haematobium is highly fragmented in thousands of scaffolds compared with the S. mansoni assembly, which comprises large chromosomal length scaffolds. However, the genomes of S. haematobium and S. mansoni are ≥89.4% syntenic (Young et al. 2012). We aligned the S. haematobium and S. mansoni assemblies to convert SNP coordinates from their position on S. haematobium contigs to the corresponding position on S. mansoni chromosomes to take advantage of the more contiguous assembly and gene annotations available for S. mansoni (Berriman et al. 2009; Protasio et al. 2012). We verified local synteny of the transposed SNP coordinates by comparing linkage disequilibrium (LD) in 1-Mb windows between the two sets of SNP positions (supplementary fig. S1, Supplementary Material online). We recovered 370,770 autosomal, exonic SNPs for all samples including outgroups after genotyping, filtering and scaffolding along the S. mansoni assembly. These included 185,601 SNPs segregating in Niger and Zanzibar S. haematobium populations, of which 35,102 were common (minor allele frequency (MAF) > 0.05). We found higher SNP diversity (paired t-test; df = 20, 825; P = 2.2 × 10−16) among common alleles in Niger (mean π = 7.97 × 10−5; interquartile range = 6.3 × 10−5) than in Zanzibar (mean π = 2.89 × 10−5; interquartile range 2.3 × 10−5).
Quantifying and Dating Admixture
We removed SNPs showing strong LD to generate a reduced SNP subset for investigating population structure. A principle component analysis (PCA) from 5,882 unlinked, autosomal SNPS MAF > 0.05) and all Schistosoma samples clearly differentiated S. haematobium from all other species along the first two PCs which accounted for 48.9% of genotypic variation observed (fig. 3A). The lack of intermediate genotypes between S. haematobium and other schistosome species suggests that we did not have early generation hybrids within our samples. We used ADMIXTURE (Alexander et al. 2009) to assign ancestry proportions to the S. haematobium, bovis, and curassoni samples. Schistosoma curassoni was included due to its close phylogenetic affiliation with S. bovis (Lockyer et al. 2003). Three distinct ancestry components were identified corresponding to S. bovis/curassoni, Nigerien, and Zanzibari S. haematobium. The S. bovis/curassoni ancestry component was present in 16 of the 48 Nigerien S. haematobium indicating low levels of potential admixture (0.1 < Q > 2.7; fig. 3B), whereas no admixture was identified in the Zanzibari S. haematobium.
Fig. 3.

Population structure in Schistosoma haematobium. (A) PCA plot shows clear distinction between the two S. haematobium populations and the rest of the species examined: we see no evidence for recent hybridization. (B) Quantification of population structure with ADMIXTURE showed low levels of admixture between S. haematobium populations and S. bovis/curassoni. (C) The three-population statistic (f3) was used to formally test admixture between each of the S. haematobium populations, S. bovis, and S. curassoni. When testing f3 (test; A, B), a negative result indicates that the test group is an admixed population from A and B. To differentiate between introgression from S. bovis and curassoni in Nigerien S. haematobium, we recalculated f3 using only derived alleles from S. bovis or curassoni. These results rule out contemporary hybridization between populations of S. haematobium and S. bovis or curassoni and indicate that low levels of introgression are restricted to the Nigerien S. haematobium population.
We used the three-population test (F3) to determine whether S. bovis and/or S. curassoni were sources of introgressed alleles in the Nigerien S. haematobium population (Reich et al. 2009). The F3 test determines whether a target population is admixed from two source populations by calculating the product of differences in allele frequencies between the target and each source population. A negative F3 indicates the target is an admixed population from the two sources. We used the Zanzibar S. haematobium population as a representative of the “source” S. haematobium population and alternated S. bovis and S. curassoni as the second source. F3 results when testing for admixture from S. bovis (F3 = −0.05, SE = 0.001, Z = −5.73) and S. curassoni (F3 = −0.05, SE = 0.01, Z = 5.28) were negative indicating admixture from both species in the Nigerien S. haematobium population (fig. 3C). Schistosoma bovis and curassoni are very closely related. The admixture signature from F3 could be driven by shared variation from the S. bovis and curassoni common ancestor. We recalculated F3 from, but only using S. bovis or curassoni derived alleles to minimize the signal from shared sites. These data provided evidence for admixture between S. haematobium and S. bovis (F3 = −0.23, SE = 0.0129, Z = −18.18) but failed to identify admixture from S. curassoni (F3 = −0.03, SE = 0.025, Z = 1.36). Incomplete lineage sorting provides a possible explanation when closely related alleles are found within different species. We confirmed that the S. bovis alleles in the Nigerien S. haematobium samples were the result of introgression as opposed to incomplete lineage sorting using Patterson’s D (D = 0.144; SE = 0.04; Patterson et al. 2012). From these results, we infer that S. bovis alleles in Nigerien S. haematobium are the result of admixture.
We used PCAdmix (Brisbin et al. 2012) to identify locally introgressed alleles in the Nigerien S. haematobium population. Ancestry was assigned to phased haplotype blocks by comparing them to a reference panel of S. bovis and Zanzibari S. haematobium. Within each Nigerien S. haematobium miracidium 3.3–8.1% (x¯ = 5.2%; fig. 4) of alleles were identified as S. bovis haplotypes. In contrast, we observed close to zero S. bovis ancestry in Zanzibari miracidia used as a control (0.004–0.2%). Combined, these results indicate at least one introgression event between Nigerien S. haematobium and S. bovis. In comparison, Zanzibari S. haematobium do not contain S. bovis alleles.
Fig. 4.

Selection on introgressed alleles in Nigerien Schistosoma haematobium. We aligned plots showing proportion of S. bovis ancestry, the α statistic from BayeScan, and xpEHH. Schistosoma bovis alleles have reached high frequencies in the Nigerien S. haematobium population. Introgressed alleles under directional selection were identified using allele frequency differences (α; alpha) between populations and using regions of extended homozygosity (xpEHH). Each test identified multiple regions under selection, but only one region was identified by both approaches. This region on Chr 4 spanned a single gene (Smp_120703, invadolysin) at which S. bovis alleles are approaching fixation in Nigerien S. haematobium.
Recombination breaks down introgressed segments over time. As a result, the length of introgressed haplotype blocks can be used to date time since admixture in number of recombination events or generations (Schumer et al. 2016). Using this method we identified 5,649 S. bovis haplotype blocks in Nigerien S. haematobium averaging 0.5 cM in length and estimated admixture to have occurred ∼240.6 generations ago (min = 107.8, max = 612.5; fig. 5). Estimates of Schistosoma generation time vary greatly: while adult worms can live up to 37 years (Chabasse et al. 1985), the minimum time for eggs to mature into adult worms is estimated to be ∼85 days in a laboratory setting (Loker 1983; Crellen et al. 2016). Additional factors including water temperature (Lawson and Wilson 1980), host availability, and drug treatment will further influence average generation times. Assuming a ∼1-year generation time (King et al. 2000), the hybridization event leading to introgression occurred ∼240.6 years ago (min = 107.8, max = 612.5), however this date should be considered as a course estimate given the complexities in estimating Schistosoma generation time in natural populations. Importantly, although all 48 Nigerien miracidia contain introgressed S. bovis DNA resulting from ancient hybridization, we observed no F1 or early generation hybrids. Given 0/48 early generation hybrids detected in our population sample, we infer that the population frequency is between (0–7.4%; 95% binomial confidence interval; Clopper and Pearson 1934).
Fig. 5.

Time since admixture. Time since admixture was estimated using introgressed haplotype block length using methods described in Schumer et al. (2016). The age of the invadolysin (Smp_127030) locus specifically (nruns = 10) was aged using startmrca (Smith et al. 2018). Age estimates for both methods overlap, but the distribution of time since introgression tends to be younger than the estimated age of the invadolysin (Smp_127030) locus. Estimates of time since introgression could be influenced by exome data, which is limited in its ability to identity small haplotype blocks and result in conservative, younger age estimates.
Selection on Introgressed Alleles
Since the admixture event(s) some S. bovis alleles are approaching or have become fixed in the Nigerien S. haematobium population (fig. 4). We used two complementary methods to examine directional selection in the Zanzibari and Nigerien S. haematobium samples. First, BayeScan (Foll and Gaggiotti 2008) was used to quantify selection (α) as a function of allele frequency differences at individual SNPs. Then large haplotype blocks under directional selection were identified using cross-population extended haplotype homozygosity (xpEHH; supplementary fig. S2, Supplementary Material online; Sabeti et al. 2007). BayeScan and xpEHH identified 2 and 17 regions under directional selection between the Zanzibari and Nigerien S. haematobium populations. The strongest signals of selection from both analyses (α > 1.75; xpEHH ≤ −3) occurred at a 23-kb locus on chromosome 4 (Chr4: 20,023,951–20,047,325; fig. 4), which also overlapped the region with the highest frequency of introgressed S. bovis alleles in the Nigerien S. haematobium population. Other regions with high frequency of introgressed alleles, including regions on Chrs. 5 and 6 (fig. 4) did not clearly indicate directional selection in Bayescan and/or xpEHH analyses. Various factors including SNP density could have influenced these findings. Future work, using whole-genome sequencing, will focus on these regions of high introgressed allele frequency.
Selection of introgressed S. bovis alleles would be expected to purge variation in the vicinity of these alleles (Smith and Haigh 1974; Stephan 2019). We therefore plotted the log ratio of diversity (π) in Niger and Zanzibar samples, to identify genome regions showing exceptionally low diversity in Niger. The 23-kb locus genome region on Chr. 4 falls within the bottom 0.2% of values genome wide, consistent with the expectations of a selective sweep (fig. 6).
Fig. 6.

Reduced diversity at the invadolysin (Smp_127030) locus in the Nigerien Schistosoma haematobium population. (A) We compared differences in nucleotide diversity (π) across the Nigerien and Zanzibari S. haematobium populations calculated as . The region containing invadolysin (shaded) shows reduced variation (in the bottom 0.2% genome wide) in the Nigerien S. haematobium population. (B) We identified 44 SNPs in Smp_127030, of which 14 resulted in nonsynonymous changes (shown) and were also shared by more than one individual.
To confirm that this locus was the product of an introgression event rather than the result of selection on standing variation, we calculated the time to the common ancestor of the 23-kb locus in the Nigerien haplotypes relative to the surrounding genomic regions using startmrca (Smith et al. 2018). Using mutation (8.1e−9/bp/generation) and recombination rates (244.2 kb/cM), reported for S. mansoni (Criscione et al. 2009; Crellen et al. 2016), we estimated the time since divergence of the 23-kb target region in the Nigerien haplotypes to be 476 generations ago (437.9–616.6; 95% CI; fig. 5), well within the range of our previous estimates of introgression from the haplotype block length analysis.
Characteristics of the Introgressed Invadolysin Gene
The 23-kb introgressed region spans a single gene alternatively referred to as Smp_127030 (S. mansoni protein ID), MS3_06416, (S. haematobium protein ID), leishmanolysin, or invadolysin. Since we are using S. mansoni genome annotations and chromosomal coordinates, we will refer to the invadolysin gene by its S. mansoni protein ID; Smp_127030. Within the invadolysin gene, we found 23 SNPs coding for 14 nonsynonymous changes (excluding singletons) two of which show fixed differences between Zanzibar and Niger populations (fig. 6). We were able to place six of the 14 nonsynonymous amino acid changes on a protein structure of Smp_127030, modeled from the Drosophila invadolysin (Protein DB: d1lmla). All were in peripheral positions on the protein (fig. 7) with little impact on the inferred protein structure (Kelley et al. 2015).
Fig. 7.
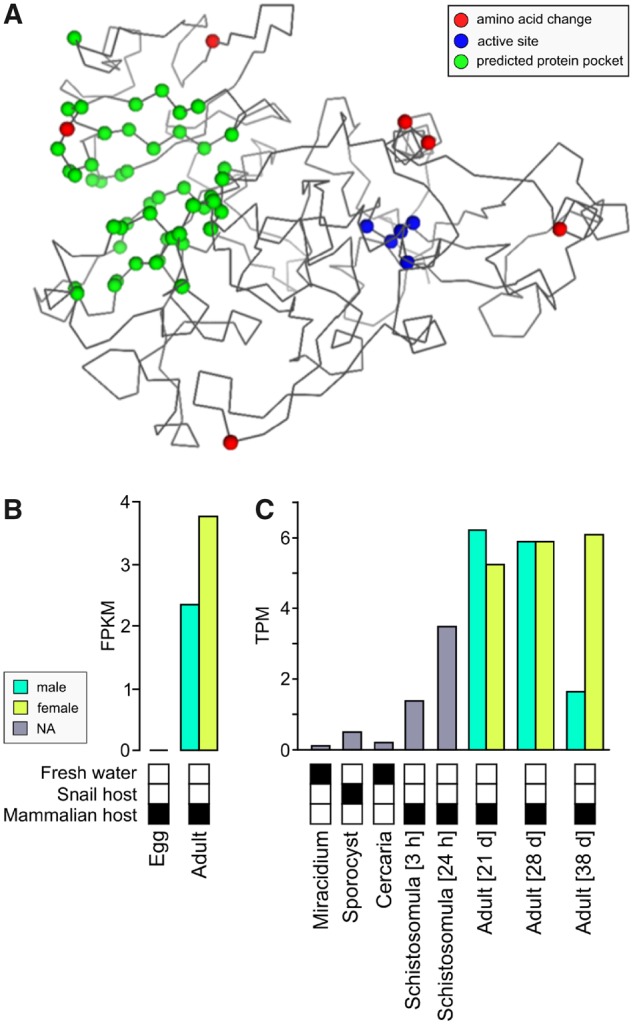
Invadolysin (Smp_127030) structure and expression. (A) The invadolysin (Smp_127030) structure was modeled based on a homologous Drosophila leishmanolysin structure. The amino acid changes are associated with the exterior of the protein and do not fall within the active site. (B) Invadolysin (Smp_127030) is expressed in adult worms in Schistosoma haematobium. (C) In S. mansoni, Smp_127030 is expressed in stages associated with the mammalian host, and most highly expressed in adult worms.
Discussion
We were unable to find evidence of contemporary hybridization between S. haematobium and other schistosome species using samples from Zanzibar and Niger. Instead our data provide evidence for an ancient hybridization event between S. haematobium and S. bovis occurring hundred(s) of generations ago. Introgression was regional occurring in Niger but not in Zanzibar. In a Niger S. haematobium population, 3.3–8.2% of the genome are introgressed S. bovis alleles. Some of the introgressed S. bovis alleles have almost reached fixation within S. haematobium and show signatures of strong selection, providing unambiguous evidence for cross species transfer of genes through hybridization and introgression.
Ancient Introgression Rather than Contemporary Hybridization
Multiple studies of West African S. haematobium miracidia have described discordant mitochondrial DNA and rDNA sequences in up to 41% of parasites sampled (table 1). These studies have been interpreted to indicate that hybridization occurs at high frequencies within many West African locations where S. haematobium and S. bovis occur sympatrically. In contrast, our exomic analyses using samples from Niger and Zanzibar reveals ancient hybridization and subsequent introgression.
Although 65% of miracidia sampled from Niger contained S. bovis-derived mtDNA, none of these 48 parasites showed evidence for recent hybridization based on analysis of the autosomal genome: we did not find any individual miracidia with 50% or 25% of their genome derived from S. bovis as would be expected for F1 or F2 hybrids. These results suggest that hybridization is either rare (0–7.4%; 95% binomial confidence interval; Clopper and Pearson 1934) in S. haematobium, or geographically localized. Broader geographical sampling and genomic characterization of miracidia will be needed to accurately determine the frequency of F1 or early generation hybrids in other S. haematobium populations.
Second, previous studies have suggested that 0–41% of parasites sampled in West African locations have hybrid origins based on mitochondrial and rDNA genotyping (table 1). This is clearly an underestimate: we found that 100% of S. haematobium miracidia sampled from Niger contain S. bovis ancestry, with 3–8% of autosomal loci derived from S. bovis. We conclude that ancient hybridization and introgression best explains the Nigerien genomic data and we predict that more extensive genomic surveys will demonstrate that recent (F1 or F2) hybrids between S. haematobium and S. bovis occur rarely in nature.
Crosses between multiple members of the S. haematobium species group, other than S. bovis and S. haematobium, have been staged in the laboratory using rodent hosts (Webster et al. 2006). Furthermore, field collected miracidia with mtDNA and rDNA markers from different species have been collected in nature. These include reports of nonviable, hybrid miracidia between S. mansoni, agent of intestinal schistosomiasis and S. haematobium (Webster et al. 1999). We emphasize that our conclusions regarding ancient introgression apply to S. haematobium and S. bovis only. Genomic analyses of putative hybrid miracidia from other schistosome species combinations or locations will determine if these reflect ancient or contemporary hybridization events and can help to understand how species barriers are maintained in schistosomes.
We estimate that the hybridization between S. haematobium and S. bovis occurred 240 (range: 108–613) generations ago, based on the size distribution of introgressed fragments. However, we emphasize that our estimate of time since introgression is likely to be conservative because exome data provides limited ability to identify small haplotype blocks. Despite these limitations the genomic data demonstrate that the introgression event occurred more than a hundred generations ago but subsequent to the divergence of the Nigerien and Zanzibari S. haematobium populations. Whole genome, rather than exome data, together with improvements in the S. haematobium genome assembly, and broader geographical sampling will improve our dating estimates of ancient hybridization with S. bovis.
Given that exome capture probes failed to capture S. bovis mtDNA, there is also a potential concern that our exome capture approach will preferentially capture S. haematobium DNA, and underestimate S. bovis alleles within the nuclear genome. We do not think this is the case for two reasons. First, S. haematobium and S. bovis genomes are ∼3% divergent (Oey et al. 2019). Exome capture effectively captures genomes at this level of divergence with minimal bias (Bi et al. 2012). Second, we examined proportions of S. bovis ancestry in 12 miracidia with both exome and genome sequence data (supplementary fig. S4, Supplementary Material online). Ancestry estimates were consistent between data types with the largest difference between estimates being 0.015 (1.5%). If exome capture was biased against detection of S. bovis admixture, then we would have expected to see much higher S. bovis ancestry estimates in the whole-genome data (e.g., 25%).
Introgression is Geographically Widespread
How widespread is this ancient introgression event in Africa? Our results indicate regional introgression in parasites from Niger, but not from Zanzibar. Introgression may extend across multiple countries in Africa. The reference S. haematobium strain originated from Egypt (Young et al. 2012) and contains introgressed S. bovis alleles. Oey et al. (2019) sequenced the S. bovis genome and compared this with the Egyptian S. haematobium reference. They noted high similarity between S. bovis and the S. haematobium reference strain in genome regions spanning up to 100 kb and hypothesized that these regions were the result of an introgression event between S. bovis and S. haematobium. Interestingly, one of these high similarity segments is on chromosome 4 and spans the invadolysin locus (Smp_127030) indicting that the S. bovis invadolysin allele is not only present in Niger but also in Egypt. Taken together, our findings, in combination with Oey et al. (2019), indicate that the common ancestor of the Nigerien and Egyptian S. haematobium reference hybridized with S. bovis, suggesting that remnants of this introgression event may be widespread in extant S. haematobium in Africa. Interestingly, S. bovis has been recorded in Zanzibar (Pennance et al. 2018), but we see no evidence for introgression of S. bovis alleles in Zanzibari S. haematobium. Further, sampling will reveal the number and geographical extent of introgression events in S. haematobium across Africa.
The Nature of Selection Driving Introgression
Invadolysin is a member of a M8 metalloprotease gene family originally identified in Leishmania (Russell and Wilhelm 1986). In Leishmania, these are surface proteins associated with degradation of collagen and fibrenonectin in the extracellular matrix and larval penetration of mammalian hosts (Yao et al. 2003). In schistosomes M8 metalloproteases have undergone a rapid gene family expansion (supplementary fig. S3, Supplementary Material online) and are among the most abundant transcripts and secreted proteins (Curwen et al. 2006; Liu et al. 2014; Coghlan et al. 2019) in larval stages. RNAi knockdowns of one invadolysin paralog in miracidia of S. mansoni led to reduced larval penetration and establishment in the snail intermediate host, reducing cercariae production (Hambrook et al. 2018).
Members of the invadolysin family are expressed in a stage specific manner, similar to globins in vertebrates (Storz et al. 2013). We suspect that the introgressed S. bovis-derived invadolysin is selected during the mammalian, rather than the snail infective stage of the lifecycle. The S. mansoni paralog of this gene (Smp_127030) is expressed during stages associated with the mammalian host and most highly expressed in adult worms (supplementary fig. S5, Supplementary Material online). Consistent with this, the Smp_127030 is expressed in adult worms in S. haematobium (fig. 7B), but expression data are not available for other life stages (except eggs). It is unclear how the S. bovis invadolysin (Smp_127030) allele benefits S. haematobium based on the currently available data.
We were able to confidently place SNPs in probed regions covering only half of the invadolysin (Smp_127030) exons due to ambiguity in lifting coordinates between the SchHae_1.0 and SMV7 S. haematobium and S. mansoni assemblies. Of those able to be transferred, fewer than half of the nonsynonymous SNPs we identified were able to be placed on the inferred protein structure. Despite these limitations, we find strong evidence of selection from the available population data. We speculate that the introgressed invadolysin is involved in tissue penetration (Wilson 2012) or immune evasion (Hambrook et al. 2018) within the mammalian host. Functional analysis of this system may be possible, since certain strains of S. haematobium can be maintained in the laboratory and will be a central priority for future work with this system.
Implications for Schistosome Control
Significant resources are being directed toward schistosome control and elimination strategies (Savioli et al. 1997; Zhou et al. 2005; King 2009; Webster et al. 2014; Webster et al. 2016; Tchuenté et al. 2017). Hybridization between human and livestock schistosomes, including S. bovis and curassoni, will complicate these efforts (King et al. 2015). The introgression of the S. bovis invadolysin allele in S. haematobium provides an example of an adaptive introgression between an animal and human parasite species. Gene exchange between these two species highlights the need for effective coordination between medical and veterinary researchers and a “one health” approach to schistosome control and management (Webster et al. 2016).
Conclusions
Hybridization between species is a powerful source of evolutionary novelty. In humans, up to 4% and 5% of the genome contains Neanderthal (Green et al. 2010) or Denisovan (Reich et al. 2010) ancestry as a direct result of admixture with these archaic species. Genes involved in immune response (Abi-Rached et al. 2011), pathogen defense (Enard and Petrov 2018), and protection from sun exposure (Dannemann and Kelso 2017) have introgressed from archaic humans as a result of these ancient hybridization events. One of the most striking conclusions from the many studies on human/Neanderthal or human/Denisovan admixture is that limited introgression can have significant genomic and phenotypic impacts.
Gene transfer across species boundaries has clearly taken place in S. haematobium. Although we see no evidence of early generation hybrids, we see a signature of ancient introgression from S. bovis in all Nigerien miracidia examined. We demonstrate that an introgressed M8 metalloprotease (invadolysin; Smp_127030) expressed in adult worms has spread to high frequencies in S. haematobium populations in Niger, and that parasites bearing introgressed S. bovis-derived alleles have a strong selective advantage over those carrying native S. haematobium alleles. Understanding the parasite phenotype conferred by the introgressed S. bovis alleles and the nature of selection driving the spread of these S. bovis alleles into S. haematobium populations is a high priority.
Materials and Methods
Ethics Statement
For the Niger sample collection, ethical clearance was obtained from the Niger National Ethical Committee, in combination with the St Mary’s Hospital Local Ethics Research Committee (part of the Imperial College London Research Ethics Committee [ICREC]; EC No. 03.36, R&D No. 03/SB/033E) in London, United Kingdom, in combination with the ongoing Schistosomiasis Control Initiative and Schistosomiasis Consortium for Operational Resaerch (SCORE) activities. For the Zanzibar sample collection, ethical approval was obtained from the Zanzibar Medical Research and Ethics Committee (ZAMREC, reference no. ZAMREC 0003/Sept/011) in Zanzibar, United Republic of Tanzania, the “Ethikkomission beider Basel” (EKBB, reference no. 236/11) in Basel, Switzerland, the Institutional Review Board of the University of Georgia (project no. 2012-10138-0), and registered at the International Standard Randomized Controlled Trial (Register Number ISRCTN48837681). Within both Niger and Zanzibar, all aspects of sample collections were carried out in the framework of the disease control activities implemented and approved by the local Ministry of Health and adopted by regional and local administrative and health authorities.
The study participants were informed about the study objectives and procedures. Written consent was obtained from parents prior to sample collection from children. Participation was voluntary and children could withdraw or be withdrawn from the study at any time without obligation. All children were offered PZQ (40 mg/kg single oral dose) treatment in the frame of the following school-based or community-wide treatment carried out by the Ministry of Health.
Sample Collection
This study used archived miracidia samples from Niger and Zanzibar (Tanzania) fixed on Whatman FTA cards archived within the SCAN (Emery et al. 2012). From Zanzibar, encompassing both the Unguja and Pemba islands, the S. haematobium miracidia were collected as part of the SCORE population genetics studies in 2011 from 26 locations spaced up to 160.9 km apart. From Niger, the S. haematobium miracidia were collected in 2013 from school-aged children from 10 locations located up to 125 km apart. These samples were also collected as part of SCORE population genetic studies within a gaining and sustaining control of schistosomiasis project in Niger and also as part of monitoring and evaluation activities carried out by the Schistosomiasis Control Initiative. To capture maximum diversity we used a single miracidium from 96 individuals (nZanzibar = 48, nNiger = 48). Schistosoma haematobium eggs were harvested from each infected urine sample by sedimentation or filtration, put into fresh water and then exposed to light to facilitate miracidial hatching. Miracidia were captured individually, under a binocular microscope, in 3 µl of water and spotted onto indicating Whatman FTA Classic Indicating cards (GE Healthcare Life Sciences, UK) using a micropipette, dried and archived in SCAN (Emery et al. 2012).
Library Prep and Sequencing
We used whole-genome amplification of single miracidia dried on FTA cards followed by exome capture and sequencing (Illumina 2500) to generate genome-wide sequence data following methods described in Le Clec’h et al. (2018). The exome capture probe set (SureSelect design ID: S0742423) was designed using the published reference genome sequence for S. haematobium (SchHae_1.0, GCA_000699445.1, lab strain, originally from Egypt). The exome capture probe set included 156,004,120-bp RNA baits from the nuclear genome and 67 from the mitochondrial genome, covering 96% (62,106/64,642) of the exons and accounting for 94% of the exome length (15,002,706 bp/15,895,612 bp). Each captured exon was covered by an average of 2.59 120-bp baits (Le Clec’h et al. 2018).
In addition to exome capture, we generated whole-genome sequence data for twelve samples, six from each population (Niger and Zanzibar). Libraries were multiplexed and paired end 150-bp reads were sequenced on a single Illumina NextSeq flowcell. In addition to the whole-genome and exome sequence data generated above, we gathered available genome sequence data from the NCBI Short Read Archives for six other species of Schistosoma in the haematobium group, including S. bovis (ERR119622, ERR103048, and ERR539853), S. curassoni (ERR310937 and ERR119623), S. haematobium (ERR084970, ERR037800, and SRR433865), S. intercalatum (ERR539854, ERR539856, and ERR119613), S. guineensis (ERR119612, ERR539850, and ERR539852), S. mattheei (ERR539851, ERR539855, ERR539857, and ERR103051), and S. margrebowiei (ERR310940) in 20 separate libraries (Young et al. 2012; Coghlan et al. 2019).
Computational Environment
We conducted all analyses on a high-performance computing cluster within a Singularity container or Conda environment. Environmental recipe files, custom programs, and shell scripts are at https://github.com/nealplatt/sH_hybridization (v1.0; doi:10.5281/zenodo.2536390; last accessed 9 July 2019).
Variant Discovery, Filtration, and Phasing
We trimmed sequence reads with Trimmomatic v0.36 (Bolger et al. 2014) so that the leading and trailing base calls had phred-scaled quality scores >10 and the phred score was >15 over a 4-nt sliding window. After trimming, we mapped paired and singleton reads to the reference S. haematobium genome (SchHae_1.0, GCA_000699445.1, lab strain, originally from Egypt). BWA v0.7.17-r1188 (Li and Durbin 2009) and merged into a single BAM file using SAMtools v1.7 (Li et al. 2009). GATK v4.0.1.1 (McKenna et al. 2010) was used to add read group information and mark duplicate reads from each library. Where possible, we added complete read group information based on information contained within the FASTQ header. In some cases (primarily the public data) pseudo read groups were created to differentiate each library.
We used GATK’s Haplotypecaller (McKenna et al. 2010) for variant discovery. Variant discovery was restricted to target regions of the S. haematobium assembly using the –L option. Target regions were identified by combining all adjacent, exome probe locations within 500 bases of one another into larger intervals with BEDtools v2.27.1 (Quinlan and Hall 2010). Each interval was genotyped using GATK’s GenotypeGVCFs and combined into a single VCF for filtering using GATK’s MergeVcfs. Low quality SNP genotypes were filtered using GATK’s VariantFiltration with the following filters: variant quality normalized by depth (QD < 2.0), strand bias (FS > 60.0), mapping quality (MQ < 40.0), mapping quality rank sum (MQRankSum < −12.5), and read position rank sum (ReadPosRankSum < −8.0). We used VCFtools v 0.1.15 (Danecek et al. 2011) to remove sites with high rates of missing data (>20%), multiallelic sites, and individuals with low call rates (>15% missing data). All indels were excluded from downstream analyses.
The published S. haematobium (SchHae_1.0) and S. mansoni (SM_V7) assemblies vary greatly in quality, despite a high degree of synteny between the two genomes (Young et al. 2012). We aligned the two genomes using progressiveCactus v0.0 (Paten et al. 2011) using default parameters to leverage the contiguity of the S. mansoni assembly. The HAL alignment file was used to lift SNP coordinates between the assemblies using progressiveCactus’ halLiftover module. We removed multiposition SNPs, those that align between assemblies in something other than a 1:1 relationship, from downstream analyses. We used LD decay curves to examine biases introduced during the coordinate liftover by comparing LD from SNPs associated with each assembly. SNPs mapping to the S. mansoni autosomes (chr1-7) were extracted using VCFtools v0.1.15. The square of the correlation coefficient (r2) was calculated for all SNPs within 1.5 Mb of each other for each data set using PLINK v1.90b4 (Purcell et al. 2007). The 1.5-Mb cutoff was chosen since it represented the largest scaffold in the S. haematobium assembly. After confirming concordance between the original and S. mansoni-lifted data sets, we used Beagle v4.1 (Browning and Browning 2016) to impute missing SNPs and phase haplotypes for each autosomal chromosome. Imputations and phasing occurred in sliding windows of 300 SNPs and a step size of 30 with 250 iterations per window.
Mitotype Assignment
Unique filters were used to manage the mitochondrial SNPs. First, the mitochondrial contig was identified from the S. haematobium assembly using the haematobium versus mansoni whole-genome alignment and confirmed using NCBI’s BLAST server. Due to low genotyping rates within the Nigerien samples, mitochondrial SNPs were filtered so that the genotyping rate per site was reduced from 85% to 25%. Putative mitochondrial haplotypes (mitotypes) were generated by removing any heterozygous sites, if present. Given previously described rates of mitochondrial introgression and divergence between S. bovis, S. curassoni and S. haematobium, we mapped filtered reads directly to the S. haematobium mitochondrial contig (AMPZ01026399.1) and a previously generated S. curassoni mitochondria sequence (AP017708.1). Full-length S. bovis mitochondrial genomes are not currently available through NCBI GenBank (last accessed April 23, 2018). Schistosoma bovis/curassoni or haematobium mitotypes were classified using the ratio of reads mapping to each mitochondrial reference and confirmed by Sanger sequencing the mitochondrial, cytochrome C oxidase 1 (cox1) gene. cox1 was amplified in a reaction containing 1× reaction buffer, 0.8 mM dNTPs, 1.5 mM MgCl2, 1 µM forward primer (Cox1_schist_5′; CTT TRG ATC ATA AGC G), and 1 µM reverse primer (Cox1_schist_3′; TAA TGC ATM GGA AAA AAA CA) and under the following reaction conditions: 2 min, 95 °C initial denaturation, 35 cycles of a 30 s, 95 °C denaturation, 30 s, 52 °C annealing phase, and a 1 min, 72 °C extension, followed by a final 7 min, 72 °C extension cycle (Lockyer et al. 2003). Amplification and fragment size were confirmed on a 1.5% agarose gel. Bidirectional sequences were generated for each fragment with the Cox1_schist_5′ and Cox1_schist_3′ primers by the Eurofins (Eurofins-MWG) sequencing service. All cox1 sequences were compared with available sequences on GenBank for species identification.
Population Structure, Admixture, and Ancestry Assignment
Summary statistics were calculated using filtered autosomal SNPs with minor allele frequency >5%. FIS was calculated for each sample using VCFtools and the “–het” option. The scikit-allel v1.1.10 (Anopheles gambiae 1000 Genomes Consortium 2017) Python library was used to calculate FST between the Zanzibari and Nigerien populations of S. haematobium. Genome-wide values for FST were averaged from blocks of 100 variants and local calculations were generated from sliding windows of 250- and 50-kb steps. Relationships between samples were visualized in genotypic space with a PCA of unlinked SNPs. Linked sites within sliding windows of 25 SNPs and a pairwise R2 value >0.2 were filtered using PLINK v1.90b4 (Purcell et al. 2007). Since comparison between species can overwhelm population level clustering, separate PCAs were generated from previously published Schistosoma species (Young et al. 2012; Coghlan et al. 2019), and the exome data from the S. haematobium miracidia (Niger and Zanzibar) presented herein. Nucleotide diversity (π) was calculated per site and in sliding windows of 50 kb with 25-kb steps using VCFtools.
Levels of admixture were calculated using ADMIXTURE v1.3.0 (Alexander et al. 2009). The number of populations (K) was explored from 1 to 20 and cross-validation between 1,000 replicates was used to identify the most robust measure of K. The 3-population test f3 as implemented in the scikit-allel was used to identify admixture in the Nigerien and Zanzibar S. haematobium populations (Reich et al. 2009). F3, standard errors, and Z-scores were calculated from block-jackknife replicates of 100 SNPs. We used the scikit-allel package to calculate D statistics and compare possible introgression from S. bovis or S. curassoni into S. haematobium. Standard error and Z-scores were calculated by block-jackknife replicates in windows of 100 SNPs. In addition to genome-wide averaged, D was calculated in local, sliding windows of 50 SNPs with 25 SNP steps. Finally, PCAdmix v1.0 (Brisbin et al. 2012) was used for SNP ancestry assignment. For individuals in the admixed Nigerien population, ancestry was assigned in sliding windows across each chromosome to one of two parental populations defined by “pure” S. haematobium (represented here by samples from Zanzibar) and S. bovis. For validation, we included five, randomly selected, Zanzibari S. haematobium samples with the assumption that the entire genome should be assigned Zanzibari ancestry. Ancestry was assigned to windows of 30 SNPs at a time, with a minimum assignment threshold of 99%.
Selection
We quantified selection across the S. haematobium exome using two independent methods. BayeScan v2.1 (Foll and Gaggiotti 2008) proposes two demographic models to explain allele frequency differences between at each locus. The first model assumes allele frequency differences are due to drift, whereas the other model adds an additional parameter (α) to account for selection. A Markov chain Monte Carlo was used to generate a posterior distribution of the alpha parameter such that significant deviations from zero indicate directional selection. We examined selection by comparing the Zanzibar and Niger S. haematobium populations using 1,000 separate BayeScan runs (chains) to guarantee convergence on posterior distributions alpha. For each chain we used 50 pilot runs of 10,000 generations to generate starting priors and burnin time of 50,000 generations. Prior odds for the neutral models (α = 0) were set at 10:1. To minimize autocorrelation, samples were thinned by 20 and 50,000 samples were taken. Convergence between chains was examined in R using the coda package v0.19-1. A Gelman-Rubin convergence diagnostic score <1.1 was defined as an acceptable level of convergence a priori. In addition to BayeScan, we identified regions under selection in both the S. haematobium populations using cross-population extended haplotype homozygosity (xpEHH; Sabeti et al. 2007) score as implemented in the rehh v2.0.2 R package (Gautier and Vitalis 2012). When possible, alleles were polarized against an outgroup (S. margrebowiei) to define the ancestral state. If sites were heterozygous or missing in S. margrebowiei they were excluded. Regions under selection were defined using a −log10(P value) = 3 an an |xpEHH| > 3. Adjacent loci ≤12.5 kb apart were combined into a single locus.
Dating Genome-Wide Introgression
Schistosoma bovis introgression tracts were identified using Python scripts available at https://github.com/nealplatt/sH_hybridization (release v1.0; doi:10.5281/zenodo.2536390). Briefly, derived (autapomorphic) S. bovis and Zanzibari S. haematobium alleles were identified and used to annotate Nigerien S. haematobium alleles. Introgression blocks were identified by finding the largest stretch of S. bovis or pleisiomorphic alleles that were unbroken by a Zanzibari S. haematobium allele. With these tracts, we estimated the number of generations since admixture (Schumer et al. 2016):
where G is generations, L is the average length of introgression tracts in Morgans, and P is the proportion of the genome from the major parent.
We dated time since divergence of the Chr4: 20,023,951–20,047,325 locus in the Nigerien S. haematobium populations using startmrca (Smith et al. 2018). We used a uniform recombination rate of 3.4 × 10−8 and mutation rate of 8.1 × 10−9. We centered the analysis on Chr4: 20,033,013 and included 1 Mb of upstream and downstream sequence. Markov chain Monte Carlo chains were run for 50,000 generations and limiting proposals to 20 standard deviations. Ten independent chains were run using the parameters described above. Estimates of divergence time were taken by discarding the first 40,000 generations of each run then thinning the remainder to 1 sample per 10 generations.
M8 Peptidase and Invadolysin Gene Family Evolution
Invadolysin (Smp_127030) is a member of the M8 peptidase family of proteins (Pfam ID: PF01457) from the Pfam database (v32.0; last accessed 28 October 2018; Finn et al. 2014). We downloaded amino acid sequences for all platyhelminths and several outgroup taxa including Homo sapiens, Mus musculus, Monodelphis domestica, Gallus gallus, Anolis carolinesis, Danio rerio, Drosophila melanogaster, Aedes aegypti, and Caenorhabditis elegans. Sequences missing the HExxH active site (Rawlings and Barrett 1995) were removed from downstream analysis. Amino acid sequences were aligned with MUSCLE v3.8.1551 (Edgar 2004), and sites with <75% coverage were removed. We used RAxML v 8.2.10 (Stamatakis 2014) to complete 100 searches for the optimal tree using the PROTGAMMAWAG substitution model. Gene duplication evens were inferred using Mega7 (Kumar et al. 2016) and the gene duplication wizard. Duplications were categorized based on their presence in lineages leading to Schistosoma paralogs.
Invadolysin (Smp_127030) Structure
We used Phyre2 (Kelley et al. 2015) in “intensive mode” to model the Nigerien S. haematobium invadolysin (Smp_127030) protein structure. We used the whole-genome resequencing data to identify SNPs at the invadolysin (Smp_127030) locus since the exome probes only targeted 76.1% of the coding region (1,722 of 2,262 bp). Pockets in the protein structure were identified with fpocket2 (Le Guilloux et al. 2009) as implemented in PHYRE2 web portal. After modeling the invadolysin (Smp_127030) protein structure, we examined the placement of amino acid differences fixed between the Zanzibari and Nigerien S. haematobium populations with PyMol v2.2.3 (DeLano 2002).
Invadolysin (Smp_127030) Expression
We quantified gene expression of Smp_127030 in S. haematobium using previously published data (Young et al. 2012) from schistosome eggs, and adult worms from both sexes (SRA accessions: SRX3632877, SRX3632879, and SRX3632881). Reads were mapped to the S. haematobium genome (SchHae_1.0) using hisat v2.1.0 (Kim et al. 2015). Transcripts were assembled and gene expression was normalized and quantified using Stringtie v1.3.4 (Pertea et al. 2015). Gene coordinates were lifted from the S. mansoni assembly to identify which of the Stringtie predicted transcripts were invadolysin (Smp_127030) using progressiveCactus’ halLiftover and the whole-genome alignment generated (described above).
Given the limited gene expression data available for S. haematobium, we examined expression of Smp_127030 and other invadolysin paralogs in S. mansoni (Berriman et al. 2009). We used RNA-seq data from miracidia (Wang et al. 2013), cercariae (Protasio et al. 2012), schistosomula (3 h, 24 h, in vitro transformation), sporocysts (48 h in vitro transformation), juveniles (single sex; 18, 21, 28 days old), and adults (single sex, mixed infections; 38 days) (Protasio et al. 2012; Wang et al. 2013; Protasio et al. 2017). We aligned the data using STAR v2.5.4b (Dobin et al. 2013). STAR references were prepared using the v7 Schistosoma mansoni genome the related v7.1 annotation (Coghlan et al. 2019) which was converted to the GTF format using gffread from cufflinks v2.2.1 (Trapnell et al. 2010) and either an overhang (-sjdbOverhang) of 75 (used for schistosomula and cercariae data) or 99 (used for all other libraries). We used RSEM (Li and Dewey 2011) to compute transcript per million counts for each isoform.
Code Availability
Scripts, code, and environmental files are available at https://github.com/nealplatt/sH_hybridization (release v1.0; doi:10.5281/zenodo.2536390; last accessed 9 July 2019).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
Sandy Smith, Richard Polich, and Roy Garcia (Texas Biomedical Research Institute) provided research support. Steffi Knopp (Swiss Tropical and Public Health Institute) Khalfan Mohammed, Mtumweni Ali Muhsin (Helminth Control Laboratory, Unguja), and Mr Haji Khatib Faki (Public Health Laboratory, Pemba) for their assistance in the collection of the Zanzibar samples. Matthew Berriman and colleagues provided genome sequence data from livestock schistosome species prior to publication. Muriel Rabone for maintaining the SCAN database. This research was funded by the National Institute of Allergy and Infectious Diseases (NIAD R01 AI097576-01 and NIAD 5R21AI096277-01), the National Center for Advancing Translational Sciences (UL1TR001120), Texas Biomedical Research Institute Forum (award 0467), the National Center for Research Resources (C06 RR013556 and RR017515), the Wellcome Trust (104958/Z/14/Z), the Gates Foundation coordinated by the University of Georgia Research Foundation Inc. (RR374-053/5054146 and RR374-053/4785426), and a ZELS research grant (combined BBSRC, MRC, ESRC, NERC, DSTL, and DFID: BB/L018985/1). W.L.C. was supported by a Cowles Fellowship. The authors declare no competing interests.
Author Contributions
Study design done by T.J.C.A., J.P.W., D.R., A.M.E., and B.L.W.; formal analysis done by R.N.P., T.J.C.A., W.L.C., and F.D.C.; writing, original draft, done by R.N.P. and T.J.C.A.; writing, review and editing, done by M.M.-W., W.L.C., F.D.C., B.L.W., J.P.W., D.R., and A.M.E.; sampling done by J.P.W., D.R., F.A., B.L.W., A.G., F.D.C., S.M.A., and A.M.E.
Sequence data are deposited in NCBI’s under BioProject accessions PRJNA508633 and PRJNA443709. Mitochondrial genomes are available in GenBank (MK253567–MK253578).
References
- Abi-Rached L, Jobin MJ, Kulkarni S, McWhinnie A, Dalva K, Gragert L, Babrzadeh F, Gharizadeh B, Luo M, Plummer FA, et al. 2011. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 3346052:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DH, Novembre J, Lange K.. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 199:1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato R, Pearson RD, Almagro-Garcia J, Amaratunga C, Lim P, Suon S, Sreng S, Drury E, Stalker J, Miotto O, et al. 2018. Origins of the current outbreak of multidrug-resistant malaria in southeast Asia: a retrospective genetic study. Lancet Infect Dis. 183:337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anopheles gambiae 1000 Genomes Consortium. 2017. Genetic diversity of the African malaria vector Anopheles gambiae. Nature 552:96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriman M, Haas BJ, LoVerde PT, Wilson RA, Dillon GP, Cerqueira GC, Mashiyama ST, Al-Lazikani B, Andrade LF, Ashton PD, et al. 2009. The genome of the blood fluke Schistosoma mansoni. Nature 4607253:352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi K, Vanderpool D, Singhal S, Linderoth T, Moritz C, Good JM.. 2012. Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales. BMC Genomics. 13:403.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissier J, Grech-Angelini S, Webster BL, Allienne J-F, Huyse T, Mas-Coma S, Toulza E, Barré-Cardi H, Rollinson D, Kincaid-Smith J, et al. 2016. Outbreak of urogenital schistosomiasis in Corsica (France): an epidemiological case study. Lancet Infect Dis. 168:971–979. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 3015:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon NA, Fannes W, Rombouts S, Polman K, Volckaert FA, Huyse T.. 2017. Detecting hybridization in African schistosome species: does egg morphology complement molecular species identification? J Parasitol. 1447:954–964. [DOI] [PubMed] [Google Scholar]
- Boon NA, Van Den Broeck F, Faye D, Volckaert FA, Mboup S, Polman K, Huyse T.. 2018. Barcoding hybrids: heterogeneous distribution of Schistosoma haematobium × Schistosoma bovis hybrids across the Senegal River Basin. J Parasitol. 1455:634–645. [DOI] [PubMed] [Google Scholar]
- Bremond P, Sellin B, Sellin E, Nameoua B, Labbo R, Theron A, Combes C.. 1993. Evidence for the introgression of the human parasite Schistosoma haematobium by genes from Schistosoma bovis, in Niger. C R Acad Sci Ser. 316:667–670. [PubMed] [Google Scholar]
- Brisbin A, Bryc K, Byrnes J, Zakharia F, Omberg L, Degenhardt J, Reynolds A, Ostrer H, Mezey JG, Bustamante CD.. 2012. PCAdmix: principal components-based assignment of ancestry along each chromosome in individuals with admixed ancestry from two or more populations. Hum Biol. 844:343–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Browning SR.. 2016. Genotype imputation with millions of reference samples. Am J Hum Genet. 981:116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano S, Sène M, Diouf ND, Fall CB, Borlase A, Léger E, Bâ K, Webster JP.. 2018. Rodents as natural hosts of zoonotic Schistosoma species and hybrids: an epidemiological and evolutionary perspective from West Africa. J Infect Dis. 2183:429–433. [DOI] [PubMed] [Google Scholar]
- Chabasse D, Bertrand G, Leroux J, Gauthey N, Hocquet P.. 1985. Developmental bilharziasis caused by Schistosoma mansoni discovered 37 years after infestation. Bull Soc Pathol Exot Ses Fil. 785:643–647. [PubMed] [Google Scholar]
- Chevalier FD, Valentim CL, LoVerde PT, Anderson TJ.. 2014. Efficient linkage mapping using exome capture and extreme QTL in schistosome parasites. BMC Genomics. 15:617.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clopper CJ, Pearson ES.. 1934. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometika 264:404–413. [Google Scholar]
- Coghlan A, Tyagi R, Cotton JA, Holroyd N, Rosa BA, Tsai IJ, Laetsch DR, Beech RN, Day TA, Hallsworth-Pepin K.. 2019. Comparative genomics of the major parasitic worms. Nat Genet. 51:163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley DG, Bustinduy AL, Secor WE, King CH.. 2014. Human schistosomiasis. Lancet 3839936:2253–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crellen T, Allan F, David S, Durrant C, Huckvale T, Holroyd N, Emery AM, Rollinson D, Aanensen DM, Berriman M, et al. 2016. Whole genome resequencing of the human parasite Schistosoma mansoni reveals population history and effects of selection. Sci Rep. 6:20954.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscione CD, Valentim CL, Hirai H, LoVerde PT, Anderson TJ.. 2009. Genomic linkage map of the human blood fluke Schistosoma mansoni. Genome Biol. 106:R71.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curwen RS, Ashton PD, Sundaralingam S, Wilson RA.. 2006. Identification of novel proteases and immunomodulators in the secretions of schistosome cercariae that facilitate host entry. Mol Cell Proteomics 55:835–844. [DOI] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 2715:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannemann M, Kelso J.. 2017. The contribution of Neanderthals to phenotypic variation in modern humans. Am J Hum Genet. 1014:578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. 2002. The PyMOL Molecular Graphics System. Delano Scientific, San Carlos.
- Djuikwo-Teukeng FF, Simo AK, Allienne J-F, Rey O, Ngapagna AN, Tchuem-Tchuente LA, Boissier J.. 2019. Population genetic structure of Schistosoma bovis in Cameroon. Parasites Vectors 12:56.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR.. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 291:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 325:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AM, Allan FE, Rabone ME, Rollinson D.. 2012. Schistosomiasis collection at NHM (SCAN). Parasites Vectors 5:185.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard D, Petrov DA.. 2018. Evidence that RNA viruses drove adaptive introgression between Neanderthals and modern humans. Cell 1752:360–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. 2014. Pfam: the protein families database. Nucl Acids Res. 42(D1):D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foll M, Gaggiotti OE.. 2008. A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 1802:977–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier M, Vitalis R.. 2012. rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 288:1176–1177. [DOI] [PubMed] [Google Scholar]
- Gittelman RM, Schraiber JG, Vernot B, Mikacenic C, Wurfel MM, Akey JM.. 2016. Archaic hominin admixture facilitated adaptation to out-of-Africa environments. Curr Biol. 2624:3375–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH-Y, et al. 2010. A draft sequence of the Neandertal genome. Science 3285979:710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambrook JR, Kaboré AL, Pila EA, Hanington PC.. 2018. A metalloprotease produced by larval Schistosoma mansoni facilitates infection establishment and maintenance in the snail host by interfering with immune cell function. PLoS Pathog. 1410:e1007393.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW. 2013. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol Ecol. 2218:4606–4618. [DOI] [PubMed] [Google Scholar]
- Huyse T, Van den Broeck F, Hellemans B, Volckaert F, Polman K.. 2013. Hybridisation between the two major African schistosome species of humans. Int J Parasitol. 438:687–689. [DOI] [PubMed] [Google Scholar]
- Huyse T, Webster BL, Geldof S, Stothard JR, Diaw OT, Polman K, Rollinson D.. 2009. Bidirectional introgressive hybridization between a cattle and human schistosome species. PLoS Pathog. 59:e1000571.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MR, Mills LS, Alves PC, Callahan CM, Alves JM, Lafferty DJ, Jiggins FM, Jensen JD, Melo-Ferreira J, Good JM.. 2018. Adaptive introgression underlies polymorphic seasonal camouflage in snowshoe hares. Science 3606395:1355–1358. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ.. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 106:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL.. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 124:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincaid-Smith J, Tracey A, de Carvalo Augusto R, Bulla I, Holroyd N, Rognon A, Rey O, Chaparro C, Oleaga A, Coma SM.. 2019. Whole genome sequencing and morphological analysis of the human-infecting schistosome emerging in Europe reveals a complex admixture between Schistosoma haematobium and Schistosoma bovis parasites. bioRxiv. doi:10.1101/387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CH. 2009. Toward the elimination of schistosomiasis. N Engl J Med. 3602:106–109. [DOI] [PubMed] [Google Scholar]
- King CH, Muchiri EM, Ouma JH.. 2000. Evidence against rapid emergence of praziquantel resistance in Schistosoma haematobium, Kenya. Emerging Infect Dis. 66:585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KC, Stelkens RB, Webster JP, Smith DF, Brockhurst MA.. 2015. Hybridization in parasites: consequences for adaptive evolution, pathogenesis, and public health in a changing world. PLoS Pathog. 119:e1005098.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K.. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 337:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson JR, Wilson R.. 1980. The survival of the cercariae of Schistosoma mansoni in relation to water temperature and glycogen utilization. Parasitology 812:337–348. [DOI] [PubMed] [Google Scholar]
- Le Clec’h W, Chevalier FD, McDew-White M, Allan F, Webster BL, Gouvras AN, Kinunghi S, Tchuenté L-A, Garba A, Mohammed KA.. 2018. Whole genome amplification and exome sequencing of archived schistosome miracidia. Parasitology 145:1739–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guilloux V, Schmidtke P, Tuffery P.. 2009. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics 10:168.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger E, Garba A, Hamidou AA, Webster BL, Pennance T, Rollinson D, Webster JP.. 2016. Introgressed animal schistosomes Schistosoma curassoni and S. bovis naturally infecting humans. Emerging Infect Dis. 2212:2212–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger E, Webster JP.. 2017. Hybridizations within the genus Schistosoma: implications for evolution, epidemiology and control. Parasitology 1441:65–80. [DOI] [PubMed] [Google Scholar]
- Li B, Dewey CN.. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2514:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R.. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 2516:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai P, Piao X, Hou N, Zhou X, Wu C, Wang H, Chen Q.. 2014. Expression profile of the Schistosoma japonicum degradome reveals differential protease expression patterns and potential anti-schistosomal intervention targets. PLoS Comput Biol. 1010:e1003856.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockyer AE, Olson PD, Ostergaard P, Rollinson D, Johnston DA, Attwood SW, Southgate VR, Horak P, Snyder SD, Le TH, et al. 2003. The phylogeny of the Schistosomatidae based on three genes with emphasis on the interrelationships of Schistosoma Weinland, 1858. Parasitology 126(Pt 3):203–224. [DOI] [PubMed] [Google Scholar]
- Loker ES. 1983. A comparative study of the life-histories of mammalian schistosomes. Parasitology 872:343–369. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 209:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moné H, Holtfreter MC, Allienne J-F, Mintsa-Nguéma R, Ibikounlé M, Boissier J, Berry A, Mitta G, Richter J, Mouahid G.. 2015. Introgressive hybridizations of Schistosoma haematobium by Schistosoma bovis at the origin of the first case report of schistosomiasis in Corsica (France, Europe). Parasitol Res. 11411:4127–4133. [DOI] [PubMed] [Google Scholar]
- Mutani A, Christensen N, Frandsen F.. 1985. A study of the biological characteristics of a hybrid line between male Schistosoma haematobium (Dar es Salaam, Tanzania) and female S. intercalatum (Edea, Cameroun). Acta Trop. 42:319–331. [PubMed] [Google Scholar]
- Norris LC, Main BJ, Lee Y, Collier TC, Fofana A, Cornel AJ, Lanzaro GC.. 2015. Adaptive introgression in an African malaria mosquito coincident with the increased usage of insecticide-treated bed nets. Proc Natl Acad Sci U S A. 1123:815–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oey H, Zakrzewski M, Gravermann K, Young ND, Korhonen PK, Gobert GN, Nawaratna S, Hasan S, Martínez DM, You H, et al. 2019. Whole-genome sequence of the bovine blood fluke Schistosoma bovis supports interspecific hybridization with S. haematobium. PLoS Pathog. 151:e1007513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paten B, Earl D, Nguyen N, Diekhans M, Zerbino D, Haussler D.. 2011. Cactus: algorithms for genome multiple sequence alignment. Genome Res. 219:1512.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson NJ, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D.. 2012. Ancient admixture in human history. Genetics 1923:1065–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson RD, Amato R, Auburn S, Miotto O, Almagro-Garcia J, Amaratunga C, Suon S, Mao S, Noviyanti R, Trimarsanto H, et al. 2016. Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nat Genet. 488:959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennance T, Ame SM, Amour AK, Suleiman KR, Allan F, Rollinson D, Webster BL.. 2018. Occurrence of Schistosoma bovis on Pemba Island, Zanzibar: implications for urogenital schistosomiasis transmission monitoring. Parasitology 14513:1727–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL.. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 333:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protasio AV, Tsai IJ, Babbage A, Nichol S, Hunt M, Aslett MA, De Silva N, Velarde GS, Anderson TJC, Clark RC, et al. 2012. A systematically improved high quality genome and transcriptome of the human blood fluke Schistosoma mansoni. PLoS Negl Trop Dis. 61:e1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protasio AV, van Dongen S, Collins J, Quintais L, Ribeiro DM, Sessler F, Hunt M, Rinaldi G, Collins JJ, Enright AJ, et al. 2017. MiR-277/4989 regulate transcriptional landscape during juvenile to adult transition in the parasitic helminth Schistosoma mansoni. PLoS Negl Trop Dis. 115:e0005559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 813:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM.. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 266:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ.. 1995. Evolutionary families of metallopeptidases In. Methods in enzymology. Amterdam, Netherlands: Elsevier; p. 183–228. [DOI] [PubMed] [Google Scholar]
- Reich D, Green RE, Kircher M, Krause J, Patterson N, Durand EY, Viola B, Briggs AW, Stenzel U, Johnson PLF, et al. 2010. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 4687327:1053–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Thangaraj K, Patterson N, Price AL, Singh L.. 2009. Reconstructing Indian population history. Nature 4617263:489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollinson D, Southgate V, Vercruysse J, Moore P.. 1990. Observations on natural and experimental interactions between Schistosoma bovis and S. curassoni from West Africa. Acta Trop. 472:101–114. [DOI] [PubMed] [Google Scholar]
- Russell D, Wilhelm H.. 1986. The involvement of the major surface glycoprotein (gp63) of Leishmania promastigotes in attachment to macrophages. J Immunol. 1367:2613–2620. [PubMed] [Google Scholar]
- Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, et al. 2007. Genome-wide detection and characterization of positive selection in human populations. Nature 4497164:913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savioli L, Renganathan E, Montresor A, Davis A, Behbehani K.. 1997. Control of schistosomiasis—a global picture. Parasitol Today 1311:444–448. [DOI] [PubMed] [Google Scholar]
- Schumer M, Cui R, Powell DL, Rosenthal GG, Andolfatto P.. 2016. Ancient hybridization and genomic stabilization in a swordtail fish. Mol Ecol. 2511:2661–2679. [DOI] [PubMed] [Google Scholar]
- Sene-Wade M, Marchand B, Rollinson D, Webster BL.. 2018. Urogenital schistosomiasis and hybridization between Schistosoma haematobium and Schistosoma bovis in adults living in Richard-Toll, Senegal. Parasitology 14513:1723–1726. [DOI] [PubMed] [Google Scholar]
- Smith J, Coop G, Stephens M, Novembre J.. 2018. Estimating time to the common ancestor for a beneficial allele. Mol Biol Evol. 354:1003–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JM, Haigh J.. 1974. The hitch-hiking effect of a favourable gene. Genet Res. 231:23–35. [PubMed] [Google Scholar]
- Soentjens P, Cnops L, Huyse T, Yansouni C, De Vos D, Bottieau E, Clerinx J, Van Esbroeck M.. 2016. Diagnosis and clinical management of Schistosoma haematobium–Schistosoma bovis hybrid infection in a cluster of travelers returning from Mali. Clin Infect Dis. 6312:1626–1629. [DOI] [PubMed] [Google Scholar]
- Song Y, Endepols S, Klemann N, Richter D, Matuschka F-R, Shih C-H, Nachman MW, Kohn MH.. 2011. Adaptive introgression of anticoagulant rodent poison resistance by hybridization between old world mice. Curr Biol. 2115:1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southgate VR, Jourdane J, Tchuenté LAT.. 1998. Recent studies on the reproductive biology of the schistosomes and their relevance to speciation in the Digenea. Int J Parasitol. 288:1159–1172. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 309:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinauer ML, Christie MR, Blouin MS, Agola LE, Mwangi IN, Maina GM, Mutuku MW, Kinuthia JM, Mkoji GM, Loker ES.. 2013. Non-invasive sampling of schistosomes from humans requires correcting for family structure. PLoS Negl Trop Dis. 79:e2456.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan W. 2019. Selective sweeps. Genetics 2111:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz JF, Opazo JC, Hoffmann FG.. 2013. Gene duplication, genome duplication, and the functional diversification of vertebrate globins. Mol Phylogenet Evol. 662:469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchuenté L-A, Rollinson D, Stothard JR, Molyneux D.. 2017. Moving from control to elimination of schistosomiasis in sub-Saharan Africa: time to change and adapt strategies. Infect Dis Poverty 61:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchuenté LT, Southgate V, Jourdane J, Kaukas A, Vercruysse J.. 1997. Hybridisation between the digeneans Schistosoma haematobium and S. mattheei: viability of hybrids and their development in sheep. Syst Parasitol. 36:123–131. [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, Pachter L.. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 285:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernot B, Akey JM.. 2014. Resurrecting surviving Neandertal lineages from modern human genomes. Science 3436174:1017–1021. [DOI] [PubMed] [Google Scholar]
- Wang B, Collins IIJJ, Newmark PA.. 2013. Functional genomic characterization of neoblast-like stem cells in larval Schistosoma mansoni. Elife 2:e00768.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster B, Southgate V, Tchuente LT.. 1999. Mating interactions between Schistosoma haematobium and S. mansoni. J Helminthol. 734:351–356. [PubMed] [Google Scholar]
- Webster BL, Diaw OT, Seye MM, Webster JP, Rollinson D.. 2013. Introgressive hybridization of Schistosoma haematobium group species in Senegal: species barrier break down between ruminant and human schistosomes. PLoS Negl Trop Dis. 74:e2110.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster BL, Southgate VR, Littlewood D.. 2006. A revision of the interrelationships of Schistosoma including the recently described Schistosoma guineensis. Int J Parasitol. 368:947–955. [DOI] [PubMed] [Google Scholar]
- Webster JP, Gower CM, Knowles SC, Molyneux DH, Fenton A.. 2016. One health—an ecological and evolutionary framework for tackling Neglected Zoonotic Diseases. Evol Appl. 92:313–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster JP, Molyneux DH, Hotez PJ, Fenwick A.. 2014. The contribution of mass drug administration to global health: past, present and future. Philos Trans R Soc B Biol Sci. 3691645:20130434.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RA. 2012. Virulence factors of schistosomes. Microbes Infect. 1415:1442–1450. [DOI] [PubMed] [Google Scholar]
- World Health Organization. 2019. Schistosomiasis Fact Sheet [Internet]. Available from: https://www.who.int/news-room/fact-sheets/detail/schistosomiasis. Geneva, Switzerland: World Health Organization (last accessed April 19, 2019).
- Wright C, Ross G.. 1980. Hybrids between Schistosoma haematobium and S. mattheei and their identification by isoelectric focusing of enzymes. J Trans R Soc Trop Med Hyg. 74:326–332. [DOI] [PubMed] [Google Scholar]
- Yao C, Donelson JE, Wilson ME.. 2003. The major surface protease (MSP or GP63) of Leishmania sp. Biosynthesis, regulation of expression, and function. Mol Biochem Parasitol. 1321:1–16. [DOI] [PubMed] [Google Scholar]
- Young ND, Jex AR, Li B, Liu S, Yang L, Xiong Z, Li Y, Cantacessi C, Hall RS, Xu X, et al. 2012. Whole-genome sequence of Schistosoma haematobium. Nat Genet. 442:221–225. [DOI] [PubMed] [Google Scholar]
- Zhou X-N, Wang L-Y, Chen M-G, Wu X-H, Jiang Q-W, Chen X-Y, Zheng J, Jürg U.. 2005. The public health significance and control of schistosomiasis in China—then and now. Acta Trop. 96(2-3):97–105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.